

Abstract
The objective of this study was to investigate the effects of insulin and insulin-like growth factor I on transepithelial Na+ transport across porcine glandular endometrial epithelial cells grown in primary culture. Insulin and insulin-like growth factor I acutely stimulated Na+ transport two- to threefold by increasing Na+-K+ ATPase transport activity and basolateral membrane K+ conductance without increasing the apical membrane amiloride-sensitive Na+ conductance. Long-term exposure to insulin for 4 d resulted in enhanced Na+ absorption with a further increase in Na+-K+ ATPase transport activity and an increase in apical membrane amiloride-sensitive Na+ conductance. The effect of insulin on the Na+-K+ ATPase was the result of an increase in Vmax for extracellular K+ and intracellular Na+, and an increase in affinity of the pump for Na+. Immunohistochemical localization along with Western blot analysis of cultured porcine endometrial epithelial cells revealed the presence of α-1 and α-2 isoforms, but not the α-3 isoform of Na+-K+ ATPase, which did not change in the presence of insulin. Insulin-stimulated Na+ transport was inhibited by hydroxy-2-naphthalenylmethylphosphonic acid tris-acetoxymethyl ester [HNMPA-(AM)3], a specific inhibitor of insulin receptor tyrosine kinase activity, suggesting that the regulation of Na+ transport by insulin involves receptor autophosphorylation. Pretreatment with wortmannin, a specific inhibitor of phosphatidylinositol 3–kinase as well as okadaic acid and calyculin A, inhibitors of protein phosphatase activity, also blocked the insulin-stimulated increase in short circuit and pump currents, suggesting that activation of phosphatidylinositol 3–kinase and subsequent stimulation of a protein phosphatase mediates the action of insulin on Na+-K+ ATPase activation.
Keywords: insulin-like growth factor I, epithelial ion transport, membrane transport, ouabain, amiloride
INTRODUCTION
The transport-related activity of the surface and glandular endometrial epithelium plays an important role in regulation of uterine lumen electrolyte composition, pH, and fluid volume, providing a suitable environment for fertilization and implantation of the developing embryo (Clemetson et al. 1972; Iritani et al. 1974; Fraser 1992, Fraser 1995). Previous studies with native porcine endometrial epithelium demonstrated that amiloride-sensitive Na+ absorption and K+ secretion were modulated by PGF2α, cAMP, and gastrin-releasing peptide (Vetter and O'Grady 1996, Vetter and O'Grady 1997). Other studies using primary cultures of rodent endometrial epithelial cells grown on permeable membrane filters demonstrated that anion secretion was stimulated by adrenergic agonists through β adrenergic receptors and by ATP through multiple subtypes of purinoceptors (Chan et al. 1996, Chan et al. 1997). In cultures of porcine glandular endometrial epithelial cells, it was reported that PGE2 also stimulates anion secretion (Deachapunya and O'Grady 1998). In human endometrial epithelial cells, bradykinin, bombesin, and gastrin-releasing peptide have all been shown to affect transepithelial electrolyte transport (Matthews et al. 1993a,Matthews et al. 1993b). Thus, the results of these studies indicate that the absorptive and secretory activities of endometrial epithelial cells are modulated in response to a variety of signaling molecules.
The first demonstration of an effect of insulin on transepithelial Na transport was reported by Herrera 1965 using isolated toad bladder epithelium. Since that time, insulin and insulin-like growth factor I (IGF-I)1 have been shown to stimulate electrogenic sodium transport in a variety of cells (Siegel and Civan 1976; Walker et al. 1984; Civan et al. 1988; Blazer-Yost et al. 1989; McGill and Guidotti 1991; Erlij et al. 1994; Feraille et al. 1994; Ewart and Klip 1995). In many cases, this increase in Na+ transport was a result of stimulation of the Na+-K+ ATPase, either directly through changes in pump concentration or kinetic properties (McGill and Guidotti 1991; Hundal et al. 1992; Marette et al. 1993; Ewart and Klip 1995) or indirectly through increases in intracellular [Na+], which in turn leads to increased substrate availability and subsequent pump stimulation (Walker et al. 1984; Marunaka et al. 1992; Erlij et al. 1994; Sargeant et al. 1995). In amphibian (Grinstein and Erlij 1974) and rodent (Hundal et al. 1992; Marette et al. 1993) skeletal muscle cells, for example, stimulation by insulin increases Na+-K+ ATPase transport activity by increasing pump translocation into the plasma membrane. This effect occurs within 15–30 min and is not blocked by inhibitors of protein synthesis. In rodent muscle, it appears that the α-2 isoform is selectively inserted into the plasma membrane (Marette et al. 1993). In another study, however, Clausen and Hansen 1977 demonstrated that the effect of insulin on [3H] ouabain binding in intact skeletal muscle was an artifact arising from stimulation of the rate of binding. Steady state binding of [3H] ouabain was unaffected, indicating that insulin does not induce translocation of the Na+-K+ ATPase to the plasma membrane. These results were confirmed in a later study and demonstrated that IGF-I also had a stimulating effect on the Na+-K+ ATPase in skeletal muscle (Dorup and Clausen 1995). In rat adipocytes, insulin does not appear to alter pump concentration in the plasma membrane or to involve elevations in intracellular [Na+] (Lytton et al. 1985; McGill and Guidotti 1991). In these cells, increased transport activity of the pump is associated with an increase in Na+ affinity for the α-1 and α-2 isoforms and an increase in Vmax associated with the α-2 isoform of the pump. In hepatocytes, skeletal muscle cells, and 3T3-L1 adipocytes, increases in Na+-K+ ATPase transport activity appear to be due to elevations in intracellular [Na+] resulting from activation of either Na+-H+ exchange (Fehlmann and Freychet 1981; Klip et al. 1986) or Na+-K+-2Cl− cotransport activity (Sargeant et al. 1995). In epithelial cells, insulin and IGF-I have also been demonstrated to stimulate Na+ transport either by increasing the Na+ affinity of the Na+-K+ ATPase (Feraille et al. 1994) or by increasing the number of amiloride-sensitive Na+ channels present within the apical membrane (Erlij et al. 1994; Record et al. 1998).
Specific binding sites for insulin and IGF-I have been identified in normal endometrium and endometrial cancer cells (Talavera et al. 1990; Nagamani et al. 1991). Thus, it was of interest to determine whether insulin and IGF-I play a role in regulation of Na+ transport across the endometrial epithelium. In this study, we report the effects of insulin and IGF-I on transepithelial Na+ transport function of cultured porcine endometrial epithelial cells under defined medium conditions. We show that insulin and IGF-I produce an acute increase in Na+ transport that involves direct regulation of the Na+-K+ ATPase and a basolateral K+ channel. In addition, we demonstrate that long-term exposure to insulin (4 d) results in enhanced Na+ absorption with a further increase in pump activity and an increase in apical membrane amiloride-sensitive Na+ conductance.
MATERIALS AND METHODS
Materials
Insulin, IGF-I, ouabain, indomethacin, nonessential amino acids, and high purity grade salts were purchased from Sigma Chemical Co. Hydroxy-2-naphthalenylmethylphosphonic acid tris-acetoxymethyl ester [HNMPA-(AM)3], wortmannin, okadaic acid, and PD-98059 (2′-amino-3′-methoxyflavone) were purchased from Biomol Research Laboratories. Amiloride, 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) and calyculin A were purchased from Research Biochemicals International, and benzamil from Molecular Probes. Dulbecco's modified Eagle's medium (DMEM), Dulbecco's phosphate buffer saline (DPBS), fetal bovine serum (FBS), collagenase (type 1), kanamycin, penicillin-streptomycin, and fungizone were purchased from GIBCO BRL.
Cell Isolation and Culture
Porcine uterine tissues were collected from 4–5-mo-old Yorkshire-Pietrain cross pigs purchased from stock herds maintained by the University of Minnesota College of Agriculture. Uterine tissues from adult, precycling animals were used to minimize variability in electrolyte transport properties at different stages of the estrus cycle. Uteri were obtained from pigs that were killed at the University of Minnesota Meat Sciences Laboratory by the captive bolt euthanasia procedure approved by the University Animal Care Committee and supervised by a USDA certified veterinarian. Uterine tissue was placed in ice cold porcine Ringer solution containing (mM): 130 NaCl, 6 KCl, 3 CaCl2, 0.7 MgCl2, 20 NaHCO3, 0.3 NaH2PO4, 1.3 Na2HPO4, gassed with 95% O2/5% CO2, pH 7.4. After removal of the serosal muscle layer, the endometrial tissue was minced into small pieces (≈1 mm3) and washed twice with Ca2+- and Mg2+-free DPBS. The tissue fragments were then subjected to collagenase digestion and the epithelial glands were isolated as described previously (Deachapunya and O'Grady 1998). The epithelial glands were suspended in DMEM supplemented with 3.7 g/liter sodium bicarbonate, 10% FBS, 5 μg/ml (850 nM) insulin, 1% nonessential amino acids, 5 μg/ml fungizone, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 μg/ml kanamycin (standard media). They were then plated onto cell culture dishes and incubated at 37°C in a humidified atmosphere of 5% CO2 in air. Culture medium was changed after 24 h, and then every 2–3 d. The epithelial cells became confluent monolayers within 3–4 d. The remaining stromal cells were removed by adding 0.02% collagenase in serum-free medium for 24 h. The epithelial cells were then trypsinized and placed on 24-mm (4.5 cm2) transparent permeable membrane filters (Corning Costar). To study the long-term effect of insulin and IGF-I, the epithelial cells were grown on permeable filters in DMEM supplemented with 10% FBS for 7 d, and then replaced with Phenol Red-free DMEM (serum-free media) supplemented with 850 nM insulin or 1.3 nM IGF-I for 4 d.
Measurement of Basal Electrical Parameters
Transepithelial resistance of the cell monolayers was measured using the EVOM epithelial voltohmmeter coupled to Ag/AgCl “chopstick” electrodes (World Precision Instruments). After 10 d, the confluent culture inserts were mounted in Ussing Chambers, bathed on both sides with standard porcine Ringer solution maintained at 37°C, and bubbled with 95% O2 /5% CO2. Transepithelial potential difference, tissue conductance, and short circuit current (Isc) were measured with the use of voltage-clamp circuitry from JWT Engineering. The data from the voltage clamp experiments was digitized, stored, and analyzed using Workbench data acquisition software (Kent Scientific Corp.), and recorded with a Compaq pentium microcomputer. All cells were pretreated with indomethacin (10 μM) added to both apical and basolateral solutions at least 10 min before the beginning of the experiment.
Measurement of Pump Current
Pump current was measured using amphotericin B (10 μM) to permeabilize the apical membrane of monolayers mounted in Ussing chambers. To determine the [K+] dependence of the pump, monolayers were bathed on both sides with NaMeSO4 Ringer solution containing (mM): 120 NaMeSO4, 30 mannitol, 3 calcium gluconate, 1 MgSO4, 20 NaHCO3, 0.3 NaH2PO4, 1.3 Na2HPO4, gassed with 95% O2/5% CO2, pH 7.4. Increasing intracellular K+ concentration was accomplished by replacement of NaMeSO4 Ringer solution with an equivalent volume of KMeSO4 Ringer solution. KMeSO4 Ringer solution contained (mM): 120 KMeSO4, 30 mannitol, 5 NaCl, 3 calcium gluconate, 1 MgSO4, 20 KHCO3, 0.3 KH2PO4, 1.3 K2HPO4, gassed with 95% O2/5% CO2, pH 7.4. To determine the [Na+] dependence of the pump, monolayers were bathed on both sides with N-methyl-d-glucamine (NMDG)–MeSO4 Ringer solution containing (mM): 130 NMDGMeSO4, 30 mannitol, 3 calcium gluconate, 1 MgSO4, 10 KHCO3, 0.3 KH2PO4, 1.3 K2HPO4, gassed with 95% O2/5% CO2, pH 7.4, and a NaMeSO4 Ringer solution containing different concentrations of Na+ was used to replace NMDGMeSO4 Ringer solution. The Na+ and K+ dependence of pump current was determined using the Hill equation: I p = I max [S]n/([S]n + K 0.5), and its linear expression: log (I p/I max − I p) = n log[S] − log K 0.5, where I p is the pump current stimulated by an increase in intracellular [Na+] or extracellular [K+], I max is the maximal pump current, S is an intracellular [Na+] or extracellular [K+], K 0.5 is the apparent dissociation constant, and n is the Hill coefficient. The kinetic parameters were determined by nonlinear regression or by linear regression analysis (Prism™ 2.0; GraphPad Software).
Measurement of Membrane Permeability
Current–voltage relationships were determined using amphotericin B–permeabilized monolayers mounted in Ussing chambers. The intracellular compartment was bathed with KMeSO4 Ringer solution and amphotericin B (10 μM) was added to permeabilize the membrane. The extracellular compartment was bathed with standard porcine Ringer solution or NaMeSO4 Ringer solution. An epithelial voltage clamp (World Precision Instruments) in combination with an LM-12 A-D interface (Dagan Corp.) were used to voltage clamp the monolayers and record the data. The voltage step commands and the resultant currents were generated using pCLAMP software (Axon Instruments). Current–voltage (I–V) relationships were obtained by a series of voltage step commands described in the figure legends. The compound-sensitive components were obtained by subtracting the currents before and after addition of the compound. The Na+:K+ selectivity ratio (P Na/P K) was calculated from reversal potential (E rev) measurements using the Goldman-Hodgkin-Katz equation {E rev = RT/zF ln (P Na [Na+])/(P K [K+])}.
Ouabain Binding
Epithelial cells (3 × 105) were subcultured onto 6.5-mm transparent permeable membranes containing 10% FBS in DMEM. After 3–4 d, the cells were placed in serum-free Phenol-Red–free DMEM for 48 h, followed by insulin treatment (850 nM) for 24 h. Specific [3H] ouabain binding was performed at 37°C in humidified incubator of 5% CO2 in air, using a procedure modified from Lobaugh and Lieberman 1987. Cell monolayers were preincubated in Phenol-Red–free DMEM for 20 min, and then the media was replaced with loading solution containing [3H] ouabain (Amersham Life Science) in DMEM. After incubation for 45 min, monolayers were rinsed with ice-cold DMEM for 10 s to remove unbound [3H] ouabain. The filters were then cut from their supports and transferred to the scintillation fluid and assayed for radioactivity. Other filters were solubilized in lysis buffer and assayed for amount of protein. Nonspecific [3H] ouabain binding was determined when an additional 500 μM ouabain was included in the loading solution. The magnitude of nonspecific ouabain binding did not exceed 30% of the total binding as determined at a concentration of 0.5 μM.
Immunocytochemistry
Epithelial cells were allowed to grow on permeable membrane filters in DMEM supplemented with 10% FBS for 5–7 d, followed by serum-free Phenol Red–free DMEM for 2 d. Insulin (850 nM) was then added to the serum-free media for 2 d. The monolayers were then washed twice in DPBS, pH 7.4, permeabilized with 0.3% Triton X-100 in DPBS at room temperature for 10 min, and then fixed in methanol at −20°C for 10 min. After fixation, filters were cut from their supports. The cells were then washed with DPBS and incubated with DPBS containing 1% bovine serum albumin and 10% normal goat serum to block nonspecific binding at room temperature for 1 h. Then the cells were incubated overnight with isoform-specific mouse monoclonal antibody against the α-1 and polyclonal antibodies to α-2 and α-3 isoforms of the rat Na+-K+ ATPase, 1:200 (Upstate Biotechnology Inc.) at 4°C. After washing, the cells were incubated with indocarbocyanine (cy3)-labeled goat anti–rabbit antibody, 1:400 (Jackson ImmunoResearch Laboratories) for 1 h at room temperature. After the final wash, the filters were placed on a glass slide. The cells and filters were embedded in fluoromount (Gallard Schlessinger) and examined by confocal microscopy, using a MRC1024 laser confocal microscope (Bio-Rad Laboratories) equipped with krypton-argon lasers.
Western Blot Analysis
Cell monolayers, as prepared for immunocytochemistry, were solubilized with lysis buffer (50 mM Tris-HCl, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 1 μg/ml aprotinin, and 1 mM NaF, pH 7.4) at 37°C for 30 min and homogenized. A protein assay was performed using a BCA Protein Assay Kit by Pierce. Proteins were separated by PAGE (8%). Electroblotting was done using Immobilon-P (Millipore Corp.). The electroblot assembly was placed into the electroblotting apparatus (Trans-Blot Cell; Bio-Rad Laboratories) and blotting was performed at 16 V overnight on ice. After the blots were removed, they were washed twice, and then blocked in freshly prepared 1× TBS-tween containing 3% nonfat dry milk (MLK) for 30 min at 20–25°C with constant agitation. After washing, blots were reacted overnight in primary antibody, 15 or 100 ml freshly prepared 1× TBS-tween containing 3% MLK with appropriate dilution of the primary antibody (anti–rat α-1 monoclonal antibody and rabbit-anti–rat α-2 polyclonal antibody from Upstate Biotechnology Inc.). The next day, blots were washed and reacted with secondary antibody, either goat anti–rabbit, alkaline phosphatase–labeled or goat anti–mouse, alkaline phosphatase–labeled. Secondary antibody was diluted 1:3,000 (33 μl into 100 ml) in 1× TBS-tween containing 3% MLK and reacted for ∼2 h. After washing, alkaline phosphatase color reagent was added to 100-ml 1× alkaline phosphatase color development buffer at room temperature. Blots were incubated in development buffer until bands were clearly developed.
Statistics
All values are presented as means ± SEM, n is the number of monolayers, and N is the number of animals in each experiment. The differences between control and treatment means were analyzed using a t test for paired or unpaired means, where appropriate. A value of P < 0.05 was considered statistically significant. The EC50 values for insulin and IGF-I and the IC50 values for benzamil and amiloride were determined using a four parameter logistic function to fit the data (Prism™ 2.0).
RESULTS
Long-Term Effects of Insulin and IGF-I on Electrolyte Transport
The basal electrical properties of cultured epithelial cells used in this study have been previously described (Deachapunya and O'Grady 1998). Our data showed that endometrial epithelial cells cultured in standard serum-containing media were capable of both Na+ absorption and Cl− secretion. To investigate acute and long-term effects of insulin and IGF-I on electrolyte transport in endometrial epithelial cells, we measured both NPPB-inhibitable Cl− secretion and benzamil-sensitive Na+ absorption as shown in Fig. 1. NPPB is an arylamino benzoate compound that has been previously shown to block apical membrane Cl− channels in cultured porcine endometrial epithelial cells (Deachapunya and O'Grady 1998). Amiloride and its more potent analogue benzamil, have been previously shown to block Na+ channels in native porcine endometrial epithelium (Vetter and O'Grady 1996). For cell monolayers cultured in standard serum-containing media, 20% of the basal Isc was blocked by 10 μM benzamil and the remaining Isc was inhibited by 100 μM NPPB. In serum-free media, there was a significant decrease in both basal Isc from 34 ± 3 μA (n = 12, N = 4) to 13 ± 2 μA (n = 9, N = 4) and NPPB-sensitive Isc from 24 ± 3 to 4 ± 1 μA without a significant change in benzamil-sensitive Isc (from 7 ± 2 to 9 ± 2 μA) compared with serum-containing media. Treatment with 850 nM (5 μg/ml) insulin or 1.3 nM (10 ng/ml) IGF-I for 4 d significantly increased basal Isc to 48 ± 3 μA (n = 23, N = 9) for insulin and 27 ± 5 μA (n = 7, N = 4) for IGF-I, and increased benzamil-sensitive Isc to 41 ± 3 μA for insulin and 23 ± 5 μA for IGF-I. However, no change in benzamil-insensitive Isc was detected after treatment with either insulin or IGF-I.
Figure 1.

Effects of insulin and IGF-I on basal, NPPB-sensitive, and benzamil-sensitive Isc. Histogram illustrating the basal Isc and the absolute decrease in Isc after apical addition of 5 μM benzamil and subsequent apical addition of 100 μM NPPB to the cell monolayers. Cells were grown in standard media (SM, n = 12, N = 4) for 1 wk and replaced with serum-free media (SFM, n = 9, N = 4) in the absence or presence of 850 nM insulin (n = 23, N = 9) or 1.3 nM IGF-I (n = 7, N = 4) for 4 d.
Short-Term Effect of Insulin on Isc
Addition of 850 nM insulin to the basolateral solution of monolayers produced an increase in Isc within 5 min that reached a maximal plateau response of 36 ± 2 μA (n = 11, N = 5) in 30–45 min, as illustrated in Fig. 2 A. The maximal Isc response was sustained for as long as 3 h. Addition of 65 nM IGF-I to the basolateral solution produced the same response as insulin with a maximal plateau response of 36 ± 3 μA (n = 6, N = 3). Subsequent addition of 10 μM benzamil to the apical solution inhibited both the basal and insulin-stimulated Isc. Concentration–response curves for benzamil and amiloride are shown in Fig. 2 B with IC50 values of 25 nM for benzamil and 194 nM for amiloride. Pretreatment with 10 μM benzamil completely inhibited the insulin-stimulated Isc (data not shown). In addition, pretreatment with 50 μM HNMPA-(AM)3, a specific inhibitor of insulin receptor tyrosine kinase activity, for 30 min inhibited the insulin-stimulated increase in Isc (7 ± 2 μA, n = 6, N = 4), as illustrated in Fig. 3 A. The concentration–response relationships for insulin and IGF-I presented in Fig. 3 B demonstrated that threshold concentrations were ∼0.85 nM for insulin and 0.13 nM for IGF-I. The EC50 values were 12 nM for insulin and 2.5 nM for IGF-I.
Figure 2.
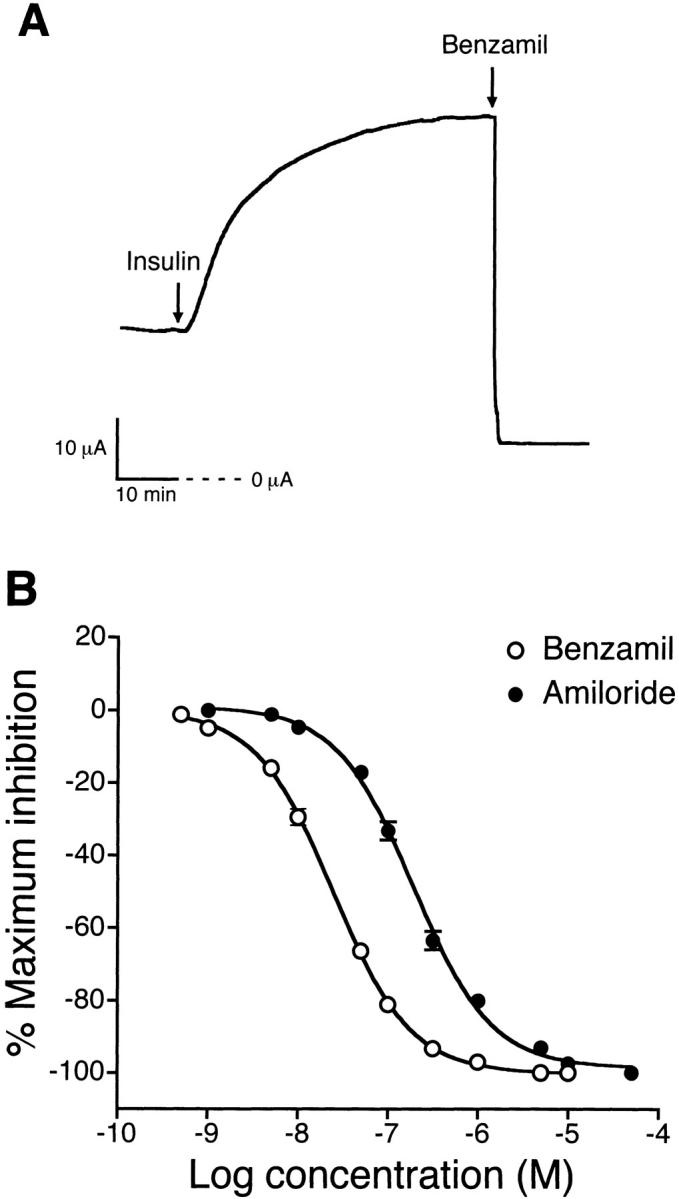
Time course of insulin-stimulated Na+ absorption and concentration–response relationships for benzamil and amiloride. (A) Representative tracing showing that addition of 850 nM insulin to the basolateral solution produced an increase in Isc within 5 min and reached a maximum plateau level within 30–45 min. The Isc stimulation produced by insulin and the basal Isc was inhibited by 10 μM benzamil added to the apical solution. Experiments were performed using cells maintained in serum-free media for 4 d (n = 11, N = 5). (B) Concentration–response relationships showing the decrease in Isc after treatment with various concentrations of benzamil or amiloride added to the apical solution of monolayers grown in serum-free media supplemented with 850 nM insulin for 4 d. The IC50 value was 25 nM for benzamil (○, n = 8, N = 4) and 194 nM for amiloride (•, n = 9, N = 4).
Figure 3.
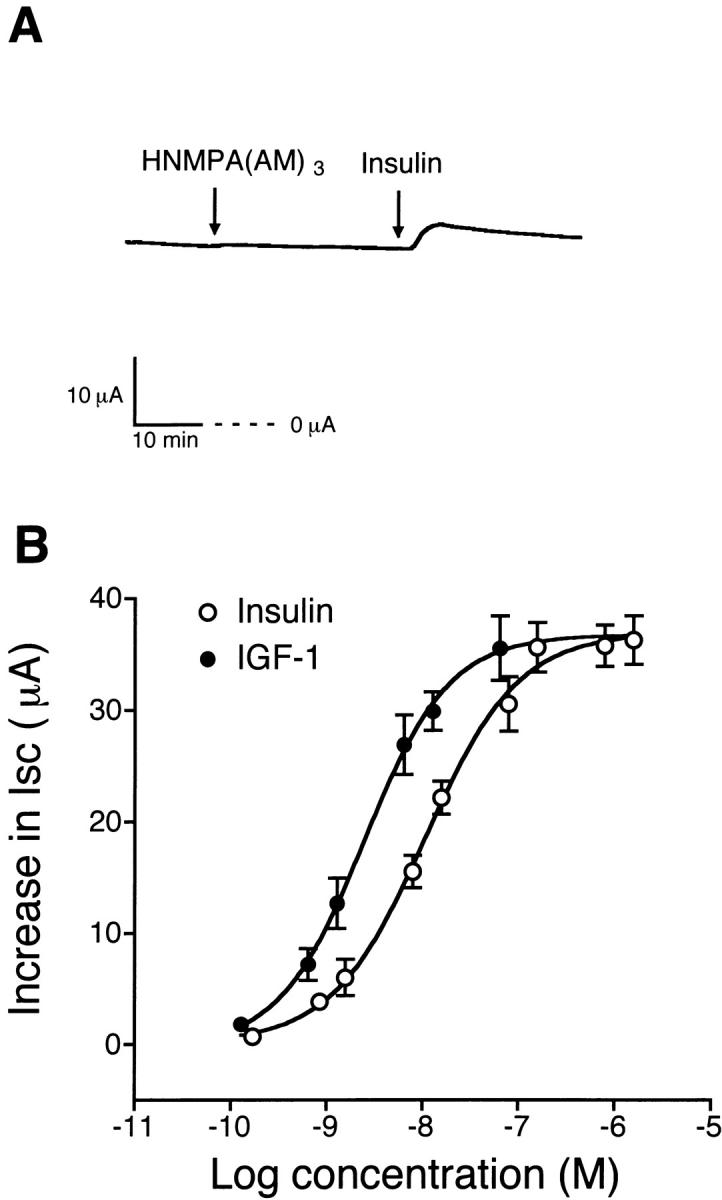
Effect of HNMPA-(AM)3 on insulin-stimulated Na+ absorption and concentration–response relationships for insulin and IGF-I. (A) Representative tracing showing that pretreatment with 25 μM HNMPA-(AM)3, an inhibitor of insulin receptor tyrosine kinase activity, added to the basolateral solution for 30 min abolished the increase in Isc produced by basolateral addition of 850 nM insulin. (B) Concentration–response relationships showing an acute increase in Isc after treatment with various concentrations of insulin or IGF-I added to the basolateral solution of monolayers. Experiments were performed using cells maintained in serum-free media for 4 d. The EC50 value was 12 nM for insulin (○) and 2.5 nM for IGF-I (•; n = at least 5, N = 3 for each concentration).
Na+ and K+ Dependence of the Na+-K+ ATPase
To determine whether the increase in Isc produced by insulin was the result of stimulation of the Na+-K+ ATPase, experiments were performed using amphotericin B–permeabilized monolayers mounted in Ussing chambers bathed on both sides with either NaMeSO4 Ringer solution containing 5 mM BaCl2 or NMDGMeSO4 Ringer solution containing 10 mM KHCO3. BaCl2 was used to inhibit basolateral K+ channels and limit the contribution of K+ recycling to pump activation. The increase in extracellular [K+] was accomplished by replacement of NaMeSO4 solution with an equal volume of KMeSO4 Ringer solution containing different concentrations of K+. To determine the Na+ dependence of the pump, NaMeSO4 Ringer solution with 10 mM KHCO3 and different concentrations of Na+ was used to replace NMDGMeSO4 Ringer solution.
Fig. 4 A shows the increase in pump current (I p) produced by increasing K+ concentrations in the basolateral solution of insulin-treated monolayers. The stimulated I p was completely inhibited by 10 μM ouabain added to the basolateral solution (data not shown). When I p was plotted as a function of basolateral [K+], it revealed that insulin treatment increased I max from 20 ± 2 μA (n = 11, N = 5) to 63 ± 6 μA (n = 9, N = 5), with an increase in the K 0.5 value from 1.8 ± 0.2 to 2.9 ± 0.2 mM. Stimulation of pump current was also dependent on intracellular [Na+], as shown in Fig. 5 A. The relationship of I p and [Na+] revealed that insulin treatment increased I max from 18 ± 1 μA (n = 8, N = 4) to 42 ± 5 μA (n = 10, N = 4) with a significant decrease in the K 0.5 value from 39 ± 2 to 24 ± 2 mM. Hill coefficients were 2.1 ± 0.3 for K+ and 1.2 ± 0.1 for Na+ under insulin treatment, which was not significantly different from corresponding control values (1.9 ± 0.2 for K+ and 1.3 ± 0.1 for Na+), as illustrated in Fig. 4 B and 5 B.
Figure 4.
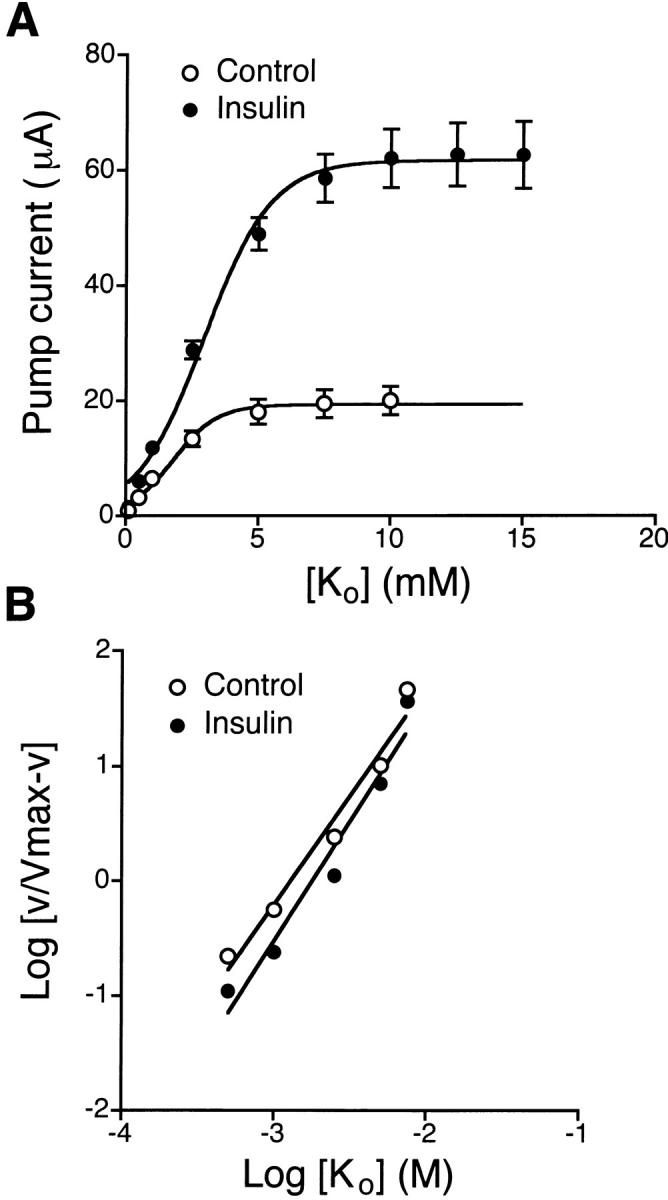
Extracellular [K+] dependence of pump current in control and insulin-treated monolayers. Experiments were performed using amphotericin B–permeabilized (apical membrane) monolayers bathed on both sides with NaMeSO4 Ringer solution. The cells were maintained in serum-free media (control) or serum-free media supplemented with 850 nM insulin for 4 d. (A) The pump current (I p) was plotted as a function of basolateral [K+]. The increase in [K+] in the basolateral solution was accomplished by replacement with KMeSO4 Ringer solution containing different concentrations of K+. Insulin treatment increased I max from 20 ± 2 μA (n = 11, N = 5) to 63 ± 6 μA (n = 9, N = 5) and increased K 0.5 from 1.8 ± 0.2 to 2.9 ± 0.2 mM (control, r 2 = 0.989; insulin, r 2 = 0.992). (B) Hill plot from the data shown in A. Insulin treatment did not significantly affect the Hill coefficient (control, 1.9 ± 0.2, r 2 = 0.971; insulin, 2.1 ± 0.3, r 2 = 0.956, estimated by linear regression analysis).
Figure 5.
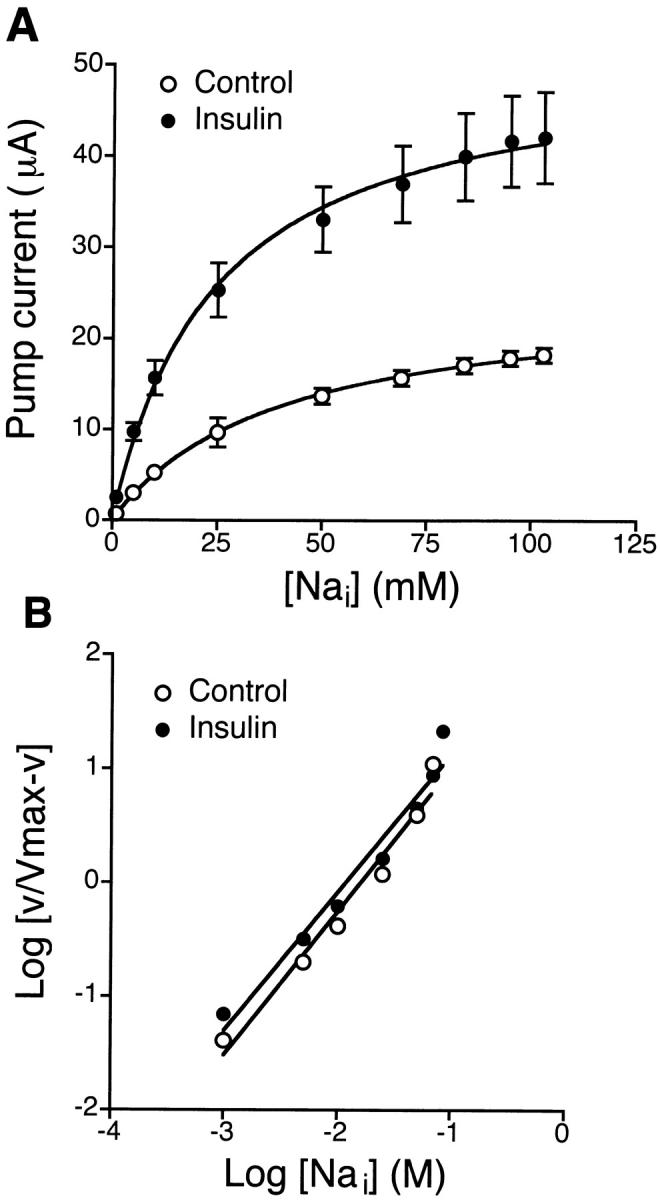
Intracellular [Na+] dependence of pump current in control and insulin-treated monolayers. Experiments were performed with amphotericin B–permeabilized (apical membrane) monolayers bathed on both sides with NMDG-MeSO4 Ringer solution with 10 mM KHCO3. The cells were maintained in serum-free media (control) or serum-free media supplemented with 850 nM insulin for 4 d. (A) Pump current (I p) was plotted as a function of apical [Na+]. The increase in [Na+] in the apical solution was accomplished by replacement with NaMeSO4 Ringer solution containing increasing concentrations of Na+. Insulin treatment increased I max from 18 ± 1 μA (n = 8, N = 4) to 42 ± 5 μA (n = 10, N = 4) and decreased K 0.5 values from 39 ± 2 to 24 ± 2 mM (control, r 2 = 0.999; insulin, r 2 = 0.996). (B) Hill plot of the data shown in A. Insulin treatment did not significantly affect the Hill coefficient (control, 1.3 ± 0.1, r 2 = 0.968; insulin, 1.2 ± 0.1, r 2 = 0.962, estimated by linear regression analysis).
Ouabain-sensitive Current Across the Basolateral Membrane
The acute (15–20 min) effects of insulin on the current–voltage relationship of the Na+-K+ ATPase is shown in Fig. 6. The experiment was performed using amphotericin B–permeabilized monolayers, as described in the previous section. The apical surface of the epithelium was bathed with KMeSO4 Ringer solution supplemented with 5 mM NaCl, and amphotericin B was used to permeabilize the apical membrane. Standard Ringer solution was used to bathe the basolateral surface of the epithelium. The I–V relationship of the pump was obtained using a voltage step protocol ranging from −90 to +90 mV (15-mV steps) at a holding potential of 0 mV. The difference currents were obtained by subtracting the current before and 10 min after basolateral addition of 100 μM ouabain. The ouabain-sensitive current after pretreatment with 850 nM insulin for 15 min was approximately twofold greater in magnitude, with no change in conductance when compared with current responses under basal conditions.
Figure 6.

I–V relationships for the basolateral ouabain-sensitive current. Ouabain-sensitive current was obtained from amphotericin B–permeabilized monolayers in response to a voltage step protocol from −90 to +90 mV in 15-mV step increments at a holding potential of 0 mV. Experiments were performed under conditions where the apical surface was bathed in KMeSO4 Ringer solution with 10 mM NaCl and permeabilized with amphotericin B. Standard Ringer solution was used to bathe the basolateral surface of the epithelium. Ouabain (100 μM) was added to the basolateral solution to inhibit the pump. Cell monolayers were maintained in serum-free media for 2 d. The ouabain-sensitive current is shown before (○, n = 5, N = 4) and after (•, n = 6, N = 4) treatment with 850 nM insulin added to the basolateral solution.
Effect of Insulin on Basolateral Membrane K+ Permeability
The effect of insulin on basolateral K+ permeability is shown in Fig. 7. The apical surface of the epithelium was permeabilized with amphotericin B and bathed with KMeSO4 Ringer solution without NaCl, while the basolateral surface was bathed with standard Ringer solution. The insulin-sensitive current obtained after basolateral addition of 850 nM insulin for 15 min exhibited slight outward rectification with a mean reversal potential of −53 ± 3 mV (n = 9, N = 4). Replacement of standard Ringer solution with KCl Ringer solution shifted the reversal potential toward zero (3 ± 2, n = 6, N = 4). Acute insulin (850 nM) treatment (15–30 min) had no effect on apical membrane conductance.
Figure 7.

I–V relationships for the insulin-sensitive K permeability in the basolateral membrane. Insulin-activated currents were obtained from amphotericin B–permeabilized monolayers in response to a voltage step protocol from −90 to +90 mV in 15-mV increments at a holding potential of 0 mV. The apical surface of the epithelium was bathed in KMeSO4 Ringer solution without NaCl and permeabilized with amphotericin B. Standard (SR) or KCl Ringer solution was used to bathe the basolateral surface of the epithelium. Insulin (850 nM) was added to the basolateral solution of the cell monolayers maintained in serum-free media for 2 d. Mean reversal potentials for the insulin-sensitive currents were −53 ± 3 mV in standard (○, n = 9, N = 4) and 3 ± 2 mV in KCl (•, n = 6, N = 4) Ringer solution.
Effect of Insulin on Benzamil-sensitive Current
To characterize the properties of the benzamil/amiloride-sensitive Na+ channel located in the apical membrane, experiments were performed with amphotericin B–permeabilized monolayers, as previously mentioned. The monolayers were bathed on the apical side with NaMeSO4 Ringer solution and on the basolateral side with KMeSO4 Ringer solution containing amphotericin B. The benzamil-sensitive current was obtained before and 2 min after apical addition of 5 μM benzamil. The current–voltage relationship of benzamil-sensitive current is shown in Fig. 8. Treatment with 850 nM insulin for 4 d showed a mean reversal potential of 65 ± 4 mV (n = 7, N = 4), which was not significantly different from control cells in serum-free media (72 ± 4 mV, n = 5, N = 3). The basal benzamil-sensitive conductance was 0.17 ± 0.05 mS. Stimulation with insulin significantly increased the conductance to 0.37 ± 0.05 mS. Interestingly, a 30-min treatment with 850 nM insulin produced no significant change in reversal potential (69 ± 8 mV, n = 6, N = 3) or conductance (0.18 ± 0.05 mS) compared with control monolayers. The Na+ to K+ selectivity ratio of the benzamil-sensitive current in the presence of insulin was calculated to be 11.6:1.
Figure 8.

I–V relationships for the benzamil-sensitive permeability in the apical membrane. Benzamil-sensitive current obtained from amphotericin B–permeabilized monolayers in response to a voltage step protocol from −90 to +165 mV in 15-mV increments from a holding potential of 0 mV. Experiments were performed under conditions where the basolateral surface was bathed in KMeSO4 Ringer solution with 5 mM NaCl and permeabilized with amphotericin B. NaMeSO4 Ringer solution with 5 mM KCl was used to bathe the apical surface of the epithelium. Benzamil (5 μM) was added to the apical solution of the cell monolayers grown in serum-free media (control) or serum-free media pretreated with 850 nM insulin for 30 min (short term) or serum-free media supplemented with 850 nM insulin for 4 d (long term). Mean reversal potentials for benzamil-sensitive currents were 72 ± 4 mV for control (○, n = 5, N = 3), 69 ± 8 mV for short-term (•, n = 6, N = 3), and 65 ± 4 mV for long-term (▵, n = 7, N = 4) insulin treatment.
Mechanism of Insulin Action
Previous studies using a rat skeletal muscle cell line demonstrated that the activation of Na+-K+ ATPase by insulin may involve the phosphatidylinositol 3-kinase (PI-3 kinase) signaling pathway (Ragolia et al. 1997). Therefore, experiments were performed to determine the signaling cascade involved in insulin-stimulated Na+ transport across endometrial epithelial cells. Incubation with 1 μM wortmannin, a PI-3 kinase inhibitor, markedly inhibited the insulin-induced increase in Isc from 47 ± 3 μA (n = 12, N = 5) to 5 ± 3 μA (n = 6, N = 4) compared with control, as shown in Fig. 9 A. Further experiments were conducted to determine the downstream components of the pathway using phosphatase inhibitors. It was demonstrated that pretreatment with 100 nM okadaic acid (Fig. 9 B) or 100 nM calyculin A, inhibitors of protein phosphatase PP-1 and PP2A, for 30 min also inhibited insulin-induced increases in Isc to 3 ± 3 μA (n = 4, N = 3) and 5 ± 3 μA (n = 4, N = 3), respectively. Previous studies of insulin signaling in adipocytes suggested the possibility that PI-3 kinase activation by the insulin receptor can stimulate mitogen-activated protein kinase (MAP kinase), so that insulin-dependent regulation of the Na+-K+ ATPase in endometrial epithelial cells might involve components of the MAP kinase signaling pathway (Suga et al. 1997). To examine this possibility, we pretreated monolayers with a MAP kinase inhibitor (5 μM PD-98059) and tested the effects of insulin. Although we observed a slight reduction in Isc stimulation produced by insulin (34 ± 4 μA, n = 5, N = 3), the results suggested that the MAP kinase signaling pathway did not play a major role in activation of the Na+-K+ ATPase.
Figure 9.
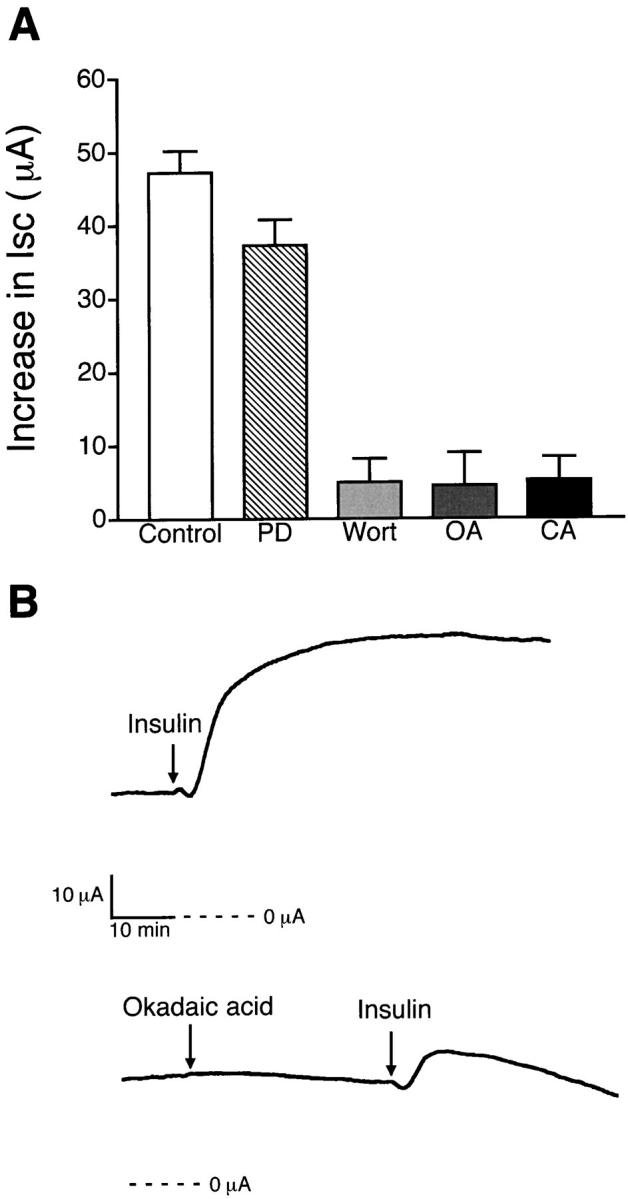
Effects of enzyme inhibitors on insulin-stimulated Isc. (A) Histogram illustrating an increase in Isc produced by 850 nM insulin when monolayers were pretreated for 30 min with 5 μM PD-98059 (PD, n = 5, N = 3), MAP kinase inhibitor; 1 μM wortmannin (Wort, n = 6, N = 4), PI-3 kinase inhibitor; 100 nM okadaic acid (OA, n = 4, N = 3) and 100 nM calyculin A (CA, n = 4, N = 3), phosphatase inhibitors, compared with insulin alone (Control, n = 12, N = 5). (B) Representative tracings showing that the increase in Isc produced by basolateral addition of 850 nM insulin is decreased after pretreatment with 100 nM okadaic acid added to the basolateral solution.
Additional experiments were performed to determine the effect of phosphatase inhibitors on insulin-stimulated pump current, as illustrated in Fig. 10. The Na-K ATPase was stimulated by basolateral addition of 5 mM KMeSO4 Ringer solution before and after treatment with insulin for 30 min. Addition of 850 nM insulin to the basolateral solution had no effects on basal current (−5 ± 1 μA, n = 25, N = 9). A subsequent addition of 5 mM KMeSO4 Ringer solution produced a rapid increase in current that was completely inhibited by 100 μM ouabain, as shown in Fig. 10 A. Compared with the control monolayers, insulin stimulated an increase in pump current from 18 ± 2 μA (n = 12, N = 5) to 32 ± 1 μA (n = 7, N = 4). Pretreatment with 100 nM okadaic acid for 30 min before addition of insulin abolished the insulin-stimulated pump current and decreased the basal pump current to 14 ± 2 μA (n = 6, N = 4), as shown in Fig. 10 B.
Figure 10.
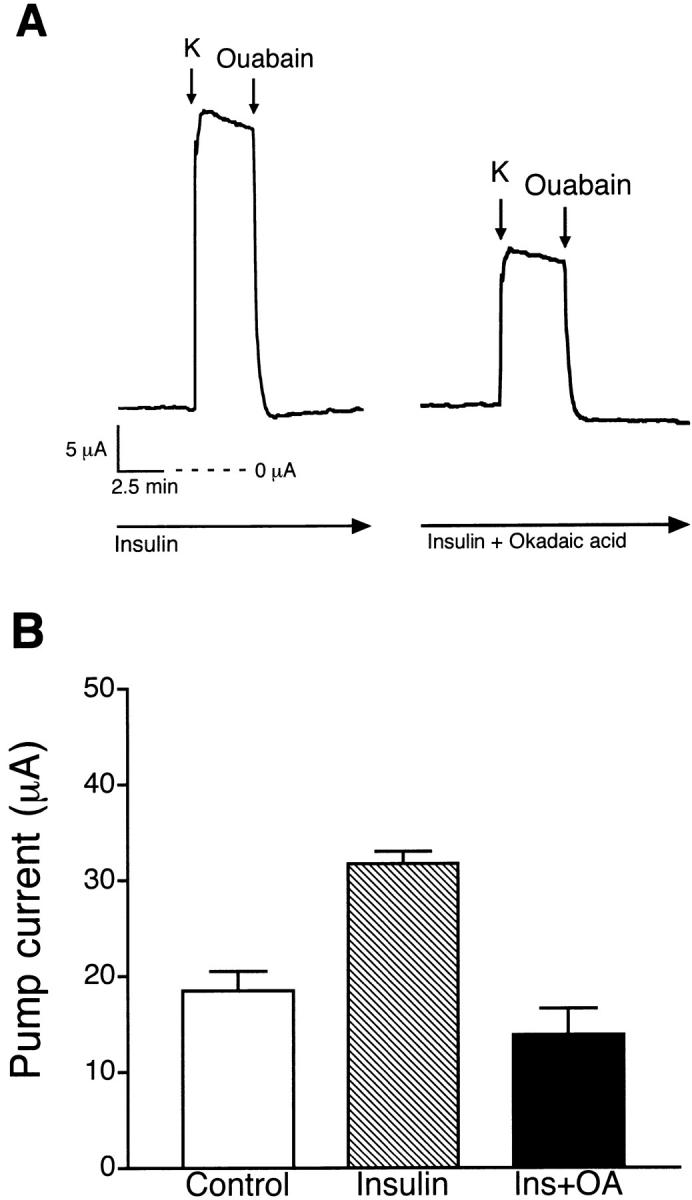
Effect of okadaic acid on insulin-stimulated pump current. (A) Representative tracing comparing pump current responses of insulin (850 nM)-treated monolayers before and after pretreatment with okadaic acid (100 nM). (B) Histogram illustrating the effect of okadaic acid on insulin activation of K+-stimulated pump current (n = 6, N = 4) compared with insulin (n = 7, N = 4) and a serum-free control group (n = 12, N = 5).
Short- and Long-term Effects of Insulin on Conductance and Pump Current
Fig. 11 compares the apical Na+ conductance (Fig. 8) and the pump current stimulated by 30-min and 4-d treatments with insulin. Administration of insulin for 30 min significantly increased K+-stimulated pump current with no effect on Na+ conductance. However, treatment with insulin for 4 d significantly increased both Na+ conductance and K+-stimulated pump current, suggesting that long-term treatment with insulin produced an increase in Na+ permeability of the apical membrane in addition to further activation of Na+-K+ ATPase.
Figure 11.
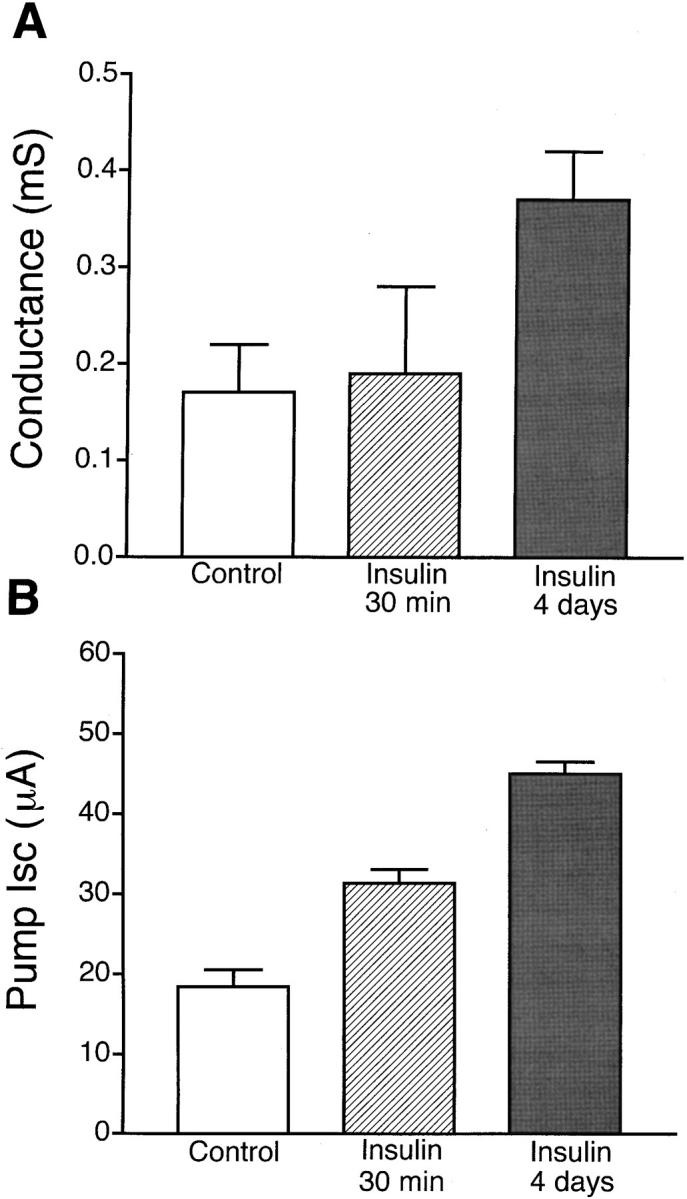
Short- and long-term effects of insulin on apical Na+ conductance and pump current. (A) Effect of 30-min and 4-d treatment with 850 nM insulin on apical membrane Na+ conductance. (B) Effect of insulin treatment on pump current induced by 5 mM extracellular K+.
Identification of Na-K-ATPase α Subunit Isoforms
The presence of α-1, α-2, and α-3 isoforms of the Na+-K+ ATPase were determined by immunofluorescence and Western blotting. Fig. 12 shows the summation of immunofluorescence images (1–3) obtained at 3-μm steps as the microscope was focused from the filter towards the apical membrane of glandular epithelial cell monolayers. The cell monolayers were labeled using a monoclonal antibody to α-1 and polyclonal antibodies to α-2 and α-3 isoforms of fusion proteins of the rat Na+-K+ ATPase. The cell monolayers were labeled with antibodies to the α-1 and α-2 isoforms, but not the α-3 isoform of the Na+-K+ ATPase. The labeling pattern of both α-1 and α-2 isoforms was intensely localized to the basolateral membrane. The cells grown in serum-free media in the absence and presence of 850 nM insulin exhibited the same pattern of labeling for both α-1 and α-2 isoforms. The control monolayers incubated with preimmune serum gave images identical to that observed with the α-3 isoform antibody (data not shown).
Figure 12.

Immunolocalization and Western blot analysis of α-1, α-2, and α-3 isoforms of Na+-K+ ATPase of cultured endometrial epithelial cells. (A) Immunofluorescence labeling of monolayers grown in the presence or absence of 850 nM insulin for 2 d. Monolayers were fixed, permeabilized, and labeled with antibodies against α-1, α-2, and α-3 isoforms of rat Na+-K+ ATPase (scale bar, 25 μm). (B) Proteins from monolayers treated with 850 nM insulin for 2 d were also extracted and analyzed by Western blot analysis using the same antibodies as indicated for immunofluorescence.
A representative Western blot is also presented in Fig. 12 and confirms the expression of α-1 and α-2 isoforms of the Na+-K+ ATPase. The monoclonal anti–Na+-K+ ATPase α-1 antibody labeled an ∼95-kD protein and the polyclonal anti–Na+-K+ ATPase antibody labeled a protein of ∼100 kD, consistent with α-1 and α-2 isoforms of the Na+-K+ ATPase.
[3H] Ouabain Binding
To determine whether insulin increased transport activity of the pump as a result of an increase in Na+-K+ ATPase concentration in the basolateral membrane, specific [3H] ouabain binding was performed with the cell monolayers cultured in serum-free media in the absence or presence of 850 nM insulin. Fig. 13 shows the specific binding of [3H] ouabain to endometrial epithelial cells as a function of ouabain concentration. Analysis of [3H] ouabain binding revealed a single class of binding sites with total receptor concentration (B max) of 13.9 ± 2.4 pmol/mg protein and K d of 252.8 ± 90.5 nM for insulin-treated cells (n = 5). The B max and K d of insulin-treated cells did not significantly differ from those of control cells (B max = 11.4 ± 3.9 pmol/mg protein and K d = 237.0 ± 173.4 nM).
Figure 13.
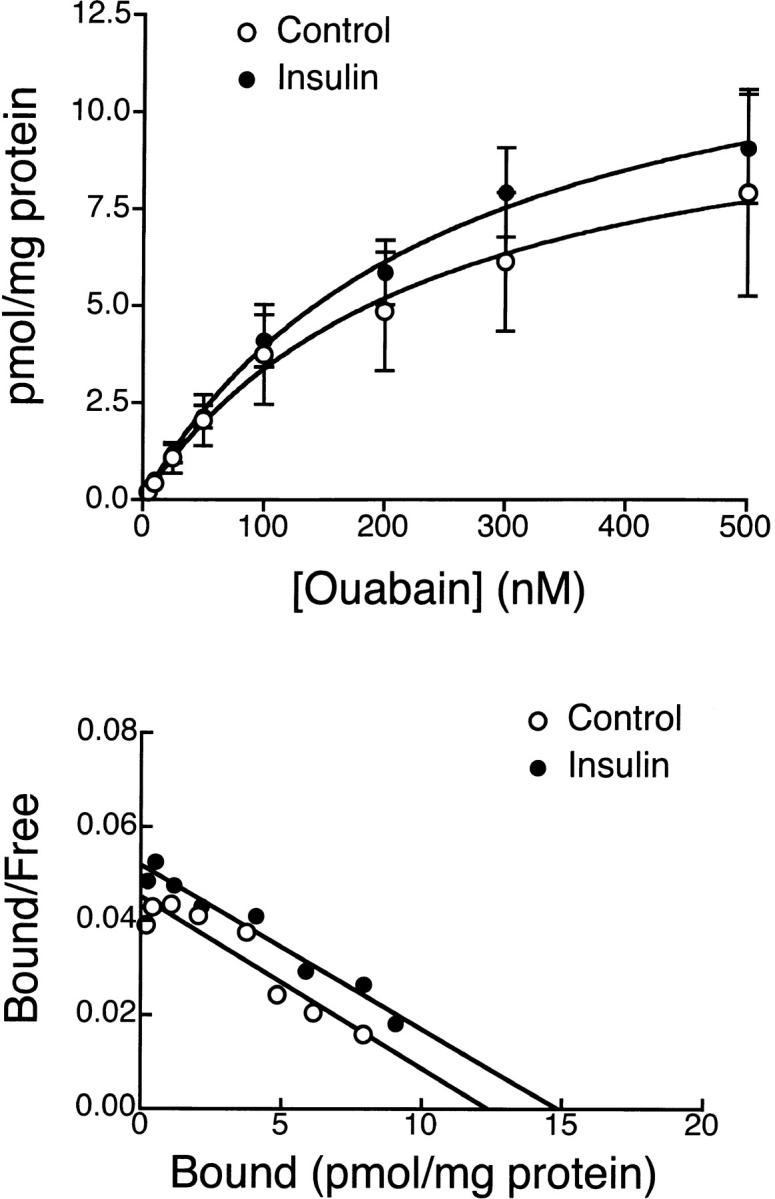
[3H] Ouabain binding of cultured endometrial epithelial cells. (A) Specific binding of [3H] ouabain to endometrial epithelial cells after incubation with 850 nM insulin for 24 h. The data were fit by nonlinear regression analysis. The B max was 13.9 ± 2.4 pmol/mg protein, and the K d was 253 nM for insulin-treated cells, which was not significantly different from values in control group (B max = 11.4 ± 3.9 pmol/mg protein, K d = 237 nM, n = 5). (B) Scatchard plot of the same data.
DISCUSSION
Early studies of toad urinary bladder demonstrated that insulin stimulates transepithelial Na+ transport by activation of the Na+-K+ ATPase without increasing apical membrane Na+ permeability (Herrera 1965). Further studies of toad bladder (Siegel and Civan 1976) and skeletal muscle (Weil et al. 1991) showed that insulin-stimulated Na+ transport resulted from activation of the Na+-K+ ATPase. More recent studies of rat proximal tubule demonstrated that insulin increases the Na+ affinity of the pump (Feraille et al. 1994) similar to that previously shown in adipocytes (McGill and Guidotti 1991). These findings are consistent with the previously mentioned study on skeletal muscle (Clausen and Hansen 1977) and with a more recent study of adipocytes where insulin stimulation did not produce any change in Na+-K+ ATPase concentration as measured using immunogold labeling (Voldstedlund et al. 1993). In contrast, studies of A6 cells demonstrated that insulin-dependent stimulation of Na+ transport was primarily due to an increase in Na+ permeability of the apical membrane (Walker et al. 1984; Marunaka et al. 1992; Erlij et al. 1994). Previous patch clamp experiments have suggested that insulin-dependent increases in Na+ permeability resulted from increased open probability of apical Na+ channels (Marunaka et al. 1992), whereas studies of intact A6 monolayers using blocker-induced noise analysis indicated that insulin increased Na+ channel density and apical membrane surface area (Erlij et al. 1994). The idea that insulin functions to increase insertion of Na+ channels into the apical membrane was further supported by recent experiments with brefeldin A, which demonstrated partial block of insulin-stimulated Na+ absorption across A6 cell monolayers (Coupaye-Gerard et al. 1994). The effect of insulin on Na+ transport in A6 cells appears to be similar to the effects of insulin on glucose transport previously reported in adipocytes where insulin enhances glucose uptake by stimulating the insertion of GLUT 4 glucose carriers into the plasma membrane (Record et al. 1998).
Unlike the results obtained using A6 cells, we find in the present study that the acute (within 30 min after stimulation) increase in Na+ transport after treatment with insulin or IGF-I is not the result of an increase in apical Na+ conductance. Insulin and IGF-I increase Na+ absorption by stimulating Na+-K+ ATPase activity and by increasing basolateral membrane K+ permeability. Stimulation of Na+-K+ ATPase activity involves increases in both Vmax and Na+ affinity, and a small decrease in K+ affinity. Previous studies of Na+-K+ ATPase stimulation by insulin in skeletal muscle indicated that increases in Vmax may be the result of insertion of pumps into the membrane (Hundal et al. 1992; Marette et al. 1993). This conclusion was supported by experiments with microtubule-disrupting agents such as colchicine, which blocked insulin-stimulated increases in Vmax of the Na+-K+ ATPase in skeletal muscle myoballs (Li and Sperelakis 1994). In the present study, insulin-stimulated increases in Isc and I P were not blocked by colchicine or brefeldin A (data not shown), suggesting that insulin-dependent increases in Vmax do not involve increases in the concentration of pumps present in the basolateral membrane. This interpretation is consistent with the results of ouabain binding experiments, which showed no significant increase in number of binding sites or K d after insulin stimulation. Immunocytochemical studies along with Western blot analysis of endometrial epithelial cells presented in this study demonstrate that α-1 and α-2 isoforms of the Na+-K+ ATPase are present in the basolateral membrane. Previous studies with adipocytes showed that insulin stimulation produced decreases in K 0.5 for Na+ of both the α-1 and α-2 isoforms of the pump (McGill and Guidotti 1991). The insulin-dependent decrease in Na+ K 0.5 (from 39 to 23 mM) for 86Rb+ uptake reported for the α-2 isoform was remarkably similar to pump current measurements reported in this study for endometrial epithelial cells (from 39 to 24 mM). Estimates of Vmax based on a theoretical fit of the 86Rb+ uptake data in adipocytes also suggested a twofold increase in Vmax, but this prediction was not experimentally confirmed. It was concluded from the studies of McGill and Guidotti 1991 that the fractional activity of the α-1 isoform was greater than that of the α-2 isoform in its contribution to basal pump activity in the absence of insulin. However, treatment with insulin produced selective activation of the α-2 isoform so that α-2 fractional activity was dominant under insulin-stimulated conditions. Stimulation of the α-2 isoform by insulin has also been reported in brain (Brodsky 1990). A similar mechanism of insulin action on activity of the α-2 isoform could account for both the increase in Na+ affinity and Vmax observed after insulin stimulation reported in this study for endometrial epithelial cells. However, it is worth noting that in renal epithelia, where only the α-1 isoform is present, insulin also produces a marked stimulation of the Na+-K+ ATPase.
Although insulin did not produce an acute increase in apical Na+ conductance, we found that longer-term treatment (4 d) with insulin resulted in a greater than twofold increase in apical Na+ conductance and an additional increase in pump current. One possible explanation for the enhanced Na+ absorption following longer term exposure to insulin may be related to the growth-stimulating effects of insulin on cultured cells. Although we did not examine this possibility directly, it is worth noting that no significant change in total protein content was observed in monolayers treated with insulin for 4 d compared with control monolayers under serum-free conditions. In addition, primary endometrial epithelial cells exhibit density-dependent arrest, making it less likely that the cell population would increase twofold after achieving confluence. Another possible explanation could involve regulation of Na+ channel expression at the transcriptional level that could result in an increase in apical membrane Na+ conductance. Regulation at this level would likely follow a time course on the order of hours or perhaps days that could be consistent with the longer-term actions of insulin on Na+ conductance. At present, evidence in support of this idea is not available, but insulin is known to stimulate DNA replication and transcription of other proteins involved in cell cycle regulation.
An interesting observation from this study, relating to the Na+ dependence of the Na+-K+ ATPase, is that the degree of cooperativity for Na+ ion binding is less than that previously reported for the Na+-K+ ATPase in adipocytes (Russo et al. 1990), proximal tubule epithelial cells (Feraille et al. 1994), and colonocytes (Halm and Dawson 1983). For most cells where the Na+ dependence of the pump has been determined, the Hill coefficient has a value near 2, whereas in endometrial epithelial cells, a value of 1.3 was obtained. This difference in cooperativity leads to a higher level of pump activity at lower Na+ concentrations in endometrial epithelial cells, where cooperativity is low. Reduced cooperativity combined with the increase in Na+ affinity of the pump after stimulation by insulin may be important in lowering intracellular Na+ activity within the cell. This would enhance the driving force for Na+ entry across the apical membrane through amiloride-sensitive Na+ channels. The electrogenic character of the Na+-K+ ATPase would also contribute to the driving force for Na+ entry by producing an increase in cell hyperpolarization after stimulation with insulin. Thus, we suggest that stimulation of the pump by insulin increases both the electrical and chemical driving forces for Na+ uptake across the apical membrane.
Experiments using amphotericin B to permeabilize the apical membrane demonstrated the presence of an insulin-activated, outwardly rectifying conductance in the basolateral membrane. Replacement of standard porcine Ringer solution with high KCl Ringer solution shifted the reversal potential to near 0 mV, suggesting K+ as the current-carrying ion. Insulin-dependent increases in basolateral K+ conductance presumably contributes to the electrical driving force for apical Na+ uptake and could serve to offset decreases in K+ recycling that would occur in the face of sustained hyperpolarization of the basolateral membrane.
It is generally accepted that the first step in the insulin/IGF-I signaling cascade involves receptor binding followed by stimulation of receptor-mediated tyrosine kinase activity. Previous studies in A6 cells demonstrated that insulin-stimulated Na+ transport was inhibited by tyrosine kinase inhibitors (Rodriguez-Commes et al. 1994; Record et al. 1996, Record et al. 1998). In the present study, we also demonstrated that a specific inhibitor of insulin receptor tyrosine kinase, HNMPA-(AM)3, blocked insulin-stimulated Na+ transport. This result supports the conclusion that an initial enzymatic step in insulin-dependent regulation of Na+ transport in endometrial cells involves receptor autophosphorylation. A model summarizing our hypothesis regarding post-receptor signaling events responsible for regulation of Na+-K+ ATPase transport activity in endometrial epithelial cells is presented in Fig. 14. We suggest that phosphorylation of PI-3 kinase either directly by the insulin/IGF-I receptor or by IRS-1 constitutes one of the early postreceptor phosphorylation steps that leads to pump activation. This suggestion is based on the observation that wortmannin, a selective inhibitor of PI-3 kinase activity, can completely abolish the stimulatory effects of insulin and IGF-I on Na+-K+ ATPase activity. We further speculate that PI-3 kinase, presumably acting through protein kinase B/AKT protein or through protein kinase C activates a protein phosphatase (presumably PP-1 or PP-2A), which in turn dephosphorylates the Na+-K+ ATPase, resulting in an increase in transport activity. Although we have no direct evidence to identify either PKB/AKT protein or PKC in this pathway, it is known in other systems (Cohen et al. 1997) that these serine-threonine kinases can be activated by PI-3 kinase and can modulate the activity of protein phosphatases, including PP-1 and PP-2A. Evidence supporting the involvement of a protein phosphatase in this signaling cascade comes from experiments showing that okadaic acid and calyculin A act to inhibit insulin/IGF-I–dependent activation of the pump at concentrations that block protein phosphatase activity. Previous studies using cultured L6 rat skeletal muscle cells demonstrated that pretreatment with okadaic acid and calyculin A blocked the effects of insulin on Na+-K+ ATPase activity and insulin-dependent dephosphorylation of the Na+-K+ ATPase. In addition, the presence of wortmannin also blocked insulin-stimulated PP-1 activation as well as dephosphorylation and activation of the pump (Ragolia et al. 1997). Thus, insulin appears to regulate Na+-K+ ATPase activity in L6 cells by promoting dephosphorylation of the α subunit through activation of PP-1, and PI-3 kinase appears to be involved in an earlier step in the signaling cascade. In addition, recent studies in A6 cells demonstrated that PI-3 kinase inhibitors blocked insulin-stimulated Na+ transport and insulin-stimulated PI-3 kinase activity (Record et al. 1998). The results of the present study demonstrate that Na+-K+ ATPase transport activity in primary cultures of endometrial epithelial cells is subject to regulation by insulin and IGF-I through a postreceptor signaling pathway that ultimately leads to dephosphorylation of resident pumps in the basolateral membrane. The signaling pathway responsible for insulin/IGF-I activation of the Na+-K+ ATPase in endometrial epithelial cells appears to follow a similar pattern previously proposed for L6 rat skeletal muscle cells and A6 epithelial cells.
Figure 14.
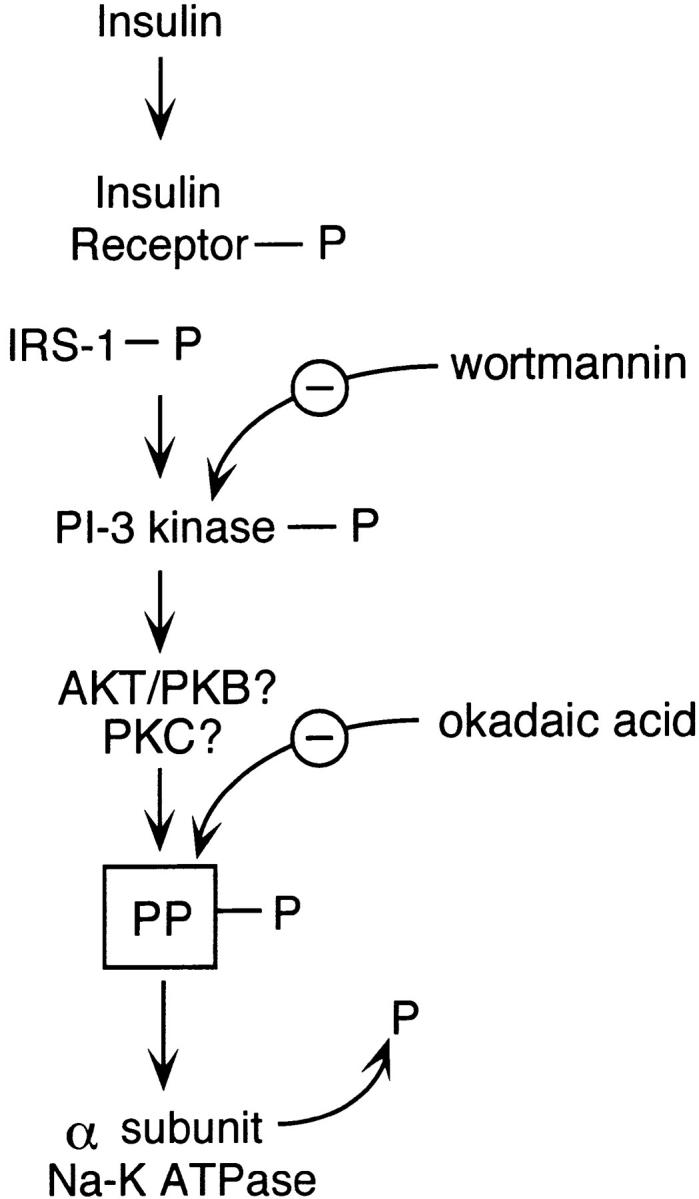
Proposed model showing the mechanism of insulin action on Na+-K+ ATPase activation in endometrial epithelial cells (discussion). IRS-1, insulin receptor substrate-1; PP, protein phosphatase; α, alpha subunit of the Na+-K+ ATPase.
Insulin and IGF-I have been previously shown to stimulate endometrial epithelial cell proliferation (Shiraga et al. 1993). Moreover, it was proposed that the growth-promoting actions of estrogen are mediated, in part, by release of IGF-I from endometrial cells (Shiraga et al. 1993). In human endometrium, IGF-I mRNA is primarily localized within stromal cells, and mRNA levels are most abundant during the proliferative phase of the cycle. IGF-I receptors are present in both stromal and epithelial cells, but are relatively more abundant in the epithelium (Zhou et al. 1994). The levels of IGF-I receptor do not appear to change during the cycle. These results suggest that the endometrial epithelium can respond to locally released IGF-I from stromal cells and that IGF-I regulation of epithelial cell growth is maximal during the proliferative phase of the cycle. In porcine uterus, IGF-I activity changes with the early development of the placenta (Persson et al. 1997). Increases in IGF-I mRNA levels within the endometrium peak at a time when conceptus estrogen secretion reaches its maximum, and decreases significantly after implantation. The role of IGF-I and insulin in endometrial epithelial cell transport function has not been previously explored. We speculate that increases in Na+ absorption in the presence of IGF-I may have some role in regulating the volume of fluid present within the uterus at the time of implantation. In porcine uterus, uterine fluid secretion is significantly stimulated just before ovulation and remains high for the next 2–3 d (Iritani et al. 1974). The increase in fluid secretion is believed to be critical in providing a fluid environment for conceptus migration and positioning along the uterine horns before implantation. Increases in IGF-I secretion before implantation may provide a means of reducing uterine fluid volume once migration is complete. Stimulation of amiloride-sensitive Na+ absorption may also account for the lower concentrations of Na+ previously measured in mammalian uterine fluid (Clemetson et al. 1972; Iritani et al. 1974). At this time, the effects of uterine fluid ionic composition on implantation are not understood, but the observation that Na+ transport can be stimulated by IGF-I suggests the possibility that regulation of uterine fluid volume and ionic composition may play some role in the process of implantation.
Footnotes
1used in this paper: IGF-I, insulin-like growth factor I; Isc, short circuit current; I–V, current–voltage; MAP, mitogen-activated protein; NMDG, N-methyl-d-glucamine; PI-3 kinase, phosphatidylinositol 3-kinase
References
- Blazer-Yost B.L., Cox M., Furlanetto J. Insulin and IGF-I receptor–mediated Na+ transport in toad urinary bladders. Am. J. Physiol. 1989;257:C612–C620. doi: 10.1152/ajpcell.1989.257.4.C612. [DOI] [PubMed] [Google Scholar]
- Brodsky J.L. Insulin activation of brain Na(+)-K(+)-ATPase is mediated by alpha 2-form of enzyme. Am. J. Physiol. 1990;258:C812–C817. doi: 10.1152/ajpcell.1990.258.5.C812. [DOI] [PubMed] [Google Scholar]
- Chan H.C., Fong S.K., Chung Y.W., Wong P.Y.D. Stimulation of anion secretion by β-adrenoreceptors in the mouse endometrial epithelium. J. Physiol. 1996;501:517–525. doi: 10.1111/j.1469-7793.1997.517bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H.C., Liu C.Q., Fong S.K., Law S.H., Wu L.J., So E., Chung Y.W., Ko W.H., Wong P.Y. Regulation of Cl secretion by ATP in cultured mouse endometrial epithelium. J. Membr. Biol. 1997;156:45–52. doi: 10.1007/s002329900186. [DOI] [PubMed] [Google Scholar]
- Civan M.M., Peterson-Yantorno K., O'Brien T.G. Insulin and phorbol ester stimulate conductive Na+ transport through a common pathway. Proc. Natl. Acad. Sci. USA. 1988;85:963–967. doi: 10.1073/pnas.85.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T., Hansen O. Active Na+–K+ transport and the rate of ouabain binding. The effect of insulin and other stimuli on skeletal muscle and adipocytes. J. Physiol. 1977;270:415–430. doi: 10.1113/jphysiol.1977.sp011959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemetson C.A.B., Kim J.K., Mallikarjuneswara V.R., Wilds J.H. The sodium and potassium concentrations in the uterine fluid of the rat at the time of implantation. J. Endocrinol. 1972;54:417–423. doi: 10.1677/joe.0.0540417. [DOI] [PubMed] [Google Scholar]
- Cohen P., Alessi D.R., Cross D.A.E. PDK1, one of the missing links in insulin signal transduction. FEBS Lett. 1997;410:3–10. doi: 10.1016/s0014-5793(97)00490-0. [DOI] [PubMed] [Google Scholar]
- Coupaye-Gerard B., Kim H.J., Singh A., Blazer-Yost B.L. Differential effects of brefeldin A on hormonally regulated Na+ transport in a model renal epithelial cell line. Biochim. Biophys. Acta. 1994;1190:449–456. doi: 10.1016/0005-2736(94)90107-4. [DOI] [PubMed] [Google Scholar]
- Deachapunya C., O'Grady S.M. Regulation of chloride secretion across porcine endometrial epithelial cells by prostaglandin E2 . J. Physiol. 1998;508:31–47. doi: 10.1111/j.1469-7793.1998.031br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorup I., Clausen T. Insulin like growth factor I stimulates active Na+–K+ transport in rat soleus muscle. Am. J. Physiol. 1995;268:E849–E857. doi: 10.1152/ajpendo.1995.268.5.E849. [DOI] [PubMed] [Google Scholar]
- Erlij D., De Smet P., Van Driessche W. Effect of insulin on area and Na+ channel density of apical membrane of cultured toad kidney cells. J. Physiol. 1994;481:533–542. doi: 10.1113/jphysiol.1994.sp020461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart N.S., Klip A. Hormonal regulation of the Na+, K+-ATPasemechanisms underlying rapid and sustained changes in pump activity. Am. J. Physiol. 1995;269:C295–C311. doi: 10.1152/ajpcell.1995.269.2.C295. [DOI] [PubMed] [Google Scholar]
- Feraille E., Carranza M.L., Rousselot M., Favre H. Insulin enhances sodium sensitivity of Na+, K+-ATPase in isolated rat proximal convoluted tubule. Am. J. Physiol. 1994;267:F55–F62. doi: 10.1152/ajprenal.1994.267.1.F55. [DOI] [PubMed] [Google Scholar]
- Fraser L.R. Requirements of successful mammalian sperm capacitation and fertilization. Arch. Pathol. Lab. Med. 1992;116:345–350. [PubMed] [Google Scholar]
- Fraser L.R. Ionic control of sperm function. Reprod. Fertil. Dev. 1995;7:905–925. doi: 10.1071/rd9950905. [DOI] [PubMed] [Google Scholar]
- Fehlmann M., Freychet P. Insulin and glucagon stimulation of Na+-K+ ATPase transport activity in isolated rat hepatocytes. J. Biol. Chem. 1981;256:7449–7453. [PubMed] [Google Scholar]
- Grinstein S., Erlij D. Unmasking of latent sodium pump sites in frog muscle by insulin. Nature. 1974;251:57–58. doi: 10.1038/251057a0. [DOI] [PubMed] [Google Scholar]
- Halm D.R., Dawson D.C. Cation activation of the basolateral sodium potassium pump in turtle colon. J. Gen. Physiol. 1983;82:315–328. doi: 10.1085/jgp.82.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera F.C. Effect of insulin on short circuit current and sodium transport across toad urinary bladder. Am. J. Physiol. 1965;209:819–824. doi: 10.1152/ajplegacy.1965.209.4.819. [DOI] [PubMed] [Google Scholar]
- Hundal H.S., Marette A., Mitsumoto Y., Ramlal R., Blostein R., Klip A. Insulin induces translocation of the α-2 and β-1 subunits of the Na+, K+-ATPase from intracellular compartments to the plasma membrane in mammalian skeletal muscle. J. Biol. Chem. 1992;267:5040–5043. [PubMed] [Google Scholar]
- Iritani A., Sato E., Nishikawa Y. Secretion rates and chemical composition of oviduct and uterine fluids in sows. J. Anim. Sci. 1974;39:582–588. doi: 10.2527/jas1974.393582x. [DOI] [PubMed] [Google Scholar]
- Klip A., Ramlal T., Cragoe E.J., Jr. Insulin-induced cytoplasmic alkalinization and glucose transport in muscle cells. Am. J. Physiol. 1986;250:C720–C728. doi: 10.1152/ajpcell.1986.250.5.C720. [DOI] [PubMed] [Google Scholar]
- Li K.-X., Sperelakis N. Electrogenic Na+-K+ pump current in rat skeletal myoballs. J. Cell. Physiol. 1994;159:181–186. doi: 10.1002/jcp.1041590122. [DOI] [PubMed] [Google Scholar]
- Lobaugh L.A., Lieberman M. Na-K pump site density and ouabain binding affinity in cultured thick heart cells. Am. J. Physiol. 1987;253:C731–C743. doi: 10.1152/ajpcell.1987.253.5.C731. [DOI] [PubMed] [Google Scholar]
- Lytton J., Lin J.C., Guidotti G. Identification of two molecular forms of Na+, K+-ATPase in rat adipocytesrelation to insulin stimulation of the enzyme. J. Biol. Chem. 1985;260:1177–1184. [PubMed] [Google Scholar]
- Matthews C.J., Redfern C.P.F., Thomas E.J., Hirst B.H. Bombesin and gastrin releasing peptide stimulate electrogenic ion transport in cultured human endometrial cell layers Exp. Physiol. 78 1993. 715 718a [DOI] [PubMed] [Google Scholar]
- Matthews C.J., McEwan G.T., Redfern C.P.F., Thomas E.J., Hirst B.H. Bradykinin stimulation of electrogenic ion transport in epithelial layers of cultured human endometrium Pflügers Arch. 422 1993. 401 403b [DOI] [PubMed] [Google Scholar]
- Marette A., Krischer J., Lavoie L., Ackerley C., Carpentier J.-L., Klip A. Insulin increases the Na+, K+-ATPase α-2 subunit in the surface of rat skeletal musclemorphological evidence. Am. J. Physiol. 1993;265:C1716–C1722. doi: 10.1152/ajpcell.1993.265.6.C1716. [DOI] [PubMed] [Google Scholar]
- Marunaka Y., Hagiwara N., Tohda H. Insulin activates single amiloride-blockable Na+ channels in a distal nephorn cell line (A6) Am. J. Physiol. 1992;263:F392–F400. doi: 10.1152/ajprenal.1992.263.3.F392. [DOI] [PubMed] [Google Scholar]
- McGill D., Guidotti G. Insulin stimulates both the α-1 and the α-2 isoforms of the rat adipocyte Na+, K+-ATPase. J. Biol. Chem. 1991;266:15824–15831. [PubMed] [Google Scholar]
- Nagamani M., Stuart C.A., Dunhardt P.A., Doherty M.G. Specific binding sites for insulin and insulin-like growth factor I in human endometrial cancer. Am. J. Obstet. Gynecol. 1991;165:1865–1871. doi: 10.1016/0002-9378(91)90047-u. [DOI] [PubMed] [Google Scholar]
- Persson E., Sahlin L., Masironi B., Dantzer V., Eriksson H., Rodriguez-Martinez H. Insulin-like growth factor-I in the porcine endometrium and placentalocalization and concentration in relation to steroid influence during early pregnancy. Anim. Reprod. Sci. 1997;46:261–281. doi: 10.1016/s0378-4320(96)01610-7. [DOI] [PubMed] [Google Scholar]
- Ragolia L., Cherpalis B., Srinivasan M., Begum N. Role of serine/threonine protein phosphatases in insulin regulation of Na+, K+-ATPase activity in cultured rat skeletal muscle cells. J. Biol. Chem. 1997;272:23653–23658. doi: 10.1074/jbc.272.38.23653. [DOI] [PubMed] [Google Scholar]
- Record R.D., Froelich L.L., Vlahos C.J., Blazer-Yost B.L. Phosphatidylinositol 3-kinase activation is required for insulin-stimulated sodium transport in A6 cells. Am. J. Physiol. 1998;274:E611–E617. doi: 10.1152/ajpendo.1998.274.4.E611. [DOI] [PubMed] [Google Scholar]
- Record R.D., Johnson M., Lee S., Blazer-Yost B.L. Aldosterone and insulin stimulate amiloride-sensitive sodium transport in A6 cells by additive mechanisms. Am. J. Physiol. 1996;271:C1079–C1084. doi: 10.1152/ajpcell.1996.271.4.C1079. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Commes J., Isales C., Kalghati L., Gasalla-Herraiz J., Hayslett J.P. Mechanism of insulin-stimulated electrogenic sodium transport. Kidney Int. 1994;46:666–674. doi: 10.1038/ki.1994.319. [DOI] [PubMed] [Google Scholar]
- Russo J.J., Manuli M.A., Ismail-Beigi F., Sweadner K.J., Edelman I.S. Na(+)-K(+)-ATPase in adipocyte differentiation in culture. Am. J. Physiol. 1990;259:C968–C977. doi: 10.1152/ajpcell.1990.259.6.C968. [DOI] [PubMed] [Google Scholar]
- Sargeant R.B., Liu Z., Klip A. Action of insulin on Na+-K+ ATPase and the Na+-K+-2Cl− cotransporter in 3T3-L1 adipocytes. Am. J. Physiol. 1995;269:C217–C225. doi: 10.1152/ajpcell.1995.269.1.C217. [DOI] [PubMed] [Google Scholar]
- Shiraga M., Takahashi S., Miyake T., Takeuchi S., Fukamachi H. Insulin-like growth factor-I stimulates proliferation of mouse uterine epithelial cells in primary culture. Proc. Soc. Exp. Biol. Med. 1993;215:412–417. doi: 10.3181/00379727-215-44152. [DOI] [PubMed] [Google Scholar]
- Siegel B., Civan M.M. Aldosterone and insulin effects on driving force of Na+ pump in toad bladder. Am. J. Physiol. 1976;230:1603–1608. doi: 10.1152/ajplegacy.1976.230.6.1603. [DOI] [PubMed] [Google Scholar]
- Suga J., Yoshimasa Y., Yamada K., Yamamoto Y., Inoue G., Okamoto M., Hayashi T., Shigemoto M, Kosaki A. Differential activation of mitogen-activated protein kinase by insulin and epidermal growth factor in 3T3-L1 adipocytesa possible involvement of PI3-kinase in the activation of the MAP kinase by insulin. Diabetes. 1997;46:735–741. doi: 10.2337/diab.46.5.735. [DOI] [PubMed] [Google Scholar]
- Talavera F., Reynolds R.K., Roberts J.A., Menon K.M.J. Insulin-like growth factor I receptors in normal and neoplastic human endometrium. Cancer Res. 1990;50:3019–3024. [PubMed] [Google Scholar]
- Walker T.C., Fidelman M.L., Watlington C.O., Biber T.U. Insulin decreases apical membrane resistance in cultured kidney cells (A6) Biochem. Biophys. Res. Commun. 1984;124:614–618. doi: 10.1016/0006-291x(84)91598-5. [DOI] [PubMed] [Google Scholar]
- Weil E., Sasson S., Gutman Y. Mechanism of insulin-induced activation of Na+-K+ ATPase in isolated rats soleus muscle. Am. J. Physiol. 1991;261:C224–C230. doi: 10.1152/ajpcell.1991.261.2.C224. [DOI] [PubMed] [Google Scholar]
- Vetter A.E., O'Grady S.M. Mechanisms of electrolyte transport across the endometrium I. Regulation by PGF2α and cAMP. Am. J. Physiol. 1996;270:663–672. doi: 10.1152/ajpcell.1996.270.2.C663. [DOI] [PubMed] [Google Scholar]
- Vetter A.E., O'Grady S.M. Mechanisms of electrolyte transport across the endometrium II. Regulation by GRP and substance P. Am. J. Physiol. 1997;273:C67–C76. doi: 10.1152/ajpcell.1997.273.1.C67. [DOI] [PubMed] [Google Scholar]
- Voldstedlund M., Tranum-Jensen J., Vinten J. Quantitation of Na+/K+ ATPase and glucose transporter isoforms in rat adipocyte plasma membrane by immunogold labeling. J. Membr. Biol. 1993;136:63–73. doi: 10.1007/BF00241490. [DOI] [PubMed] [Google Scholar]
- Zhou J., Dsupin B.A., Giudice L.C., Bondy C.A. Insulin-like growth factor system gene expression in human endometrium during the menstrual cycle. J. Clin. Endocrinol. Metab. 1994;79:1723–1734. doi: 10.1210/jcem.79.6.7527408. [DOI] [PubMed] [Google Scholar]