

The difficulties with and controversies surrounding the interpretation of electrophysiological data were highlighted in the recent Perspectives on Ion Permeation through membrane-spanning channels. The main issue was whether such data are more appropriately analyzed within the framework of chemical kinetics (Eyring's Transition State Theory; Eyring 1935) or the continuum electrodiffusional approach (Nernst-Planck Theory and its more modern extensions; see, e.g., Nonner et al. 1998). Cogent arguments were presented for both viewpoints. Their importance is in the physical insights each can generate.
In the most commonly used treatments of kinetic modeling found in the physiological literature, the Eyring expression for the rate constant is presented as:
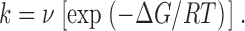 |
1 |
Here, ν is identified with the Eyring “frequency factor” (kT/h) and ΔG is the “energy barrier” associated with an elementary step in the reaction mechanism. To see why even this can be misleading, it is worthwhile to refer to Eyring's original formulation (Eyring 1935), which, for the pseudo first-order process of an ion traversing an intermediate barrier to permeation, can be written as:
 |
2 |
The unfamiliar quantities in this equation are:
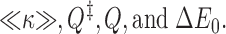 |
3 |
Here, ΔE 0 is the minimum barrier on the potential energy surface (the energy difference between a well and an adjacent peak) and 〈κ〉 is the mean transmission coefficient. 〈κ〉 measures the average likelihood that the trajectories for which the ionic energy exceeds ΔE 0 are actually reactive. Because chemical reaction is fundamentally a quantum mechanical process, there is no guarantee that, even when the ion is sufficiently energetic, it actually reacts (see, e.g., Jordan 1979). Q ‡ and Q are partition functions for the ion near a peak and a well in the potential energy surface. They measure the number (thermally weighted) of accessible states in these two separable configurations. The “reaction coordinate” has been separated out of Q ‡; it accounts for the “Eyring frequency,” kT/h. A comparison of and demonstrates that the empirical quantity ΔG really is an amalgamation of the four more basic quantities of . There is nothing wrong with defining ΔG in this way, the difficulty is in how to construe it. If an energy diagram such as that of Figure 3 A of McCleskey 1999 is one's primary interpretive guide, it can easily seduce one into equating ΔG with ΔE 0, and then quantifying it. This is clearly wrong. The kinetic barrier for an elementary step in the permeation process is a composite quantity, not simply reflecting the lowest energy required to surmount an intermediate barrier, ΔE 0. While the pre-exponential factor in has not been directly measured for ion channel kinetics (what is available are a few rough estimates of Q 10), it has been extensively studied in the chemical literature. In an illustrative series of simple binary gas phase reactions involving bi- and triatomic species, its value ranged between 100 and 0.01× the Eyring frequency (Herschbach et al. 1956), factors that translate into huge energy differences, ∼10 kT. With more complex reactants, the pre-exponential factor can be further reduced. Diagrams such as Figure 2 of McCleskey 1999 are common throughout the biochemical literature (Jencks 1969). They are not quantitated, but provide useful qualitative insights with respect to a reaction free energy; however, they cannot be reliably translated into plots of potential energy vs. reaction coordinate (in electrophysiology, the electrical distance).
There is another, more phenomenological, way to look at rate data. The Eyring expression for the rate constant can be written in an alternative form by thermodynamic analogy (see e.g., Espenson 1981):
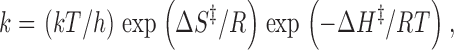 |
4 |
where ΔS ‡ and ΔH ‡ are the entropy and enthalpy of activation for the primary process. ΔG of is then recognized as ΔH ‡ − TΔS ‡. Comparing , , and , the enthalpy term is identified with ΔE 0 and the entropy term is determined by the other three quantities of . ΔS ‡ can be quite negative if the ion trajectories, even though sufficiently energetic, are predominantly nontransmittive (〈κ〉<< 1) or if there are very few thermally accessible states in the transition state near the peak in the reaction path (Q ‡ << Q, a bottleneck).
Considering the known structure for the selectivity filter in the KCsA K channel (Doyle et al. 1998), it is clear that the entropic term could be quite important indeed. In either of the two wells, the ion can rattle around quite easily because the carbonyl oxygens have formed rather nice binding pockets. For transport to occur, the carbonyl oxygens would seem to have to undergo some sort of highly concerted, relatively improbable, rearrangement. This really means a substantial reorganization, with a large amount of intermediate ordering; i.e., a large negative ΔS ‡. The associated ΔH ‡ might be quite minimal, especially as everywhere in the binding domain an ion is in fairly close contact with many carbonyl oxygens.
It should be noted in this context that translocation in gramicidin probably has a large entropic component as well. The single file involves six to eight water molecules, which must move concertedly for translocation to occur. The probability of such an event is ∼1/64 to ∼1/256 (Jordan 1984), equivalent to an energy of ∼4–6 kT. Put differently, the Eyring frequency for translocation in gramicidin is ∼100-fold less than (kT/h). To see how this would affect energy profiles, consider Na+ translocation in various gramicidin analogues; modeling the kinetics with a wells and barriers model, the rate constants are ∼107 s−1 (Becker et al. 1992). Taking at face value, this would translate into free energy barriers of ∼13 kT, of which almost half would be entropic. Model calculations on an analogue of the gramicidin interior, a periodic poly-(l,d)-alanine β helix (Roux and Karplus 1991), provide a further cautionary lesson. Na+ translocation (7.5 kT barrier) is kinetically limited, while K+ motion (1.5 kT barrier) is essentially diffusive. Their effective diffusion constants differ by ∼100, even though when gramicidin is kinetically modeled their translocation rate constants are essentially equal (Urban et al. 1980).
There is of course a “straightforward” way to distinguish entropy and enthalpy—by measuring the temperature dependence of the rate constants. This sort of experiment is called for, as it would separate the entropic and enthalpic effects. Even though it might be difficult to do (conceivably a gross understatement coming from someone who hasn't done experiments, other than culinary ones, for 40 yr), it would provide a basis for evaluating the energy scales in mechanistic schemes and truly separate the energetic and configurational terms of .
There is another issue in the kinetics-diffusion dichotomy that, while commonly accepted, may be overstated. It's not immediately obvious that the “hopping” and “diffusion” pictures can always be made equivalent by breaking a diffusive barrier into sufficiently small wells and barriers ala Läuger 1973. Maybe it is possible mathematically, but not physically. There is actually a minimum diffusional step size, on the order of the ion's mean free path. This is typically ∼0.1 Å in channels. If, anywhere along the reaction coordinate, the ion is subject to an abrupt change in energy in effecting such a differential step, it's not clear that diffusion is a good model for that piece of the path. Similarly, in regions of the reaction path where the energy profile is slowly varying, there can be genuine entropic (ordering) effects, or changes in the basic “Eyring-like” frequency. The deviation of the associated frequency factors from (kT/h) doesn't map into an energy (it does map into a free energy, but this begs the question of whether the plots are just tautologies). And the hopping analysis will be misleading.
Finally, in the diffusion picture, entropic changes along the permeation path are analogous to local variation of the diffusion constants; it is therefore a matter of concern that the PNP fits appear insensitive to local variation in D (molecular dynamics modeling suggests such variability may be substantial; Smith and Sansom 1999). In the hopping picture, if each elementary process is assigned the frequency factor kT/h, the associated energy barriers are arbitrary. Under conditions where the energy profile is flat, and entropic effects dominate (or are significant), a process with a large negative entropy of activation would equate to a very low local diffusion constant. Without measuring the temperature dependence, it is unclear how entropy and enthalpy are to be separated.
References
- Becker M.D., Koeppe R.E., II, Andersen O.S. Amino acid substitutions and ion channel function. Model-dependent conclusions. Biophys. J. 1992;62:25–27. doi: 10.1016/S0006-3495(92)81767-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle D.A., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Espenson J.H. Chemical Kinetics and Reaction Mechanisms 1981. McGraw Hill Book Co; New York, NY: pp. 218 [Google Scholar]
- Eyring H. The activated complex in chemical reactions. J. Chem. Phys. 1935;3:107–115. [Google Scholar]
- Herschbach D.R., Johnston H.S., Pitzer K.S., Powell R.E. Theoretical pre-exponential factors for twelve bimolecular reactions. J. Chem. Phys. 1956;25:736–741. [Google Scholar]
- Jencks W.P. Catalysis in Chemistry and Enzymology 1969. McGraw Hill Book Co; New York, NY: pp. 644 [Google Scholar]
- Jordan P.C. Chemical Kinetics and Transport 1979. Plenum Publishing Corp; New York, NY: pp. 368 [Google Scholar]
- Jordan P.C. The total electrostatic potential in a gramicidin channel. J. Membr. Biol. 1984;78:91–102. [Google Scholar]
- Läuger P. Ion transport through poresa rate theory analysis. Biochim. Biophys. Acta. 1973;311:423–441. doi: 10.1016/0005-2736(73)90323-4. [DOI] [PubMed] [Google Scholar]
- McCleskey E.M. Calcium channel permeationa field in flux. J. Gen. Physiol. 1999;113:765–772. doi: 10.1085/jgp.113.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonner W., Chen D.P., Eisenberg B. Anomalous mole fraction effect, electrostatics, and binding in ionic channels. Biophys. J. 1998;74:2327–2334. doi: 10.1016/S0006-3495(98)77942-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B., Karplus M. Ion transport in a gramicidin-like channeldynamics and mobility. J. Phys. Chem. 1991;95:4856–4868. [Google Scholar]
- Smith G.R., Sansom M.S.P. Effective diffusion coefficients of K+ and Cl− ions in ion channel models. Biophys. Chem. 1999;79:129–151. doi: 10.1016/s0301-4622(99)00052-6. [DOI] [PubMed] [Google Scholar]
- Urban B.W., Hladky S.B., Haydon D.A. Ion movements in gramicidin pores. An example of single-file transport. Biochim. Biophys. Acta. 1980;602:331–354. doi: 10.1016/0005-2736(80)90316-8. [DOI] [PubMed] [Google Scholar]