

Abstract
Phosphatidylinositol 4,5-bisphosphate (PIP2) activates KATP and other inward rectifier (Kir) channels. To determine residues important for PIP2 regulation, we have systematically mutated each positive charge in the COOH terminus of Kir6.2 to alanine. The effects of these mutations on channel function were examined using 86Rb efflux assays on intact cells and inside-out patch-clamp methods. Both methods identify essentially the same basic residues in two narrow regions (176–222 and 301–314) in the COOH terminus that are important for the maintenance of channel function and interaction with PIP2. Only one residue (R201A) simultaneously affected ATP and PIP2 sensitivity, which is consistent with the notion that these ligands, while functionally competitive, are unlikely to bind to identical sites. Strikingly, none of 13 basic residues in the terminal portion (residues 315–390) of the COOH terminus affected channel function when neutralized. The data help to define the structural requirements for PIP2 sensitivity of KATP channels. Moreover, the regions and residues defined in this study parallel those uncovered in recent studies of PIP2 sensitivity in other inward rectifier channels, indicating a common structural basis for PIP2 regulation.
Keywords: potassium channel, ATP, PH domain, Kir6.2, phospholipid
INTRODUCTION
ATP-sensitive potassium (KATP) channels couple cell metabolism to excitability in many tissues (Ashcroft 1998; Nichols and Lederer 1991). Normally, reconstitution of KATP current requires coexpression of a sulfonylurea receptor (SUR) subunit and an inward rectifier (Kir6) subunit (Aguilar-Bryan et al. 1995; Inagaki et al. 1995), assembled into an octameric complex of four SUR and four Kir6 subunits (Clement et al. 1997; Inagaki et al. 1997; Shyng and Nichols 1997). However, truncation of the Kir6.2 COOH terminus by 26 or 36 amino acids (Kir6.2[ΔC]) enables the formation of functional channels in the absence of SUR (Tucker et al. 1997). The Kir6.2 COOH terminus contains a signal (-RKR-) that prevents exit from the endoplasmic reticulum; SUR1 shields this retention signal and chaperones the complex to the plasma membrane (Zerangue et al. 1999).
ATP inhibits KATP channels with half-maximal inhibitory concentration (K1/2,ATP) of ∼10 μM in excised patches (Ashcroft 1998), and MgADP antagonizes this inhibition by an action on the SUR subunit (Ashcroft 1998; Nichols et al. 1996; Gribble et al. 1997; Shyng et al. 1997b). Activation of KATP channels occurs under conditions where cytoplasmic [ATP] (3–5 mM) is much higher than that required to inhibit channels in excised patches (Niki et al. 1989). The physiological regulation is mediated, at least in part, by changes in the intracellular [ATP]/[ADP] ratio, since mutations in SUR1 that abolish stimulation by MgADP remain silent, even after glucose starvation (Nichols et al. 1996; Shyng et al. 1998). Negatively charged phospholipids, particularly phosphatidylinositol-4,5-bisphosphate (PIP2), activate many, if not all, inward rectifier K channels (Hilgemann and Ball 1996; Fan and Makielski 1997; Huang et al. 1998) by increasing the open state stability of the channel. In KATP channels, application of PIP2 leads to an increase in open probability and a decrease of ATP sensitivity (Baukrowitz et al. 1998; Shyng and Nichols 1998; Fan and Makielski 1999). If membrane PIP2 levels increase, KATP channels will be rendered less sensitive to ATP, providing another potential mechanism for channel activation in physiological [ATP] (Xie et al. 1999; Shyng et al. 2000).
The antagonistic effect of PIP2 on ATP inhibition suggests that the two ligands compete functionally for interaction with the channel. The Kir6.2 subunit is primarily responsible for these interactions, since Kir6.2[ΔC] expressed without SUR1 (Tucker et al. 1997) is still inhibited by ATP and stimulated by PIP2 (Baukrowitz et al. 1998; Enkvetchakul et al. 2000). Binding of [32P]azido-ATP to Kir6.2 has been demonstrated (Tanabe et al. 1999) and involves residues in the cytoplasmic NH2 and COOH termini (Drain et al. 1998; Tucker et al. 1998; Koster et al. 1999; Tanabe et al. 1999). The molecular determinants of PIP2 activation of KATP and other Kir channels remain unclear, although several conserved COOH terminus residues are involved (Baukrowitz et al. 1998; Huang et al. 1998; Shyng and Nichols 1998; Zhang et al. 1999). The isolated COOH terminus of Kir6.2 inhibits KATP channel activity, possibly by competing for PIP2 binding (Shyng and Nichols 1998). The isolated COOH terminus of Kir1.1 has also been shown to bind to PIP2, and this binding is reduced when a putative PIP2 binding residue (K188) is neutralized (Huang et al. 1998). Mutation of the adjacent residue (R176A) in Kir6.2 dramatically reduces channel activity (Fan and Makielski 1997), and there is a much slower and more significant response to PIP2 (Baukrowitz et al. 1998; Shyng and Nichols 1998), an effect which is consistent with this mutation causing reduced affinity for PIP2.
We have now performed systematic mutagenesis and electrophysiological analysis to determine the positively charged residues in the Kir6.2 COOH terminus that are critical for PIP2 interaction. We report multiple such residues clustered in the proximal COOH terminus, and consider the possible structural basis for channel regulation by phospholipids.
MATERIALS AND METHODS
Molecular Biology
Constructs containing point mutations were prepared by overlap extension at the junctions of the relevant residues by sequential PCR. The resulting PCR products were subcloned into the pCMV6b vector. Before transfection, the constructs were sequenced to verify the correct mutations.
Expression of KATP channels in COSm6 Cells
COSm6 cells were plated at a density of ∼2.5 × 105 cells per well (30-mm six-well dishes) and cultured in Dulbecco's Modified Eagle Medium plus 10 mM glucose (DMEM-HG) supplemented with 10% FCS. The next day, cells were transfected by incubation for 4 h at 37°C in DMEM medium containing 10% Nuserum, 0.4 mg/ml diethylaminoethyl-dextran, 100 μM chloroquine, and 5 μg each of pCMV6b-Kir6.2 or mutant isoforms, pECE-SUR1 cDNA, and pECE-GFP (green fluorescent protein). Cells were subsequently incubated for 2 min in HEPES-buffered salt solution containing 10% DMSO, and returned to DMEM-HG plus 10% FCS.
86Rb+ Efflux Assay
Cells were incubated for 24 h in culture medium containing 86RbCl (1 μCi/ml) for 2–3 d after transfection. Before measurement of Rb efflux, cells were incubated for 30 min at 25°C in Krebs' Ringer solution, with metabolic inhibitors (2.5 μg/ml oligomycin plus 1mM 2-deoxy-d-glucose). At selected time points, the solution was aspirated from the cells and replaced with fresh solution. At the end of the 40-min period, cells were lysed in 2% SDS-Ringer's solution. The 86Rb+ in the aspirated solution and the cell lysates were counted. The percent efflux at each time point was calculated as the cumulative counts in the aspirated solution divided by the total counts from the solutions and the cell lysates.
Patch-Clamp Measurements
Patch-clamp experiments were made at room temperature in a chamber that allowed rapid exchange of bathing solution. Micropipettes were pulled from thin-walled glass (WPI Inc.) on a horizontal puller (Sutter Instrument, Co.). Electrode resistance was typically 0.5–1 MΩ when filled with K-INT solution (below). Inside-out patches were voltage-clamped with an Axopatch 1B amplifier (Axon Inc.). The standard bath (intracellular) and pipette (extracellular) solution (K-INT) had the following composition: 140 mM KCl, 10 mM K-HEPES, and 1 mM K-EGTA, pH 7.3. PIP2 was bath-sonicated in ice for 30 min before use. ATP was added as the potassium salt. All currents were measured at a membrane potential of −50 mV (pipette voltage = +50 mV), and inward currents at this voltage are shown as upward deflections. Data were filtered at 0.5–3 kHz, digitized at 22 kHz (Neurocorder; Neurodata), and stored on videotape. Experiments were replayed onto a chart recorder or digitized into a microcomputer using Axotape software (Axon Inc.). Offline analysis was performed using Microsoft Excel programs. Wherever possible, data are presented as mean ± SEM. Microsoft Solver was used to fit data by a least-square algorithm.
Interpretation of PIP2 Response Data
Wild-type KATP (Kir6.2 + SUR1) channels have an intrinsic open probability in the absence of ATP (Po,zero) of ∼0.4 and are inhibited by ATP with K1/2,ATP of ∼10 μM (Inagaki et al. 1995; Enkvetchakul et al. 2000). Many mutations of Kir6.2, or exposure to cytoplasmic PIP2, cause changes in Po,zero and ATP sensitivity. In almost all cases, Po,zero and K1/2,ATP are strongly correlated, and this can be explained by assuming that the action of ATP is on the closed channel, such that both Po,zero and K1/2,ATP are increased when open state stability is increased by addition of PIP2 (Shyng et al. 1997a; Baukrowitz et al. 1998; Shyng and Nichols 1998; Enkvetchakul et al. 2000). PIP2 might bind to a specific state of the channel, and mutations could affect PIP2 response by altering either the affinity or availability of a PIP2 binding site. These possibilities are experimentally unresolvable. Therefore, in the present experiments, we conclude that cytoplasmic domain mutations that reduce intrinsic activity and cause a slower and greater response to PIP2 are likely to be either involved in PIP2 binding directly or in the translation of this binding into an effect on channel open state stability.
RESULTS
Residues Critical for Regulation of Kir6.2 by PIP2
In the Kir6.2 COOH terminus (residues 173–390), there are 23 basic residues (Fig. 1 A), any or all of which might contribute electrostatically to PIP2 binding. We performed alanine scanning mutagenesis of these basic residues to determine their involvement in PIP2 sensitivity and/or ATP sensitivity. Channel activity of all mutants was initially assessed using a 86Rb+ efflux assay (Fig. 1 B). In addition to the previously recognized R176 and R177 residues (Fan and Makielski 1997; Baukrowitz et al. 1998; Shyng and Nichols 1998), the efflux was virtually abolished in three additional mutants (R206A, K222A, and R301A), and significantly reduced in the R195A and R314A mutants. The affected mutants clustered in two regions of the near COOH terminus (R176–K222 and R301–R314). There was no significant reduction of efflux in any mutants downstream of R314.
Figure 1.

Alanine scanning mutagenesis of COOH-terminal basic residues. (A) Primary sequence of Kir6.2, highlighted to show transmembrane M1 and M2 domains together with the P-loop (gray), and regions that enclose PIP2-sensitive residues (pink). All residues that were mutated in this study are indicated (bold), including those that alter PIP2 response in this study (red) or ATP sensitivity (blue). Also indicated are the PIP2-sensitive residues identified in Kir2.1/Kir3.1 by Zhang et al. 1999(orange). (B) 86Rb efflux in 40 min from COSm6 cells cotransfected with mutant Kir6.2 subunits (as indicated) + SUR1, expressed as a percentage of the efflux from cotransfected wild-type Kir6.2 + SUR1 subunits in parallel transfections (mean ± SEM, n = 3 in each case). See materials and methods for details.
Mutant channel activity was examined in more detail in inside-out membrane patches. Wild-type Kir6.2 + SUR1 channels have an intrinsic Po,zero of ∼0.4 (Inagaki et al. 1995; Enkvetchakul et al. 2000), and increasing PIP2 in the membrane increases the open state stability. In wild-type channels, PIP2 application leads to an approximately twofold increase in macroscopic channel activity as Po,zero saturates at ∼0.9 (Fig. 2A and Fig. C). As considered above, mutations that reduce apparent PIP2 affinity, either by real changes in PIP2 binding affinity or by lowering the intrinsic stability of the open pore, will lower the intrinsic Po,zero. When membrane PIP2 is increased by cytoplasmic exposure, the increase in current in these mutants should occur more slowly than wild-type, and to a relatively greater extent. The sensitivity of mutant channels to PIP2 stimulation was estimated from the time course (Fig. 2 B) and the extent (Fig. 2 C) of increase in relative current in response to cytoplasmic application of 5 μg/ml PIP2. Nine mutations were identified as having altered sensitivity to PIP2 in this assay (R176A, R177A, R195A, R206A, K222A, R301A, and R314A [identified above], plus R192A and R201A, which also show a nonsignificant reduction in Rb flux compared with wild-type). Again, there was no apparent effect of mutations downstream of R314 on PIP2 sensitivity. The correlation between mutant effects on Rb efflux and response to PIP2 indicates that, in each case, reduction in Rb efflux is likely due to a reduced sensitivity to the ambient phospholipid level in the intact cell.
Figure 2.

Mutation of certain basic residues affects channel response to PIP2. (A) Representative current recorded from inside-out membrane patch containing wild-type Kir6.2 + SUR1 subunits (WT). In this and subsequent figures, the patch was excised at the arrow, and the bars indicate the application of PIP2 (5 μg/ml, unless indicated) or ATP (as shown). The dashed line indicates zero current. Inward currents are shown as upward deflections in this and subsequent figures. (B) Time taken for 95% response to PIP2 for mutant Kir6.2 + SUR1 channels. (C) Fold increase in steady state patch current after addition of PIP2 (mean ± SEM, n = 3–9 in each case). (D) Mean K1/2,ATP for mutant Kir6.2 + SUR1 channels after patch excision (shaded) and after treatment with PIP2 (n = 2–7 in each case, mean ± SEM for mean of 3 or more). Asterisks in B–D indicate mutations for which there was no channel activity in excised patches; plus signs indicate where K1/2,ATP could not be measured reliably on inactivating mutants in D. Dashed lines in B–D indicate wild-type mean.
As shown previously (Fan and Makielski 1997; Shyng and Nichols 1998; Baukrowitz et al. 1998), the R176A mutation greatly reduces intrinsic channel activity. The response to PIP2 is markedly slower, and the relative current increase is much greater, than wild-type (Fig. 2B and Fig. C), which is consistent with reduced PIP2 affinity. The R177A mutation abolishes channel activity (Shyng and Nichols 1998). However, as shown in Fig. 3, the R177A mutant subunits can be functionally rescued by coexpression with active Kir6.2 subunits. Kir6.2 [L157C] mutants (Loussouarn et al. 2000) have high intrinsic open state stability, and are relatively insensitive to ATP, with K1/2,ATP ∼1 mM (Enkvetchakul et al. 2000). Coexpression of L157C and R177A subunits with SUR1 generated channels that were much more sensitive to ATP (Fig. 3 B; mean K1/2,ATP = 0.18 mM) than L157C + SUR1 alone. This rescue of channel activity by coexpression confirms that the R177A mutation results in channels that are closed, which is consistent with reduced PIP2 affinity, and not in any gross structural defect.
Figure 3.
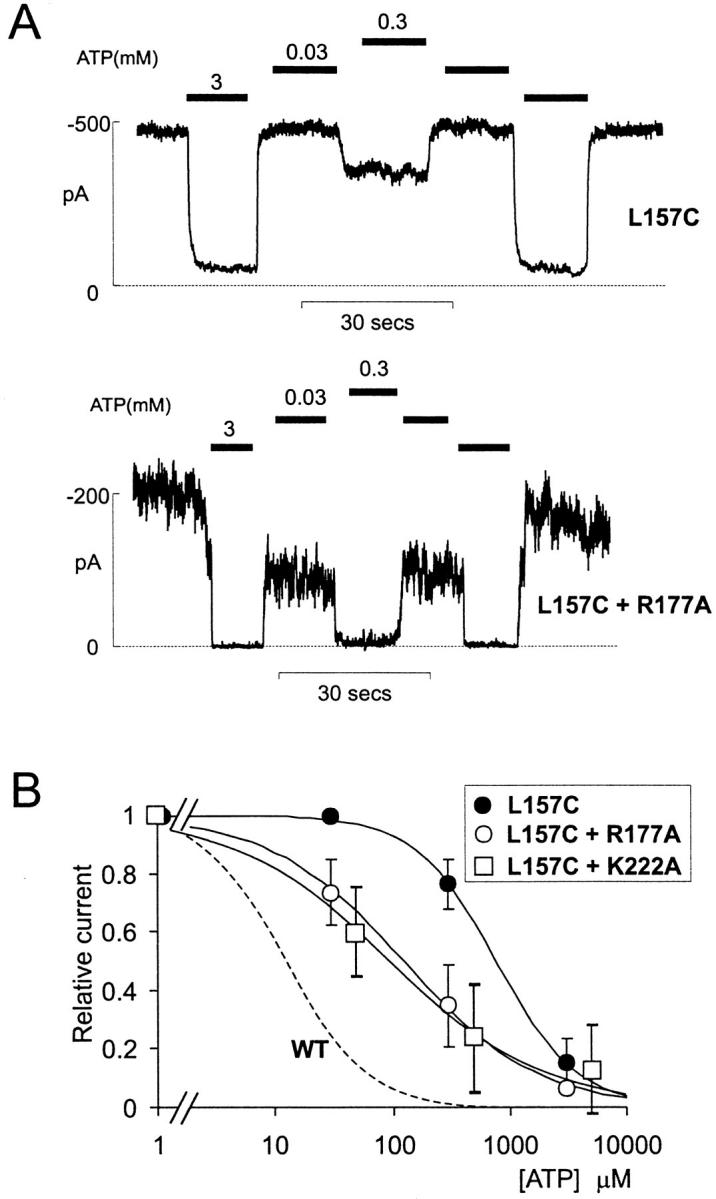
Inactive mutants can be rescued by coexpression. (A) Representative currents recorded from an inside-out membrane patch containing Kir6.2[L157C] + SUR1 channels (L157C, above) and from a patch containing mutant Kir6.2[L157C] and Kir6.2[R177A] subunits coexpressed with SUR1 (L157C + R177A, below). (B) Steady state dependence of membrane current on [ATP] (relative to current in zero ATP) for patches as in A and for coexpressed Kir6.2[L157C] and Kir6.2[K222A] subunits, with SUR1 (mean ± SEM, n = 5–7). The data were fit using a Hill equation (relative current=11+ATPK12 H), with K1/2,ATP = [ATP] causing half-maximal inhibition and H free to vary. Dashed line indicates relationship for wild-type Kir6.2 + SUR1 (WT).
Nonidentical Residues Control ATP and PIP2 Sensitivity
All expressed mutants had comparable intrinsic ATP sensitivity to wild-type channels (Fig. 2 D) except K185Q and R201A. After PIP2 stimulation, K1/2,ATP increased to between 1 and 10 mM for all mutants. Of the PIP2-sensitive residues, only R201A (Fig. 4) also affects sensitivity to ATP. Compared with wild-type channels, R201A mutant channels actually showed only moderately increased stimulation by PIP2 (Fig. 2) and no significant reduction of 86Rb efflux in intact cells (Fig. 1 B). However, R201A channels showed a significantly decreased ATP sensitivity (K i ∼115 μM; Fig. 4 B). Since the mutation reduces ATP sensitivity without increasing Po,zero, R201 is a candidate ATP binding site residue. A change in ATP binding affinity might also result in altered cooperativity between subunits, which could be an explanation for the experimentally significant reduction of the Hill coefficient (H) for inhibition by ATP (Fig. 4 B). The shift of ATP sensitivity in this mutation is similar to that observed for the K185Q mutant (Fig. 2 D), which has lower affinity for ATP binding (Tanabe et al. 1999; Tucker et al. 1998), but wild-type open state stability and PIP2 response (Fig. 2).
Figure 4.

R201A mutants alter sensitivity to PIP2 and ATP. (A and C) Representative currents recorded from inside-out membrane patch containing Kir6.2[R201A] and Kir6.2[R206A] subunits coexpressed with SUR1 subunits. (B) Steady state dependence of membrane current on [ATP] (relative to current in zero ATP) for Kir6.2[R201A] or wild-type Kir6.2 subunits coexpressed with SUR1 (mean ± SEM, n = 5–7). The data were fitted using the Hill equation, with K1/2,ATP = 12 μM (WT) and 115 μM (R201A), and H = 1.9.(WT) and 0.9 (R201A).
Like R177A, the R206A and K222A mutant channels displayed no activity after membrane excision. There was no detectable response of either mutant to a 10-min exposure to 5 μg/ml PIP2 (not shown), but R206A channel activity did appear after several minutes of exposure to a much higher concentration of PIP2 (100 μg/ml; Fig. 4 C), indicating that the lack of current after patch excision reflects a much lower open state stability. The K222A mutant showed no activity even on exposure to 100 μg/ml PIP2. However, like the R177A mutant, K222A channels could be rescued by coexpression with L157C mutants (Fig. 3 B), generating channels that were much more sensitive to ATP (Fig. 3 B; K1/2,ATP = 0.085 mM) than L157C + SUR1 alone. Again, this result indicates that the lack of current in the K222A mutant results from channels being closed, which is consistent with reduced PIP2 affinity, and not from any gross structural defect.
Some Mutants Display Prominent PIP2-sensitive Inactivation
Interestingly, three of the six mutations (R192A, R301A, and R314A) showed inactivation after removal of ATP, and this inactivation was most pronounced in the R301A mutant (Fig. 5). After patch excision, the estimated time constant of inactivation, after a step from 1 mM ATP to zero ATP, was 10.9 ± 0.5 s for R192A, 2.7 ± 0.3 s for R301A, and 19.5 ± 4.7 s for R314A (n = 4–10 patches). We previously observed similar inactivation in an M2 pore mutation (N160Q; Shyng et al. 1997a). The underlying mechanism of such ATP-dependent inactivation remains unclear, but may represent a time-dependent change in open state stability after ATP removal (i.e., a transient high stability of the open state, followed by a subsequent conformation rearrangement that leads to a reduction of open state stability and a low steady state open probability). In every case, after application of PIP2, this inactivation disappears (Fig. 5), as open state stability increases and ATP sensitivity decreases. Interestingly, the 86Rb efflux from mutants that showed inactivation in ATP-free solutions correlated with the severity of the inactivation. Efflux through the very rapidly inactivating R301A mutant was almost background, whereas efflux through R192A and R314A mutants was ∼85 and ∼70% of wild-type, respectively (Fig. 1 B). Given the stimulatory effect of MgADP in intact cells, and that PIP2 reverses the inactivation process, it is conceivable that these agents sustain activity of the less severe inactivation mutants (R192A and R314A) in intact cells.
Figure 5.

Inactivation is induced by some neutralization mutations. Representative currents recorded from inside-out membrane patch containing R192A, R301A, and R314A mutant Kir6.2 subunits coexpressed with SUR1 subunits (see Fig. 2 legend). Note that PIP2 treatment abolishes inactivation.
DISCUSSION
Distinct Residues Are Involved in Channel Gating of Kir6.2 Channels by ATP and PIP2
The mechanism of PIP2 activation of inward rectifier channels, and of ATP inhibition of KATP channels, remain elusive. It has been suggested previously that electrostatic interaction of the Kir subunit cytoplasmic domain with phospholipids in the membrane stabilizes the open state of the channel (Fan and Makielski 1997). We propose that ATP and PIP2 are negative heterotropic regulators of the Kir6.2 channels, such that binding of ATP at a nonidentical site stabilizes the closed channel (Shyng and Nichols 1998; Enkvetchakul et al. 2000). No high resolution structures are available for the cytoplasmic domains of Kir channels, and there is no homology of these domains to any known structures. Extensive mutagenesis of the Kir6.2 subunit reveals very few residues that are likely to be involved in binding ATP (Drain et al. 1998; Tucker et al. 1997; Enkvetchakul et al. 2000). In the NH2 terminus of Kir6.2, R50 is one such residue (Tucker et al. 1998) and, in the COOH terminus of Kir6.2, one particularly basic residue (K185) has been shown to affect ATP binding (Tucker et al. 1997; Koster et al. 1999; Tanabe et al. 1999). Neutralization of this residue (K185Q) had no effect on channel response to PIP2 (Fig. 2). Mutagenesis of Kir1.1, 2.1, and 3.1 channels highlights specific regions of the Kir COOH termini that are significant determinants of PIP2 sensitivity (Huang et al. 1998; Zhang et al. 1999), and the present study indicates that distinct sets of basic residues regulate the channel response to ATP and PIP2. Seven mutations caused a significant reduction of channel activity in intact cells. All of these, plus two additional mutations, were shown to reduce the sensitivity to activation by PIP2 in excised membrane patches. In every case, even when no channel activity was present in excised patches (R177A, R206A, and K222A), channel activity could be detected in coexpression or after prolonged PIP2 treatment. Thus, none of the mutations caused global structural changes that made subunits nonfunctional, instead the results are consistent with lower open state stability due to reduced PIP2 sensitivity. Only the R201A mutation affected both ATP and PIP2 interaction, reducing the sensitivity to both ligands (Fig. 2), which is consistent with the idea that each ligand interacts with separate, but possibly overlapping, sites on the same domain to stabilize either the closed (ATP) or open (PIP2) states.
Regulation of Other Inwardly Rectifying Potassium Channels by Membrane PIP2
Other inward rectifier channels in the Kir family are regulated by membrane PIP2 (Hilgemann and Ball 1996; Fan and Makielski 1997; Huang et al. 1998; Zhang et al. 1999). Using a PIP2 antibody sensitivity assay, Huang et al. 1998 showed that neutralization of R188 in Kir1.1 (equivalent to R177 in Kir6.2), enhanced sensitivity of channel activity to PIP2 antibodies and reduced PIP2 binding to the purified Kir1.1 COOH terminus. Several other residues contribute to differential interaction of PIP2 with Kir3.4 (GIRK4) and Kir2.1 (IRK1), including the K207-L245 region of Kir2.1 (corresponding to R196-L233 in Kir6.2). When this segment of Kir2.1 is introduced in place of the corresponding segment into Kir3.4, the resultant chimera shows Kir2.1-like sensitivity to PIP2 antibodies (Zhang et al. 1999).
The regions equivalent to residues 206–222 in Kir6.2 have been mutated in several studies on various Kir channels. E224 in Kir2.1 (S212 in Kir6.2) is a major determinant of the affinity for pore-blocking polyamines (Yang et al. 1995). The neighboring residue (H216) in Kir6.2 also controls pH dependence of polyamine block (Baukrowitz et al. 1999), and a recent cysteine scanning mutagenesis of this region in Kir2.1 (Lu et al. 1999) indicates that several residues within it are accessible to methyl-thio-sulfhydryl reagents. Together, these results are consistent with this region being accessible to permeant ions and forming part of the cytoplasmic entrance to the pore. However, many mutations in the pore-lining M2 region also cause changes in Po,zero (Loussouarn et al. 2000) and alter the responsiveness to PIP2 (Enkvetchakul et al. 2000). Since these residues are clearly lining the pore within the membrane itself, they presumably cannot interact with phospholipid headgroups. In the latter case, we can reasonably conclude that the effect of the mutation is on intrinsic pore stability (i.e., transduction of PIP2 action) rather than on PIP2 binding. Based on the above evidence that the 206–222 region is likely also to be pore-lining, and therefore, unlikely to be interacting directly with membrane PIP2 headgroups, we speculate that the effects of mutations (e.g., R206A and R222A) in this region on PIP2 sensitivity also result from alterations in open pore stability rather than on PIP2 binding.
Critical residues involved in PIP2 regulation are conserved, or appropriate changes in PIP2 sensitivity are observed, when such residues are introduced to different Kir channels (Huang et al. 1998; Zhang et al. 1999). Although the physiological behaviors of Kir channel family members appear quite different (e.g., strong inward rectifiers in the Kir2 subfamily, G-protein–gated channels in the Kir3 family, and KATP channels in the Kir6 family), there is a growing convergence of their fundamental molecular nature. All contain essentially similar pore structures, and strong versus weak rectification can be conferred or removed by one or two point mutations within the pore. All are activated by PIP2, and homologous residues control open state stability and PIP2 sensitivity. One question that arises is: Why are only Kir6 subfamily members ATP-sensitive? Recent studies demonstrate that the relatively high ATP sensitivity of Kir6.2 channels is labile; point mutations can render the channel very ATP-insensitive (Tucker et al. 1997, Tucker et al. 1998; Drain et al. 1998; Trapp et al. 1998; Enkvetchakul et al. 2000; Loussouarn et al. 2000), and any mutation, or manipulation, that increases open state stability reduces the apparent ATP sensitivity. Accordingly, while the lack of ATP sensitivity in other inward rectifiers might be due to the lack of an appropriate binding site, it may be simply a consequence of altered pore stability. Interestingly, there have been reports of weak ATP sensitivity of several Kir subfamily members, including Kir2.3 (Collins et al. 1996), Kir1.1 (McNicholas et al. 1996), and Kir4.1 (Bredt et al. 1995).
The present study was undertaken without any knowledge, or presupposition, of the overall structure of the cytoplasmic domain. In the absence of any homology to known structures, we performed a prediction of the secondary structure of the COOH terminus of Kir channels using multiple alignments of the primary sequences (PHD program [Rost 1996]). This prediction indicates that the first ∼150 amino acids of the COOH termini of all Kir channels are likely to contain seven antiparallel β-strands with an α-helix at the COOH-terminal end. These are characteristics of pleckstrin homology (PH) domains (Lemmon et al. 1996; Shaw 1996; Rebecchi and Scarlata 1998), which are found in many PIP2-interacting proteins. In such proteins, multiple positively charged residues in the loops between the β-strands are responsible for the interaction with PIP2. Although present in a large number of signaling molecules, PH domains are elusive and difficult to recognize on the basis of sequence homology, and have not been reported in any ion channels. A high resolution structure of Kir channel cytoplasmic domains may well soon appear. However, until such a time, although mere speculation, the possibility that the PIP2 sensitivity of inward rectifiers arises from a PH-like domain should perhaps be considered.
Acknowledgments
We are grateful to the Washington University Diabetes Research and Training Center for continued molecular biology supplies.
This work was supported by a career development grant from the American Diabetes Association (to S.L. Shyng) and grants HL45742 and HL54171 from the National Institutes of Health (to C.G. Nichols).
Footnotes
Abbreviations used in this paper: KATP, ATP-sensitive potassium; PH, pleckstrin homology; PIP2, phosphatidylinositol 4,5-bisphosphate; SUR, sulfonylurea receptor.
Wild-type channels have a Po,zero of ∼0.45 under normal conditions, and this rises to a maximum of ∼0.9 after addition of PIP2, so the macroscopic relative current approximately doubles. Mutations that reduce the apparent PIP2 affinity will reduce the ambient Po,zero, and this means that the potential increase of Po,zero, after addition of PIP2, is greater. However, unless PIP2 efficacy is reduced, or the affinity is reduced so far that it becomes impossible to add sufficient PIP2 (as may in fact be the case for R206A), Po,zero would still rise eventually to the same saturating value (∼0.9). Hence, the increase in the relative current will be greater, but take longer, than in wild-type channels.
References
- Aguilar-Bryan L., Nichols C.G., Wechsler S.W., Clement J.P., IV, Boyd A.E., III, Gonzalez G., Herrera-Sosa H., Nguy K., Bryan J., Nelson D.A. Cloning of the β-cell high affinity sulfonylurea receptora regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Ashcroft F.M. Adenosine 5′-triphosphate-sensitive potassium channels. Annu. Rev. Neurosci. 1998;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T., Schulte U., Oliver D., Herlitze S., Krauter T., Tucker S.J., Ruppersberg J.P., Fakler B. PIP2 and PIP as determinants for ATP inhibition of K-ATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T., Tucker S.J., Schulte U., Benndorf K., Ruppersberg J.P., Fakler B. Inward rectification in KATP channelsa pH switch in the pore. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:847–853. doi: 10.1093/emboj/18.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt D.S., Wang T.L., Cohen N.A., Guggino W.B., Snyder S.H. Cloning and expression of two brain-specific inwardly rectifying potassium channels. Proc. Natl. Acad. Sci. USA. 1995;92:6753–6757. doi: 10.1073/pnas.92.15.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement J.P., IV, Kunjilwar K., Gonzalez G., Schwanstecher M., Panten U., Aguilar-Bryan L., Bryan J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Collins A., German M.S., Jan Y.N., Jan L.Y., Zhao B. A strongly inwardly rectifying K+ channel that is sensitive to ATP. J. Neurosci. 1996;16:1–9. doi: 10.1523/JNEUROSCI.16-01-00001.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain P., Li L.H., Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C-terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul D., Loussouarn G., Makhina E., Shyng S.-L., Nichols C.G. The kinetic and physical basis of KATP channel gatingtowards a unified molecular understanding. Biophys. J. 2000;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z., Makielski J.C. Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 1997;272:5388–5395. doi: 10.1074/jbc.272.9.5388. [DOI] [PubMed] [Google Scholar]
- Fan Z., Makielski J.C. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J. Gen. Physiol. 1999;114:251–269. doi: 10.1085/jgp.114.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble F.M., Tucker S.J., Ashcroft F.M. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D.W., Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2 . Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- Huang C.L., Feng S.Y., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by G-beta-gamma. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Inagaki N., Gonoi T., Clement J.P., IV, Namba N., Inazawa J., Gonzales G., Aguilar-Bryan L., Seino S., Bryan J. Reconstitution of IKATPan inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N., Gonoi T., Seino S. Subunit stoichiometry of the pancreatic β-cell ATP-sensitive K+ channel. FEBS Lett. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- Koster J.C., Sha Q., Shyng S.-L., Nichols C.G. ATP inhibition of KATP channelscontrol of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J. Physiol. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M.A., Ferguson K.M., Schlessinger J. PH domainsdiverse sequences with a common fold recruit signaling molecules to the cell surface. Cell. 1996;85:621–624. doi: 10.1016/s0092-8674(00)81022-3. [DOI] [PubMed] [Google Scholar]
- Loussouarn G., Makhina E.N., Rose T., Nichols C.G. Structure and dynamics of the pore of inward rectifier KATP channels. J. Biol. Chem. 2000;275:1137–1144. doi: 10.1074/jbc.275.2.1137. [DOI] [PubMed] [Google Scholar]
- Lu T., Zhu Y.G., Yang J. Cytoplasmic amino and carboxyl domains form a wide intracellular vestibule in an inwardly rectifying potassium channel. Proc. Natl. Acad. Sci. USA. 1999;96:9926–9931. doi: 10.1073/pnas.96.17.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas C.M., Yang Y., Giebisch G., Hebert S.C. Molecular site for nucleotide binding on an ATP-sensitive renal K+ channel (ROMK2) Am. J. Physiol. 1996;271:F275–F285. doi: 10.1152/ajprenal.1996.271.2.F275. [DOI] [PubMed] [Google Scholar]
- Nichols C.G., Lederer W.J. ATP-sensitive potassium channels in the cardiovascular system. Am. J. Physiol. 1991;261:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- Nichols C.G., Shyng S.-L., Nestorowicz A., Glaser B., Clement J., IV, Gonzalez G., Aguilar-Bryan L., Permutt A.M., Bryan J.P. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Niki I., Ashcroft F.M., Ashcroft S.J. The dependence on intracellular ATP concentration of ATP-sensitive K-channels and of Na,K-ATPase in intact HIT-T15 beta-cells. FEBS Lett. 1989;257:361–364. doi: 10.1016/0014-5793(89)81572-8. [DOI] [PubMed] [Google Scholar]
- Rebecchi M.J., Scarlata S. Pleckstrin homology domainsa common fold with diverse functions. Annu. Rev. Biophys. Biomol. Struct. 1998;27:503–528. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- Rost B. PHDpredicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol. 1996;266:525–539. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- Shaw G. The pleckstrin homology domainan intriguing multifunctional protein module. Bioessays. 1996;18:35–46. doi: 10.1002/bies.950180109. [DOI] [PubMed] [Google Scholar]
- Shyng S.-L., Nichols C.G. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S.-L., Nichols C.G. Phosphatidyl inositol phosphates control of nucleotide-sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Shyng S.-L., Ferrigni T., Nichols C.G. Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit J. Gen. Physiol. 110 1997. 141 153a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S.-L., Ferrigni T., Nichols C.G. Regulation of KATP channel activity by diazoxide and MgADPdistinct functions of the two nucleotide binding folds of the sulfonylurea receptor J. Gen. Physiol. 110 1997. 643 654b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S.-L., Ferrigni T., Shepard J.B., Nestorowicz A., Glaser B., Permutt M.A., Nichols C.G. Functional analyses of novel mutations of the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes. 1998;47:1145–1151. doi: 10.2337/diabetes.47.7.1145. [DOI] [PubMed] [Google Scholar]
- Shyng S.-L., Barbieri A., Gumusboga A., Cukras C., Pike L., Davis J.N., Stahl P.D., Nichols C.G. Modulation of nucleotide sensitivity of KATP channels by PI-4-P 5-kinase. Proc. Natl. Acad. Sci. USA. 2000;97:937–941. doi: 10.1073/pnas.97.2.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K., Tucker S.J., Matsuo M., Proks P., Ashcroft F.M., Seino S., Amachi T., Ueda K. Direct photoaffinity labeling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. J. Biol. Chem. 1999;274:3931–3933. doi: 10.1074/jbc.274.7.3931. [DOI] [PubMed] [Google Scholar]
- Trapp S., Proks P., Tucker S.J., Ashcroft F.M. Molecular analysis of KATP channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 1998;112:333–350. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker S.J., Gribble F.M., Zhao C., Trapp S., Ashcroft F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Tucker S.J., Gribble F.M., Proks P., Trapp S., Ryder T.J., Haug T., Reimann F., Ashcroft F.M. Molecular determinants of K-ATP channel inhibition by ATP. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L.-H., Horie M., Takano M. Phospholipase C-linked receptors regulate the ATP-sensitive potassium channel by means of phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. USA. 1999;96:15292–15297. doi: 10.1073/pnas.96.26.15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Jan Y.N., Jan L.Y. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 1995;14:1047–1054. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]
- Zerangue N., Schwappach B., Jan Y.N., Jan L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- Zhang H.L., He C., Yan X.X., Mirshahi T., Logothetis D.E. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P-2 interactions. Nat. Cell Biol. 1999;1:183–188. doi: 10.1038/11103. [DOI] [PubMed] [Google Scholar]