

Abstract
The human and shark Na-K-Cl cotransporters (NKCCs) are 74% identical in amino acid sequence yet they display marked differences in apparent affinities for the ions and bumetanide. In this study, we have used chimeras and point mutations to determine which transmembrane domains (tm's) are responsible for the differences in ion transport and in inhibitor binding kinetics. When expressed in HEK-293 cells, all the mutants carry out bumetanide-sensitive 86Rb influx. The kinetic behavior of these constructs demonstrates that the first seven tm's contain all of the residues conferring affinity differences. In conjunction with our previous finding that tm 2 plays an important role in cation transport, the present observations implicate the fourth and seventh tm helices in anion transport. Thus, it appears that tm's 2, 4, and 7 contain the essential affinity-modifying residues accounting for the human–shark differences with regard to cation and anion transport. Point mutations have narrowed the list of candidates to 13 residues within the three tm's. The affinity for bumetanide was found to be affected by residues in the same tm 2–7 region, and also by residues in tm's 11 and 12. Unlike for the ions, changes in bumetanide affinity were nonlinear and difficult to interpret: the K i(bumetanide) of a number of the constructs was outside the range of sNKCC1 and hNKCC1 K is.
Keywords: cotransporter, kinetics, binding, mutations, Na-K-Cl
introduction
The Na-K-Cl cotransporter (NKCC; also called BSC)1 is an integral membrane protein that facilitates the passage of the ions Na, K, and Cl across cell membranes. The transporter has been identified in a wide variety of cell types from vertebrate as well as invertebrate species (Xu et al., 1994; Delpire et al., 1994; Payne et al., 1995; Payne and Forbush, 1994). NKCC belongs to the cation-chloride cotransporters (CCC; Haas, 1989; Gillen et al., 1996), a family that also includes the Na-Cl cotransporter (NCC or TSC; Gamba et al., 1993) and the K-Cl cotransporter (KCC; Gillen et al., 1996; Payne et al., 1996). Two types of Na-K-Cl cotransporters have been identified. NKCC1 is a widely distributed isoform (Xu et al., 1994); it is found in nonpolarized cells where it plays an important role in cell volume regulation (Haas, 1989), and it is also prominent in certain secretory epithelia where it drives net transport of fluid and electrolytes (Xu et al., 1994). NKCC2 is a kidney-specific isoform that occurs as three splice variants called A, B, and F; these variants are distributed along the thick ascending limb of Henle and play an important role in electrolyte absorption (Payne and Forbush, 1994; Igarashi et al., 1995).
All NKCCs described to date are characterized by a strict dependence of transport on the three ions, Na, K, and Cl (Forbush and Palfrey, 1983; Lytle et al., 1992; Moore et al., 1995). In most tissues, the binding stoichiometry is 1Na:1K:2Cl per transporter and, therefore, the transport process is not associated with net movement of charge (Haas, 1989). Loop diuretics such as furosemide, bumetanide, and benzmetanide inhibit cotransporter function by interacting with the protein at an extracellular site (Forbush and Palfrey, 1983; Payne and Forbush, 1995; Isenring and Forbush, 1997). This process also requires the presence of the ions Na, K, and Cl in the binding media. Inhibition of transport activity by bumetanide occurs with a stoichiometry of one inhibitor molecule per transporter protein (Forbush and Palfrey, 1983).
Members of the Na-K-Cl cotransporter family exhibit similar hydropathy profiles, delineating three broad regions: large NH2 and COOH termini, and a 500-residue central domain that is predicted to contain 12 transmembrane domains (tm's). Sites of regulatory phosphorylation have been identified in the NH2 and COOH termini, demonstrating that part of these domains are intracellular. The central domain of the CCC proteins exhibits the highest sequence conservation. For example, the NH2 terminus, the COOH terminus, and the tm's of the shark and human cotransporters are 55, 90, and 85% identical, respectively.
Despite a high level of amino acid sequence conservation in the NKCC1, apparent affinities for the ions and for bumetanide vary as functions of the species (Payne and Forbush, 1995; Isenring and Forbush, 1997; Isenring et al., 1998b ). For example, the K m for all the ions and the K i(bumetanide) are four- to sixfold lower in the human cotransporter compared with the shark protein. In previous studies, we took advantage of these differences to identify regions that are responsible for mediating transport and binding characteristics in the two species. By interchanging the NH2 and COOH termini between the shark and human carriers, we were able to demonstrate that the large hydrophobic central domain encodes all of the information involved in specifying the kinetic singularities of the wild-type proteins (Isenring and Forbush, 1997). Further experiments showed that the second tm plays an important role in the species differences in both cation and bumetanide affinities (Isenring et al., 1998a ). Interestingly, this domain is not involved in determining Cl affinity, suggesting that residues beyond tm 3 encode cotransporter behavior for anion transport.
In this study, we have pursued the chimera approach and examined the role of tm's 4–12. We find that variant residues between tm's 4 and 7 also account for human–shark differences in ion transport. Further analysis using site-specific mutagenesis confirms the results obtained with the chimeras, and in conjunction with the results described above, shows that tm's 2, 4, and 7 contain the essential residues that determine the kinetic behavior of the NKCC1s. On the other hand, we find that bumetanide binding involves multiple residues localized before as well as beyond tm 7.
methods
Nomenclature for cDNA Constructs
As previously described (Isenring and Forbush, 1997), we use single letters in lowercase to designate domains from an NKCC species (h, human; s, shark) that are included in a chimera. A number 1.x–12.x refers to the position of the chimeric junctions in the 12 putative tm's, and a number 1L.x–11L.x refers to the connecting loops after each tm α helix. Each domain is further divided into 10 subdomains from x.0 to x.9. The application of this nomenclature is illustrated in Fig. 1, which presents models of the NKCC1 constructs used in these studies.
Figure 1.

Models for sNKCC1, hNKCC1, five chimeras, and three point mutations in tm's of sNKCC1 based on proposed structure (Payne et al., 1995). Circles symbolize amino acid residues (gray, human NKCC1; black, sNKCC1). AI-V-I-L(4.5) is a tm-4 sNKCC1 mutant with the substitutions S386, V387, I389, V391, and F393 → A414, 415, V417, I419, and L421. In the mutant GD-VL (5.3), V419, N420, T422, and V423 in tm 5 of sNKCC1 are replaced by the corresponding G447, D448, V450, and L451 of hNKCC1. In the shark L-V-I-A (7.7) mutant, the substitutions are I504, A507, A509, and V511 → L532, V535, I537, and A539 in the tm 7 of the sNKCC1 protein.
All the point mutations described here are in the sNKCC1 sequence and consist of sNKCC1 → hNKCC1 replacements. A number in parenthesis indicates the site of the mutations following the nomenclature described above. In GD-VL (5.3) for example, the GD and VL residues from hNKCC1 replace the corresponding VN and TV residues in the sNKCC1 sequence; the substitutions are located near the middle of tm 5 (site 5.3). These point mutations are shown in Fig. 1.
Vectors
A mutant p-Bluescript (pBSM-SK−) containing additional restriction sites in the polylinker (Isenring and Forbush, 1997) was used to facilitate various subcloning steps. The wild-type sNKCC1 was subcloned in the vector pTZ18U (Bio-Rad Laboratories, Richmond, CA) at XbaI-KpnI sites to serve as a template for site-directed mutagenesis. The vector used for expression of wild-type and mutant cDNA (pJB20M) was as in Isenring and Forbush (1997). The vector pJB20 is derived from pCB6 and contains additional intronic sequences in the 3′ linker to improve transcript stability. The vector also contains the strong cytomegalovirus promoter and a G418 resistance cassette for selection of transfectants. In pJB20M, additional restriction sites were also introduced in the polylinker to facilitate subcloning.
Chimeras and Mutations
The chimeras hsh5L.7/13.1 and shs5L.7/13.1 (shown in Fig. 1, A and B, respectively) were generated by fragment exchange at the restriction site ApaI situated at bp 1596 of hNKCC1. This site occurs at three other locations in the hNKCC1 cDNA (bp 341, 381, and 660), but is absent in sNKCC1. We used the Kunkel method to introduce an ApaI site (sNKCC1APA/pTZ18U) at the junction 5L.7 in the sNKCC1 cDNA (bp 1785); the mutagenic oligonucleotide, shown in Table I (A), changes the valine at position 450 in sNKCC1 to a glycine in the extracellular loop. The junction corresponds to residues V450 → G/P451 in sNKCC1 and G478/P479 in hNKCC1. The other junction site at 13.1 corresponds to residues H756/V757 in sNKCC1 and H783/V784 in hNKCC1. After creating the ApaI site in sNKCC1, hsh5L.7/13.1 was generated in two separate steps. First, the Bst1107I-ApaI fragment of sNKCC1APA/pTZ18U (600 bp) was ligated to the Bst1107I-ApaI fragment of hNKCC1/ pJB20M (9,600 bp) to generate shs0.9/5L.7 in pTZ18U. In the second step, the following fragments were ligated: KpnI-Bst1107I from hNKCC1/pJB20M (1,100 bp), Bst1107I-PmlI from shs0.9/ 5L.7 in pTZ18U (1,500 bp), and PmlI-KpnI from hNKCC1/ pJB20M (7,800 bp). The chimera shs5L.7/13.1 was generated in a single step ligation using the following fragments: KpnI-ApaI from sNKCC1APA/pTZ18U (1,800 bp), ApaI-PmlI from hNKCC1/ pJB20M (900 bp), and PmlI-KpnI from sNKCC1/pJB20M (8,800 bp). Representations of the chimeras are shown in Fig. 1, A and B.
Table I.
Oligonucleotides Used to Generate the Five Point Mutations in sNKCC1 or hNKCC1
| A | sNKCC1APA | ct gaa aac ttt ggg ccc gat ttt cgt gat g | ||
| B | sNKCC1SNAB | ca att gtc tac gta gga ata gct gct tct gta gtt tc | ||
| C | sNKCC1NDE | c ctg gct tca tat gct ttg att ttc | ||
| D | AI-V-I-L (4.5) | att ata gga gcc att act gtc gtg atc ctg ctt ggc ata tc | ||
| E | GD-VL (5.3) | cta tta gct ata ggt gac ttc gtc ata gga act ttc atc cca g | ||
| F | L-I-V-A (7.5) | same as B |
For the chimeras hsh7.9/13.1 and shs7.4/13.1 (shown in Fig. 1, C and D, respectively), the junction points are in tm 7 (Y506/A507 → V in sNKCC1 and Y534/V535 in hNKCC1) and at 13.1 (as described above). For the tm 7 junction, a SnaBI restriction site is present in hNKCC1 (bp 1761) but absent in the shark sequence. We used the Kunkel method to obtain a matching site in sNKCC1 at bp 1950. The SnaBI-containing oligonucleotide, shown in Table I (B), also introduces four substitutions in sNKCC1; the residues I504, A507, A509, and V511 are replaced by their counterparts L532, V535, I537, and A539 from hNKCC1. The resulting mutant, L-V-I-A (7.7)/pTZ18U or sNKCC1SNAB/pTZ18U, was used to generate hsh7.9/13.1 and shs7.4/13.1. The KpnI-SnaBI fragments of hNKCC1/pBSM-SK− (1,800 and 5,300 bp; see Isenring and Forbush, 1997) and of sNKCC1SNAB /pTZ18U (2,000 and 6,200 bp) were cross-ligated to generate hs7.9/pTZ18U and sh7.4/pBSM-SK−; the KpnI-PmlI fragment of each mutant (2,500 and 2,700 bp) was religated to the KpnI-PmlI fragment of hNKCC1/pJB20M or sNKCC1/pJB20M to generate hsh7.9/13.1 and shs7.4/13.1. As illustrated in Fig. 1, C and D, these two chimeras are not perfect complements of one another since they share human residues in tm 7.
The chimera hsh10.5/13.1 (see Fig. 1 E) was produced by exchanging human/shark fragments at NdeI and PmlI restriction sites after creating a NdeI site in sNKCC1 at bp 2410 (sNKCC1NDE/ pTZ18U) to match the human site at bp 2218 [see oligonucleotide in Table I (C)]. The junction point 10.5 corresponds to Y686/A687 in hNKCC1 and to Y659/A660 in sNKCC1. Chimeras hs3.1 and sh3.1 were available from the studies in Isenring et al., 1998a .
The point mutations AI-V-I-L (4.5), GD-VL (5.3), and L-V-I-A (7.7), shown in Fig. 1, F–H, were created with the Kunkel method using mutagenic oligonucleotides [see Table I (D–F)]. Residues in sNKCC1 were replaced with corresponding residues from hNKCC1: AI-V-I-L (4.5) was generated by replacing the S386, V387, I389, V391, and F393 residues in the tm 4 sNKCC1 sequence by the A414, I415, V417, I419, and L421 residue counterparts from hNKCC1; for GD-VL (5.3), V419, N420, T422, and V423 in tm 5 were replaced by G447, D448, V450, and L451; for L-V-I-A (7.7), I504, A507, A509, and V511 in tm 7 were replaced by L532, V535, I537, and A539.
Silent restriction sites or residue substitutions were confirmed by partial automated sequencing (W.M. Keck Foundation Biotechnology Resource Lab DNA Sequencing Group, Yale University) and/or by extensive restriction analyses.
Cell Lines
Lines stably expressing hNKCC1and sNKCC1 were the same as in Isenring and Forbush, 1997. The five chimeras and three point mutations were transfected into HEK-293 cells by calcium phosphate precipitation and stable lines (generally 12–16) were isolated by G-418 resistance after screening with 86Rb influx assays, Western blotting, or immunofluorescence. Transfected HEK-293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Life Technologies, Inc., Grand Island, NY), penicillin, streptomycin, and G-418 (950 μg/ml). The level of functional expression achieved with six of eight constructs was comparable to the wild-type hNKCC1 and sNKCC1 (Isenring and Forbush, 1997), approximately six- to eightfold above the endogenous cotransport activity of mock-transfected HEK-293 cells (results not shown). The chimeras shs5L.7/13.1 and shs7.4/13.1 (see Fig. 1, B and D) were found to transport 86Rb at a lower rate, approximately threefold above the background. When cotransporter expression was tested by immunofluorescence using J3 and J7 antibodies, predominant cell surface expression as well as perinuclear staining were observed (Isenring et al. 1998a ).
Protein Analysis
Experiments were performed as previously described (Lytle et al., 1992). In brief, transfected HEK-293 cells were allowed to grow to near confluence and solubilized in 3% CHAPS {3-[(3-chiolamido-propyl)dimethylammonio]-1-propanesulfonate} with protease inhibitors. Whole cell lysates were assayed for protein content by the Micro BCA method (Pierce Chemical Co., Rockford, IL). Western blots (2–5-μg protein samples) were carried out using the sNKCC1 monoclonal antibodies J3 or J7 (Lytle et al., 1992). As previously reported (Isenring and Forbush, 1997), J3 recognizes a segment of the NH2 terminus located between residues 49 and 196, and J7 recognizes a COOH terminal segment situated between residues 1050 and 1168. As shown in Fig. 2, the level of cotransporter synthesis and the pattern of glycosylation were comparable for all the cell lines studied in this project. Recognition by J3 and J7 was also consistent with whether the NH2 or COOH terminus was derived from the shark NKCC1.
Figure 2.
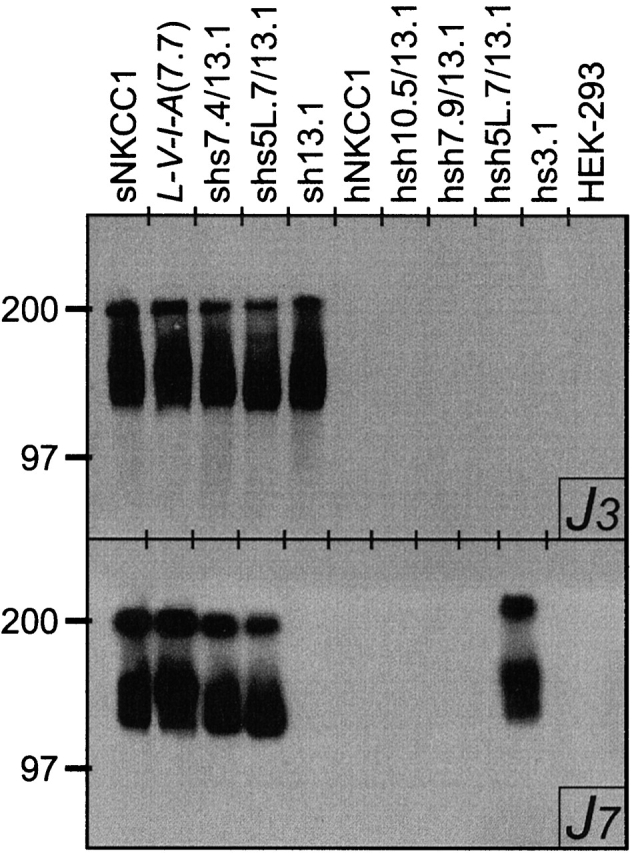
Western blot analyses of cDNA-transfected HEK-293 cells using the shark NKCC1 monoclonal antibodies J3 (top) and J7 (bottom).
Flux Studies
As previously described (Isenring and Forbush, 1997), HEK-293 cells were subcultured onto 96-well plates precoated with polylysine and grown to confluence (6–8 d). Ion transport rates and bumetanide binding were determined by 86Rb influx measurements. All experiments were done at room temperature (≃22°C) and the solutions were at pH 7.4. The cells were preincubated in hypotonic low Cl (163 mosM, 2 mM Cl) medium for 1 h (67.5 mM Na gluconate, 2.5 mM K gluconate, 0.5 mM CaCl2, 0.5 mM MgCl2, 0.5 mM Na2HPO4, 0.5 mM Na2SO4, and 7.5 mM Na HEPES) to activate cotransporter activity. For bumetanide inhibition studies, the cells were also preincubated 15 min with the inhibitor diluted in a 20-mM Cl solution (154 mM Na and 144 mM Cl). The influxes were performed for 1 min in a medium containing 135 mM NaCl, 5 mM RbCl (2 μCi/ml 86RbCl), 1 mM CaCl2, 1 mM MgCl2, 1 mM Na2HPO4, 1 mM Na2SO4, 15 mM Na HEPES, and 0.1 mM ouabain; when necessary, Na and Rb were replaced with equimolar amounts of N-methyl glucamine, and Cl was replaced with gluconate. To determine bumetanide-sensitive influx, 250 μM bumetanide was added directly to the media. 86Rb uptake was terminated by addition of, and three rinses in, ice-cold stop solution (135 mM K gluconate, 5 mM Na gluconate, 1 mM CaCl2, 1 mM MgCl2, 1 mM Na2HPO4, 1 mM Na2SO4, 15 mM Na HEPES, 250 μM bumetanide, and 0.1 mM ouabain). Cells were solubilized in 2% sodium dodecylsulfate, and assayed for protein content and for 86Rb by counting Cerenkov radiation.
Each concentration curve was carried out in a single row of the 96-well plate, and in a typical experiment there were two to six replicate rows. Data were handled on a per-row basis and counts were normalized to the value at the highest ion concentration, or to the value of uninhibited flux in inhibition studies. K m and K i values were obtained by nonlinear least-squares curve fitting using the Simplex algorithm (program PLOT, B. Forbush), and are expressed as means ± SEM among all rows in 4–15 experiments. Rows with obvious rogue values (data sets for which least squares fits did not converge, or for which individual data points were >2.0 or <0.5 of the value of the fit line) were omitted from averages, in all cases less than one row in eight.
Chemicals and Reagents
Where not otherwise noted, general chemicals and reagents were from Sigma Chemical Co. (St. Louis, MO), and reagents for molecular biology and tissue culture were from New England Biolabs, Inc. (Beverly, MA), FMC Bioproducts (Rockland, ME), and GIBCO BRL (Gaithersburg, MD).
Calculation of Kinetic Parameters for the Ordered Binding Model
Predictions of kinetic parameters of the ordered binding model of Miyamoto et al. (1986) and Lytle et al. (1998) were obtained by solving the set of simultaneous rate equations describing the transport model (see Benjamin and Johnson, 1997). Isotopic exchange was included by explicitly considering labeled and unlabeled Rb as independently transported species. Solutions were obtained at various concentrations of a test ion, and K ms were obtained from fits of these curves (for Cl, a fixed Hill coefficient, n = 2, was used for parallelism to our treatment of data; actually, best fits are obtained with Hill 7 ≃ 1,6; as discussed in a footnote in Isenring and Forbush, 1997). For simplicity, the constants are symmetrical across the membrane; constants were chosen to produce K ms near those of hNKCC1. All association rate constants were 107 s−1 M−1, translocation rate constants for unloaded and loaded carrier were 40,000 and 10,000 s−1, respectively, and affinities (1/K d) for Na, Rb, and Cl (at each of two sites) were 127, 54, and 6 mM, respectively (program PLOT, B. Forbush).
results
Role of tm's 10–12 in Ion Transport Compared with That of tm's 1–9
To determine if the last two tm's of NKCC1 contain affinity-specifying residues, we replaced these segments by the corresponding shark sequence (hsh10.5/13.1; see Fig. 1 E). Between the species, these last two tm's differ by six residues, while predicted α helix 10 is fully conserved. The dependence of 86Rb influx on [Na], [Rb], and [Cl] is illustrated in Fig. 3. The data demonstrate that shark residues in tm's 11 and 12 have no effect on the kinetic behavior of hNKCC1. The K m(Na), K m(Rb), and K m(Cl) are 14, 1.9, and 34 mM, similar to those reported for the hNKCC1-transfected cells (15, 1.7, and 31 mM, respectively).
Figure 3.

Dependence of 86Rb influx on [Na], [Rb], and [Cl] for the chimera hsh10.5/13.1. The data are shown as averages of 30– 40 flux rows in 7–10 experiments.
Role of tm's 7–9 Compared with That of tm's 1–6
To next determine the contribution of domains 7–9 in ion transport by the Na-K-Cl protein, we examined two other human–shark chimeras with a junction located in tm 7 (hsh7.9/13.1 and the nearly complementary shs7.4/ 13.1; see Fig. 1, C and D). The K m values derived from the dependence of 86Rb transport on [Na], [Rb], and [Cl] are summarized in Fig. 4. They show that the hsh7.9/13.1 chimera has a phenotype that is indistinguishable from hNKCC1; the K m(Na), K m(Rb), and K m(Cl) are 19, 1.8, and 31 mM, respectively. Thus, it appears that tm's 8 and 9, as shown above for tm's 10–12, do not confer differences in ion affinities between human and shark. These results, in conjunction with previous observations that the NH2 and COOH termini are devoid of affinity-modifying properties (Isenring and Forbush, 1997), indicate that differences in apparent binding parameters between the two cotransporters are specified by residues found in the first seven tm's.
Figure 4.

K m(Na), K m(Rb), and K m(Cl) for the hsh7.9/13.1 and the shs7.4/13.1 chimeras, and for the sNKCC1 point mutant L-V-I-A compared with the wild-type proteins (as described above, the residue replacements in L-V-I-A (7.7) are I504, A507, A509, and V511 → L532, V535, I537, and A539). The values for sNKCC1 and hNKCC1 are from Isenring et al. (1998). For the other constructs, the data are shown as averages of 10–40 flux rows in 4–15 experiments for Na, 25–34 flux rows in 8–14 experiments for Rb, and 16–37 flux rows in 4–7 experiments for Cl.
Interestingly, ion constants for the nearly complementary chimera shs7.4/13.1 are all intermediate between human and shark. Since the results described above rule out a role for tm's 8–12, tm 7 could contain all of the variant residues accounting for the behavior of shs7.4/13.1. To test this hypothesis, we characterized the ion dependence of 86Rb influx in L-V-I-A (7.7)-transfected cells. In this sNKCC1 mutant (see Fig. 1 H), all the tm-7 residues were replaced by their human residue counterparts. It is seen in Fig. 4 that the ion constants for L-V-I-A (7.7) are very similar to those of shs7.4/13.1. This result confirms that the behavior of shs7.4/13.1, relative to that of sNKCC1, was mediated through substituted residues in tm 7. The data is also consistent with previous observations in showing that residues in tm's 8–12 do not encode variant transport behaviors.
Role of tm's 4–5
Of tm's 1 to 6, only 2 and 4 or 5 could potentially mediate differences in ion affinities between the human and shark cotransporters; as shown in Fig. 7 A, tm's 1, 3, and 6 are fully conserved. We have previously determined that tm 2 plays a role in cation transport, but is not involved in Cl affinity differences (Isenring et al., 1998a ). Specifically, the chimera hs3.1 had cation affinities that were intermediate between sNKCC1 and hNKCC1, and a Cl affinity that was similar to that of sNKCC1 (results from Isenring et al. [1998a] are reproduced in Fig. 5). Thus, with regard to cations, tm's 2 and 7 could contain all of the information that defines the kinetic behavior of NKCC1: each domain endows approximately half the affinity difference between the species. For the anions, on the other hand, tm's 4 or 5 are likely to play an important role given that variant residues in tm 7, as shown above with L-V-I-A (7.7), account for only part of the differences in K m(Cl) between sNKCC1 and hNKCC1.
Figure 7.
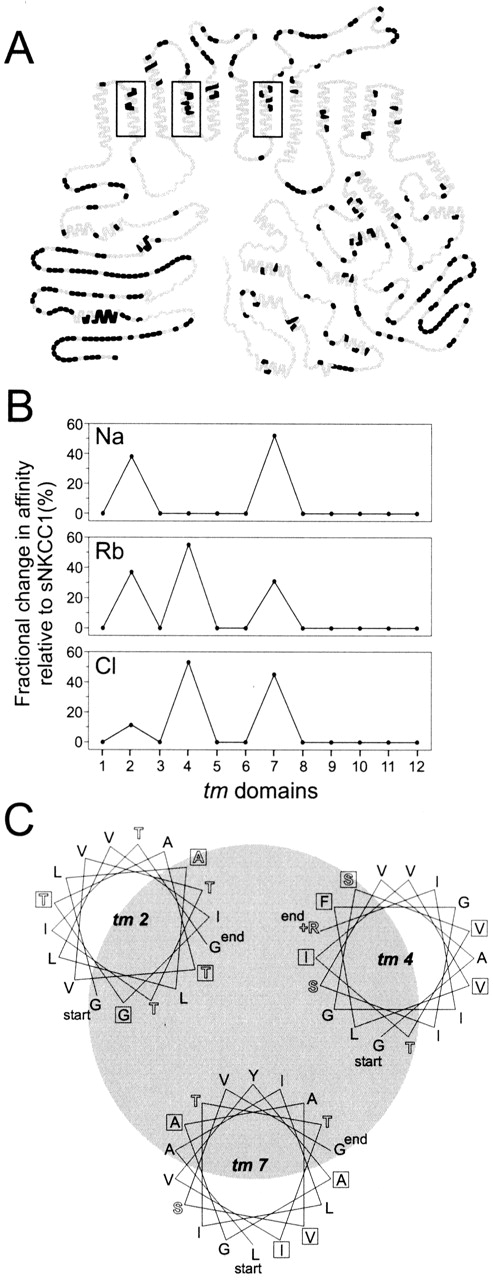
(A) Identity plot between the human and shark cotransporters. Here, residues that are identical between the species are represented by gray symbols, and residues that are unique to the shark cotransporter are represented by black symbols. (B) Fractional change in affinity relative to sNKCC1 as contributed by each tm (see text). (C) Helical wheels of tm's 2, 4, and 7. Using an angle of 100°, wheels are drawn so the most hydrophilic poles face each other. Hydrophilic amino acids are represented by solid letters, and hydrophobic residues by open letters. Boxed letters indicate candidate affinity-modifying residues as determined by mutational analyses.
Figure 5.

K m(Na), K m(Rb), and K m(Cl) for the 5L.7/ 13.1 chimeras and the sNKCC1 point mutant GD-VL compared with hs3.1 (see Isenring et al., 1998) and the wild-type proteins (as described above, the residue replacements in GD-VL (5.3) are V419, N420, T422 and V423 → G447, D448, V450, and L451 of hNKCC1). The values for hs3.1, sNKCC1, and hNKCC1 are also from Isenring et al. (1998). For the other constructs, the data are shown as averages of 24–43 flux rows in 6–11 experiments for Na, 29–48 flux rows in 7–10 experiments for Rb, and 13–35 flux rows in 3–7 experiments for Cl.
To test these predictions, we analyzed the role of tm's 4 and 5 in Na transport by using two additional chimeras (hsh5L.7/13.1 and shs5L.7/13.1) with a junction point situated in the connecting loop after the fifth tm helix (see NKCC1 in Fig. 1, A and B). The dependence of 86Rb influx on [Na] translates into similar K ms for both chimeras, as depicted in Fig. 5 A, and these constant values are not significantly different from the K m(Na) previously reported for hs3.1. The results confirm that the differences in Na affinity between shark and human are not specified by residues located in tm's 4 and 5.
Unexpectedly, analysis of the same chimeras demonstrates that tm's 4 or 5 are possibly involved in the Rb affinity difference. The data in Fig. 5 B show that when tm's 2, 4, and 5 are from hNKCC1, and 7 is from sNKCC1 (chimera hsh5L.7/13.1; see Fig. 1 A), the K m(Rb) is intermediate between human and shark, and lower than the constants obtained for L-V-I-A (7.7) and hs3.1. In conjunction with our previous observations, these results indicate that three tm's (2, 4 or 5, and 7) may be involved in determining Rb affinity. The behavior of the complementary chimera shs5L.7/13.1 (see Fig. 1 B) is consistent with expectation in showing a Rb affinity that is intermediate between human and shark (Fig. 5 B), and close to that of L-V-I-A (7.7) (6.3 mM compared with 4.8 mM). This result confirms the role of tm 7 as a Rb affinity-modifying domain, and is concordant with the fact that tm 6 is fully conserved between human and shark.
The kinetic behavior of the 5L.7/13.1 chimeras with regard to the Cl dependence of 86Rb influx is summarized in Fig. 5 C. It is seen that the K m(Cl) for hsh5L.7/ 13.1 is intermediate between human and shark (54 mM, compared with 31 and 107 mM for hNKCC1 and sNKCC1, respectively), and that it is similar to the value obtained for L-V-I-A (7.7). The K m(Cl) for the reverse chimera shs5L.7/13.1 is also similar to that of L-V-I-A (7.7)-transfected cells. These results confirm our hypothesis above that tm's 4 or 5, along with tm 7, must account for differences in anion affinity between the human and shark carriers.
To further determine which of tm's 4 and 5 mediated affinity behaviors with regard to Rb and Cl, we analyzed the point mutation GD-VL (5.3) (see Fig. 1 G), in which residues in the tm 5 shark sequence (n = 4) were substituted by corresponding residues from hNKCC1 (a similar tm 4 shark mutant, AI-V-I-L(4.5), was nonfunctional; see Fig. 1 F ). The ion dependence of 86Rb influx for GD-VL (5.3)-transfected HEK cells (Fig. 5) yields constants for Na, Rb, and Cl that are very similar to sNKCC1: 91, 11, and 108 mM, compared with 109, 12, and 107 mM for the wild-type sNKCC1. This finding suggests that tm 5 does not contain affinity-modifying residues; hence, some residues in tm 4 must account for the higher Rb-Cl affinity obtained for hsh5L.7/13.1 compared with hs3.1.
Regions Involved in Bumetanide Binding
sNKCC1 has an apparent affinity for bumetanide that is three- to fourfold lower than that of hNKCC1 (Payne et al., 1995; Isenring and Forbush, 1997). Our previous studies have demonstrated that these differences are encoded by the 500-residue central domain of the protein (Isenring and Forbush, 1997), and that residues in or near the second tm appear to be responsible for part of the effect. To extend this analysis, we measured bumetanide inhibition of 86Rb influx in the five chimeras discussed above, as summarized in Fig. 6.
Figure 6.

Bumetanide inhibition constants for the five chimeras compared with those of sNKCC1 and hNKCC1. K i(bu) for the wild-type proteins are from Isenring et al. (1998).
Looking first at the results with hsh7.9/13.1 (see Fig. 1 C), we observe that the bumetanide affinity of this chimera is 1.5-fold lower than that of the parent hNKCC1. This indicates that, unlike the situation for ions, residues beyond the seventh tm do have an effect on bumetanide binding. This fact is also illustrated by the behavior of chimera hsh10.5/13.1, which is 2.5-fold more sensitive to bumetanide than is hNKCC1; this shows that residues in tm 11 or 12 affect bumetanide binding.
Surprisingly, the bumetanide-binding behavior illustrated in Fig. 6 does not fit the principle of additivity that seems to apply to the ion transport properties of the chimeras. For instance, three chimeras exhibit bumetanide affinities that are even higher than that of hNKCC1, and one chimera (hsh5L.7/13.1; Fig. 1 A) exhibits an affinity that is even lower than that of sNKCC1. Although, interestingly, the 5L.7/13.1 chimeras exhibit reciprocal forms of this “misbehavior,” we have been unable to develop a simple model to explain the data. One possibility is that conformational interactions between extracellular domains affect the structure of the bumetanide binding site.
discussion
In this study, we have identified regions in NKCC1 that confer the differences between shark and human NKCC1 in the kinetics of ion transport. By studying chimeric constructs of shark and human cotransporters, we have been able to demonstrate that these differences are caused by variant residues found exclusively in a region that includes the first seven tm's (see Fig. 7 A). Using site-directed mutagenesis, we further determined that the residues involved are localized in the predicted tm α helices, but not in the predicted intracellular or extracellular loops. In the current work, we have identified tm's 4 and 7 as determining part of the differences in K ms: in conjunction with our previous findings (Isenring and Forbush, 1997; Isenring et al., 1998a ), the data demonstrate that residues in tm's 2, 4, and 7 contain all of the information specifying shark–human kinetic differences in ion transport.
A summary of the mutagenic mapping studies is shown in Fig. 7 B. Plotted are approximate percent changes in apparent affinity contributed by each tm, inferred from differences in K m values resulting from substitution of residues in the shark sequence. The current studies show that changes in tm 7 affect the K m for all three ions, and that changes in tm 4 impact mainly Rb and Cl affinities. On the other hand, tm 2 residues selectively affect K ms for cations. Two residues have been implicated in determination of Na affinity, and two other residues in the determination of Rb affinity. To a first approximation, the affinity changes that occur in the three individual tm's are additive in accounting for the full difference between shark and human cotransporter kinetics.
We have used site-specific mutagenesis to localize pairs or groups of residues that account for human– shark differences in ion affinities. We have narrowed the search to 13 candidate residues in three tm's, and it is possible that as few as four of these are important: two in tm 2 and one each in tm's 4 and 7. These residues may be within the translocation pocket, interacting directly with transported ions. A helical-wheel diagram of the three tm's is shown in Fig. 7 C, highlighting the candidate residues in boxes. Each helix is oriented with the most hydrophilic side towards the hypothetical pore, and it is seen that in each of the tm's, at least one to two of the candidate residues are on this face.
Interestingly, in predicted tm's 2, 4, and 7, there is only one charged residue, an arginine at the extracellular end of tm 4. In the Na-K-Cl transporter in which both anions and cations are proposed to pass single file through a translocation pocket (Lytle et al., 1998), it might be expected that charged groups would pose significant barriers to ion entry into or exit from the pocket. An alternative possibility is that a significant amount of ion coordination is through oxygen atoms in serine and threonine side chains as well as in the peptide backbone (Kaplan et al., 1994; Doyle et al., 1998). Serine and threonine side chains have been implicated in several ion binding sites both indirectly (Arguello and Lingrel, 1995) and through crystallographic analysis (Toney et al., 1995). Interestingly, the ser/thr composition of tm's 2 and 8 of NKCC is remarkably high, particularly near the intracellular end of the predicted helices, suggesting a role in ion coordination (the number of threonine residues in tm 2 is even higher if the helix is one turn longer at the cytoplasmic end).
As we have previously discussed (Isenring and Forbush, 1997; Isenring et al., 1998a ), regions that confer species differences in the kinetics of ion translocation are possibly not the same residues that are essential for ion binding. Some of the essential residues must be fully conserved through evolution and these would not be identified by a technique aimed at correlating functional differences with structural differences. Nonetheless, in the example recently provided by the structure of a K channel (Doyle et al., 1998), residues that affect K selectivity have been shown to be those that are directly involved in ion coordination. This supports our hypothesis that the identified NKCC1 residues are closely involved with the ion binding sites and that some of them may indeed directly participate in ion binding.
The measurable kinetic behavior of an ion transporter is determined by a number of kinetic constants, including all of the rate constants for the various steps in the transport cycle. In the case of a multi-ion cotransporter such as NKCC, this is a particularly complex system in which a change in affinity for one ion may change the K m (“apparent affinity”) for other ions. Fortunately, the way in which these constants are interrelated has been determined experimentally for NKCC1 (Miyamoto et al., 1986; also Isenring, P., S.C. Jacoby, and B. Forbush, unpublished data, for sNKCC1 and hNKCC1). These data, as well as transport data for various cis and trans ions (Lytle et al., 1998), support an ordered binding model for Na-K-Cl transport in which extracellular ions bind in the order Na+-Cl−-K+-Cl− and are released in the same order on the inside of the membrane (Miyamoto et al., 1986; Lytle et al., 1998). For the present work, we are able to infer the significance of K m changes based directly on the available kinetic data and from predictions of the ordered model. Such relationships are illustrated in Fig. 8 by kinetic predictions for a particular set of model parameters (this is only an example: the quantitative behavior depends on the specific choice of parameters, and there is currently too little information to determine these uniquely). The interdependence of kinetic constants is illustrated by the fact that every K m changes in response to a change in binding site affinity for any of the ions. However, as a general rule, the apparent affinity of one ion in the ordered binding scheme is most affected by changes in the true affinities of ions that bind subsequently, particularly by the affinity of the ion immediately following.
Figure 8.

Kinetic parameters of 86Rb influx through the Na-K-Cl cotransporter based on the ordered binding model of Lytle et al. (1998) and Miyamoto et al. (1986). Plotted are predicted values of observed 1/K m for Na (left), Rb (center), or Cl (right) as a function of change of Na or Rb affinities (top), of Cl affinities at two sites (center), or of the rate constants for translocation of loaded or unloaded carriers (bottom). Values of the model parameters are given in methods; predicted K m values are relative to 14.2, 1.5, and 32 mM for Na, Rb, and 2Cl, respectively.
With regard to the effect of variant residues in tm 2, K m(Na) is affected by changes in one pair of residues and K m(Rb) by another pair; there is, however, a smaller change in K m(Cl) (Isenring et al., 1998a ). These results are probably attributable to changes in the real affinity for Na and Rb at the two sites, since no other change in kinetic constants would result in this behavior in the ordered binding model. This interpretation is consistent with the expectation of the model that, at high Na and Rb concentrations, changes in either Na or Rb affinity have smaller effects on K ms for the other ions; they are also consistent with the experimental observations of Miyamoto et al. (1988) and with our own observations (Jacoby, S.C., P. Isenring, and B. Forbush, unpublished data) that changes in [Na]ext or [K]ext have sublinear effects on K m(Na) and K m(K), respectively.
The kinetic changes associated with tm 4 substitutions affect K m(Rb) and K m(Cl), but not K m(Na). This behavior is most consistent with a tm 4–related change in the true affinity for Cl at the second of two Cl binding sites (see Fig. 8, middle row, ○). Differences in tm 7 affect the affinity for all three ions: this result could reflect a change in true affinity at the first Cl binding site, but it could also be explained by a change in multiple kinetic parameters or in the rate constant for movement of the loaded carrier (Fig. 8, bottom).
This work has also offered considerable insight into the regions of NKCC involved in loop-diuretic binding. (a) Expanding on our earlier report (Isenring et al., 1998a ), we find that the pattern of changes that affect bumetanide binding is not the same as the pattern of those changes affecting kinetics of ion translocation. This finding is inconsistent with the hypothesis that the bumetanide binding site is identical with one of the Cl binding sites (Haas and McManus, 1983; Turner and George, 1988), but it does not rule out the possibility that some of the residues involved in bumetanide binding are part of the Cl translocation site. (b) In contrast to our observation that changes in ion translocation involves only the first seven tm's, changes in tm 11 and 12 residues are found to alter the kinetics of bumetanide inhibition. (c) Many of the chimeras exhibit bumetanide affinities that are outside the range of the parent NKCC molecules, and we have not found a simple model that can explain the results. One possible explanation is that the bumetanide binding site spans multiple domains on NKCC and that conformational interactions, possibly within extracellular loops, give rise to complexities in the behavior of chimeric constructs.
In conclusion, we have found that tm's 2, 4, and 7 contain the residues that confer the differences between shark and human NKCC1 with regard to ion translocation kinetics, and we propose that these helices form part of the Na-Cl-K-Cl binding pocket. Structural considerations suggest that at least four helices would be involved in forming a translocation “pore” (Doyle et al., 1998); other candidate helices for the Na-K-Cl cotransporter include the S/T-rich tm 8 and the very highly conserved tm's 1 and 6. Parameters affecting bumetanide binding appear to be more complex, indicating that loop-diuretic binding involves interactions with multiple regions of the transporter in the large central domain.
Acknowledgments
We gratefully acknowledge the technical assistance of Mrs. Grace Jones. We also thank Rachel Behnke, Susan Brill, Deborah Lynn, Iñaki Giménez, and Andreas Flemmer for reading the manuscript.
This study was supported by National Institutes of Health grant DK-47661. Paul Isenring was supported by a Clinician Scientist Fellowship from the Medical Research Council of Canada.
Abbreviations used in this paper
- NKCC
Na-K-Cl cotransporter
- tm
transmembrane domain
Footnotes
Portions of this work were previously published in abstract form (Isenring, P., and B. Forbush III. 1996. J. Am. Soc. Nephrol. 7:1282).
references
- Arguello JM, Lingrel JB. Substitutions of serine 775 in the alpha subunit of the Na,K-ATPase selectively disrupt K+ high affinity activation without affecting Na+interaction. J Biol Chem. 1995;270:22764–22771. doi: 10.1074/jbc.270.39.22764. [DOI] [PubMed] [Google Scholar]
- Benjamin BA, Johnson EA. A quantitative description of the Na,K,2Cl cotransporter and its conformity with experimental data. Am J Physiol. 1997;273:F473–F482. doi: 10.1152/ajprenal.1997.273.3.F473. [DOI] [PubMed] [Google Scholar]
- Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na(+)-K(+)-2Cl−cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem. 1994;269:25677–25683. [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Forbush B, III, Palfrey HC. 3H-Bumetanide binding to membranes isolated from dog kidney outer medulla. Relationship to the Na,K,Cl co-transport system. J Biol Chem. 1983;258:11787–11792. [PubMed] [Google Scholar]
- Gamba G, Saltzberg SN, Lombardi M, Miyanoshita A, Lytton J, Hediger MA, Brenner BM, Hebert SC. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc Natl Acad Sci USA. 1993;90:2749–2753. doi: 10.1073/pnas.90.7.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillen CM, Brill S, Payne JA, Forbush B., III Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem. 1996;271:16237–16244. doi: 10.1074/jbc.271.27.16237. [DOI] [PubMed] [Google Scholar]
- Haas M. Properties and diversity of (Na-K-Cl) cotransporters. Annu Rev Physiol. 1989;51:443–457. doi: 10.1146/annurev.ph.51.030189.002303. [DOI] [PubMed] [Google Scholar]
- Haas M, McManus TJ. Bumetanide inhibits (Na + K + 2Cl) co-transport at a chloride site. Am J Physiol. 1983;245:C235–C240. doi: 10.1152/ajpcell.1983.245.3.C235. [DOI] [PubMed] [Google Scholar]
- Igarashi P, Vanden GB, Heuvel, Payne JA, Forbush B., III Cloning, embryonic expression, and alternative splicing of a murine kidney-specific Na-K-Cl cotransporter. Am J Physiol. 1995;269:F405–F418. doi: 10.1152/ajprenal.1995.269.3.F405. [DOI] [PubMed] [Google Scholar]
- Isenring P, Forbush B., III Ion and bumetanide binding by the Na-K-Cl cotransporter. Importance of transmembrane domains. J Biol Chem. 1997;272:24556–24562. doi: 10.1074/jbc.272.39.24556. [DOI] [PubMed] [Google Scholar]
- Isenring P, Jacoby SC, Forbush B., III The role of transmembrane domain 2 in cation transport by the Na-K-Cl cotransporter. Proc Natl Acad Sci USA. 1998a;95:7179–7184. doi: 10.1073/pnas.95.12.7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenring P, Jacoby SC, Payne JA, Forbush B., III Comparison of Na-K-Cl cotransporters: NKCC1, NKCC2 and the HEK cell Na-K-Cl cotransporter. J Biol Chem. 1998b;273:11295–11301. doi: 10.1074/jbc.273.18.11295. [DOI] [PubMed] [Google Scholar]
- Kaplan, J.H., J.M. Arguello, G.C.R. Ellis-Davies, and S. Lutsenko. 1994. The stabilization of cation binding and its relation to Na+/ K+-ATPase structure and function in the sodium pump. B. Bamberg and W. Schoner, editors. Steinkopff, Darmstadt, Germany. 321–325.
- Lytle C, McManus TJ, Haas M. A model of Na-K-2Cl cotransport based on ordered ion binding and glide symmetry. Am J Physiol. 1998;274:C299–C309. doi: 10.1152/ajpcell.1998.274.2.C299. [DOI] [PubMed] [Google Scholar]
- Lytle C, Xu J-C, Biemesderfer D, Haas M, Forbush B., III The Na-K-Cl cotransport protein of shark rectal gland. I. Development of monoclonal antibodies, immunoaffinity purification, and partial biochemical characterization. J Biol Chem. 1992;267:25428–25437. [PubMed] [Google Scholar]
- Miyamoto H, Ikehara T, Yamaguchi H, Hosokawa K, Yonezu T, Masuya T. Kinetic mechanism of Na+, K+, Cl−-cotransport as studied by Rb+ influx into HeLa cells: effects of extracellular monovalent ions. J Membr Biol. 1986;92:135–150. doi: 10.1007/BF01870703. [DOI] [PubMed] [Google Scholar]
- Moore ML, George JN, Turner RJ. Anion dependence of bumetanide binding and ion transport by the rabbit parotid Na+-K+-2Cl−co-transporter: evidence of an intracellular anion modifier site. Biochem J. 1995;309:637–642. doi: 10.1042/bj3090637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Forbush B., III Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differentially distributed within the rabbit kidney. Proc Natl Acad Sci USA. 1994;91:4544–4548. doi: 10.1073/pnas.91.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Forbush B., III Molecular characterization of the epithelial Na-K-Cl cotransporter isoforms. Curr Opin Cell Biol. 1995;7:493–503. doi: 10.1016/0955-0674(95)80005-0. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- Payne JA, Xu JC, Haas M, Lytle CY, Ward D, Forbush B., III Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J Biol Chem. 1995;270:17977–17985. doi: 10.1074/jbc.270.30.17977. [DOI] [PubMed] [Google Scholar]
- Toney MD, Hohenester E, Keller JW, Jansonius JN. Structural and mechanistic analysis of two refined crystal structures of the pyridoxal phosphate-dependent enzyme dialkylglycine decarboxylase. J Mol Biol. 1995;245:151–179. doi: 10.1006/jmbi.1994.0014. [DOI] [PubMed] [Google Scholar]
- Turner RJ, George JN. Ionic dependence of bumetanide binding to the rabbit parotid Na/K/Cl cotransporter. J Membr Biol. 1988;102:71–77. doi: 10.1007/BF01875354. [DOI] [PubMed] [Google Scholar]
- Xu J, Lytle C, Zhu TT, Payne JA, Benz E, Jr, Forbush B., III Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc Natl Acad Sci USA. 1994;91:2201–2205. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]