

Abstract
The triethylammonium QX-314 and the trimethylammonium QX-222 are lidocaine derivatives that act as open-channel blockers of the acetylcholine (ACh) receptor. When bound, these blockers should occlude some of the residues lining the channel. Eight residues in the second membrane-spanning segment (M2) of the mouse-muscle α subunit were mutated one at a time to cysteine and expressed together with wild-type β, γ, and δ subunits in Xenopus oocytes. The rate constant for the reaction of each substituted cysteine with 2-aminoethyl methanethiosulfonate (MTSEA) was determined from the time course of the irreversible effect of MTSEA on the ACh-induced current. The reactions were carried out in the presence and absence of ACh and in the presence and absence of QX-314 and QX-222. These blockers had no effect on the reactions in the absence of ACh. In the presence of ACh, both blockers retarded the reaction of extracellularly applied MTSEA with cysteine substituted for residues from αVal255, one third of the distance in from the extracellular end of M2, to αGlu241, flanking the intracellular end of M2, but not with cysteine substituted for αLeu258 or αGlu262, at the extracellular end of M2. The reactions of MTSEA with cysteines substituted for αLeu258 and αGlu262 were considerably faster in the presence of ACh than in its absence. That QX-314 and QX-222 did not protect αL258C and αE262C against reaction with MTSEA in the presence of ACh implies that protection of the other residues was due to occlusion of the channel and not to the promotion of a less reactive state from a remote site. Given the 12-Å overall length of the blockers and the α-helical conformation of M2 in the open state, the binding site for both blockers extends from αVal255 down to αSer248.
Keywords: cysteine mutagenesis, reaction kinetics, methanethiosulfonate, open-channel block
introduction
There is considerable evidence that certain noncompetitive inhibitors (NCIs)1 of the acetylcholine receptor act as open-channel blockers, binding within the open channel and occluding it (Adams, 1976; Ruff, 1977; Ascher et al., 1978; Neher and Steinbach, 1978; Colquhoun and Sheridan, 1981; Farley et al., 1981). NCIs, binding either within the channel or at lower affinity sites (Krodel et al., 1979; Heidmann et al., 1983; Herz et al., 1987; Lurtz et al., 1997), can also interact with closed states of the receptor (Adams, 1976; Farley et al., 1981; Neher, 1983) and promote desensitization (Magleby and Pallotta, 1981; Karpen et al., 1982; Maleque et al., 1982; Sine and Taylor, 1982; Neher, 1983; Oswald et al., 1983; Boyd and Cohen, 1984; Clapham and Neher, 1984; Gage and Wachtel, 1984). Based on the likelihood that NCIs bind within the channel, reactive NCI derivatives were used to identify channel-lining residues. Chlorpromazine (Revah et al., 1990), triphenylmethylphosphonium (Hucho et al., 1986), and meproadifen mustard (Pedersen et al., 1992) all labeled residues in or flanking the predicted second membrane-spanning segment (M2) (Fig. 1) of one or more Torpedo species acetylcholine (ACh)-receptor subunits (Fig. 2). In particular, the labeling of αS248 and the aligned residues in β, γ, and δ by chlorpromazine and triphenylmethylphosphonium (Fig. 2) provided evidence that the channel was on the axis of the pentameric complex (α2βγδ) surrounded by the five M2 segments.
Figure 1.
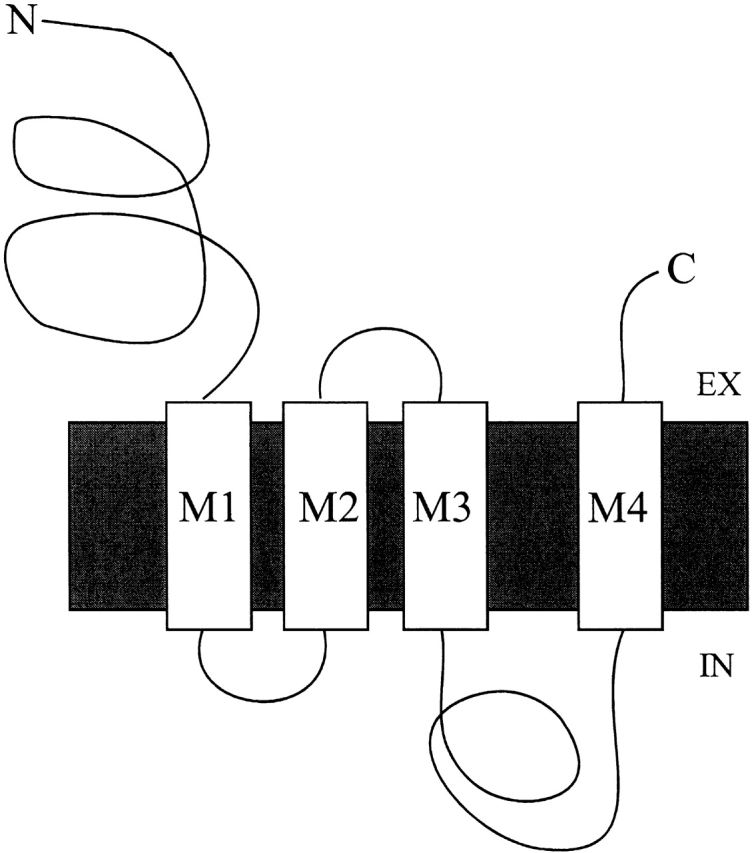
Topology of the ACh receptor subunits. M1, M2, M3, and M4 are the membrane-spanning segments. EX, extracellular; IN, intracellular.
Figure 2.

Aligned sequences of the M2 segments of mouse-muscle ACh receptor subunits. The numbering is that of the α subunit. The predicted membrane-spanning segments correspond to αM243 to αV261. The intracellular (IN) and extracellular (EX) ends are indicated. *Residues are aligned with residues in the ACh receptor from Torpedo electric tissue that were labeled by noncompetitive inhibitor derivatives. Mutations of the boxed-in residues affected channel block by QX-222. Cysteines substituted for the residues in bold italics are exposed in the channel in the presence of ACh (tested only in α and β). See text for references.
Other crucial evidence for the topology of M2 and for its central role in the channel was provided by the effects on conductance and selectivity of mutating charged residues flanking M2 in each of the subunits (Imoto et al., 1988). In addition, the effects of mutations of the M2 residues αS248 and αS252 and of the aligned residues in the other subunits (Fig. 2) on the kinetics of QX-222 block (Fig. 3) provided further evidence that open-channel blockers bind in the channel in the vicinity of these M2 residues (Charnet et al., 1990; Leonard et al., 1988). Furthermore, the mutation L247T in the homopentameric neuronal ACh receptor formed by the α7 subunit (Anand et al., 1991; Cooper et al., 1991) resulted in the loss of sensitivity to block by QX-222 (Revah et al., 1991). (α7)L247 aligns with mouse-muscle αL251 (Fig. 2).
Figure 3.
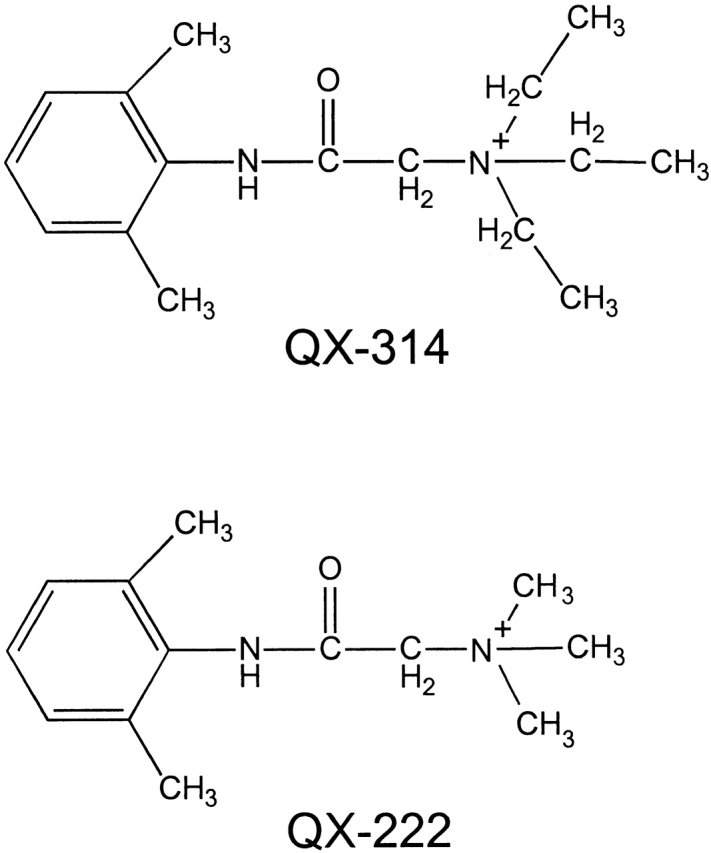
Structures of QX-314 and QX-222.
Not all evidence, however, points to the region between αS248 and αS252 as the sole binding site in the channel. Quinacrine azide photolabeled residues at the extracellular end of αM1, specifically in the open state of the channel (Karlin, 1989; DiPaola et al., 1990), and mutations at the extracellular end of αM1 affected quinacrine but not chlorpromazine binding (Tamamizu et al., 1995). Nevertheless, chlorpromazine and a number of other NCIs retarded the labeling by quinacrine azide (DiPaola et al., 1990) and competed with the binding of quinacrine (Lurtz et al., 1997). In addition, meproadifen mustard labeled αE262 at the extracellular end of M2 (Pedersen et al., 1992). Different NCIs may bind to nonidentical, possibly overlapping sites in the channel.
We report here on another approach to the localization of the sites of action of channel-blocking NCIs, using them in conjunction with the substituted-cysteine-accessibility method (SCAM). SCAM was applied previously to the α and β subunits, identifying residues exposed in the channel along the entire length of M2 (Fig. 2), in the M1–M2 loop, and in the extracellular third of M1 (Akabas et al., 1992, 1994; Akabas and Karlin, 1995; Zhang and Karlin, 1997, 1998; Wilson and Karlin, 1998). These residues, when substituted by cysteine, reacted with small, positively charged sulfhydryl-specific methanethiosulfonate reagents such as 2-aminoethyl methanethiosulfonate (MTSEA). The rate constants for the reactions with the different residues depended on the state of the receptor (Pascual and Karlin, 1998) and on the side of application of the reagent (Wilson and Karlin, 1998). In this paper, we test to what extent the channel blockers QX-222 and QX-314 (Fig. 3; Neher and Steinbach, 1978; Horn et al., 1980; Neher, 1983; Charnet et al., 1990) protect Cys-substituted channel-lining residues in αM2 from reaction with extracellularly applied MTSEA. Previously, Cys-substituted residues in the M2 segment of the GABAA receptor α1 subunit were protected by picrotoxin (Xu et al., 1995), Cys-substituted residues in the Na channel were protected by tetrodotoxin (Kirsch et al., 1994), Cys-substituted residues in Kv2.1 potassium channel were protected by tetraethylammonium (Pascual et al., 1995), and Cys-substituted residues in Shaker potassium channel were protected by agitoxin (Gross and MacKinnon, 1996). We find that QX-222 and QX-314, in the presence of ACh, retard the reactions of extracellularly added MTSEA with Cys-substituted residues from αV255C (Fig. 2) down to the cytoplasmic end of αM2.
methods
Materials
The quaternary lidocaine derivative 2-(triethylammonio)-N-(2,6-dimethylphenyl)acetamide bromide (QX-314) was from Alomone Laboratories (Jerusalem, Israel) and from Astra (Westborough, MA), and 2-(trimethylammonio)-N-(2,6-dimethylphenyl)acetamide chloride (QX-222) was from Astra. MTSEA was synthesized as previously described (Stauffer and Karlin, 1994) and purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). 2-Hydroxyethyl methanethiosulfonate (MTSEH) was synthesized as described previously (Pascual and Karlin, 1998).
Mutagenesis and Expression
All mutations were introduced in the M2 segment of the mouse muscle α subunit, and capped, runoff cRNA transcripts were obtained for the α-subunit mutants and for wild-type α, β, γ, and δ subunits after linearization of the plasmid cDNA, using the mMessage mMachine kit (Ambion Inc., Austin, TX). cRNAs at a concentration of 1 mg/ml in water were stored at −80°C. They were diluted and mixed for injection at a ratio of 2α:1β:1γ:1δ. Stage V and VI Xenopus laevis oocytes were collected and defolliculated in collagenase following standard procedures (Akabas et al., 1992). Oocytes were injected with 60 nl of cRNA diluted to 1–100 ng/μl, depending on desired current expression levels. Cells were kept in culture and used for recording on days 1–10.
Two-Electrode Voltage Clamp Recording
Currents were recorded under two-electrode voltage clamp. The oocyte bath solution contained (mM): 115 NaCl, 2.5 KCl, 1.8 MgCl2, 10 HEPES, pH 7.2, except where indicated otherwise. Solutions flowed at 7 ml/min first through a stainless steel coil immersed in a thermostat at 18.0°C, and then past the oocyte, which was held in a rectangular chamber with a cross-section normal to the direction of solution flow of 4 mm2. An agar bridge connected a Ag:AgCl reference electrode to the bath and was placed as close as possible to the oocyte. The bath was clamped at ground potential. We used beveled agarose-cushion (Schreibmayer et al., 1994) glass micropipettes filled with 3 M KCl, of resistance ∼0.2–0.5 MΩ, for both current-passing and voltage- recording electrodes. A few uninjected oocytes from each batch were tested for the presence of endogenous ACh-induced currents, which were never found. The function of wild-type and mutant receptors was assayed as the ACh-induced current elicited by the application of brief (3–25 s) pulses of ACh, at a concentration 10× the EC50, as determined for each mutant, and at a holding potential of −50 mV, except where indicated otherwise. ACh-induced currents ranged from 1 to 25 μA at −50 mV.
Characterizing Inhibition by QX-314 and QX-222
The concentrations (IC50) of QX-314 and of QX-222 that inhibited the ACh-induced current by 50% were determined at several holding potentials (see Fig. 4). ACh was applied at 10× its EC50 for each mutant. Leak currents were measured at holding potentials of −50, −125, −75, and −25 mV, each held for 400 ms. ACh was added and, at the peak of the current, the holding potential was stepped through the same four values. After a 5-min wash, QX-222 or QX-314 was applied continuously for 2 min, during which time leak and ACh-induced currents were determined as before. This protocol was repeated with increasing concentrations of blocker. We calculated the net ACh-induced currents in the presence (I ACh, QX) and absence (I ACh) of blocker, and we fit the data at each membrane potential to the equation I ACh, QX/I ACh = 1/{1+([QX]/IC50)}, where [QX] is the blocker concentration. The IC50 values obtained from the fit were themselves fit to the Woodhull (1973) equation: IC50 = IC50(0 mV) exp(−zδFψ/RT), where z is the charge on the blocker, F is the Faraday constant, ψ is the transmembrane potential difference (inside–outside), and δ is the apparent electrical distance from the extracellular medium to the blocker binding site.
Figure 4.

Inhibition of ACh-induced current in αL251C by QX-314 at different holding potentials. The top trace shows the holding potential. Both before and ∼3 s after the application of 25 μM ACh, the holding potential was stepped from −50 mV to −125, −75, −25, and back to −50 mV. The bottom trace shows the current. (inset) The peak ACh-induced current during the voltage steps minus the current during the voltage steps before ACh was added, with an expanded time axis. (A) No QX-314, (B) 0.1 mM QX-314, (C) 0.3 mM QX-314, (D) 1 mM QX-314. The four sections are part of a single experiment. Between the sections shown, the oocyte was washed and QX-314 at the indicated concentration was applied for 45 s before, and in addition to, the time indicated in the traces.
Protection
The protection of substituted Cys against reaction with MTSEA afforded by bound blocker was estimated from the second-order rate constants of the reaction in the presence and absence of blocker. The second-order rate constants (k) were determined as previously described (Pascual and Karlin, 1998). Oocytes were superfused for 20 s with ACh (at 10× the EC50 as determined for each mutant), for 3 min with bath solution, for 2–25 s with MTSEA in the presence of ACh, or for 1–4 min with MTSEA in the absence of ACh, for 3 min with bath solution, for 20 s with ACh, and for 3 min with bath solution; this sequence was repeated several times. MTSEA was applied at concentrations ranging from 5 μM for the rapidly reacting residues up to 10 mM for the slowly reacting residues. MTSEA was applied also in the presence of QX-314 and QX-222, both in the presence and absence of ACh. The peaks of the ACh-induced currents obtained before and after the exposure to MTSEA were fit to I t = I ∞ + (I 0 − I ∞) exp(−kxt), where I 0 is the initial ACh-induced peak current, I t is the ACh-induced current after a cumulative time, t, of application of MTSEA at concentration, x, and I ∞ is the ACh-induced current after complete reaction of the Cys. Note that the fraction of unmodified receptors = (I t − I ∞)/(I 0 − I ∞) (Pascual and Karlin, 1998).
In the presence of ACh, the receptors are distributed among a number of states: closed, open, and desensitized. During the 2–25 s that MTSEA and ACh (at 10× EC50) were applied, the receptor was predominantly in the open state or the rapid-onset desensitized state. There was little slow desensitization during this time, and in any case the rate constants for the reactions of two mutants in the slow-onset desensitized state were small (Pascual and Karlin, 1998; Wilson and Karlin, 1998). The rate constant for the reaction in the mixed open state and rapid-onset desensitized state we call k open. When blocker was also added, it could have bound to the open state and to the rapid-onset desensitized state (Neher, 1983). To compare the effects of QX-314 and QX-222, at different concentrations, on the rate constants for the reactions of MTSEA with the different mutants, we estimated the rate constants for the reactions of MTSEA with the blocked state(s), k blocked, from the observed rate constants in the presence of blocker, k obs, and from the IC50 for the particular blocker and mutant. We assumed that the observed rate constant, k obs, is approximated by k obs = k open(1 − y) + k blocked y, where y, the fraction of channels that was blocked, is given by: y = 1/(1 + IC50/[QX]).
With each oocyte, we first applied MTSEA (in the presence of ACh) several times in the absence of blocker to determine k open, and then applied MTSEA several times in the presence of blocker to determine k obs. The concentrations of blocker used were in the range of 2–5× IC50 for the given blocker and mutant, so that y ranged from 2/3 to 5/6. If we take the extent of protection against reaction to be [1 − (k blocked/k open)], then 1 − (k blocked/k open) = (1/y)[1 − (k obs/k open)]; i.e., the (corrected) extent of protection was 1.2–1.5× the observed extent of protection.
results
Eight residues in αM2 were substituted, one at a time, by Cys and expressed in Xenopus oocytes together with wild-type β, γ, and δ subunits. In each case, the current elicited by ACh was inhibited by QX-314 and QX-222, and the inhibition increased with increasing concentration of blocker. This inhibition is illustrated for αL251C and QX-314 in Fig. 4. We derived the IC50s for the inhibition by the blockers of the ACh-induced currents at each holding potential from such experiments. The IC50 for inhibition of the mutants by QX-314 ranged from 0.4× (αT244C) to 4.6× (αV255C) the IC50 for wild type (Table I). The range for QX-222 was from 0.05× (αT244C) to 1× (αS252C) the IC50 for wild type. Typically, when QX-314 and ACh were washed out, there was an immediate increase in the amplitude of the current before it returned to the baseline (Fig. 4, B–D). This increased current resulted either from the blocker dissociating from its site of inhibition before ACh dissociated from its binding sites or from the faster dilution below effective concentration of the blocker (initially at 2–5× IC50) than of ACh (initially at 10× EC50).
Table I.
The Characteristics of QX-314 and QX-222 Inhibition of the ACh-induced Currents Mediated by Wild-Type and Mutant ACh Receptors
| Mutant | QX-314 | QX-222 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 | δ | IC50 | δ | |||||||
| μM | μM | |||||||||
| Wild type | 78 ± 12 (4) | 0.35 ± 0.02 | 2780 ± 230 (7) | 0.80 ± 0.01 | ||||||
| αE262C | 92 ± 11 (4) | 0.39 ± 0.03 | 765 ± 80 (4) | 0.55 ± 0.05 | ||||||
| αL258C | 284 ± 21 (4) | 0.29 ± 0.03 | 558 ± 112 (4) | 0.34 ± 0.02 | ||||||
| αV255C | 363 ± 51 (3) | 0.28 ± 0.01 | 426 ± 93 (4) | 0.47 ± 0.07 | ||||||
| αS252C | 175 ± 15 (3) | 0.45 ± 0.03 | 2900 ± 294 (4) | 0.66 ± 0.08 | ||||||
| αL251C | 297 ± 48 (4) | 0.57 ± 0.02 | 530 ± 9 (5) | 0.53 ± 0.03 | ||||||
| αS248C | 84 ± 8 (3) | 0.38 ± 0.03 | 1339 ± 131 (4) | 0.68 ± 0.04 | ||||||
| αT244C | 30 ± 2 (4) | 0.51 ± 0.02 | 140 ± 14 (4) | 0.68 ± 0.03 | ||||||
| αE241C | 256 ± 12 (4) | 0.37 ± 0.01 | 1862 ± 299 (4) | 0.42 ± 0.04 | ||||||
The IC50s were determined at holding potentials of −25, −50, −75, and −125 mV (see Fig. 4 and methods). The means and SEMs of the IC50s at −50 mV are presented. The voltage dependence of the block is characterized by the apparent electrical distance, δ, estimated from the fit of the Woodhull equation to the IC50 as a function of holding potential.
For both blockers and for all mutants, the more negative the holding potential, the greater the inhibition of current (Fig. 4). The voltage dependence of the inhibition was consistent with the site of inhibition being in the open channel. The voltage dependence of the IC50 was fit by the Woodhull (1973) equation to yield the blocker charge times the apparent electrical distance, zδ. For these quaternary ammonium blockers, z = 1. For QX-314, δ was 0.35 for wild type and ranged from 0.28 for αV255C to 0.57 for αL251C (Table I). For QX-222, δ was 0.80 for wild type and ranged from 0.34 for αL258C to 0.68 for αT244C and αS248C. The effects of the mutations on the apparent electrical distances for QX-314 and for QX-222 were not correlated. The interpretation of the apparent electrical distances is difficult because the IC50s are dependent not only on the equilibrium dissociation constants of the blockers, but also on the kinetics of the transitions between functional states.
The reactions of MTSEA with engineered Cys exposed in the channel were manifested by irreversible effects on the ACh-induced current. We determined the rates of the irreversible changes in the response to ACh and the effects of QX-314 and QX-222 on these rates. These blockers had no effect on the rates of reaction of the two Cys-substituted residues closest to the extracellular end of M2: the reactions of MTSEA in the presence of ACh with αL258C (Fig. 5, A and B) and αE262C were unchanged by the addition of QX-314 or QX-222 at concentrations several times their IC50s. By contrast, the reactions of MTSEA in the presence of ACh with Cys-substituted residues deeper in the channel were markedly slowed by the addition of these blockers. For example, the irreversible reaction of MTSEA in the presence of ACh with αS248C was nearly stopped by the addition of QX-314 (Fig. 5, C and D).
Figure 5.

The effect of QX-314 on the irreversible inhibition of the ACh-induced current in two mutants. (A and B) αL258C. (C and D) αS248C. A and C show the test responses to ACh between applications of MTSEA in the presence of ACh and in the presence or absence of QX-314. B and D show the test currents as a function of cumulative exposure time to MTSEA. (▪) Initial responses and, in C and D, also extra final responses without any further application of MTSEA. (▵) MTSEA in the presence of ACh. (○) MTSEA in the presence of both ACh and QX-314. ACh was applied at 25 μM to αL258C and at 100 μM to αS248C. 1 mM MTSEA was applied in 3-s pulses to αL258C, and 1.5 mM MTSEA was applied in 15-s pulses to αS248C. QX-314 was applied at 1 mM to αL258C and at 0.2 mM to αS248C. The calibration bars are 3 s and 5 μA.
QX-314 retarded the reactions of MTSEA only in the presence of ACh at the two residues on which this was tested, αT244C at the intracellular end of the channel and αL251C in the middle of the channel. The time course of the reaction of αT244C with MTSEA in the presence of ACh, first with, and then without, QX-314 is shown in Fig. 6 A. The rate of reaction was faster after QX-314 was removed. On the other hand, the rate of reaction of MTSEA in the absence of ACh was not changed by the addition of QX-314 (Fig. 6 B). Similar results were obtained with αL251C.
Figure 6.

The dependence on ACh of the protection by QX-314 against the irreversible inhibition by MTSEA. The mutant was αT244C. The log of the ACh-induced current is plotted against the cumulative time of application of MTSEA. (A) QX-314 added in the presence of ACh. (B) QX-314 added in the absence of ACh. (▪) Responses to 60 μM ACh before MTSEA was added. (○) Responses to ACh after 25-s applications of 2.5 μM MTSEA plus 60 μM ACh plus 0.1 mM QX-314. (▵) Responses to ACh after 25-s applications of 2.5 μM MTSEA plus 60 μM ACh. (▴) Responses to ACh after 25-s applications of 250 μM MTSEA. (•) Responses to ACh after 25-s applications of 250 μM MTSEA plus 1 mM QX-314.
The extent of the retardation of the MTSEA reaction depended on the concentration of blocker. This is illustrated by the rate constants of the reactions of MTSEA in the presence of ACh with αS248C and αV255C, in the additional presence of different concentrations of QX-314 (Fig. 7). To compare the effects of QX-314 and QX-222 on the rates of reaction of the different mutants, we estimated the rate constants for the reactions of MTSEA with the blocked state from the observed rate constants in the presence and absence of blocker and from the IC50 for the particular blocker and mutant (see methods). The IC50 for a particular blocker and mutant is a function not only of the equilibrium dissociation constant for the blocker binding to the open channel, but also of kinetic constants characterizing the isomerizations of the receptor among closed, open, and desensitized states. Although the IC50s (Fig. 7, arrows) were two to four times smaller than the concentrations of blocker giving half of the maximum retardation of rate,2 the use of the IC50 to estimate the fraction of a given mutant blocked at different concentrations of blocker yielded consistent rate constants for the blocked state.
Figure 7.

The dependence of protection on blocker concentration. The rate constant for the reaction of MTSEA in the presence of ACh at 10× EC50 and of the indicated concentration of QX-314 is divided by the rate constant in the presence of ACh but absence of QX-314. The mutants are αV255C (•) and αS248C (▪). The arrows mark the interpolated values of the relative rate constants at the IC50s for QX-314 inhibition of the ACh-induced current (Table I).
We take as the extent of protection by blocker, 1 − (k blocked/k open). At the extracellular end of M2, αL258C and αE262C were not protected by QX-314 or QX-222 (Fig. 8). At the intracellular end of M2, αE241C, αT244C, and αS248C were nearly completely protected. Between the fully protected and the unprotected residues, αL251C was protected ∼70% and αV258C ∼60% by both blockers. αS252C was protected 58% by QX-222 and not at all by QX-314.
Figure 8.

The protection by QX-314 and QX-222 of Cys-substituted residues in M2 against reaction with MTSEA. For each mutant, the rate constant of the reaction in the presence of channel blocker and ACh (k blocked) and the rate constant of the reaction in the presence of ACh alone (k open) were determined as described in methods. The extent of protection is taken as 1 − (k blocked/k open). The means of two to four determinations and average errors or SEM are plotted. The lighter bars represent the protection by QX-314, and the darker bars represent the protection by QX-222. *ND.
The reaction of αT244C with an uncharged reagent, MTSEH, was also retarded by QX-314 in the presence of ACh: the protection was 88% ± 2% (n = 5). QX-314 had no effect on the rate of the reaction of MTSEH in the absence of ACh.
discussion
We found that QX-314 and QX-222, two quaternary ammonium lidocaine derivatives that act as open-channel blockers, protected channel-lining residues substituted by Cys from reaction with positively charged MTSEA. The protection was partial from αV255C to αL251C and was nearly complete from αS248C to αE241C (Fig. 8). There are a number of possible mechanisms for this protection, including steric hindrance, charge repulsion, and allosteric stabilization of unreactive receptor states.
The last mechanism is not a likely explanation for the observed protection. The rate constants for the reactions of some of the mutants are quite different in different functional states of the receptor, so that the blockers could retard the reactions of these mutants by promoting the less reactive state. Such a mechanism, however, would affect all of the state-dependent and none of the state-independent reaction rates, contrary to our results. The rate constants for the reactions of αE241C, αT244C, αL251C, and αV255C with extracellularly applied MTSEA were much larger in the presence of ACh (in the open and rapid-onset desensitized states) than in the absence of ACh (in the predominantly closed state) (Pascual and Karlin, 1998), and these mutants were protected by QX-314 and QX-222 (Fig. 8). On the other hand, the rate constants for the reactions of αS248C and αS252C with MTSEA were not much different in the presence and absence of ACh, and these mutants were also protected. Conversely, αL258C and αE262C were not protected, even though they reacted much faster in the presence of ACh than in its absence. Thus, the blockers did not act by stabilizing an unreactive state from a remote site.
It is also unlikely that the blockers protected the Cys-substituted residues by promotion of the closed state through direct competition with ACh. Although the concentrations of QX-314 and QX-222 used to protect against MTSEA were high enough that some binding to sites in addition to the open channel was possible (Neher, 1983), there was no protection of αL258C or αE262C, which were far less reactive in the absence than presence of ACh.
The mechanism of protection also could not have been solely electrostatic repulsion. QX-314 protected αT244C against the reaction of the neutral reagent MTSEH. Furthermore, the protection of αS252C by QX-222 and not by QX-314 indicates that the mechanism of protection was not primarily electrostatic.
The most likely mechanism of protection was the steric hindrance of access to the substituted Cys by QX-314 and QX-222. Since αV255C was the protected residue closest to the extracellular end of M2, this residue marks the extracellular end of the binding site for the two blockers. If we compare the dimensions of QX-314 and αM2, configured in the open state as an α helix (Karlin and Akabas, 1995), and if the xylyl moiety of QX-314 overlaps αV255, the triethylammonium group overlaps αS248 (Fig. 9). QX-314 or QX-222 bound in this location would protect the residues from αV255C to αS248C by occluding them directly and would protect αT244C and αE241C by blocking the pathway to these residues from the extracellular medium.
Figure 9.
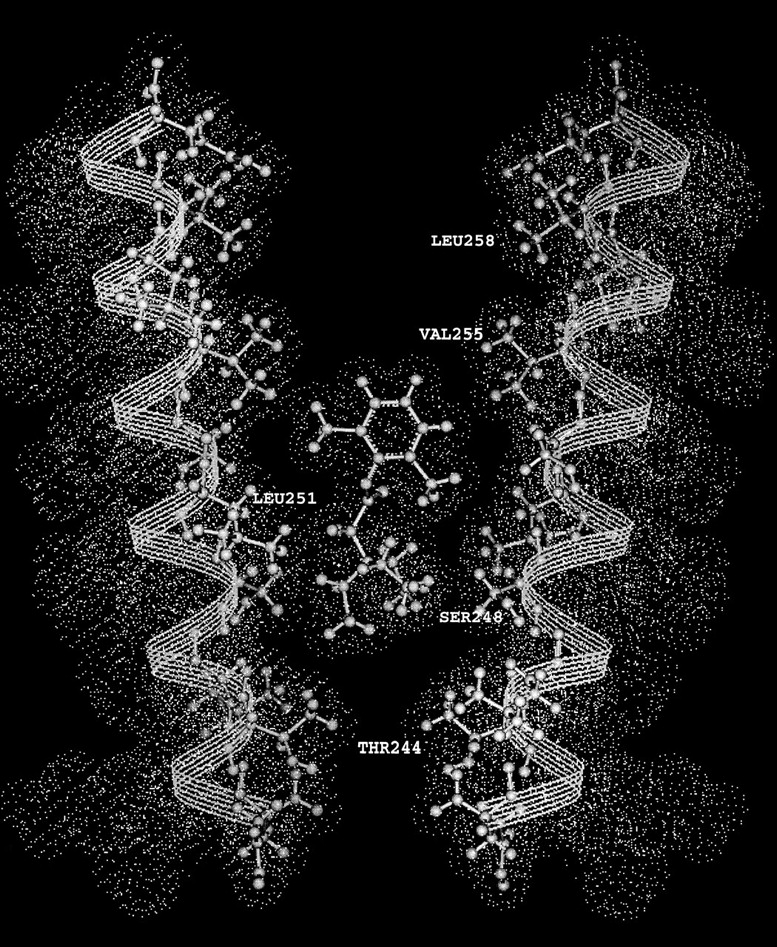
A model of QX-314 binding in the channel between two αM2 segments. The M2 segments are drawn as α helices. The dots represent the van der Waals surfaces. The drawing was made on a Silicon Graphics workstation running Insight II.
The orientation of the blocker with its positively charged quaternary ammonium group toward the intracellular end of the channel would be favored by the electrostatic potential profile in the channel. The net electrostatic potential at each point in the channel is the sum of the electrical distance times the transmembrane potential and of the intrinsic potential due to fixed charges and dipoles in the protein and lumen of channel. The electrostatic potential in the open channel is ∼75 mV more negative at αS248 than at αV255 (Pascual and Karlin, 1998), so that QX-314 or QX-222 would bind ∼20× more tightly with its charged ammonium group near αS248 than near αV255. The proposed location and orientation of the blocker in the channel (Fig. 9) are the same as those proposed by Charnet et al. (1990).
Assuming the location and orientation of the blocker is as shown in Fig. 9, we can estimate the electrostatic contribution to the binding energy. The net electrostatic potential in the open channel in the vicinity of αS248, with a transmembrane potential of −50 mV, was estimated to be ∼−100 mV relative to the extracellular medium (Pascual and Karlin, 1998). Therefore, the electrostatic contribution to the binding energy per mole due to the quaternary ammonium group of QX-222 or QX-314 binding close to αS248 would be approximately four RT.
The apparent affinity constant for QX-222 estimated from burst durations and open times (Neher, 1983) can be extrapolated to ∼2.5 × 104 M−1 at −50 mV. The affinity of QX-314 is considerably greater than that of QX-222 (Neher and Steinbach, 1978). The free energy per mole of QX-222 binding would be ∼10 RT. Thus, hydrophobic and van der Waals interactions would contribute approximately six RT to the binding of QX-222 and more to the binding of QX-314. The side chains of αL251 and αV255, the aligned side chains in β, and possibly the aligned side chains in γ and δ, are all more exposed in the open than in the closed channel (Akabas et al., 1994; Pascual and Karlin, 1998; Zhang and Karlin, 1998). These hydrophobic side chains are likely to interact with bound blocker (Fig. 9).
Although the mutation of (α7)L247 (aligned with αL251) to Thr rendered the neuronal, homopentameric α7 complex insensitive to QX-222 (Revah et al., 1991), the mutation in mouse-muscle receptor of the two αL251 to Cys, and the mutation of the two αV255 to Cys, increased the IC50 for QX-314 but decreased the IC50 for QX-222 (Table I). The effects of these mutations on IC50 may have been confounded by underlying effects of the mutations on the kinetics of state transitions.
The patterns of protection indicate that QX-314 and QX-222 bind at the same depth in the channel. Yet for wild-type receptor, the electrical distances estimated from the fits of the IC50s to the Woodhull equation were quite different, 0.35 for QX-314 and 0.80 for QX-222. The IC50, however, depends not only on the kinetics of binding to the open channel but also on the kinetics of the transitions of the receptor among states. Also, the IC50 was determined after considerable fast desensitization had occurred. Thus, the voltage dependence of the IC50 does not have a simple interpretation. The complexity of the IC50 could partly account for the variation we observed in electrical distance from 0.28 to 0.57 for QX-314 and from 0.34 to 0.68 for QX-222 among the Cys-substitution mutants. Two different single-channel analyses of QX-222 blocking gave electrical distances of 0.78 (Neher and Steinbach, 1978) and ∼0.4 (Neher, 1983). Voltage-jump–relaxation analysis yielded electrical distances from 0.65 to 0.75 for QX-222 (Charnet et al., 1990) and 0.72 for QX-314 (Horn and Brodwick, 1980). In general, quite disparate electrical distances for different ACh receptor channel blockers have been reported (e.g., Table I in Sanchez et al., 1986); whether these reflect different binding sites in the channel or other complications is not known. The electrical distance estimated by the Woodhull (1973) equation may not be a consistent marker of position in the channel.
The protection of αS252C by QX-222 and not by QX-314 (Fig. 8) was unexpected and may indicate that the two blockers do not bind in exactly the same configuration. The orientation of the xylyl ring structure shown in Fig. 9 is drawn roughly in the plane containing the two αM2 segments; the M2 segments and β, γ, and δ subunits are not shown. Because the quaternary ammonium group is symmetrical, the somewhat rigid blocker molecule can rotate around its axis, which would maintain similar interactions of the ammonium group but change the subunit interactions of the xylyl ring. Different distributions of axial rotation for the two blockers could be the basis for the different protection of αS252C. In addition, the rotation of the xylyl group and the presumed widening of the channel toward the extracellular end could account for the incompleteness of the protection of αL251C and αV255C, as well as of αS252C.
In the absence of ACh, QX-314 did not protect αT244C or αL251C. αT244C is the mutant that reacts most rapidly with MTSEA, both in the presence and absence of ACh, and the inhibition due to the reaction is complete (Pascual and Karlin, 1998). αT244C is at the intracellular end of the channel, so that QX-314 binding above would protect αT244C from extracellular MTSEA coming through the channel. Thus, αT244C affords a sensitive test for protection. The lack of protection in the absence of ACh indicates that QX-314 did not bind in the closed channel at the concentrations tested.
The poor binding of QX-314 to the closed channel is not due to the obstruction of the channel by the activation gate, which we have located between αG240 and αT244, at the intracellular end of the channel (Wilson and Karlin, 1998). Notwithstanding, there could be a partial obstruction at the extracellular end of the closed channel that would limit access by channel blockers but not by the smaller methanethiosulfonates used to locate the gate or by inorganic cations. Alternatively, the blockers could bind poorly in the closed channel because the net electrostatic potential is ∼100 mV less negative in the closed than in the open channel (Pascual and Karlin, 1998), and the side chains of αL251 and αV255 and of the aligned residues in the other subunits are less exposed (Akabas et al., 1994; Pascual and Karlin, 1998; Zhang and Karlin, 1998). Restating this, we suggest that more favorable electrostatic and hydrophobic interactions, rather than greater accessibility, could account for the stronger binding of blockers in the open than in the closed channel. Some of the complexities of local anesthetic action on the voltage-gated Na+ channel have been explained on the basis of different intrinsic local-anesthetic binding affinities in different states (the modulated receptor model); however, in contrast to the ACh receptor, in the Na+ channel, accessibility to the binding site controlled by the activation and inactivation gates is crucial for the binding of quaternary ammonium derivatives of local anesthetics like QX-314 (Hille, 1977; Ragsdale et al., 1994).
Acknowledgments
We thank Drs. Myles Akabas, Jonathan Javitch, and Gary Wilson for comments on the manuscript. We are also grateful to Gilda Salazar-Jimenez for oocyte culture.
Abbreviations used in this paper
- ACh
acetylcholine
- M2
second membrane-spanning segment
- MTSEA
2-aminoethyl methanethiosulfonate
- MTSEH
2-hydroxyethyl ethanethiosulfonate
- NCI
noncompetitive inhibitor
Footnotes
This research was supported by research grants to A. Karlin from the National Institutes of Health (NIH) (NS-07065), the Muscular Dystrophy Association, and the McKnight Endowment Fund for Neuroscience. During part of this work, J.M. Pascual was supported by NIH Neurological Sciences Academic Development Award NS01698. We thank Dr. Rune Sandberg of Astra for gifts of QX-222 and QX-314.
If MTSEA itself first bound reversibly at its site of reaction, and then reacted, the discrepancy between the IC50 for QX-314 and the QX-314 concentration that gave half-maximal retardation of the reaction of MTSEA could be explained by the competition of QX-314 and MTSEA for reversible binding in the channel. However, even at 10 mM MTSEA, the highest concentration tested, MTSEA had no effect on the ACh-induced current mediated by wild-type receptor. In addition, there was no rapidly reversible component of the inhibition by MTSEA of the ACh-induced current mediated by any of the mutants. Hence, MTSEA does not reversibly bind in the channel to an appreciable extent, and thus, before its covalent reaction in the channel, MTSEA is unlikely to compete with the binding of QX-314.
references
- Adams PR. Drug blockade of open end-plate channels. J Physiol (Camb) 1976;260:532–552. doi: 10.1113/jphysiol.1976.sp011530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akabas MH, Karlin A. Identification of acetylcholine receptor channel-lining residues in the M1 segment of the alpha subunit. Biochemistry. 1995;34:12496–12500. doi: 10.1021/bi00039a002. [DOI] [PubMed] [Google Scholar]
- Akabas MH, Kaufmann C, Archdeacon P, Karlin A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit. Neuron. 1994;13:919–927. doi: 10.1016/0896-6273(94)90257-7. [DOI] [PubMed] [Google Scholar]
- Akabas MH, Stauffer DA, Xu M, Karlin A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- Anand R, Conroy WG, Schoepfer R, Whiting P, Lindstrom J. Neuronal nicotinic acetylcholine receptors expressed in Xenopusoocytes have a pentameric quaternary structure. J Biol Chem. 1991;266:11192–11198. [PubMed] [Google Scholar]
- Ascher PR, Marty A, Neild TO. The mode of action of antagonists of the excitatory response to acetylcholine in Aplysia neurones. J Physiol (Camb) 1978;278:207–235. doi: 10.1113/jphysiol.1978.sp012300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd ND, Cohen JB. Desensitization of membrane-bound Torpedo acetylcholine receptor by amine noncompetitive antagonists and aliphatic alcohols: studies of [3H]acetylcholine binding and 22Na+ion fluxes. Biochemistry. 1984;23:4023–4033. doi: 10.1021/bi00313a003. [DOI] [PubMed] [Google Scholar]
- Charnet P, Labarca C, Leonard RJ, Vogelaar NJ, Czyzyk L, Gouin A, Davidson N, Lester HA. An open-channel blocker interacts with adjacent turns of alpha-helices in the nicotinic acetylcholine receptor. Neuron. 1990;4:87–95. doi: 10.1016/0896-6273(90)90445-l. [DOI] [PubMed] [Google Scholar]
- Clapham DE, Neher E. Trifluoperazine reduces inward ionic currents and secretion by separate mechanisms in bovine chromaffin cells. J Physiol (Camb) 1984;353:541–564. doi: 10.1113/jphysiol.1984.sp015350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sheridan RE. The modes of action of gallamine. Proc R Soc B Biol Sci. 1981;211:181–203. doi: 10.1098/rspb.1981.0002. [DOI] [PubMed] [Google Scholar]
- Cooper E, Couturier S, Ballivet M. Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature. 1991;350:235–238. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]
- DiPaola M, Kao PN, Karlin A. Mapping the alpha- subunit site photolabeled by the noncompetitive inhibitor [3H]quinacrine azide in the active state of the nicotinic acetylcholine receptor. J Biol Chem. 1990;265:11017–11029. [PubMed] [Google Scholar]
- Farley JM, Yeh JZ, Watanabe S, Narahashi T. Endplate channel block by guanidine derivatives. J Gen Physiol. 1981;77:273–293. doi: 10.1085/jgp.77.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage PW, Wachtel RE. Some effects of procaine at the toad end-plate are not consistent with a simple channel-blocking model. J Physiol (Camb) 1984;346:331–339. doi: 10.1113/jphysiol.1984.sp015025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, MacKinnon R. Agitoxin footprinting the Shakerpotassium channel pore. Neuron. 1996;16:399–406. doi: 10.1016/s0896-6273(00)80057-4. [DOI] [PubMed] [Google Scholar]
- Heidmann T, Oswald RE, Changeux JP. Multiple sites of action for noncompetitive blockers on acetylcholine receptor rich membrane fragments from torpedo marmorata. Biochemistry. 1983;22:3112–3127. doi: 10.1021/bi00282a014. [DOI] [PubMed] [Google Scholar]
- Herz JM, Johnson DA, Taylor P. Interaction of noncompetitive inhibitors with the acetylcholine receptor. The site specificity and spectroscopic properties of ethidium binding. J Biol Chem. 1987;262:7238–7247. [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Brodwick MS. Acetylcholine-induced current in perfused rat myoballs. J Gen Physiol. 1980;75:297–321. doi: 10.1085/jgp.75.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Brodwick MS, Dickey WD. Asymmetry of the acetylcholine channel revealed by quaternary anesthetics. Science. 1980;210:205–207. doi: 10.1126/science.6251552. [DOI] [PubMed] [Google Scholar]
- Hucho F, Oberthur W, Lottspeich F. The ion channel of the nicotinic acetylcholine receptor is formed by the homologous helices M II of the receptor subunits. FEBS Lett. 1986;205:137–142. doi: 10.1016/0014-5793(86)80881-x. [DOI] [PubMed] [Google Scholar]
- Imoto K, Busch C, Sakmann B, Mishina M, Konno T, Nakai J, Bujo H, Mori Y, Fukuda K, Numa S. Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature. 1988;335:645–648. doi: 10.1038/335645a0. [DOI] [PubMed] [Google Scholar]
- Karlin A. Explorations of the nicotinic acetylcholine receptor. Harvey Lect. 1989;85:71–107. [PubMed] [Google Scholar]
- Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- Karpen JW, Aoshima H, Abood LG, Hess GP. Cocaine and phencyclidine inhibition of the acetylcholine receptor: analysis of the mechanisms of action based on measurements of ion flux in the millisecond-to-minute time region. Proc Natl Acad Sci USA. 1982;79:2509–2513. doi: 10.1073/pnas.79.8.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch GE, Alam M, Hartmann HA. Differential effects of sulfhydryl reagents on saxitoxin and tetrodotoxin block of voltage-dependent Na channels. Biophys J. 1994;67:2305–2315. doi: 10.1016/S0006-3495(94)80716-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krodel EK, Beckman RA, Cohen JB. Identification of a local anesthetic binding site in nicotinic post-synaptic membranes isolated from Torpedo marmorata electric tissue. Mol Pharmacol. 1979;15:294–312. [PubMed] [Google Scholar]
- Leonard RJ, Labarca CG, Charnet P, Davidson N, Lester HA. Evidence that the M2 membrane-spanning region lines the ion channel pore of the nicotinic receptor. Science. 1988;242:1578–1581. doi: 10.1126/science.2462281. [DOI] [PubMed] [Google Scholar]
- Lurtz MM, Hareland ML, Pedersen SE. Quinacrine and ethidium bromide bind the same locus on the nicotinic acetylcholine receptor from Torpedo californica. . Biochemistry. 1997;36:2068–2075. doi: 10.1021/bi962547p. [DOI] [PubMed] [Google Scholar]
- Magleby KL, Pallotta BS. A study of desensitization of acetylcholine receptors using nerve-released transmitter in the frog. J Physiol (Camb) 1981;316:225–250. doi: 10.1113/jphysiol.1981.sp013784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleque MA, Souccar C, Cohen JB, Albuquerque EX. Meproadifen reaction with the ionic channel of the acetylcholine receptor: potentiation of agonist-induced desensitization at the frog neuromuscular junction. Mol Pharmacol. 1982;22:636–647. [PubMed] [Google Scholar]
- Neher E. The charge carried by single-channel currents of rat cultured muscle cells in the presence of local anaesthetics. J Physiol (Camb) 1983;339:663–678. doi: 10.1113/jphysiol.1983.sp014741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Steinbach JH. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J Physiol (Camb) 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald RE, Heidmann T, Changeux JP. Multiple affinity states for noncompetitive blockers revealed by [3H]phencyclidine binding to acetylcholine receptor rich membrane fragments from Torpedo marmorata. Biochemistry. 1983;22:3128–3136. doi: 10.1021/bi00282a015. [DOI] [PubMed] [Google Scholar]
- Pascual JM, Karlin A. State-dependent accessibility and electrostatic potential in the channel of the acetylcholine receptor: inferences from rates of reaction of thiosulfonates with substituted cysteines in the M2 segment of the alpha subunit. J Gen Physiol. 1998;111:717–739. doi: 10.1085/jgp.111.6.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual JM, Shieh CC, Kirsch GE, Brown AM. Multiple residues specify external tetraethylammonium blockade in voltage-gated potassium channels. Biophys J. 1995;69:428–434. doi: 10.1016/S0006-3495(95)79915-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SE, Sharp SD, Liu WS, Cohen JB. Structure of the noncompetitive antagonist-binding site of the Torpedo nicotinic acetylcholine receptor. [3H]Meproadifen mustard reacts selectively with alpha-subunit Glu-262. J Biol Chem. 1992;267:10489–10499. [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Revah F, Galzi JL, Giraudat J, Haumont PY, Lederer F, Changeux JP. The noncompetitive blocker [3H]chlorpromazine labels three amino acids of the acetylcholine receptor gamma subunit: implications for the alpha-helical organization of regions MII and for the structure of the ion channel. Proc Natl Acad Sci USA. 1990;87:4675–4679. doi: 10.1073/pnas.87.12.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff RL. A quantitative analysis of local anaesthetic alteration of miniature end-plate currents and end-plate current fluctuations. J Physiol (Camb) 1977;264:89–124. doi: 10.1113/jphysiol.1977.sp011659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JA, Dani JA, Siemen D, Hille B. Slow permeation of organic cations in acetylcholine receptor channels. J Gen Physiol. 1986;87:985–1001. doi: 10.1085/jgp.87.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreibmayer W, Lester HA, Dascal N. Voltage clamping of Xenopus laevisoocytes utilizing agarose-cushion electrodes. Pflügers Arch. 1994;426:453–458. doi: 10.1007/BF00388310. [DOI] [PubMed] [Google Scholar]
- Sine SM, Taylor P. Local anesthetics and histrionicotoxin are allosteric inhibitors of the acetylcholine receptor. Studies of clonal muscle cells. J Biol Chem. 1982;257:8106–8114. [PubMed] [Google Scholar]
- Stauffer DA, Karlin A. Electrostatic potential of the acetylcholine binding sites in the nicotinic receptor probed by reactions of binding-site cysteines with charged methanethiosulfonates. Biochemistry. 1994;33:6840–6849. doi: 10.1021/bi00188a013. [DOI] [PubMed] [Google Scholar]
- Tamamizu S, Todd AP, McNamee MG. Mutations in the M1 region of the nicotinic acetylcholine receptor alter the sensitivity to inhibition by quinacrine. Cell Mol Neurobiol. 1995;15:427–438. doi: 10.1007/BF02071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GG, Karlin A. The location of the gate in the acetylcholine receptor channel. Neuron. 1998;20:1269–1281. doi: 10.1016/s0896-6273(00)80506-1. [DOI] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Covey DF, Akabas MH. Interaction of picrotoxin with GABA(A) receptor channel-lining residues probed in cysteine mutants. Biophys J. 1995;69:1858–1867. doi: 10.1016/S0006-3495(95)80056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Karlin A. Identification of acetylcholine receptor channel-lining residues in the M1 segment of the beta subunit. Biochemistry. 1997;36:15856–15864. doi: 10.1021/bi972357u. [DOI] [PubMed] [Google Scholar]
- Zhang H, Karlin A. Contribution of the beta subunit M2 segment to the ion-conducting pathway of the acetylcholine receptor. Biochemistry. 1998;37:7952–7964. doi: 10.1021/bi980143m. [DOI] [PubMed] [Google Scholar]