

Abstract
The mechanism by which physiological signals regulate the conformation of molecular gates that open and close ion channels is poorly understood. Voltage clamp fluorometry was used to ask how the voltage-sensing S4 transmembrane domain is coupled to the slow inactivation gate in the pore domain of the Shaker K+ channel. Fluorophores attached at several sites in S4 indicate that the voltage-sensing rearrangements are followed by an additional inactivation motion. Fluorophores attached at the perimeter of the pore domain indicate that the inactivation rearrangement projects from the selectivity filter out to the interface with the voltage-sensing domain. Some of the pore domain sites also sense activation, and this appears to be due to a direct interaction with S4 based on the finding that S4 comes into close enough proximity to the pore domain for a pore mutation to alter the nanoenvironment of an S4-attached fluorophore. We propose that activation produces an S4–pore domain interaction that disrupts a bond between the S4 contact site on the pore domain and the outer end of S6. Our results indicate that this bond holds the slow inactivation gate open and, therefore, we propose that this S4-induced bond disruption triggers inactivation.
Keywords: Shaker, rearrangement, voltage, gating, fluorescence
INTRODUCTION
After depolarization of the membrane, voltage-dependent ion channels undergo a series of conformational changes that open and close several distinct molecular gates that regulate transmembrane ion flux. All of these gates must be open for ions to flow through the pore and cross the membrane. Depolarization first activates channels by displacing the positively charged S4, the fourth transmembrane segment, of each of the channels' four subunits. This displacement generates the gating current by carrying basic residues outward across the membrane electric field (Yang and Horn 1995; Aggarwal and MacKinnon 1996; Larsson et al. 1996; Seoh et al. 1996; Yusaf et al. 1996; Starace et al. 1997; Baker et al. 1998), while undergoing what appears to be a 180° helical twist (Cha et al. 1999; Glauner et al. 1999). Activation is followed by the opening of the activation gate and the closure of one or more inactivation gates (for review see Yellen 1998). In most channels, the activation gate opens more quickly than the inactivation gates close, leading to a transient current, or, if the inactivation gates do not close, to a current that is sustained for the duration of the depolarization. For the gates to return to their resting states (closed activation gate and open inactivation gates) the membrane must be repolarized.
Inactivation occurs via two distinct mechanisms. Fast (N-type) inactivation occurs in milliseconds after the opening of the activation gate because of a block of the internal mouth of the pore by the NH2-terminal domain of the protein (Hoshi et al. 1990; Zagotta et al. 1990; Isacoff et al. 1991; Timpe et al. 1988). Slow inactivation usually occurs over a much longer time scale than N-type inactivation. It takes place in at least two steps, one which closes the gate (P-type inactivation), and a subsequent step which stabilizes the closed conformation (C-type inactivation) by shifting the voltage dependence of recovery from inactivation and the return of S4 to its resting conformation (De Biasi et al. 1993; Olcese et al. 1997; Yang et al. 1997; Loots and Isacoff 1998)
Slow inactivation is sensitive to mutation of the pore region (P) and S6, as well as S4 (Timpe et al. 1988; Iverson and Rudy 1990; Hoshi et al. 1991; Lopez-Barneo et al. 1993; Boland et al. 1994; Kupper et al. 1995; Olcese et al. 1997; Yang et al. 1997; Molina et al. 1998; Ogielska and Aldrich 1998; Perez-Cornejo 1999; Mitrovic et al. 2000), and appears to involve protein motions in and around those domains (Boland et al. 1994; Liu et al. 1996; Cha and Bezanilla 1997; Basso et al. 1998; Loots and Isacoff 1998). The gate appears to close by pinching shut the outer mouth of the pore at the end of the selectivity region of P (Boland et al. 1994; Liu et al. 1996; Doyle et al. 1998; Harris et al. 1998). For the slow inactivation gate to close, a K+ binding site near the outer end of the selectivity filter must first be evacuated (Lopez-Barneo et al. 1993; Baukrowitz and Yellen 1996). The subsequent closure does not prevent conductance entirely, but severely attenuates it and alters its selectivity, which is consistent with a rearrangement at one or more K+ binding sites (Starkus et al. 1997, Starkus et al. 1998; Harris et al. 1998; Kiss et al. 1999; Ogielska and Aldrich 1999).
Although the mechanism of slow inactivation is inherently voltage-independent, it follows voltage because of its dependence on activation (Olcese et al. 1997; Loots and Isacoff 1998). The mechanism of coupling of voltage sensing to the conformation of the gate is not understood. Does S4 interact directly with the pore domain (S5, P-region, and S6)? Does S4 participate in inactivation? How can S4 motion, presumably occurring far from the central axis of the pore, permit closure of the selectivity filter?
Recent work has used scanning mutagenesis to identify protein–protein interaction surfaces in voltage-gated K+ channels via perturbation of the voltage dependence of activation (Li-Smerin et al. 2000a,Li-Smerin et al. 2000b; Monk and Miller, 1999). One of the studies (Li-Smerin et al. 2000b) identified a surface on the outer end of the pore domain that appears to interact with the voltage-sensing domain without, however, determining whether the interaction is with S4 or with S1, S2, or S3, which is the other membrane segments that form the voltage-sensing domain along with S4. Other work has shown that S4 is located close enough to the pore to be in direct contact with the outer edge of the pore domain (Blaustein et al. 2000), indicating that part of the interaction surface identified by Li-Smerin et al. 2000b likely does contact S4.
Our present results provide two new classes of evidence that S4 and the pore domain interact directly and show that this is a dynamic interaction that changes with gating. The results demonstrate that fluorophores attached to both S4 and the pore domain change fluorescence in parallel with both activation and slow inactivation. To determine whether S4 and the pore domain interact directly, and where, we mutated residues in one domain to see if this influenced the environment of a fluorophore attached to the other domain, a method we call environment scanning. This technique is similar to that described by Sorensen et al. 2000, in which they characterize two quenching groups in the S3–S4 linker of a fluorophore attached to S4. However, in our study, instead of using protein deletions, we used size-conserving neutralizations of single residues and, thus, reduced the likelihood of large-scale perturbations of channel structure that would confound the interpretation. Our results are consistent with a direct S4–pore domain interaction. What is the functional consequence of S4–pore domain interaction? We examined as a possible target of S4 interaction candidate bonds based on the KcsA crystal structure, which involve one residue located near the apparent site on the pore domain of S4 contact in S5 and partner residues in S6. We found that closure of the inactivation gate occurs under two different conditions: (a) with the specific mutation of either the S5 or S6 residue; or (b) by the addition of a side chain adduct that binds specifically to one of the interaction partners, inhibiting formation of the bond. This suggests that S4 motion directly triggers inactivation by destabilizing specific S5-S6 bonds, leading to the closure of the outer mouth of the pore.
MATERIALS AND METHODS
Molecular Biology
Site-directed mutagenesis was done by the use of Quick Change mutagenesis kits from Stratagene and examined either by 35S or fluorescence sequencing. Unless otherwise denoted, the standard composition of the channel was Shaker H4 (Δ6-46/W434/C245V/C462A) (Kamb et al. 1987; Hoshi et al. 1990; Mannuzzu et al. 1996).
cRNA was transcribed using T7 Ambion mMessage mMachine. Injection of the oocytes (50 nl mRNA at 1 ng/nl), native cysteine blocking with tetraglycine maleimide, and the attachment of the fluorescent fluorophore 6′-tetramethylrhodamine maleimide (TMRM) were performed as previously described (Mannuzzu et al. 1996). In brief, 3–4 d after injection and incubation at 12°C, oocytes were incubated for 1 h at 22°C in tetraglycine maleimide to block native cysteines at room temperature. After 14 h of incubation at room temperature, oocytes were washed and labeled with 50 μM TMRM in a high potassium solution (in mM: 92 KCl, 0.75 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.5; 30 min on ice) and kept in this solution (in the dark at 12°C) until being voltage-clamped at room temperature.
Voltage Clamp Fluorometry and Analysis
Two-electrode voltage clamp fluorometry was performed as described previously (Mannuzzu et al. 1996) using a Dagan CA-1 amplifier (Dagan Corporation), which was illuminated with a 100-W mercury arc lamp, on a Zeiss IM35 microscope, using a 20× 0.75 NA fluorescence objective (Nikon). Photometry was performed with a Hamamatsu HC120-05 photomultiplier tube. The voltage clamp, photomultiplier, and Uniblitz shutter (Vincent Associates) were digitized and controlled by a Digidata-2000 board and PClamp7 or a Beta version of PClamp8 software, respectively (Axon Instruments).
The bath solutions consisted of the following (in mM): 110 NaMes, 2 KMes, 2 CaMES2, 10 HEPES, pH 7.5; or 110 KMes, 2 CaMES2, and 10 HEPES, pH 7.5. Oocytes were washed before being placed in the bath, and the bath solution was constantly perfused through the chamber during recording if kinetics were required. The excitation light was reduced by neutral density filters (Carl Zeiss) with either a 5% transmission or with two 5% neutral density filters in series resulting in 1.25% transmission. Light was filtered with an HQ 535/50 excitor, an HQ 610/75 emitter, and a 565LP dichroic (Chroma Technology). The voltage output of the photomultiplier was low pass–filtered at one quarter the sampling frequency with an 8-pole Bessel filter (Frequency Devices). A minimum of a 2-min rest interval at the holding potential of −80 mV was given between steps to +40 mV. Solutions were exchanged until current–voltage relations indicated no further change in reversal potential (usually 10 min). All measurements on conducting (424C-TMRM/W434) channels were made under continuous perfusion. Data analysis was done with the Clampfit programs of PClamp6 and the Beta version of PClamp8 (Axon Instruments) and, in some cases, analyzed further and plotted with Origin (Microcal). Fits to the ionic current were made starting after the capacitive transient (∼1.5 ms after the start of the step) for current activation and partway into inactivation (∼50–100 ms after the start of the step). Fluorescence and current traces were normalized and were not averaged unless so noted. All values are mean ± SEM.
Normalization of Fluorescence Change
Normalizing to account for differing levels of channel expression is a potentially difficult process. To take into account differences in the level of expression of various mutants, changes in the ΔF (ΔΔFs) because of mutations in the protein environment of the fluorophore were normalized by two different methods. The methods gave similar and consistent ΔΔFs. All channel constructs were examined in the W434F background (Perozo et al. 1993) and ΔFs were normalized by their gating charge (integrated IgOFF). Normalization by the conductance of low numbers of W434 conducting channels yielded similar numbers (data not shown). ΔFs were also normalized by comparing the baseline fluorescence at –80 mV after subtracting the background fluorescence of an oocyte that had been injected with conducting wild-type Shaker channels without any introduced cysteines. Normalizing by gating charge (or low level of conducting channels) for a fluorophore at position 359C (359C-TMRM) yielded a change of fluorescence of 28 ± 8.2%, 22 ± 2.2%, 6.98 ± 3.2%, and 16.9 ± 2.3% for E418Q, E422Q, D431N, and D447N, respectively; normalization by baseline fluorescence yielded ΔFs of 23 ± 4.7%, 23 ± 4.8%, 6.3 ± 0.37%, and 11.6 ± 1.6%. Normalization by the gating charge of nonconducting (W434F) versions of 424C-TMRM yielded ΔFs of 6.25 ± 1.1%, 10.2 ± 5.9%, 0.8 ± 0.18%, and 7.63 ± 0.5% for E418,Q E422Q, D431N, and D447N, respectively, whereas normalizing by baseline fluorescence gave ΔFs of 5.98 ± 1.4%, 8.06 ± 3.5%, 0.42 ± 0.2%, and 5.68 ± 1.1%, respectively.
RESULTS
We set out to understand how voltage sensing and gating are coupled at the molecular level. We used fluorescence to examine protein motions in S4 and the pore domain (S5, P-region, and S6). We reasoned that if a site in S4 couples to inactivation then it may experience a change in its local environment not only when S4 moves through the surrounding protein during its transmembrane charge translocation (voltage sensing) step, but also during the structural changes that underlie the slow closure of the inactivation gate. Although previous studies have suggested a role for S4 in slow inactivation, they have been restricted to either functional effects of mutations (Mitrovic et al. 2000) or to observations of inactivation-correlated fluorescence changes at a single attachment site that could not distinguish between S4 motion and motion of a neighboring segment (Loots and Isacoff 1998). Here, we examined fluorescence at a series of sites and, for the first time, we measured protein motions with fluorophores attached at the perimeter of the pore domain, where S4 has been suggested to contact the pore domain (Blaustein et al. 2000; Li-Smerin et al. 2000b). We used the fluorophore TMRM, which we attached site-specifically to cysteines introduced singly into either S4 or the pore domain. TMRM senses the chemical nature of its environment and rapidly responds to changes in local structure by changing its fluorescence (Mannuzzu et al. 1996).
All of the experiments were carried out in the NH2-terminal ball-deleted and external cysteine-removed channel, so that the only form of inactivation was slow inactivation, and where the only TMRM attachment site was the introduced cysteine (see materials and methods). The depolarizing steps were long enough to close the inactivation gate (P-type inactivation), but not long enough to stabilize the closed state (C-type inactivation; see materials and methods).
Fluorescence Analysis of the Outer End of S4
We began with an examination of a series of positions (359–362) at the outer end of S4. The most NH2-terminal (and external) of these sites (359) is buried in the gating canal in the resting state when S4 is retracted into the cell (Larsson et al. 1996; Yusaf et al. 1996; Baker et al. 1998). This entire NH2-terminal end of S4 appears to be exposed to the extracellular solution in the activated state, and probably is already exposed by the first outward step of S4 (Baker et al. 1998).
A previous characterization of the fluorescence of TMRM attached to one position (359C) demonstrated that the fluorophore's environment undergoes two very different phases of change after membrane depolarization: one that correlates with fast gating charge movement and a second that correlates with the slow closure of the external inactivation gate (Loots and Isacoff 1998). From the observation at a single site, the earlier study could not distinguish between a possible inactivation motion of S4 itself or motion of the domains surrounding S4 during inactivation. We now reexamined the fluorescence of TMRM attached to 359 and also tested each of the next three positions (360, 361, and 362). The idea was that whatever the secondary structure of S4 (for evidence of a helical structure see Peled-Zehavi et al. 1996; Li-Smerin, et al. 2000a) TMRM, attached to a series of consecutive positions, should sample the environment all around S4. If S4 undergoes an inactivation motion then positions projecting in every direction from S4 would be expected to sense an inactivation environment change; however, if the readout of the fluorophore at 359C is due to a motion of a neighboring protein segment then only fluorophores attached to S4 sites that point at that neighboring segment should display a slow inactivation component.
Fluorescence traces for the four consecutive TMRM attachment sites are shown in Fig. 1. At each of the sites, a depolarizing step evoked a large fluorescence change (ΔF) with an onset (ΔFon) that paralleled the onset of inactivation of the ionic current (Fig. 1; also see legend). At 359 and 362, the inactivation component of the ΔFon was preceded by a prominent fast component that paralleled the opening of the activation gate (Fig. 1 B). In nonconducting channels, the fast ΔFon for these sites has been shown to follow the gating current (Cha and Bezanilla 1997). This is consistent with S4 extrusion being rate-limiting for channel opening at moderate depolarizations, like the voltage tested here.
Figure 1.

A fluorophore attached to S4 reports on both activation and inactivation rearrangements. (A) TMRM at four consecutive sites in S4 undergoes one or two steps of change in environment resulting in a ΔF. The ΔF at 359 and 362, has both fast and slow components, whereas at 360 and 361, it is purely slow. Cartoon illustrates the relative location of each side chain for an α-helix. The apparent delay in ΔFon of 360 is due to a small downward transient. (B) Comparison of ionic current (I) and fluorescence (F) reveals a correlation of the fast ΔF with channel opening, and the slow ΔF with inactivation. Black traces superimposed onto the F traces have the time constants of single exponential fits to the rise or inactivation of the current in response to steps to +40 mV. The correlation between the rate of inactivation (Inactrate) and rate of slow ΔFon (ΔFon-rate) was high, with a ratio of nearly 1.00 for all of the sites: [(Inactrate)/(slow ΔFon-rate)] = 1.06 ± 0.06 (n = 8); 0.97 ± 0.08 (n = 7); 0.89 ± 0.12 (n = 9); 1.00 ± 0.09 (n = 8); for sites 359, 360, 361, and 362, respectively (mean ± SEM [number of oocytes]). Note that the differences in inactivation kinetics are due to site-specific effects of the cysteine substitution and TMRM attachment.
Surprisingly, at the intervening sites (360 and 361), ΔFon was dominated by an inactivation-coupled component. The activation component was less than signal noise in a single sweep and was difficult to detect without averaging. In addition, at 361, the foot of the ΔFon was distorted by a component with intermediate kinetics, revealing an intermediary conformational step between activation and closure of the gate.
The common feature of all of the sites is that TMRM experiences a change in environment with closure of the inactivation gate. The detection by the fluorophore of the inactivation motion in all directions projecting from S4 is consistent with either a rigid body motion of S4 occurring during inactivation or a cooperative inactivation rearrangement of the entire protein surrounding S4.
The difference between the ΔFs for these four neighboring sites indicates that TMRM occupies a distinct molecular environment at each residue. The rough resemblance in kinetics between 359 and 362, on one hand, and 360 and 361, on the other (Fig. 1 A), suggests that TMRM faces the same direction, which is consistent with an α-helical structure for S4.
Fluorescence Analysis of an Outer Edge of the Pore Domain
Is there a functional tertiary interaction between S4 and the perimeter of the pore domain? We examined as a possible contact region the outer end of S5. Based on the KcsA crystal structure (Doyle et al. 1998), this region is predicted to be helical and to lie at the farthest point from the central axis, about midway along the perimeter of the pore domain from the subunit interfaces (Li-Smerin et al. 2000a; see Fig. 2B and Fig. C). As with S4, we examined four consecutive positions: 416–419.
Figure 2.

A fluorophore attached to the pore domain reports on both activation and inactivation rearrangements. (A) TMRM at four consecutive sites in the pore domain undergoes one or two steps of ΔF. The ΔFon at 418 and 419 has both fast and slow components, whereas at 416 and 417, it is purely slow. (B) Top view of KcsA crystal structure with backbone atoms of residues 416–419 (progressing counterclockwise) space filled at their homologous KcsA positions in one subunit. (C) Side view of one KcsA subunit's turret structure with residues 416–419 and side chains space-filled to indicate the possible direction of fluorophore projection. (D) Comparison of ionic current (I) and fluorescence (F) reveals a correlation of the fast ΔF with channel opening and the slow ΔF with inactivation. Black traces superimposed onto the F traces have the time constants of single exponential fits to the rise or inactivation of the current in response to steps to 140 mV. [(Inactrate)/(slow ΔFon-rate)] = 1.00 ± 0.07 (n = 4); 0.99 ± 0.05 (n = 5); 1.04 ± 0.06 (n = 3); for sites 416, 417, and 419, respectively (mean ± SEM [number of oocytes]). (E) Homologous location in KcsA of Shaker's acidic residues, which could interact with a fluorophore attached to 424C, was tested in the environmental scan.
All four sites displayed a ΔF (Fig. 2). The magnitude of the ΔF was largest at 418 and 419 and smallest at 416. As with S4, the fluorescence behavior of TMRM at each position was unique, confirming the very local nature of the environmental detection seen at consecutive positions in S4. At 416 and 417, the ΔFon was slow (Fig. 2 A) and paralleled the onset of inactivation (Fig. 2 legend). At 418 and 419, the ΔFon had a fast component that paralleled activation, and 419 also had a slow inactivation component (Fig. 2A and Fig. D, and legend). This is the first detection of an inactivation-correlated protein motion in the pore domain at a location so far from the selectivity filter (the site where closure of the pore is thought to occur), indicating that the slow inactivation rearrangement is more widespread than previously known. Equally striking is the detection of an activation motion by a fluorophore at pore domain residue 419, but not at neighboring residues 416 and 417 (418 also follows activation, see discussion). This finding suggests that 419 interacts with S4 during its transmembrane motion, in keeping with earlier work (Elinder and Arhem 1999), which indicated that 418 and 419 interact electrostatically with S4.
Environmental Scanning Detects Direct S4–Pore Domain Interaction
The kinetics of the ΔF that we observe for TMRM attached to both S4 and the pore domain suggest that these domains are both involved in slow inactivation gating. Could this match between the fluorescence kinetics be due to a direct interaction between the domains? To address this question, we took advantage of the evidence, described above (Fig. 1 and Fig. 2), that the spatial extent over which the fluorophore senses its environment is so confined that even neighboring residues have distinct fluorescence reports of protein motion. This means that a selective change in the protein nanoenvironment surrounding the fluorophore attachment site should alter the fluorescence report. We illustrate this idea in Fig. 3 A, where we show how the fluorophore (represented by an asterisk) is confined to a small space around its site of attachment (represented by a small circle). Mutation of a residue that is in the nanoenvironment in one state of the channel (illustrated as a change in the circle from black to white) will alter both the fluorescence of that state and the change in fluorescence that occurs upon transition to other states (in this case shown as when the inactivation gate closes and the fluorophore moves into another environment). The significance is that this effect can be used to identify amino acids in one part of the protein that are located so close to a fluorophore attached elsewhere in the protein as to contribute to it's nanoenvironment and, thus, influence its fluorescence (see Sorensen et al. 2000 who used this idea with protein deletions in the S3-S4 of Shaker).
Figure 3.

Environmental scan identifies direct S4–pore domain interaction. (A) Rationale for identifying domain interactions by environment scanning. Membrane topology model (left) and cartoon of the open to inactivated rearrangement (right). Fluorophore is attached to one site (either 424C in the pore domain or 359C in S4) and a comparison is made between the wild-type protein (top) and point mutation (bottom). If the mutation alters the nanoenvironment of the fluorophore in any channel conformation this will be detected as a change in the ΔF. (B) Representative fluorescence (F) and current (I) traces from channels with fluorophore attached to either 359C or S424C. Comparison between wild-type (WT) and the single charge neutralizations E418Q, E422Q, D431N, or D447N. (Responses to 5-s steps to +40 mV.) (C) Summary of the change in ΔF (ΔΔF) due to neutralization of the acidic residues. Significant difference (P < 0.05) from wild type (WT) in the t test.
With this idea in mind, we carried out an environment scan in the pore region for two fluorophore attachment sites, one at position 359 in S4 and another at position 424 in the pore region (Fig. 3 A). To limit the number of permutations in the scan to manageable proportions, we confined ourselves to one possible cause of fluorescence change in a fluorophore's nanoenvironment, changing proximity to a charged residue. The possibility of interaction with acidic residues was particularly attractive to explore since the ΔF of TMRM at both 359 and 424 was found to be pH-dependent, with a midpoint at approximately pH 4–5 (data not shown; Cha and Bezanilla 1998). We identified four candidate acidic residues in the pore region (E418, E422, D431, and D447), which the KcsA crystal structure indicated to lie on the protein surface. These acidic residues were neutralized individually in either the 424C or 359C background so that the fluorophore could be selectively attached to the pore domain or S4 site.
We first examined the effect of neutralizations on the fluorescence of TMRM attached to position 424 in the pore region so that the results could be interpreted in light of the KcsA crystal structure (Fig. 2 E). Two of the neutralizations, E418Q and D447N, eliminated ionic current but retained gating current (Fig. 3 B). This effect could be explained by the stabilization of the closed conformation of the inactivation gate, as described below (Fig. 4 and Fig. 6) and as shown earlier for D447N by Olcese et al. 1997. The other two neutralizations, E422Q and D431N, had little functional effect (Fig. 3 B). All four mutants expressed well enough to give substantial membrane fluorescence at the holding potential of –80 mV. Expression levels were matched (see materials and methods) and the effect of the neutralizations on the percent ΔF was examined. We found that three of the four neutralizations (E418Q, E422Q, and D447N) had no effect on the magnitude of the percent ΔF for a fluorophore attached to 424C (Fig. 3B and Fig. C). However, the neutralization D431N virtually eliminated the ΔF of 424-TMRM (Fig. 3B and Fig. C).
Figure 4.

The E418Q mutation locks the inactivation gate closed into the P-type–inactivated state. The charge–voltage relations of 359C-TMRM/W434F and 359C-TMRM/E418Q shift to the left by ∼15 mV when the holding potential is switched from −80 to +20 mV (with a prehold at +20 mV for 2 min). This shift is characteristic of the predepolarization dependence of C-type inactivation, indicating that E418Q channels are not C-type–inactivated at negative voltage, and that they do not conduct because they are locked into the P-type–inactivated state, as shown earlier for W434F (Loots and Isacoff 1998).
Figure 6.

Putative bonding partner for E418. Mutation of a residue predicted from the KcsA crystal structure to hydrogen bond with E418 has the same effect as mutation of E418. (Left) Top view of crystal structure of a single KcsA subunit. Shaker residues E418, its possible bonding partners V451 and G452, a control neighboring residue V453, and the fluorophore attachment site S424C are displayed with side chains at their homologous KcsA positions. Hydrogen bonds are possible between the E418 side chain and the backbone amino groups of either 451V and/or 452G, as defined by the Swiss Prot program. (Right) Ionic currents and fluorescence traces for 424C-TMRM (inset, right) are shown for proline substitutions. V451P is similar to the W434F, both in current (I) and fluorescence (F), which is consistent with a relative destabilization of the open conformation of the inactivation gate. Mutation of G452P prevented channel expression. V453P did not affect either the ionic current or the ΔF, indicating that there is no major structural perturbation because of proline substitution in this region.
We next examined the effect of the same four pore domain neutralizations on the fluorescence of TMRM attached at position 359 in S4. Again, there was no significant effect of E418Q and D422N on the ΔF, and again D431N had the greatest reduction of the ΔF (Fig. 3B and Fig. C). In addition, D447N, which had no effect on 424C-TMRM, significantly reduced the ΔF of 359C-TMRM.
The effect of the neutralization mutations could be explained in one of two ways. The COOH group of the side chain may directly contribute to the nanoenvironment of fluorophore, so that its removal alters the fluorescence report. Alternatively, the mutation could disrupt channel structure in a manner that propagates for some distance from the site of mutation, and, thus, indirectly alters the nanoenvironment of the fluorophore. The perturbation of function by the D447N mutation indicates that this mutation may indeed produce a global distortion of channel structure, although it may also maintain normal structure, but simply favor the native inactivated conformation. Therefore, to be safe, we refrain from interpreting the effect of this mutation.
By contrast, the case of D431N is much clearer. Two sets of factors argue against a large-scale disruption of channel structure for D431N and in favor of a direct interaction of D431 with both 424C-TMRM and 359C-TMRM. First, in KcsA, the residue at the 431 position is located at the outermost extremity of the protein surface, where the side chain points out into the water (Doyle et al. 1998), suggesting that it plays no essential role in channel structure. In keeping with this superficial location, D431 in Shaker has been shown to interact electrostatically with the recessed edge of the pore-blocking toxin AgTx2 (Ranganathan et al. 1996). Second, the D to N substitution had only one small effect on channel function: the rate of inactivation was slightly accelerated (431N, τ = 45 ± 0.8 ms; and 431D, τ = 51 ± 2 ms for oocytes with equal expression). Activation kinetics remained normal, and there was no detectable effect on voltage dependence, sensitivity of inactivation to external K+, or ionic selectivity (data not shown). The lack of a significant functional effect of D431N is consistent with an absence of structural perturbation of the mutation.
To further test the idea that side chain substitution at D431 does not perturb channel structure, we examined the effect on channel function of other, far less conservative substitutions at D431. Substitutions with H, C, and C-TMRM, which changed the charge and bulk of the side chain considerably, had no significant effect on either the conductance–voltage relation or the kinetics of channel activation (data not shown), adding further support to the idea that the side chain points away from the protein and into the external solution, so that the effect of substitution remains confined to a local effect on the chemical nanoenvironment of 431.
The results indicate that the influence of the D431N mutation on the fluorescence of TMRM at 359C and 424C is due to the entry of the fluorophore into the 431 side chain environment. This leads us to the functional conclusion that site 359 in S4 comes into close proximity to the pore domain during inactivation. This is consistent with direct S4–pore domain interaction, supporting recent work by Blaustein et al. 2000 and Li-Smerin et al. 2000b.
S4–Pore Interaction May Trigger Inactivation: The Role of E418
Could interaction of S4 with the pore domain be responsible for triggering inactivation? Stabilization of the closed conformation of the inactivation gate by W434F and D447N (Perozo et al. 1993; Olcese et al. 1997; Loots and Isacoff 1998) suggests that wild-type residues at these positions make bonds that hold the inactivation gate open. Closure of the gate may require breakage of those bonds, as suggested earlier for the W434 analogue in KcsA (Doyle et al. 1998).
One residue that may play such a role is E418. Like W434F and D447N, we found the E418Q mutant to be nonconducting (Fig. 3 B), to have normal gating currents (data not shown), and to convert the ΔF of 359-TMRM and 424-TMRM from slow inactivation into fast activation kinetics while maintaining the size of the ΔF (Fig. 3 B). All of these features are consistent with closure of the inactivation gate by the E418Q mutation (Loots and Isacoff 1998). As with both W434F and D447N (Olcese et al. 1997), the E418Q mutation closes the gate, but it does not consolidate into the C-type–inactivated state. This can be seen from the fact that the charge–voltage (Q-V) relation is shifted to the left by predepolarization (Fig. 4), as channels go from chronic P-type inactivation at negative voltage to C-type inactivation at positive voltage (Olcese et al. 1997; Loots and Isacoff 1998).
The implication of these results is that E418 forms a bond that contributes energy to the open conformation of the inactivation gate, leading to the prediction that other changes of the side-chain at this position would also alter the rate of closure of the gate. Results consistent with this were obtained. First, the E418C mutation inactivated more quickly than wild type and destabilized the open state (Fig. 5 A). Second, conjugation of TMRM to 418C completely abolished ionic current, leaving only gating current (Fig. 5 B), indicating an even larger destabilization of the open conformation of the inactivation gate. Interestingly, while this point mutation dramatically alters the conducting properties of the pore, it doesn't seem to alter the structure of the outer mouth of the protein since AgTx2 is still able to readily block K+ conduction. (Fig. 5 C).
Figure 5.

The open conformation of the inactivation gate is destabilized by the E418C mutation and even more so by TMRM conjugation. (A) Ionic currents from a series of steps (−120 to +60 mV, increments of 10 mV) generate rapidly inactivating currents despite a high concentration (110 mM) of external K+. (B) Same oocyte as in A, with ionic currents blocked by 1 μM of AgTx-2, displays only gating currents. (C) Ionic currents are also eliminated (leaving only gating currents) by conjugation of 418C with TMRM, which is consistent with stabilization of the closed conformation of the inactivation gate.
S4–Pore Interaction May Trigger Inactivation: Possible Bonding Partner of E418
We attempted to identify the bonding partner of E418. By analogy with the KcsA crystal structure, it appears that E418 may interact with the backbone amino groups of two residues at the beginning of S6: V451 and G452 (Fig. 6 A). Although the resolution of the structure is not refined enough to reveal many of the possible hydrogen bonds, these hydrogen bonds are the only ones that appear to connect S5 to S6 in this area of the crystal structure.
Mutations of positions 451 and 452 would not be expected to alter the protein backbone, except in the case of a proline substitution. Therefore, we tested the idea that E418 interacts with the backbone amino of V451 and G452 by making several substitutions including proline. We found that substitution with glutamate or cysteine labeled with fluorophore (V451C-TMRM, G452E, and G452C-TMRM), which are side chains of very distinct bulk and polarity, had no effect on the conductance–voltage relation or activation kinetics (data not shown). We substituted proline, whose side chain protects the backbone amino group. V451, G452, and V453 were individually mutated to proline with the prediction that if 451 and 452 interact with E418 then the proline mutations should disrupt the bond and shut the inactivation gate. The mutation V453P served as a control for the effect of a proline substitution at this general location on the global structure. G452P did not express as functional channels, however, other point mutations at this residue (452E, 452C-TMRM) showed no effect on channel activation or inactivation kinetics arguing for the importance of the amino group for bond formation (data not shown). However, V451P channels did express. They lacked ionic current, which is consistent with the stabilization of the closed conformation of the inactivation gate. To test this, V451P channels were labeled with TMRM at 424C and measured photometrically. These channels showed a fast ΔF, typical of permanent P-type inactivation (Fig. 6). V453P channels were fully functional, conducting ions, inactivating slowly, and generating ΔF from 424C-TMRM that correlated with inactivation in the manner of wild-type channels (Fig. 6). This wild-type behavior of V453P suggests that kinking of the S6 α-helix, which could displace it along most of its length from its wild-type angle, is probably not responsible for effect of the V451P mutation. Instead, we propose that closure of the inactivation gate by the V451P mutation is due to a block of the backbone amino, which prevents formation of the hydrogen bond with E418 that normally holds the inactivation gate open.
DISCUSSION
Membrane depolarization drives voltage-gated ion channels through a series of functional transitions. The process for the Shaker K+ channel starts with transmembrane displacement of charged residues on the voltage sensor in each of the four subunits, followed by the opening of the internal activation gate, finally followed by the closure of the external slow inactivation gate. This is not an obligatory sequence. The slow inactivation gate can close before the activation gate opens, and even in channels where slow inactivation follows opening, it can take place more rapidly from activated closed states (Marom and Levitan 1994; Smith et al. 1996; Spector et al. 1996; Klemic et al. 1998; Fleischhauer et al. 2000). Thus, each of the gates appears to be separately controlled by the voltage-sensing apparatus.
To determine how the voltage-sensing apparatus could control the inactivation gate, we studied the interaction between S4 and the pore domain, where the inactivation gate appears to reside (Iverson and Rudy 1990; Hoshi et al. 1991; Lopez-Barneo et al. 1993; Olcese et al. 1997; Yang et al. 1997). The inactivation gate was shown earlier to close via a rearrangement of the outer mouth of the pore and selectivity filter (Baukrowitz and Yellen 1996; Starkus et al. 1997, Starkus et al. 1998; Harris et al. 1998; Kiss and Korn 1998; Immke et al. 1999; Kiss et al. 1999). Here, we provide evidence that direct interaction between S4 and the pore domain couples voltage sensing to gating of the slow inactivation gate.
Fluorescence Evidence for Direct Interaction Between S4 and the Pore Domain
We obtained several pieces of evidence that S4 and the pore domain interact. The interpretation of these findings depends on how big the environment of a fluorophore really is. If the fluorophore is large, or is attached by a long flexible linker, it could sweep out an extensive volume of space and interact with many residues scattered over large parts of the protein. In this case, we would expect there to be little difference in fluorescence between nearby attachment sites since they would occupy virtually the same large space. In fact, our observations indicate that the contrary is true, and that a fluorophore senses only a very confined local environment specific to its attachment site.
The local nature of the fluorescence report means that we have three pieces of evidence for direct dynamic interaction between S4 and the pore domain. First, fluorophores attached to S4 sense a molecular motion that closes the inactivation gate (an event known to involve the pore domain); however, this change in environment occurs at sites that point in every direction from S4, which is consistent with an inactivation rearrangement of S4 itself, and suggestive of an integral role for S4 in slow inactivation, an idea consistent with earlier evidence that mutation of S4 alters inactivation (Mitrovic et al. 2000). Second, a fluorophore attached to the pore domain senses channel activation, an event known to involve the transmembrane motion of basic residues on S4 (Yang and Horn 1995; Aggarwal and MacKinnon 1996; Larsson et al. 1996; Seoh et al. 1996; Yang et al. 1996; Starace et al. 1997). Third, environmental scanning identifies a residue in the pore domain that appears to interact directly with a fluorophore attached to the outer end of S4. These results lead us to the conclusion that S4 undergoes a dynamic interaction with the edge of the pore domain. This conclusion leads us to explore the possible role of such S4–pore domain interaction in coupling voltage sensing to inactivation gating.
Proposed Mechanism by which S4 Movement Triggers Inactivation
Among the sites in the pore domain that have been examined with fluorescence, sites 416, 417, and 419 near the outer end of S5, which we examined here, as well as turret residues 424 and 425, which were examined earlier (Cha and Bezanilla 1997; Loots and Isacoff 1998), had a fluorescence change that correlated with inactivation. One of these pore domain sites, 419, also had an activation fluorescence change when the inactivation gate was open, suggesting that during activation S4 interacts with the pore at or near position 419. This interpretation is supported by the finding by others that mutations that alter the charge of 418 or 419 affect the voltage dependence of gating charge movement, which is consistent with close-range electrostatic interactions between S4 and the 418/419 region of the pore domain (Elinder and Arhem 1999). A more extensive fluorescence scan of many additional positions in the pore domain suggests that the 418/419 region actually lies within a stripe of pore domain residues that S4 contacts (Gandhi et al. 2000).
How could an interaction between S4 and 418/419 trigger inactivation? We observe here, in agreement with recent work from two other groups (Larsson and Elinder 2000; Ortega-Sáenz et al. 2000), that three substitutions at 418 (glutamine substitution, cysteine substitution, and TMRM conjugation) all reduce the relative stability of the open conformation of the gate. Furthermore, mutation of V451, a predicted hydrogen-bonding partner of E418, has the same destabilizing effect, but only when substituted with proline, which is consistent with a prediction based on the KcsA crystal structure that E418 would interact with the backbone amino of V451. The idea that position 418 comes into close enough proximity to position 451 for them to interact, and that this interaction changes when channels inactivate, is supported by Larsson and Elinder 2000 who showed that disulfide bond formation between E418C and 451C or 452C stabilizes the open conformation of the gate in one case and the closed conformation in the other.
Our finding that the E418C mutation produces a weaker destabilization of the open conformation than E418Q indicates that glutamine is less well accommodated than the smaller cysteine, and suggests that there is a steric hindrance to the packing of the native glutamate, which is compensated for by the greater ability of its charged oxygen to hydrogen bond. Together, these results are consistent with an S4–pore domain interaction that triggers inactivation with E418 acting as a latch. In this model, S4 extrusion and/or twist breaks the 418–451 bond, leading to a rearrangement that propagates from the perimeter of the pore domain to the central pore axis, where it closes the gate. Larsson and Elinder 2000 and Ortega-Sáenz et al. 2000 have proposed similar models.
The Inactivation Rearrangement Involves Multiple Bonds
The propagation of the inactivation rearrangement from the perimeter of the pore domain, at the S4 contact site, to the central axis of the pore appears to involve at least three residues other than 418–451. Residues W434 in the pore helix, D447 and T449 just outside the selectivity filter, and Y445 in the selectivity filter were shown here, and earlier, to stabilize the open conformation of the gate (Perozo et al. 1993; Hurst et al. 1996; Olcese et al. 1997; Harris et al. 1998; Loots and Isacoff 1998). The implication is that rupture of a bond between 418 and 451 can trigger rearrangements of bonds involving each of these residues.
We suggest that the role of K+ in stabilizing the open state of the slow inactivation gate derives from a stabilization by K+ occupancy of the selectivity filter of the backbone orientation of the outer end of S6, favoring the 418–451 interaction even when S4 is in its activated conformation, thus, slowing the onset of inactivation. Thus, external K+ may accelerate recovery from inactivation (Levy and Deutsch 1996) because of short-lived, spontaneous reopenings of the gate that provide access to K+, whose entry to its binding site would accelerate the reformation of the resting bonds that stabilize the open conformation of the slow inactivation gate. In this way, control would be exerted at the two physical ends of the inactivation mechanism: the contact site of the pore domain with S4, where voltage is sensed; and the selectivity filter, where the accumulation of external K+ ions is sensed.
A model for the mechanism of how S4's activation movement may be linked to slow inactivation is presented in Fig. 7. Underlying the model is the finding that S4 appears to have an intimate dynamic interaction with the pore domain, based on fluorescence reports from both the pore domain and S4, and supported by the present environment scan, as well as by recent work by Li-Smerin et al. 2000b and Blaustein et al. 2000. Interaction of S4 and the pore domain is proposed to trigger inactivation by breaking a bond between 418, in the turret near the S4 contact site, and 451, at the external end of S6. The rearrangement at 418 is proposed to propagate via the pore loop, which is stabilized in the open conformation by pore helix site W434, to the inactivation gate, near 445 and 447 in the selectivity filter.
Figure 7.
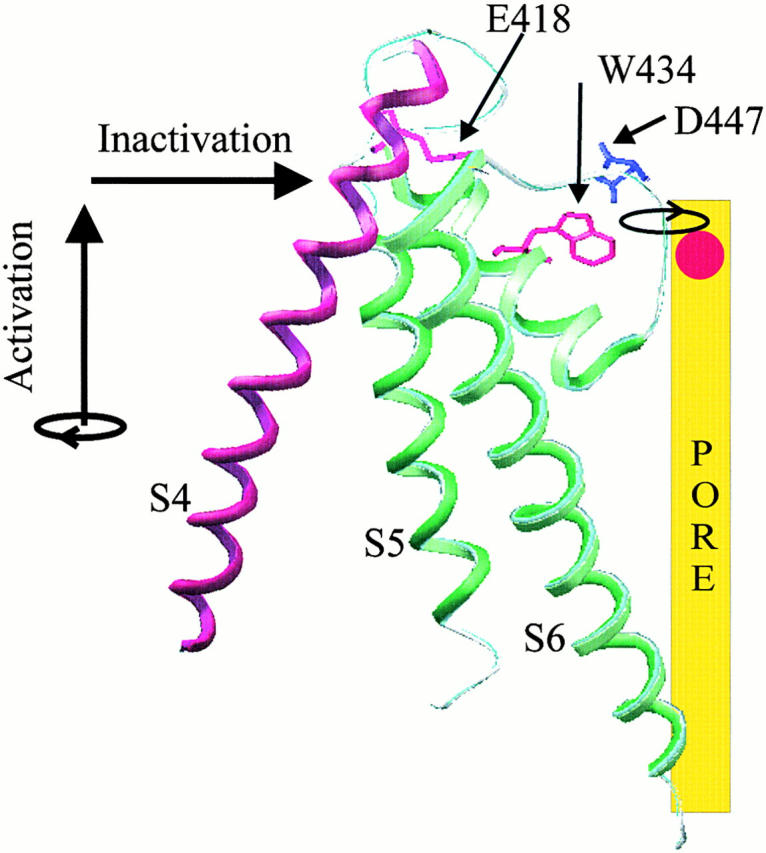
Model of proposed interaction between S4 and the pore domain that triggers inactivation. S4 twist or extrusion (or the combination of the two) directly destabilizes a hydrogen bond between E418, at the perimeter of the pore domain, and positions 451 and 452, at the outer end of S5. Breakage of the E418-451/452 hydrogen bond results in conformational changes throughout the outer sections of S5 and S6, which propagate to the selectivity filter and close the inactivation gate. The E418-451/452 bond is predicted from the KcsA crystal structure and from the similar effects of mutations at 418 and 451. The tilt of S4 relative to the pore is based upon previous FRET data on the proximal S3-S4 (Glauner et al. 1999) and on the simplifying assumption that the proximal S3-S4 and S4 form a continuous helix. An inactivation motion is proposed for S4 (depicted in one possible version as a tilt by an arrow parallel to the membrane), based on the finding that fluorophores, expected to point in all directions from S4, sense inactivation, an indication of an inactivation rearrangement of S4 with respect to its entire surround.
Conclusion
In summary, our results show that the inactivation rearrangement projects much further than previously known from the central axis, all the way to the edge of the pore domain and into the voltage-sensing domain to S4. We also find that the edge of the pore domain senses the activation rearrangement, providing evidence for direct interaction between the pore domain and S4, which is supported by our environmental scanning mutagenesis. These findings support recent evidence by others for contact between S4 and the pore domain, provide a first indication of the location on the pore domain of the S4 contact site, and demonstrate that this contact is dynamic, changing during gating. We explore the possible functional significance of the S4–pore domain interaction and find that the open conformation of the slow inactivation gate is destabilized by mutation of a pore domain edge residue, E418, near a site of apparent S4 contact. In addition, the open conformation of the slow inactivation gate is also destabilized by a mutation of residue V451 in S6, whose backbone amino group is predicted to interact with E418 based on the KcsA crystal structure. This destabilization only occurs when V451 is substituted with a proline, which eliminates the ability of its backbone amino group to form a hydrogen bond. Together, the results lead to an explicit structural model for how the voltage-sensing apparatus controls the conformation of the inactivation gate: S4 activation motion changes S4 contact with the edge of the pore domain, and breaks a key interaction of the edge of the pore domain with S6, which is required to hold the inactivation gate open, thus triggering the slow inactivation closure.
Acknowledgments
We would like to thank Chris Gandhi, Lidia Mannuzzu and other members of our lab, as well as John Ngai and members of his lab for helpful discussion.
This study was funded by the National Institutes of Health (grant No. NS35549), by the Department of Energy, and by a Laboratory Directed Research and Development Grant from the Lawrence Berkeley National Lab.
Footnotes
Abbreviation used in this paper: TMRM, 6′-tetramethylrhodamine maleimide.
References
- Aggarwal S.K., MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- Baker O.S., Larsson H.P., Mannuzzu L.M, Isacoff E.Y. Three transmembrane conformations and sequence-dependent displacement of the S4 domain in Shaker K+ channel gating. Neuron. 1998;20:1283–1294. doi: 10.1016/s0896-6273(00)80507-3. [DOI] [PubMed] [Google Scholar]
- Basso C., Labarca P., Stefani E., Alvarez O., Latorre R. Pore accessibility during c-type inactivation in Shaker K channels. FEBS Lett. 1998;429:375–380. doi: 10.1016/s0014-5793(98)00635-8. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T., Yellen G. Modulation of K1 current by frequency and external [K+]a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T., Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Boland L.M., Jurman M.E., Yellen G. Cysteines in the Shaker K+ channel are not essential for channel activity or zinc modulation. Biophys. J. 1994;66:694–699. doi: 10.1016/s0006-3495(94)80843-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein R., Cole P., Williams C., Miller C. Tethered blockers as molecular tape measures for a voltage-gated K+ channel. Nat. Struct. Biol. 2000;7:309–311. doi: 10.1038/74076. [DOI] [PubMed] [Google Scholar]
- Cha A., Bezanilla F. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 1997;19:1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Cha A., Bezanilla F. Structural implications of fluorescence quenching in the Shaker K+ channel. J. Gen. Physiol. 1998;112:391–408. doi: 10.1085/jgp.112.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A., Snyder G.E., Selvin P.R., Bezanilla F. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature. 1999;402:809–813. doi: 10.1038/45552. [DOI] [PubMed] [Google Scholar]
- De Biasi M., Hartman H.A., Drewe J.A., Tagialatela M., Brown A.M., Kirsh G.E. Inactivation determined by a single site in K+ pores. Pflügers Arch. 1993;422:354–363. doi: 10.1007/BF00374291. [DOI] [PubMed] [Google Scholar]
- Doyle D.A., Morais Cabral J., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., Mackinnon R. The structure of the potassium channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Elinder F., Arhem P. Role of individual surface charges of voltage-gated K+ channels. Biophys. J. 1999;77:1358–1362. doi: 10.1016/S0006-3495(99)76984-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischhauer R., Davis M., Dzhura I., Neely A., Avery L., Joho R. Ultrafast inactivation causes inward rectification in a voltage-gated K+ channel from Caenorhabditis elegans . J. Neurosci. 2000;20:511–520. doi: 10.1523/JNEUROSCI.20-02-00511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi C.S., Loots E., Isacoff E.Y. Reconstructing maps of K+ channel rearrangements from local protein motions. Neuron. Neuron. 2000;27:585–595. doi: 10.1016/s0896-6273(00)00068-4. [DOI] [PubMed] [Google Scholar]
- Glauner K.S., Mannuzu L.M., Gandhi C.S., Isacoff E.Y. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 1999;402:813–817. doi: 10.1038/45561. [DOI] [PubMed] [Google Scholar]
- Gross A., MacKinnon R. Agitoxin footprinting the Shaker potassium channel pore. Neuron. 1996;16:399–406. doi: 10.1016/s0896-6273(00)80057-4. [DOI] [PubMed] [Google Scholar]
- Harris R.E., Larsson H.P., Isacoff E.Y. A permeant ion binding site located between two gates of the Shaker K+ channel. Biophys. J. 1998;74:1808–1820. doi: 10.1016/s0006-3495(98)77891-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W., Aldrich R. Two types of inactivation in Shaker K+ channelseffect of alterations in the carboxyterminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Hurst R.S., Toro L., Stefani E. Molecular determinants of external barium block in Shaker potassium channels. FEBS Lett. 1996;388:59–65. doi: 10.1016/0014-5793(96)00516-9. [DOI] [PubMed] [Google Scholar]
- Immke D., Wood M., Kiss L., Korn S. Potassium-dependent changes in the conformation of the Kv2.1 potassium channel pore. J. Gen. Physiol. 1999;113:819–836. doi: 10.1085/jgp.113.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isacoff E., Jan Y., Jan L. Evidence for the formation of heteromultimeric potassium channels in Xenopus oocytes. Nature. 1990;345:530–534. doi: 10.1038/345530a0. [DOI] [PubMed] [Google Scholar]
- Isacoff E., Jan Y., Jan L. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+ channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- Iverson L.E., Rudy B. The role of the divergent amino and carboxyl domains on the inactivation properties of potassium channels derived from the Shaker gene of Drosophila . J. Neurosci. 1990;10:2903–2916. doi: 10.1523/JNEUROSCI.10-09-02903.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamb A., Iverson L.E., Tanouye M.A. Molecular characterization of Shaker a Drosophila gene that encodes a potassium channel. Cell. 1987;50:405–413. doi: 10.1016/0092-8674(87)90494-6. [DOI] [PubMed] [Google Scholar]
- Kiss L., Korn S. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys. J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss L., LoTurco J., Korn S.J. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 1999;76:253–263. doi: 10.1016/S0006-3495(99)77194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic K., Shieh C., Kirsch G., Jones S. Inactivation of Kv2.1 potassium channels. Biophys. J. 1998;74:1779–1789. doi: 10.1016/S0006-3495(98)77888-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y., Baldwin T.J., Jan Y.N., Jan L.Y. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Kupper J., Bowlby M.R., Marom S., Levitan I. Intracellular and extracellular amino acids that influence C-type inactivation and its modulation in a voltage-dependent potassium channel. Pflügers Arch. 1995;430:1–11. doi: 10.1007/BF00373833. [DOI] [PubMed] [Google Scholar]
- Larsson H.P., Baker O.S., Dhillon D.S., Isacoff E.Y. Transmembrane movement of the Shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Larsson H.P., Elinder F. A conserved glutamate is important for slow inactivation in K+ channels. Neuron. 2000;27:573–583. doi: 10.1016/s0896-6273(00)00067-2. [DOI] [PubMed] [Google Scholar]
- Levy D., Deutsch C. Recovery from C-type inactivation is modulated by extracellular potassium. Biophys. J. 1996;70:798–805. doi: 10.1016/S0006-3495(96)79619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y., Hackos D., Swartz K.J. α-Helical structural elements within the voltage-sensing domains of a K+ channel J. Gen. Physiol 115 2000. 33 49a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y., Hackos D., Swartz K.J. A localized interaction surface for voltage sensing domains on the pore domain of a K+ channel Neuron 25 2000. 411 423b [DOI] [PubMed] [Google Scholar]
- Liu Y., Jurman M., Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- Loots E., Isacoff E.Y. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. J. Gen. Phys. 1998;112:377–389. doi: 10.1085/jgp.112.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J., Hoshi T., Heinemann S., Aldrich R. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Mannuzzu L.M., Moronne M.M., Isacoff E.Y. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- Marom S., Levitan I. State-dependent inactivation of the Kv3 potassium channel. Biophys. J. 1994;67:579–589. doi: 10.1016/S0006-3495(94)80517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N., George A., Horn R. Role of domain 4 in sodium channel slow inactivation. J. Gen. Physiol. 2000;115:707–718. doi: 10.1085/jgp.115.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina A., Ortega-Saenz P., Lopez-Barneo J. Pore mutations alter closing and opening kinetics in Shaker K+ channels. J. Physiol. 1998;509:327–337. doi: 10.1111/j.1469-7793.1998.327bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk S., Needelman D.J., Miller C. Helical structure and packing orientation of the S2 segment in the Shaker K+ channel. J. Gen. Physiol. 1999;113:415–423. doi: 10.1085/jgp.113.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska E.M., Aldrich R.W. A mutation in the S6 of Shaker potassium channels decreases the K+ affinity of an ion binding site revealing ion–ion interactions in the pore. J. Gen. Phys. 1998;112:243–257. doi: 10.1085/jgp.112.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska, E.M., and R.W. Aldrich. 1999. Functional consequences of a decreased potassium affinity in a potassium channel pore. Ion interactions and C-type inactivation. J. Gen. Physiol. 113:347–358. [DOI] [PMC free article] [PubMed]
- Olcese R., Latorre R., Toro L., Bezanilla F., Stefani E. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J. Gen. Physiol. 1997;110:579–589. doi: 10.1085/jgp.110.5.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Sáenz P., Pardal R., Castellano A., López-Barneo J. Collapse of conductance is prevented by a glutamate residue conserved in voltage-dependent K+ channels. J. Gen. Physiol. 2000;116:181–190. doi: 10.1085/jgp.116.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled-Zehavi H., Arkin I.T., Engleman D.M., Shai Y. Coassembly of synthetic segments of Shaker K+ channel within phospholipid membranes. Biochemistry. 1996;35:6828–6838. doi: 10.1021/bi952988t. [DOI] [PubMed] [Google Scholar]
- Perez-Cornejo P. H ion modulation of C-type inactivation of Shaker K+ channels. Pflügers Arch. 1999;437:25–33. doi: 10.1007/s004240050856. [DOI] [PubMed] [Google Scholar]
- Perozo E., MacKinnon R., Bezanilla F., Stefani E. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 1993;11:353–358. doi: 10.1016/0896-6273(93)90190-3. [DOI] [PubMed] [Google Scholar]
- Ranganathan R., Lewis J.H., MacKinnon R. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 1996;16:131–139. doi: 10.1016/s0896-6273(00)80030-6. [DOI] [PubMed] [Google Scholar]
- Seoh S.A., Sigg D., Papazian D.M., Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- Smith P., Baukrowitz T., Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–866. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Sorensen J.B., Cha A., Latorre R., Roseman E., Bezanilla F. Deletion of the S3-S4 linker in the Shaker K+ channel reveals two quenching groups near the outside of S4. J. Gen. Physiol. 2000;115:209–221. doi: 10.1085/jgp.115.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector P., Curran M., Zou A., Keating M., Sanguinetti M. Fast inactivation causes rectification of the Ikr channel. J. Gen. Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace D.M., Stefani E., Bezanilla F. Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron. 1997;19:1319–1327. doi: 10.1016/s0896-6273(00)80422-5. [DOI] [PubMed] [Google Scholar]
- Starkus J.G., Kuschel L., Rayner M., Heinemann S. Ion conduction through C-type inactivated Shaker channels. J. Gen. Physiol. 1997;110:539–550. doi: 10.1085/jgp.110.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus J.G., Kuschell L., Rayner M., Heinemann S. Macroscopic Na+ currents in the “nonconducting” Shaker potassium channel mutant W434F. J. Gen. Phys. 1998;112:85–93. doi: 10.1085/jgp.112.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpe L., Jan Y., Jan L. Four cDNA clones from the shaker locus of Drosophila induce kinetically distinct A-type potassium currents in Xenopus oocytes. Neuron. 1988;1:659–667. doi: 10.1016/0896-6273(88)90165-1. [DOI] [PubMed] [Google Scholar]
- Yang N., Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Yang N., George A., Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Yang Y., Yan Y., Sigworth F. How does the W434F mutation block current in the Shaker potassium channels? J. Gen. Physiol. 1997;109:779–789. doi: 10.1085/jgp.109.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. THe moving parts of voltage-gated ion channels. Q. Rev. Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yellen G., Sodickson D., Chen T., Jurman E. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys. J. 1994;66:1068–1075. doi: 10.1016/S0006-3495(94)80888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yool A.J., Schwarz T.L. Alterations of ionic selectivity of a K+ channel by mutation of the H5 region. Nature. 1991;349:700–704. doi: 10.1038/349700a0. [DOI] [PubMed] [Google Scholar]
- Yusaf S.P., Wray D., Sivaprasadarao A. Measurement of the movement of the S4 segment during the activation of a voltage-gated potassium channel. Pflügers Arch. 1996;433:91–97. doi: 10.1007/s004240050253. [DOI] [PubMed] [Google Scholar]
- Zagotta W.N., Hoshi T., Aldrich W. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]