

Abstract
The skeletal and cardiac muscle dihydropyridine receptors (DHPRs) differ with respect to their rates of channel activation and in the means by which they control Ca2+ release from the sarcoplasmic reticulum (Adams, B.A., and K.G. Beam. 1990. FASEB J. 4:2809–2816). We have examined the functional properties of skeletal (SkEIIIK) and cardiac (CEIIIK) DHPRs in which a highly conserved glutamate residue in the pore region of repeat III was mutated to a positively charged lysine residue. Using expression in dysgenic myotubes, we have characterized macroscopic ionic currents, intramembrane gating currents, and intracellular Ca2+ transients attributable to these two mutant DHPRs. CEIIIK supported very small inward Ca2+ currents at a few potentials (from −20 to +20 mV) and large outward cesium currents at potentials greater than +20 mV. SkEIIIK failed to support inward Ca2+ flux at any potential. However, large, slowly activating outward cesium currents were observed at all potentials greater than + 20 mV. The difference in skeletal and cardiac Ca2+ channel activation kinetics was conserved for outward currents through CEIIIK and SkEIIIK, even at very depolarized potentials (at +100 mV; SkEIIIK: τact = 30.7 ± 1.9 ms, n = 11; CEIIIK: τact = 2.9 ± 0.5 ms, n = 7). Expression of SkEIIIK in dysgenic myotubes restored both evoked contractions and depolarization-dependent intracellular Ca2+ transients with parameters of voltage dependence (V0.5 = 6.5 ± 3.2 mV and k = 9.3 ± 0.7 mV, n = 5) similar to those for the wild-type DHPR (Garcia, J., T. Tanabe, and K.G. Beam. 1994. J. Gen. Physiol. 103:125–147). However, CEIIIK-expressing myotubes never contracted and failed to exhibit depolarization-dependent intracellular Ca2+ transients at any potential. Thus, high Ca2+ permeation is required for cardiac-type excitation–contraction coupling reconstituted in dysgenic myotubes, but not skeletal-type. The strong rectification of the EIIIK channels made it possible to obtain measurements of gating currents upon repolarization to −50 mV (Qoff) following either brief (20 ms) or long (200 ms) depolarizing pulses to various test potentials. For SkEIIIK, and not CEIIK, Qoff was significantly (P < 0.001) larger after longer depolarizations to +60 mV (121.4 ± 2.0%, n = 6). The increase in Qoff for long depolarizations exhibited a voltage dependence similar to that of channel activation. Thus, the increase in Qoff may reflect a voltage sensor movement required for activation of L-type Ca2+ current and suggests that most DHPRs in skeletal muscle undergo this voltage-dependent transition.
Keywords: voltage-dependant calcium channels, skeletal muscle, calcium transients, charge movement, ion channel
introduction
Skeletal and cardiac muscle dihydropyridine receptors (DHPRs)1 are highly homologous proteins that function both as voltage-gated L-type Ca2+ channels (L-channels) and as links between sarcolemmal depolarization and the release of Ca2+ from the sarcoplasmic reticulum (SR). Nonetheless, there are important differences between the two proteins. Cardiac L-channels activate 10-fold more rapidly upon depolarization (Tanabe et al. 1991) and the resulting Ca2+ influx triggers the intracellular release of Ca2+ via ryanodine receptors (RyRs) located nearby in the SR (Nabauer et al. 1989). In contrast to this “cardiac-type” excitation–contraction (EC) coupling, it is generally accepted that activation of RyRs in skeletal muscle is coupled to conformational changes of the skeletal DHPR via a mechanical interaction (Chandler et al. 1976; Ríos and Brum 1987; Tanabe et al. 1987) that is independent of the entry of extracellular Ca2+ (Armstrong et al. 1972; Gonzalez-Serratos et al. 1982). Studies of heterologously expressed Ca2+ channels in dysgenic myotubes, which lack endogenous skeletal L-channels, are consistent with this difference in type of EC coupling: removal of external Ca2+, or addition of Cd2+ and La3+ to block Ca2+ entry, abolishes EC coupling in dysgenic myotubes expressing cardiac L-channels (Tanabe et al. 1990b; Garcia et al. 1994), but not in dysgenic myotubes expressing skeletal L-channels (Tanabe et al. 1988; Garcia et al. 1994). However, although there is little doubt that Ca2+-entry–induced Ca2+ release is involved in cardiac EC coupling, recent work suggests that a mechanical coupling may occur between cardiac L-channels and RyRs when intracellular cAMP levels are elevated (Hobai et al. 1997; Santana et al. 1998; Howlett et al. 1998).
In spite of these distinct functional properties, the selectivity of both skeletal (Almers and McCleskey 1984) and cardiac (Hess and Tsien 1984; Hess et al. 1986) L-channels for Ca2+ over monovalent cations appears to involve intrapore binding of Ca2+ with high affinity (Dang and McCleskey 1998). Mutational analysis of the cardiac L-channel indicates that this intrapore Ca2+ binding is coordinated by a cage of four conserved glutamate residues residing in corresponding positions within each of the pore-lining regions of repeats I–IV of the α1 subunit (Kim et al. 1993; Yang et al. 1993). In fact, a single mutation of the glutamate residue in repeat III to a lysine residue is sufficient to alter the cardiac L-channel such that it exhibits minimal divalent permeability, large monovalent permeability, and a >1,000-fold decrease in high affinity Ca2+ block of monovalent currents (Yang et al. 1993). The effects of mutating pore region glutamates on the Ca2+ permeability of skeletal L-channels have yet to be elucidated.
Here, we have used expression in dysgenic myotubes to compare permeation and EC coupling in skeletal and cardiac L-channels after substitution of lysine for the repeat III glutamate. Compared with the wild-type channel, the mutant cardiac L-channel (CEIIIK) conducted small inward currents carried by Ca2+ and large outward currents carried by monovalent cations. The mutant skeletal L-channel (SkEIIIK) conducted large outward currents, but no detectable inward Ca2+ current. As a result of the greatly reduced Ca2+ entry, CEIIIK lost the ability of wild-type cardiac L-channels to trigger the release of SR Ca2+ in dysgenic myotubes. By contrast, SkEIIIK channels were able, despite the complete loss of Ca2+ entry, to trigger the release of SR Ca2+ with a voltage dependence similar to that of the wild type skeletal DHPR.
The production of large outward currents by the SkEIIIK and CEIIIK channels allowed us to compare activation rates over a much broader range of voltages than wild type channels, which can only be compared for test potentials that are both sufficiently positive to cause activation and sufficiently negative to provide a significant driving force for inward Ca2+ current. We found that the activation rate of both channels was very weakly voltage dependent and that the activation of SkEIIIK was >10-fold slower than that of CEIIIK, even at +100 mV.
Skeletal L-channels are unusual in that channel activation is both slow (τact > 50 ms; Dirksen and Beam 1995) and weakly voltage dependent, while channel deactivation is strongly voltage dependent. This behavior can be accounted for by a linear reaction scheme in which the rate-limiting transition has an asymmetric voltage dependence (Dirksen and Beam 1996). Because activation is slow, this asymmetric transition would generate only a very small “ON” gating current (Qon). However, because deactivation is fast at negative voltages, repolarization should produce a component of “OFF” gating current (Qoff) with a magnitude that is dependent on the extent of activation occurring during a preceding depolarization. An unambiguous test of this prediction is difficult for wild-type channels since any apparent increase in Qoff might simply be due to the presence of incompletely blocked ionic tail current. However, SkEIIIK, which did not permit inward Ca2+ current, provided a powerful tool for testing this prediction. A comparison of Qoff for SkEIIIK after strong depolarizations of 20 or 200 ms showed that the longer depolarizations recruited additional gating current, which increased Qoff by >20%. The size of the recruitable SkEIIIK Qoff and the similarity of its voltage dependence to that of channel conductance indicates that the majority of skeletal muscle DHPRs not only undergo the voltage-dependent conformational changes that trigger SR Ca2+ release, but also the voltage-dependent transitions that are required for L-channel opening.
methods
Preparation of Dysgenic Myotubes
Primary cultures of myotubes were prepared from skeletal muscle of newborn dysgenic mice, as described previously (Beam and Knudson 1988). All experiments were performed 7–11 d after the initial plating of myoblasts and were carried out at room temperature (20–22°C). Numerical figures are presented in the text and figures as mean ± SEM. Data were taken to be statistically significant at P < 0.001.
Preparation and Expression of cDNAs
A glutamate-to-lysine mutation in the repeat III pore region of the skeletal muscle dihydropyridine receptor was constructed using PCR. Two initial PCR fragments were produced in separate but parallel reactions. Each initial fragment was generated by amplifying pCAC6 (Tanabe et al. 1988) with an “outer” primer and a mutating primer. The final mutated fragment was obtained by mixing the two initial PCR products and amplification in a third reaction with the outer primers (Garcia et al. 1997). The composition of the two mutating primers was, sense: 5′-GTGTCCACCTTTAAAGGATGGCCCCAG-3′ and antisense: 5′-CTGGGGCCATCCTTTAAAGGTGGACAC-3′, where underlining indicates the mutated region. The composition of the outer primers was, sense (starting at base 2700): 5′-GTCCGTGAGGAATCAGATCCTTGG-3′ and antisense (starting at base 4000): 5′-TCCACAGCAGCGTGCGCACGCCCT-3′. This mutagenesis strategy not only substituted a lysine codon (AAA) for the glutamate codon (GAG) in the pore region of repeat III, but also exchanged phenylalanine codons (TTT replaced TTC) so as to introduce a novel DraI restriction site (TTTAAA) that was used to screen clones for the mutation. Each PCR reaction contained template DNA, 100 pmol of each primer, 100 μM of each deoxyribonuclease triphosphate (GIBCO-BRL), 2 U Vent DNA polymerase, 10 μl polymerase buffer (10×), and sterilized water sufficient to bring the final volume to 100 μl. 35 amplification cycles were performed with a PCR thermal cycler (Perkin-Elmer Corp.); cycle 1, 95°C for 4 min, 60°C for 2 min, and 72°C for 2 min; cycles 2–35, 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min. After the last cycle, an additional 5-min extension period at 72°C was used. This PCR mutagenesis strategy generated a 1.3-kB mutant fragment that was subsequently digested with XhoI and HincII to generate a 1.0-kB mutant fragment that was used to replace the corresponding fragment from the 2.7-kb XhoI–SacI fragment of pCAC6 subcloned into pBluescript (Stratagene Inc.). Finally, the 1.7-kB XhoI–DraIII fragment of the mutant subclone was used to replace the corresponding fragment from pCAC6 (Tanabe et al. 1987) to generate SkEIIIK. The entire 1.0-kB XhoI–HincII region of SkEIIIK was ultimately sequenced using an ABI 377 automatic sequencer to verify sequence integrity. The cDNA encoding CEIIIK was a generous gift form Dr. William Sather. Both SkEIIIK and CEIIIK use the mammalian expression vector pKCRH2 (Tanabe et al. 1988).
Approximately 1 wk after plating, myotubes were microinjected (Tanabe et al. 1988; Beam and Franzini-Armstrong 1997) into a single nucleus with expression plasmid for either CEIIIK or SkEIIIK (200–500 ng/μl). Myotubes expressing SkEIIIK were identified by contraction in response to extracellular electrical stimulation (80 V, 10–30 ms). Myotubes expressing CEIIIK never exhibited electrically evoked contractions and were usually identified by coinjecting 20 ng/μl of a cDNA expression plasmid (Grabner et al. 1998) encoding an enhanced green fluorescent protein (GFP). In control experiments, coexpression of the GFP cDNA did not alter the function of expressed L-channels (data not shown). Coinjection of GFP cDNA was omitted when cells were to be used for measurement of intracellular Ca2+ transients and the expression of CEIIIK was established on the basis of large, rapidly activating outward ionic currents.
Measurements of Ionic Currents
The whole cell variant of the patch clamp technique (Hamill et al. 1981) was used to measure ionic currents, intramembrane charge movements, and intracellular calcium transients. Pipettes were fabricated from borosilicate glass and had resistances of 1.8–2.2 MΩ when filled with the internal solution. Linear capacitative and leakage currents were determined by averaging the currents elicited by multiple (usually 10) 20-mV hyperpolarizing pulses from a holding potential of −80 mV. This control current was then scaled appropriately and used to correct test currents for linear components of capacitative and leakage currents. Electronic compensation was used to reduce the effective series resistance (usually to ∼1 MΩ) and the time constant for charging the linear cell capacitance to <0.5 ms. Cell capacitance was determined by integration of the capacity transient resulting from the control pulse and was used to normalize currents (pA/pF) or charge movements (nC/μF) obtained from different myotubes. Ionic currents were filtered at 2 kHz and digitized at 1 kHz. To measure macroscopic L-current in isolation, a 1-s prepulse to −30 mV followed by a 25-ms repolarization to −50 mV was administered before the test pulse (prepulse protocol) to inactivate T-type Ca2+ currents.
The activation phase of L-currents was fitted by the following exponential function:
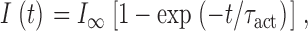 |
1 |
where I(t) is the current at time t after the depolarization, I ∞ is the steady state current, and τact is the time constant of activation. The voltage dependence of SkEIIIK L-channel activation was obtained by tail-current analysis. In brief, myotubes were stepped for 200 ms to test potentials (V) ranging from −50 to +80 mV; the instantaneous current (I T) was then measured immediately after stepping to +40 mV. These data were then normalized to I max (the maximal I T) and fitted according to:
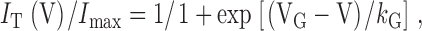 |
2 |
where VG is the potential causing half-maximal activation of L-type conductance and k G is a slope parameter.
Measurements of Gating Currents
For measurement of intramembrane charge movements, filtering was at 2 kHz (eight pole Bessel filter; Frequency Devices Inc.) and digitization was at 10 kHz. Voltage clamp command pulses were exponentially rounded with a time constant of 50–300 μs and the prepulse protocol (see above) was used to reduce the contribution of gating currents from sodium channels and T-type Ca2+ channels. “OFF” transients of charge movement were measured for repolarization to −50 mV after test pulses to various test potentials (−50 to +60 mV, in 10-mV increments). The rectification of the mutant L-channels meant that influx of Ca2+ ions was minimal for CEIIIK and nonexistent for SkEIIIK (see Fig. 1 and Fig. 5). However, since some dysgenic myotubes exhibit a small, endogenous, rapidly activating L-current, residual Ca2+ channel ionic currents were blocked by the addition of 2.0 mM CdCl2 + 0.2 mM LaCl3 to the extracellular recording solution (see Solutions). The integral of the OFF transient for each test potential (V) was normalized by the maximal value of Qoff (Qmax) and fitted according to:
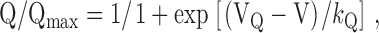 |
3 |
where VQ is the potential causing movement of half the maximal charge, and k Q is a slope parameter.
Figure 1.

A glutamate-to-lysine mutation in the pore region of repeat III of the skeletal (SkEIIIK) and cardiac (CEIIIK) DHPRs alters channel permeation, but not the rate of channel activation. (A) Representative whole-cell ionic currents obtained from dysgenic myotubes expressing either SkEIIIK (left) or CEIIIK (right). Currents were elicited by 200-ms depolarizations to the indicated potentials after a prepulse protocol (see methods) used to inactivate T-type Ca2+ currents (Adams et al. 1990). Inward currents were not observed at any potential for the SkEIIIK-expressing myotube and minimal inward currents were observed at a few potentials for the CEIIIK-expressing myotube. The dashed lines represent zero current levels. (B) Superimposed current–voltage relationships for the two experiments shown in A. Compared with their wild-type counterparts (Tanabe et al. 1988, 1990), the potential at which SkEIIIK and CEIIIK permit outward current flux is shifted by ∼60 mV in the hyperpolarized direction. (C) The difference between the rate of skeletal and cardiac L-current activation persists in the SkEIIIK and CEIIIK mutants (data from 11 SkEIIIK- and 7 CEIIIK-expressing myotubes). Note that the rate of skeletal L-current activation is nearly independent of voltage and remains slow even at +100 mV.
Figure 5.

Lack of inward ionic tail currents under the recording conditions used to measure Qoff. Representative data collected from a SkEIIIK-expressing dysgenic myotube obtained under both ionic (10 mM Ca2+, ○) and gating current recording conditions (8 mM Ca2+, 2.0 mM Cd2+, and 0.2 mM La3+, •). Cells were depolarized for 200 ms to +60 mV (to fully activate SkEIIIK channels) and subsequently repolarized to the potentials indicated on the abscissa. Currents during the repolarization period (20 ms) were subsequently integrated and plotted against the repolarization potential. Outward ionic currents through SkEIIIK (seen as positive integrals at potentials >0 mV) were significantly attenuated by Cd2+ and La3+. At potentials hyperpolarized to −20 mV (inset), negative integrals were not largely affected by either the Cd2+ and La3+ solution or by the increase in driving force, indicating a lack of inward ionic currents through SkEIIIK channels. Similar results were obtained from four separate experiments.
Measurements of Intracellular Ca2+ Transients
Changes in intracellular Ca2+ were recorded with Fluo-3, as described previously (Garcia et al. 1994; Garcia and Beam 1994). In brief, the salt form of the dye was added to the internal solution. After rupture of the cell membrane and entry into the whole-cell mode, a waiting period of >5 min was used to allow the dye to diffuse into the cell interior. A 75W xenon illuminator and a set of fluorescein filters were used to excite the dye present in a small rectangular region of the voltage-clamped myotube. A computer-controlled shutter was used to block illumination in the intervals between test pulses. Fluorescence emission was measured by means of a fluorometer apparatus (Biomedical Instrumentation Group, University of Pennsylvania, Philadelphia, PA). The background fluorescence was measured for each myotube before membrane rupture and cancelled by analogue subtraction. Fluorescence data are expressed as ΔF/F b, where ΔF represents the change in fluorescence from baseline (ΔF = F − F b) and F b represents the average myotube fluorescence immediately before depolarization.
Solutions
For measurements of macroscopic ionic and gating currents, the internal solution consisted of (mM): 140 Cs-aspartate, 10 Cs2-EGTA, 5 MgCl2, and 10 HEPES, pH 7.40 with CsOH. For the measurements of depolarization-induced Ca2+ transients (Garcia and Beam 1994), the patch pipettes contained an internal solution composed of (mM): 110 Cs-aspartate, 5 MgATP, 22.8 EGTA, 2.66 CaCl2, 1.2 MgCl2, 0.2 K5 Fluo-3 (Molecular Probes, Inc.), and 10 HEPES, pH 7.40 with CsOH. Ionic currents and intracellular Ca2+ transients were recorded in an external solution containing (mM): 145 TEA-Cl, 10 CaCl2, 0.003 tetrodotoxin (TTX), and 10 HEPES, pH 7.40 with tetraethylammonium (TEA)-OH. Gating currents were recorded in an external solution containing (mM): 145 TEA-Cl, 8 CaCl2, 2 CdCl2, 0.2 LaCl3, 0.003 TTX, and 10 HEPES, pH 7.40 with TEA-OH. The dihydropyridine antagonist, (+)-PN 200-110, was kindly provided by Drs. E. Rossi and A. Lindenmann of Sandoz Ltd. (Basel, Switzerland).
results
A Glutamate-to-Lysine Mutation in the Pore Region of Repeat III Alters L-Channel Permeability, but Not the Rate of Channel Activation
Fig. 1 shows ionic currents in dysgenic myotubes expressing SkEIIIK and CEIIIK, the skeletal and cardiac DHPRs, respectively, in which a highly conserved glutamate residue in the pore region of repeat III was mutated to a positively charged lysine residue. Under the recording conditions used, the wild-type skeletal and cardiac L-channels support inward Ca2+ currents for test potentials ranging from about −20 (cardiac) or 0 (skeletal) mV to +60 mV or greater, with the currents reversing to outward at an extrapolated potential of more than +70 mV (Tanabe et al. 1988, Tanabe et al. 1990b; Garcia and Beam 1994). As reported previously for expression in Xenopus oocytes (Yang et al. 1993), CEIIIK channels expressed in dysgenic myotubes had dramatically altered divalent permeability and mediated inward Ca2+ currents for only a few potentials (−20 to +10 mV; Fig. 1 A, right). At potentials greater than +20 mV, dysgenic myotubes expressing CEIIIK exhibited rapidly activating outward currents (presumably carried by Cs+, the predominant intracellular cation). Dysgenic myotubes expressing SkEIIIK L-channels did not exhibit inward currents at any potential, but did give rise to large, slowly activating outward currents at all potentials greater than +20 mV (Fig. 1 A, left). The outward currents for both SkEIIIK and CEIIIK were blocked by a DHP antagonist, confirming that they were produced by expressed L-channels (at +100 mV, 1 μM PN 200-110 reduced the SkEIIIK current by 90.0 ± 3.6%, n = 3, and CEIIIK current by 93.5%, n = 2).
Fig. 1 B illustrates peak current–voltage relationships determined from the cells illustrated in Fig. 1 A. Compared with wild-type cardiac channels, the reversal potential for CEIIIK was negatively shifted by ∼60 mV, indicative of a large change in the permeability of Ca2+ relative to Cs+. The negative shift in reversal potential appeared to have been still larger for SkEIIIK since inward Ca2+ currents were not elicited at any test potential, even though depolarizations >10 mV caused significant activation of SkEIIIK channels (see Fig. 2 B). Furthermore, when outward currents via SkEIIIK were elicited by strong depolarizations, subsequent repolarization to negative potentials failed to cause inward ionic tail currents (see below). Thus, the conserved glutamate in the pore region of repeat III appears to play a role in controlling Ca2+ permeation through the skeletal L-channel, which is even more pivotal than its role in the cardiac L-channel.
Figure 2.

Skeletal-type EC coupling is unaffected, and cardiac-type EC coupling is abolished, by the repeat III glutamate-to-lysine mutations in the skeletal and cardiac DHPRs, respectively. (A) Simultaneously recorded intracellular Ca2+ transients (top) and ionic currents (bottom) elicited by test pulses to the indicated potentials. The dysgenic myotube expressing SkEIIIK (left) exhibited depolarization-induced intracellular Ca2+ transients at potentials greater than −10 mV, whereas the CEIIIK-expressing myotube (right) failed to exhibit Ca2+ transients at any potential. The dashed lines represent basal fluorescence (top) and zero current (bottom) levels (B) Comparison of the voltage dependence of the normalized peak intracellular Ca2+ transient (•, n = 5) and peak conductance (▴, n = 16) obtained from dysgenic myotubes expressing SkEIIIK L-channels. The smooth solid curves through the data were obtained by Boltzmann fitting of the averaged data points. These fits yielded the following values: F V; V0.5 = +6.9 mV and k = 10.7 mV and GV; V0.5 = +21.8 mV and k = 10.5.
Like wild-type skeletal L-channels, the SkEIIIK channels activated slowly. At test potentials where both produce appreciable currents (e.g., +60 mV), the SkEIIIK and native skeletal L-channels both display slow activation (native: 52.4 ± 2.0 ms, n = 6; SkEIIIK: 40.8 ± 1.7 ms, n = 11). Thus, the identity of the permeating ion has little effect on activation and the EIIIK channels provide a convenient tool for extending the comparison of skeletal and cardiac activation to more positive potentials (Fig. 1 C). Even at +100 mV, activation of SkEIIIK is more than 10-fold slower than that of CEIIIK (SkEIIIK: 30.7 ± 1.9 ms, n = 11; CEIIIK: 2.9 ± 0.5 ms, n = 7). Because activation of the skeletal L-channel is slow over a broad range of test potentials (+20 to +100 mV), the rate-limiting event for the transition of the channel from closed to open must be very weakly voltage dependent, as we have suggested previously from single channel analyses (Dirksen and Beam 1996).
L-Channel Permeability to Ca2+ Is Required for Cardiac-type, but Not Skeletal-type, EC Coupling
Controversy remains as to whether Ca2+ permeation and/or intrapore Ca2+ binding are necessary for the ability of the cardiac and skeletal L-channels to mediate EC coupling. We reexamined this issue with the CEIIIK and SkEIIIK L-channels since they have a profoundly altered intrapore binding site for Ca2+. In initial experiments, contractions were observed in response to extracellular electrical stimulation (80 V, 10–30 ms) of dysgenic myotubes injected with cDNA for SkEIIIK but not in myotubes injected with CEIIIK cDNA. Thus, the substantial loss of Ca2+ permeation caused by the EIIIK mutation appeared to have eliminated the ability of the cardiac L-channel, but not the skeletal L-channel, to mediate EC coupling. However, to determine whether CEIIIK supports Ca2+ release too small to be detected by contractions, we also used whole-cell voltage clamping to measure membrane currents and intracellular Ca2+ transients with patch pipettes containing the pentapotassium salt of Fluo-3 (Garcia et al. 1994; Garcia and Beam 1994).
Fig. 2 A illustrates Ca2+ transients and membrane currents in dysgenic myotubes expressing either SkEIIIK (left) or CEIIIK (right). Large Ca2+ transients were present in the myotube expressing SkEIIIK (left), but not in the one expressing CEIIIK, despite the presence of large ionic currents (right). Similar results were obtained in five myotubes expressing SkEIIIK, all of which displayed depolarization-evoked Ca2+ transients, and five myotubes expressing CEIIIK, none of which displayed Ca2+ transients. These data lend strong support to the notion that Ca2+ permeation through skeletal muscle DHPRs is not required to trigger the release of Ca2+ from the SR (Garcia et al. 1989; Garcia and Beam 1994). The lack of SR Ca2+ release in CEIIIK-expressing myotubes strongly argues against a Ca2+-independent interaction between the cardiac DHPR and the skeletal SR Ca2+ release channel.
Fig. 2 B plots both normalized conductance (G) and the amplitude of the Ca2+ transient (ΔF/F) as a function of test potential for SkEIIIK-expressing myotubes. As in myotubes expressing the wild-type skeletal DHPR (Garcia and Beam 1994), the conductance of SkEIIIK L-channels is positively shifted by ∼15 mV compared with that of Ca2+ release. Moreover, the average voltage dependencies (i.e., averages of Boltzmann fits obtained from multiple experiments) of G (VG = 21.6 ± 2.3 mV, k G = 8.8 ± 0.4 mV, n = 16) and of ΔF/F (VF = 6.5 ± 3.2 mV, k F = 9.3 ± 0.7 mV, n = 5) for SkEIIIK L-channels are also similar to the values reported previously (Garcia et al. 1994) for dysgenic myotubes expressing the wild-type skeletal DHPR (VG = 24.0 mV, k G = 8.2 mV; VF = 6.6 mV, k F = 9.0 mV). The qualitative and quantitative similarity of Ca2+ release caused by wild-type and SkEIIIK L-channels indicates that neither Ca2+ permeation nor high affinity binding of Ca2+ within the L-channel pore play an important role in skeletal-type EC coupling.
An Increase in Qoff Correlates with Slow Activation of Skeletal L-Current
In comparison with other voltage-gated Ca2+ channels, skeletal L-channels have extremely slow (and relatively voltage-independent) activation kinetics. Despite this slow activation, repolarization to negative potentials causes rapid deactivation, a behavior that can be explained by a single transition, with an asymmetric voltage dependence causing it to be slow in the opening direction and fast in the closing direction (Dirksen and Beam 1996). Accordingly, such a transition would produce only a small Qon but a significant Qoff gating current. We tested this prediction by comparing Qoff gating currents for SkEIIIK and CEIIIK.
Fig. 3 illustrates representative Qoff gating currents generated upon repolarization to −50 mV for dysgenic myotubes expressing either SkEIIIK (left) or CEIIIK (right). Data were obtained in the presence of 2.0 mM Cd2+ and 0.2 mM La3+ (which was required to block the small inward currents supported by CEIIIK). Qoff is shown after either brief (20 ms, light traces) or long (200 ms, dark traces) depolarizing pulses to the indicated potentials. These two pulse durations were used because 20 ms is long enough to open cardiac L-channels fully and short enough to only marginally open skeletal L-channels, whereas 200 ms is sufficiently long to open both cardiac and skeletal L-channels. At potentials less than +10 mV, Qoff was independent of the test pulse duration for both SkEIIIK and CEIIIK. However, at potentials that typically activate ionic current (greater than or equal to +10 mV), Qoff was significantly larger only for SkEIIIK L-channels after the 200-ms test pulses. The additional Qoff for SkEIIIK L-channels was manifested as a prolongation of the total gating current rather than simply as an increase in peak of the “OFF” gating current. Thus, the increase in SkEIIIK Qoff caused by longer depolarizations is inconsistent with a contamination from an ionic tail current and presumably arises from an additional amount of gating current recruited during slow channel activation. In contrast to SkEIIIK, the magnitude of Qoff for CEIIIK was not increased by the longer test pulse duration. Rather, a slight decrease in Qoff was found for CEIIIK after the largest 200-ms depolarizations (to potentials greater than +30 mV), most likely owing to a small degree of inactivation during the 200-ms pulses.
Figure 3.

Long depolarizations recruit additional OFF gating current for dysgenic myotubes expressing SkEIIIK, but not CEIIIK. Representative Qoff recordings obtained from dysgenic myotubes expressing either SkEIIIK (left) or CEIIIK (right). Data were obtained in the presence of 2.0 mM Cd2+ and 0.2 mM La3+. Expression of constructs was confirmed in these experiments by the presence of large outward ionic currents (>40 pA/pF at +100 mV) under control conditions (i.e., in the absence of Cd2+/La3+). Qoff gating currents were elicited by repolarization to −50 mV after either brief (20 ms, light traces) or long (200 ms, dark traces) depolarizing pulses to the indicated potentials. The 20-ms pulse was sufficient to fully activate current through CEIIIK channels, but too brief to significantly activate current through SkEIIIK L-channels (see Fig. 1). The 200-ms pulse fully activated both SkEIIIK and CEIIIK channels. The dashed lines represent zero current levels. A significant increase in Qoff was only observed at potentials >0 mV for dysgenic myotubes expressing the SkEIIIK L-channel. A prepulse protocol (see methods) preceded all test depolarizations in order to immobilize gating currents due to sodium and T-type Ca2+ channels.
Fig. 4 illustrates the voltage dependence of Qoff for SkEIIIK (A) and CEIIIK (B) after 20-ms test pulses (Q20; •) and 200-ms test pules (Q200; ▪), and the difference between the two (Q200–Q20; ▴). For potentials >0 mV, Qoff SkEIIIK was larger after 200-ms test pulses than after 20-ms test pulses. For CEIIIK, no such increase in Qoff occurred after longer duration pulses. The normalized voltage dependence of Q20, Q200, Q200–Q20, and channel conductance for the SkEIIIK-expressing myotube shown in Fig. 3 are compared in Fig. 4 C. As reported previously for Qon (Adams et al. 1990; Garcia et al. 1994; Dirksen and Beam 1995), the voltage dependence of Qoff after brief depolarizations (Q20) was leftward shifted and shallower than that of channel conductance. However, the additional Qoff recruited by long depolarizations of SkEIIIK L-channels (Q200–Q20; ▴) exhibited a voltage dependence nearly identical to that of channel conductance (○). In a total of four experiments, good agreement was found between the voltage dependence of conductance (VG = 24.2 ± 6.3 mV and k G = 7.5 ± 1.0 mV) and that of Q200–Q20 (VQ = 31.4 ± 6.7 mV and k Q = 7.5 ± 0.7 mV). Fig. 4 D summarizes the results on maximal Qoff obtained from multiple experiments. On average, the maximal value of Qoff for SkEIIIK after 200-ms test pulses was significantly greater (121.4 ± 2.0%, n = 6, P < 0.001) than that after 20-ms test pulses. For CEIIIK, Qoff after 200 ms was not significantly different from that after 20 ms (P > 0.3). Additionally, for both SkEIIIK and CEIIIK, Qoff after 20 ms had essentially the same magnitude as Qon (data not shown). Thus, the voltage and time dependence of the Q200–Q20 gating charge, coupled with its presence only in the SkEIIIK channels, suggests that this extra gating charge underlies the rate-limiting transition of slow skeletal channel activation.
Figure 4.

Long depolarizations increase Qoff in dysgenic expressing SkEIIIK, and not CEIIIK, with a voltage dependence that mirrors that of channel conductance. (A) Dependence of the absolute magnitude of Qoff for SkEIIIK on voltage during test pulses of 20- (Q20, •) or 200-ms (Q200, ▪) duration. The voltage dependence of the additional Qoff gating current recruited by the 200-ms depolarization pulses was determined by subtracting the value of Q20 from the value of Q200 for each voltage (Q200–Q20, ▪). (B) Dependence of the absolute magnitude of Qoff for CEIIIK on test potential of either 20- (Q20, •) or 200-ms (Q200, ▪) duration, together with the voltage dependence of Q200–Q20 (▪). Data in A and B are taken from the same myotubes as those shown in Fig. 3. (C) Comparisons of the voltage dependence of the normalized values for Q20, Q200, Q200–Q20, and channel conductance (G) for SkEIIIK-expressing myotubes. Each data set was fitted by a Boltzmann distribution which yielded values of Q20, V0.5 = −20.3 mV and k = 11.6 mV; Q200: V0.5 = −16.0 mV and k = 13.4 mV; Q200–Q20: V0.5 = +13.0 mV and k = 8.0 mV; and G: V0.5 = +10.6 mV and k = 8.5 mV. Similar results were obtained from a total of six different SkEIIIK-expressing myotubes. (D) Effect of depolarization duration on maximal Qoff recorded from dysgenic myotubes expressing either SkEIIIK (left, n = 6) or CEIIIK (right, n = 3). A significant (P < 0.001) increase in maximal Qoff was observed only for dysgenic myotubes expressing SkEIIIK.
Since contamination by inward ionic tail current would artifactually increase Qoff, it was important to ensure that no such contamination was present under our recording conditions. As one approach, we attempted to reduce ionic currents by adding Cd2+ and La3+ to the external bath. At a test potential of +60 mV, these blockers reduced the outward current by 75.3 ± 2.6% (n = 5) for SkEIIIK and 82.0 ± 2.7 (n = 5) for CEIIIK. To determine the effectiveness of this block at more negative potentials, cells were depolarized to +60 mV for 200 ms to cause maximal activation of channels and then either maintained at +60 mV or repolarized to varying potentials. As shown for a SkEIIIK-expressing cell in Fig. 5, the addition of Cd2+ and La3+ caused a large reduction at +60 mV, where the integrated current was entirely ionic, but only a small reduction at more negative potentials. The small effect of the blockers at negative potentials strengthens the conclusion that SkEIIIK has a very low permeability to Ca2+. Moreover, it seems likely that the block of this already small Ca2+ permeability by Cd2+ and La3+ should have been sufficient to eliminate any ionic contamination of the Qoff charge.
discussion
Mutation of Glutamate to Lysine in the Pore Region of Repeat III Eliminates Ca2+ Permeation through the Skeletal L-Type Ca2+ Channel
In this paper, we have characterized the effects of mutational alteration of Ca2+ permeability on the behavior of skeletal and cardiac L-channels expressed in dysgenic myotubes. Because there had been no previous descriptions of mutations affecting Ca2+ permeability of skeletal L-channels, we chose to substitute lysine for glutamate in the repeat III pore region (EIIIK) since the corresponding mutation had already been shown to produce a dramatic alteration in the divalent selectivity of the cardiac L-channel expressed in Xenopus oocytes (Yang et al. 1993). Similarly, depolarization of dysgenic myotubes expressing the CEIIIK channel elicited inward Ca2+ currents only for a few test potentials (<20 mV), while stronger depolarizations elicited outward currents. Thus, introduction of the EIIIK mutation into the cardiac L-channel caused a large (>60 mV) hyperpolarizing shift in reversal potential compared with wild-type cardiac L-channels under the same recording conditions (Tanabe et al. 1990b; Garcia et al. 1994). Unlike the CEIIIK channel, which retained a small Ca2+ permeability, the SkEIIIK channel completely lost permeability to Ca2+ as judged by the absence of inward ionic currents either during depolarizing steps (Fig. 1) or after repolarization to negative potentials (Fig. 2 and Fig. 5). The greater effect of the EIIIK mutation on the selectivity of skeletal L-channels suggests functional differences between the geometric arrangement of the conserved pore region glutamates in skeletal and cardiac L-channels. To test this hypothesis, it will be necessary to carry out a more systematic analysis of mutations of the glutamates in each of the four repeats of the skeletal DHPR.
Reconstitution of EC Coupling in Dysgenic Myotubes: Ca2+ Permeation and Skeletal and Cardiac EC Coupling
Previous studies have used expression in dysgenic myotubes of skeletal, cardiac, and chimeric DHPRs to dissect mechanisms of EC coupling. With this approach, it was shown that substitution of skeletal sequence for all (Tanabe et al. 1990a) or part (Nakai et al. 1999) of the II–III loop of an otherwise entirely cardiac DHPR was sufficient to convert the reconstituted EC coupling from cardiac- to skeletal-type. Subsequently, a number of laboratories have tested whether peptides derived from the II–III loop can activate reconstituted RyRs and/or Ca2+ release from SR vesicles. This approach has revealed that either the entire cardiac II–III loop (Lu et al. 1994) or peptides corresponding to part of the loop (El-Hayek and Ikemoto 1998) are capable of causing activation of RyRs in these reduced systems. Furthermore, it has been reported that a skeletal-type EC coupling mechanism independent of Ca2+ influx can contribute to SR Ca2+ release in cardiac muscle cells, particularly when intracellular cAMP is elevated (Hobai et al. 1997; Howlett et al. 1998). Thus, it is important to be certain that the EC coupling mechanism truly differs when cardiac and skeletal DHPRs are expressed in dysgenic myotubes. This is particularly important since earlier assessments of cardiac-type coupling in injected dysgenic myotubes relied, in part, on the loss of Ca2+ release after the addition of Cd2+ to the external medium. However, in addition to blocking entry of external Ca2+, any Cd2+ entering the cells could have interfered with the measurement of Ca2+ by quenching the Ca2+-sensitive dye and/or have directly blocked RyRs (Xu et al. 1999). We have now shown that CEIIIK in dysgenic myotubes is unable to trigger the release of SR Ca2+, even in the absence of inorganic Ca2+ channel blockers. Specifically, myotubes with robust CEIIIK expression never contracted in response to strong electrical stimuli and never exhibited depolarization-induced intracellular Ca2+ transients. Thus, large Ca2+ flux through cardiac L-channels appears to be required for reconstitution of cardiac-type SR Ca2+ release in dysgenic myotubes. Moreover, the distinct behaviors of CEIIIK and SkEIIIK in triggering the release of SR Ca2+ support the use of skeletal/cardiac DHPR chimeras for identifying regions important for skeletal-type EC coupling. However, since the RyR isoform in dysgenic myotubes (i.e., RyR1) differs from that of cardiac muscle (RyR2), our results do not rule out the possibility that under some conditions a skeletal-type mechanism of EC coupling may contribute to SR Ca2+ release in native cardiac myocytes.
Occupancy of a metal cation binding site, which is accessible from the extracellular space, has been reported to be required for the conformational changes of the skeletal L-channel that result in the activation of RyR1s (Pizzaro et al. 1989). This site has a binding selectivity and a micromolar Ca2+ affinity similar to that of the site governing ion permeability of L-channels (Hess and Tsien 1984; Almers and McCleskey 1984; Hess et al. 1986; Dang and McCleskey 1998), suggesting that this cation-binding site corresponds to the structures governing Ca2+ selectivity of the skeletal L-channel. Our data demonstrate that SkEIIIK retains the ability of the wild-type channel to control the release of SR Ca2+ in spite of a complete loss of Ca2+ permeability. In fact, the voltage dependence of SR Ca2+ release in dysgenic myotubes expressing SkEIIIK L-channels was nearly identical to that of the wild-type skeletal DHPR (Garcia et al. 1994). Thus, the capability of the skeletal L-channel to mediate EC coupling does not depend on Ca2+ permeation. Despite the lack of Ca2+ permeation, it is possible that a low affinity site in the pore of SkEIIIK is appreciably occupied by Ca2+ under our experimental conditions (10 mM external Ca2+). Alternatively, the presence of large outward Cs+ currents, which should have been blocked by binding of Ca2+, may indicate that there is very little residual binding of Ca2+ within the SkEIIIK pore. If the latter is indeed the case, it would imply that the metal cation site described by Pizzaro et al. 1989 does not lie within the pore. However, a direct evaluation of the contribution of the pore to the metal binding site involved in EC coupling will require measurement of SkEIIIK monovalent ionic currents, intramembrane charge movements, and SR Ca2+ release over a range of extracellular Ca2+ concentrations.
The results with SkEIIIK raise the obvious question of what function is played by the slowly activating L-type Ca2+ current in skeletal muscle. Clearly, this current plays little or no role in contractions elicited by relatively brief depolarizations. Perhaps the Ca2+ current becomes important for contraction only during tetanic stimulation (Sculptoreanu et al. 1993), or has other functions such as the regulation of acetylcholine receptor gene expression (Shainberg et al. 1976; Klarsfeld and Changeux 1985) or the metabolic stabilization of acetylcholine receptors (Rotzler et al. 1991). One approach for identifying these or other roles of the slow L-type Ca2+ current would be to construct mice that carry a transgene for the SkEIIIK channel and lack the wild-type skeletal L-channel.
Gating Charge Associated with Slow Activation of Skeletal L-Current
As in the case for inward Ca2+ currents via wild-type skeletal L-channels, outward currents carried by SkEIIIK L-channels also activate slowly. Moreover, because they are slow over a broad range of test potentials, the SkEIIIK L-currents extend previous conclusions that activation of skeletal L-channels is only very weakly voltage dependent (Fig. 1 C). Nonetheless, steady state activation is strongly voltage dependent for wild-type (Garcia et al. 1994) and SkEIIIK channels (Fig. 2 B), with Boltzmann fits of conductance–voltage (G–V) relationships yielding a value of k ≈ 8 mV, which corresponds to an effective valence of approximately three electronic charges. This effective valence represents ∼25% of the total gating charge expected for voltage-gated calcium channels (Noceti et al. 1996). However, the G–V relationship for wild-type skeletal DHPRs expressed in dysgenic myotubes is rightward shifted ∼30 mV with respect to the Qon–V relationship (Adams et al. 1990). Thus, nearly all of the measured gating current moves at voltages more negative than those over which the conductance increase occurs. One possibility consistent with this result is that there may be two separate populations of DHPRs in skeletal muscle: one population related to EC coupling that produces nearly all of the measured charge movement and a second population that functions as Ca2+ channels and produces negligible gating current. An alternative possibility is that all of the DHPRs may undergo gating transitions that lead to the activation of L-channels, but that one of these transitions moves appreciable charge (e.g., approximately three electronic charges) too slowly to be detected during brief depolarizations (as is typically used in measurements of Qon).
A simple linear kinetic scheme consistent with the second possibility above, as well as with measurements of unitary currents through skeletal L-channels (Dirksen and Beam 1996) is shown in Fig. 1.
Scheme S1.

In this model, rapid gating currents are produced by closed–closed transitions, which for simplicity are indicated by the single C0–C1 transition. These gating transitions could be important for triggering release of Ca2+, since Ca2+ release has a voltage dependence much closer to that of Qon than does L-channel conductance (Garcia et al. 1994). In the model, the rate constant β must be large because single channel open times are brief (∼1 ms; Dirksen and Beam 1996), and slow activation of L-type conductance is a consequence of the C1–C2 transition, with forward and reverse rate constants δ and ∈, respectively. The rate constant δ must be small and not strongly voltage dependent because activation remains slow for SkEIIIK even at +100 mV. By contrast, the rate constant ∈ must be strongly voltage dependent such that it is small at the depolarized potentials causing activation of current and large at negative voltages (e.g., −50 mV) causing deactivation. This behavior, which suggests that the energy barrier separating C1 and C2 is asymmetrically located with respect to the transmembrane potential drop, is required because activation is slow, whereas deactivation is rapid (Dirksen and Beam 1996). An explicit prediction of this reaction scheme is that the component of gating charge that is associated with the C1–C2 transition would move slowly outward during depolarization and rapidly inward upon repolarization. The rectification of SkEIIIK under our experimental conditions (TEA+ and Ca2+ as external cations) allowed a clear test of this prediction and revealed that additional Qoff is recruited by depolarizations that are sufficient to activate ionic current. The voltage dependence of this additional charge is nearly identical to that of channel conductance, consistent with the hypothesis that this charge moves during a transition required for channel activation.
Based on chimeras of the skeletal and cardiac L-channels, it was shown that the sequence of IS3 and the IS3–IS4 linker determines whether activation is fast or slow (Nakai et al. 1994), perhaps by governing the rate of movement of the nearby voltage-sensing IS4 segment. Thus, it is tempting to speculate that repeat I produces the additional Qoff that we found to be recruited by long depolarizations. If each of the four repeats contributed equivalent gating charge, then the long depolarizations should have caused a 33% (1/4 ÷ 3/4) increase in Qoff, somewhat larger than the observed value of ∼20%. However, it seems likely that our measurement of the difference in Qoff after 20- and 200-ms depolarizations underestimated the gating charge associated closely with channel activation, because 20-ms depolarizations cause some activation (see Fig. 1 A and 2 A). In any case, the size of the recruitable Qoff suggests that all DHPRs undergo the conformational change required for activation of L-type conductance. Neutralization of basic residues in the S4 segments (Garcia et al. 1997) would provide one test for the idea that this recruitable charge represents movements within IS4.
Acknowledgments
We thank Dr. William Sather for providing us with the cDNA for CEIIIK. We also thank Robin Morris, Kim Lopez-Jones, and Katherine Parsons for expert technical assistance.
This study was supported by the Muscular Dystrophy Association (to R.T. Dirksen) and National Institutes of Health grants NS-24444, AR-44750 (to K.G. Beam), and AR44657 (to R.T. Dirksen).
Footnotes
1used in this paper: DHPR, dihydropyridine receptor; EC, excitation–contraction; L-channel, L-type Ca2+ channel; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; TEA, tetraethylammonium
Portions of this work have been previously published in abstract form (Dirksen, R.T., and K.G. Beam. 1996. Biophys. J. 70:A146).
References
- Adams B.A., Tanabe T., Mikami A., Numa S., Beam K.G. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 1990;346:569–572. doi: 10.1038/346569a0. [DOI] [PubMed] [Google Scholar]
- Almers W., McCleskey E.W. Non-selective conductance in calcium channels of frog musclecalcium selectivity in a single-file pore. J. Physiol. 1984;353:585–608. doi: 10.1113/jphysiol.1984.sp015352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C.M., Bezanilla F.M., Horowicz P. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N′-tetraacetic acid. Biochim. Biophys. Acta. 1972;267:605–608. doi: 10.1016/0005-2728(72)90194-6. [DOI] [PubMed] [Google Scholar]
- Beam K.G., Franzini-Armstrong C. Functional and structural approaches to the study of excitation–contraction coupling. Methods Cell Biol. 1997;52:283–306. doi: 10.1016/s0091-679x(08)60384-2. [DOI] [PubMed] [Google Scholar]
- Beam K.G., Knudson C.M. Calcium currents in embryonic and neonatal mammalian skeletal muscle. J. Gen. Physiol. 1988;91:781–798. doi: 10.1085/jgp.91.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler W.K., Rakowski R.F., Schneider M.F. Effects of glycerol treatment and maintained depolarization on charge movement in skeletal muscle. J. Physiol. 1976;254:285–316. doi: 10.1113/jphysiol.1976.sp011233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang T.X., McCleskey E.W. Ion channel selectivity through stepwise changes in binding affinity. J. Gen. Physiol. 1998;111:185–193. doi: 10.1085/jgp.111.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen R.T., Beam K.G. Single calcium channel behavior in native skeletal muscle. J. Gen. Physiol. 1995;105:227–247. doi: 10.1085/jgp.105.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen R.T., Beam K.G. Unitary behavior of skeletal, cardiac, and chimeric L-type Ca2+ channels expressed in dysgenic myotubes. J. Gen. Physiol. 1996;107:731–742. doi: 10.1085/jgp.107.6.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hayek R., Ikemoto N. Identification of the minimum essential region in the II–III loop of the dihydropyridine receptor alpha 1 subunit required for activation of skeletal muscle-type excitation–contraction coupling. Biochemistry. 1998;37:7015–7020. doi: 10.1021/bi972907o. [DOI] [PubMed] [Google Scholar]
- Garcia J., Amador M., Stefani E. Relationship between myoplasmic calcium transients and calcium currents in frog skeletal muscle. J. Gen. Physiol. 1989;94:973–986. doi: 10.1085/jgp.94.6.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J., Beam K.G. Measurement of calcium transients and slow calcium current in myotubes. J. Gen. Physiol. 1994;103:107–123. doi: 10.1085/jgp.103.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J., Nakai J., Imoto K., Beam K.G. Role of S4 segments and the leucine heptad motif in the activation of an L-type calcium channel. Biophys. J. 1997;72:2515–2523. doi: 10.1016/S0006-3495(97)78896-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J., Tanabe T., Beam K.G. Relationship of calcium transients to calcium currents and charge movements in myotubes expressing skeletal and cardiac dihydropyridine receptors. J. Gen. Physiol. 1994;103:125–147. doi: 10.1085/jgp.103.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Serratos H., Valle-Aguilera R., Lathrop D.A., Garcia M.C. Slow inward calcium currents have no obvious role in muscle excitation–contraction coupling. Nature. 1982;298:292–294. doi: 10.1038/298292a0. [DOI] [PubMed] [Google Scholar]
- Grabner M., Dirksen R.T, Beam K.G. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non–L-type Ca2+ channels expressed in dysgenic myotubes. Proc. Natl. Acad. Sci. USA. 1998;95:1903–1908. doi: 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hess P., Lansman J.B., Tsien R.W. Calcium channel selectivity for divalent and monovalent cations. Voltage and concentration dependence of single channel current in ventricular heart cells. J. Gen. Physiol. 1986;88:293–319. doi: 10.1085/jgp.88.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess P., Tsien R.W. Mechanism of ion permeation through calcium channels. Nature. 1984;309:453–456. doi: 10.1038/309453a0. [DOI] [PubMed] [Google Scholar]
- Hobai I.A., Howarth F.C., Pabbathi V.K., Dalton G.R., Hancox J.C., Zhu J.Q., Howlett S.E., Ferrier G.R., Levi A.J. “Voltage-activated Ca release” in rabbit, rat and guinea-pig cardiac myocytes, and modulation by internal cAMP. Pflügers Arch. 1997;435:164–173. doi: 10.1007/s004240050496. [DOI] [PubMed] [Google Scholar]
- Howlett S.E., Zhu J.Q., Ferrier G.R. Contribution of a voltage-sensitive calcium release mechanism to contraction in cardiac ventricular myocytes. Am. J. Physiol. 1998;274:H155–H170. doi: 10.1152/ajpheart.1998.274.1.H155. [DOI] [PubMed] [Google Scholar]
- Kim M.S., Morii T., Sun L.X., Imoto K., Mori Y. Structural determinants of ion selectivity in brain calcium channel. FEBS Lett. 1993;318:145–148. doi: 10.1016/0014-5793(93)80009-j. [DOI] [PubMed] [Google Scholar]
- Klarsfeld A., Changeux J.P. Activity regulates the levels of acetylcholine receptor alpha-subunit mRNA in cultured chicken myotubes. Proc. Natl. Acad. Sci. USA. 1985;82:4558–4562. doi: 10.1073/pnas.82.13.4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Xu L., Meissner G. Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. J. Biol. Chem. 1994;269:6511–6516. [PubMed] [Google Scholar]
- Nabauer M., Callewaert G., Cleemann L., Morad M. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. 1989;244:800–803. doi: 10.1126/science.2543067. [DOI] [PubMed] [Google Scholar]
- Nakai J., Adams B.A., Imoto K., Beam K.G. Critical roles of the S3 segment and S3–S4 linker of repeat I in activation of L-type calcium channels. Proc. Natl. Acad. Sci. USA. 1994;91:1014–1018. doi: 10.1073/pnas.91.3.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J., Tanabe T., Konno T., Adams B., Beam K.G. Localization in the II–III loop of the dihydropyridine receptor of a sequence critical for excitation–contraction coupling. J. Biol. Chem. 1999;273:24983–24986. doi: 10.1074/jbc.273.39.24983. [DOI] [PubMed] [Google Scholar]
- Noceti F., Baldelli P., Wei X., Qin N., Toro L., Birnbaumer L., Stefani E. Effective gating charges per channel in voltage-dependent K+ and Ca2+ channels. J. Gen. Physiol. 1996;108:143–155. doi: 10.1085/jgp.108.3.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzaro G., Fitts R., Uribe I., Ríos E. The voltage sensor of excitation–contraction couplingion dependence and selectivity. J. Gen. Physiol. 1989;94:405–428. doi: 10.1085/jgp.94.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos E., Brum G. Involvement of dihydropyridine receptors in excitation–contraction coupling in skeletal muscle. Nature. 1987;325:717–720. doi: 10.1038/325717a0. [DOI] [PubMed] [Google Scholar]
- Rotzler S., Schramek H., Brenner H.R. Metabolic stabilization of endplate acetylcholine receptors regulated by Ca2+ influx associated with muscle activity. Nature. 1991;349:337–339. doi: 10.1038/349337a0. [DOI] [PubMed] [Google Scholar]
- Santana L.F., Gomez A.M., Lederer W.J. Ca2+ flux through promiscuous cardiac Na+ channelsslip-mode conductance. Science. 1998;279:1027–1033. doi: 10.1126/science.279.5353.1027. [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A., Scheuer T., Catterall W.A. Voltage-dependent potentiation of L-type Ca2+ channels due to phosphorylation by cAMP-dependent protein kinase. Nature. 1993;364:240–243. doi: 10.1038/364240a0. [DOI] [PubMed] [Google Scholar]
- Shainberg A., Cohen S.A., Nelson P.G. Induction of acetylcholine receptors in muscle cultures. Pflügers Arch. 1976;361:255–261. doi: 10.1007/BF00587290. [DOI] [PubMed] [Google Scholar]
- Tanabe T., Adams B.A., Numa S., Beam K.G. Repeat I of the dihydropyridine receptor is critical in determining calcium channel activation kinetics. Nature. 1991;352:800–803. doi: 10.1038/352800a0. [DOI] [PubMed] [Google Scholar]
- Tanabe T., Beam K.G., Adams B.A., Niidome T., Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation–contraction coupling Nature. 346 1990. 567 569a [DOI] [PubMed] [Google Scholar]
- Tanabe T., Beam K.G., Powell J.A., Numa S. Restoration of excitation–contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- Tanabe T., Mikami A., Numa S., Beam K.G. Cardiac-type excitation–contraction coupling in dysgenic skeletal muscle injected with cardiac dihydropyridine receptor cDNA Nature. 344 1990. 451 453b [DOI] [PubMed] [Google Scholar]
- Tanabe T., Takeshima H., Mikami A., Flockerzi V., Takahashi H., Kangawa K., Kojima M., Matsuo H., Hirose T., Numa S. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature. 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- Xu L., Pasek D.A., Meissner G. Zn2+ and Cd2+ inactivate the cardiac and skeletal muscle Ca2+ release channels. Biophys. J. 1999;76:A303. [Google Scholar]
- Yang J., Ellinor P.T., Sather W.A., Zhang J.F., Tsien R.W. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature. 1993;366:158–161. doi: 10.1038/366158a0. [DOI] [PubMed] [Google Scholar]