

Abstract
Negative regulation of the heartbeat rate involves the activation of an inwardly rectifying potassium current (IKACh) by G protein–coupled receptors such as the m2 muscarinic acetylcholine receptor. Recent studies have shown that this process involves the direct binding of Gβγ subunits to the NH2- and COOH-terminal cytoplasmic domains of the proteins termed GIRK1 and GIRK4 (Kir3.1 and Kir3.4/CIR), which mediate IKACh. Because of the very low basal activity of native IKACh, it has been difficult to determine the single channel effect of Gβγ subunit binding on IKACh activity. Through analysis of a novel G protein–activated chimeric inward rectifier channel that displays increased basal activity relative to IKACh, we find that single channel activation can be explained by a G protein–dependent shift in the equilibrium of open channel transitions in favor of a bursting state of channel activity over a long-lived closed state.
Keywords: ion channel, patch-clamp, G protein, gating, acetylcholine
INTRODUCTION
Secretion of acetylcholine (ACh) by the vagus nerve slows the heartbeat rate by activating an inward rectifier potassium current (IKACh) in the pacemaker cells of the sinoatrial node (Trautwein and Dudel 1958; Hille 1992). Inward rectifier potassium currents flow preferentially when the membrane potential is negative or slightly positive relative to the potassium equilibrium potential (EK). This increase in potassium permeability at the cell's resting potential strengthens the contribution of the EK to the overall membrane potential. As a result, it takes more time for depolarizing pacemaker currents to bring the membrane potential to the activation threshold of voltage-gated currents that mediate the action potentials, which ultimately trigger heart muscle contraction.
The biochemical events underlying the stimulation of IKACh have been well studied. ACh activates the m2 muscarinic acetylcholine receptor (mAChR) which, in turn, catalyzes the exchange of GDP for GTP on the α subunit of the heterotrimeric G protein Gi. The dimer of the G protein β and γ subunits becomes dissociated from the GTP-bound α subunit and activates IKACh by a direct, membrane-delimited mechanism (Soejima and Noma 1984; Breitwieser and Szabo 1985; Pfaffinger et al. 1985; Logothetis et al. 1987).
The molecular cloning of genes encoding the proteins that form inward rectifier potassium channels has allowed a more detailed understanding of how Gβγ activates IKACh (Kubo et al. 1993a,Kubo et al. 1993b). Inward rectifier potassium channel proteins are divided into six subfamilies (Doupnik et al. 1995); all share a similar topology including two membrane spanning regions and a pore domain, which is homologous to that of the voltage-gated potassium channels. The subfamilies are distinguished by differing cytoplasmic NH2- and COOH-terminal domains, which confer different sensitivities to a variety of intracellular signals. IKACh is mediated by heterotetramers of the GIRK1 and GIRK4 (G protein–activated inward rectifier potassium channel) proteins (Krapivinsky et al. 1995). The channels formed by these proteins are very inactive in the absence of G protein signaling and are strongly activated by stimulation of the m2 mAChR. In contrast, members of the Kir2 (inward rectifier potassium channel [IRK]) subfamily can function as homotetramers, are constitutively active, and are unaffected by m2 mAChR stimulation.
In a previous study (Kunkel and Peralta 1995), we used chimeric fusion proteins of GIRK1 and RB-IRK2 (a member of the Kir2 subfamily) to define the sequences within GIRK1 that are required for Gβγ activation. We constructed a chimeric molecule (termed GR7.1) in which the NH2 terminus and the distal portion of the COOH-terminal cytoplasmic domains of GIRK1 were fused to the pore region and proximal COOH-terminal cytoplasmic domain of RB-IRK2 (see Fig. 1). The homomeric channels formed by GR7.1 have a high basal activity similar to that of RB-IRK2, but are further activated by stimulation of m2 mAChRs. Furthermore, G protein βγ subunits bind directly to bacterially expressed glutathione–S-transferase fusion proteins of the GIRK1 domains included in the chimera, suggesting that activation occurs through a direct interaction between the G protein and the channel.
Figure 1.

Schematic representation of inward rectifier channel topology. Sequence from GIRK1 is in light gray. Sequence from RB-IRK2 is in black. Chimera GR7.1 contains amino acids 1–84 and 290–501 of GIRK1 fused to amino acids 85–289 of RB-IRK2.
The link between binding events involving the intracellular domain of the channel and gating of the channel pore remains unclear. One approach to this problem is to measure the properties of single channels before and after activation to see which parameters are affected. This has not been possible for GIRK1/GIRK4 channels because of their very low basal activity. In this study, we take advantage of the relatively high basal activity of GR7.1 to determine the single channel effect of G protein binding. We find that activation of single channels by mAChR stimulation leads to an increase in the mean duration of bursts of channel openings. These observations suggest a simple molecular model of G protein activation of inward rectifier potassium channel activity.
MATERIALS AND METHODS
Xenopus oocytes were injected with cRNA for the m2 mAChR (∼10 ng) and either a mixture of GIRK1 (∼10 ng) and GIRK4 (∼1 ng), GR7.1 (∼10 ng), or RB-IRK2 (∼0.5 ng; Kunkel and Peralta 1995). Cells were checked for the expression of receptor and channels after 12–72 h using two-electrode voltage clamp whole-cell recordings. Patch-clamp studies were done using a pipet holder that had been drilled to accept a tube through which solutions could be delivered to the inside of the pipet while recording (Soejima and Noma 1984; Tang et al. 1992). Pipettes were pulled from glass capillaries (model 8161; Corning) and had tip resistances between 0.8 and 4 ΜΩ.
Recordings of Patches Containing Multiple Channels
Cell-attached patches having many channels were subjected to a voltage protocol consisting of a pulse to −60 mV for 800 ms followed by a pulse to +60 mV for 800 ms; both were repeated every 5 s. 3 μl of 10 mM carbachol was added to a 3-ml bath volume after ∼100 s and allowed to mix by diffusion. In control whole cell experiments, this generally led to activation within 2 min. After 100 episodes, the perfusion tube was transferred to a reservoir containing 20 μM carbachol and the pipet was perfused by suction until the volume of the pipet solution had doubled. The observed delay in activation reflects the time required for the drug to diffuse to the tip of the pipet and varied considerably with pipet geometry.
Recordings of Patches Containing Single Channels
Patches that appeared to contain only one channel were subjected to voltage ramp protocols to determine rectification properties, and were recorded continuously for 500 s at −60 mV. The data were filtered at 1 kHz, and the sampling frequency was 10 kHz. After the initial recording, the pipet was perfused as before. After several minutes, during which the drug was allowed to diffuse to the tip of the pipet, another 500 s of data were acquired. Single channel data were idealized using Fetchan (Axon Instruments) with a filter frequency of 150 Hz for GR7.1 and RB-IRK2; no filter was applied for the GIRK1/GIRK4 analysis. Analysis of the dwell-time histograms was performed using Pstat (Axon Instruments). The burst delimiter for burst analysis was determined by finding the minimum of the fit between the second and third exponential terms. It was generally between 175 and 300 ms.
RESULTS
To use GR7.1 as a model channel for G protein activation, we first needed to demonstrate that this chimeric channel is activated through a similar mechanism to that of IKACh. Membrane-delimited activation by G protein–coupled receptors is a hallmark property of IKACh. In cell-attached patches from rabbit atrial cells, the addition of a mAChR agonist to the recording pipet activates the current, whereas the addition of the same agonist to the bath does not (Soejima and Noma 1984). This implies that cytoplasmic kinases, phosphatases, or second messengers, which can diffuse across the boundary of the patch pipet, are not involved in channel activation.
To test whether activation of GIRK1/GIRK4 and chimera GR7.1 occurs by a similar membrane-delimited mechanism, we recorded from patches containing multiple channels while adding the mAChR agonist carbachol first to the bath and then to the pipet. Oocytes expressing GIRK1/GIRK4 and m2 mAChRs showed very little activity in the absence of carbachol or when carbachol was added to the bath. However, when carbachol was added to the pipet, there was a large increase in activity (Fig. 2 A). Patches from cells expressing RB-IRK2 or GR7.1 showed significant activity in the absence of carbachol. This activity did not increase upon addition of the agonist to the bath. Importantly, the addition of carbachol to the pipet had no effect on RB-IRK2, but significantly increased the activity of GR7.1 (Fig. 2, B–D). These results show that activation of GR7.1 and GIRK1/GIRK4 share the feature of membrane-delimited regulation as originally defined for IKACh.
Figure 2.

GIRK1/GIRK4 and GR7.1 currents are activated via similar membrane-delimited pathways. Cell-attached patches containing multiple channels from oocytes expressing GIRK1/GIRK4 (A), RB-IRK2 (B), or chimera GR7.1 (C), were held at −60 mV for 800 ms, and then depolarized to +60 mV for 800 ms every 5 s for the duration of the experiment. Carbachol was added to the bath at the indicated time by hand pipetting. Carbachol was perfused into the pipet by suction (see Materials and Methods) over the indicated time period. Each point on the larger plot represents the integrated current over the portion of the trace at one holding potential for that time point. The insets show representative traces from before the addition of carbachol to the bath (left), after addition to the bath (center), and after addition to the pipet (right). The transient increase in outward current during the perfusion reflects activation of the oocyte's endogenous stretch-activated channels by the suction applied to the pipet. D shows the average percent change in integrated current after carbachol addition to the bath and to the pipet for GR7.1 (filled bars, n = 5) and RB-IRK2 (open bars, n = 5); error bars represent SEM. Because there is virtually no current before addition of carbachol to the pipet for GIRK1/GIRK4, the percent change is undefined.
To examine the mechanism of channel activation by G protein binding, we analyzed recordings of individual GR7.1 channels before and after perfusion of the patch pipet with carbachol. The trace in Fig. 3 A shows the activity of a single GR7.1 channel before addition of agonist to the pipet. Bursts of activity are separated by long inactive periods of up to several seconds. The trace in Fig. 3 B shows the activity of the same channel after addition of carbachol to the pipet. For this channel, there was a 60% increase in open probability (0.20 before carbachol and 0.32 after carbachol), which is wholly accounted for by a similar increase in the burst duration (1.2 s before carbachol and 2.1 s after carbachol; see Fig. 4E and Fig. F). This enhanced open probability results both from an increase in the duration of openings within bursts (94 ms before carbachol and 126 ms after carbachol; Fig. 4A and Fig. B) and an increase in the number of openings per burst (13.2 before carbachol and 16.2 after carbachol). Since each opening within a burst is preceded and followed by a closing, an increase in the number of openings per burst is reflected in the closed time distribution as an increased population of the region of the distribution corresponding to short closings that occur during bursts (Fig. 4C and Fig. D, leftmost peaks). Importantly, the interburst interval remains unchanged after stimulation implying that bursts do not occur more frequently after stimulation (Fig. 4G and Fig. H). Similar activation was seen in five out of seven single channel experiments with GR7.1; it may be that there were no mAChRs in those patches where no activation was observed. Except for a small change in open duration, no significant changes in single channel properties were observed in control experiments with RB-IRK2 (Fig. 4 I).
Figure 3.

Single channel recordings of GR7.1. (A) Excerpt from a recording of a single GR7.1 channel before addition of carbachol to the pipet. (B) Excerpt from a recording of the same patch after addition of carbachol to the pipet. Bars to the left of traces indicate the current level when the channel is closed. Insets show the indicated region of the trace with a magnified time scale.
Figure 4.
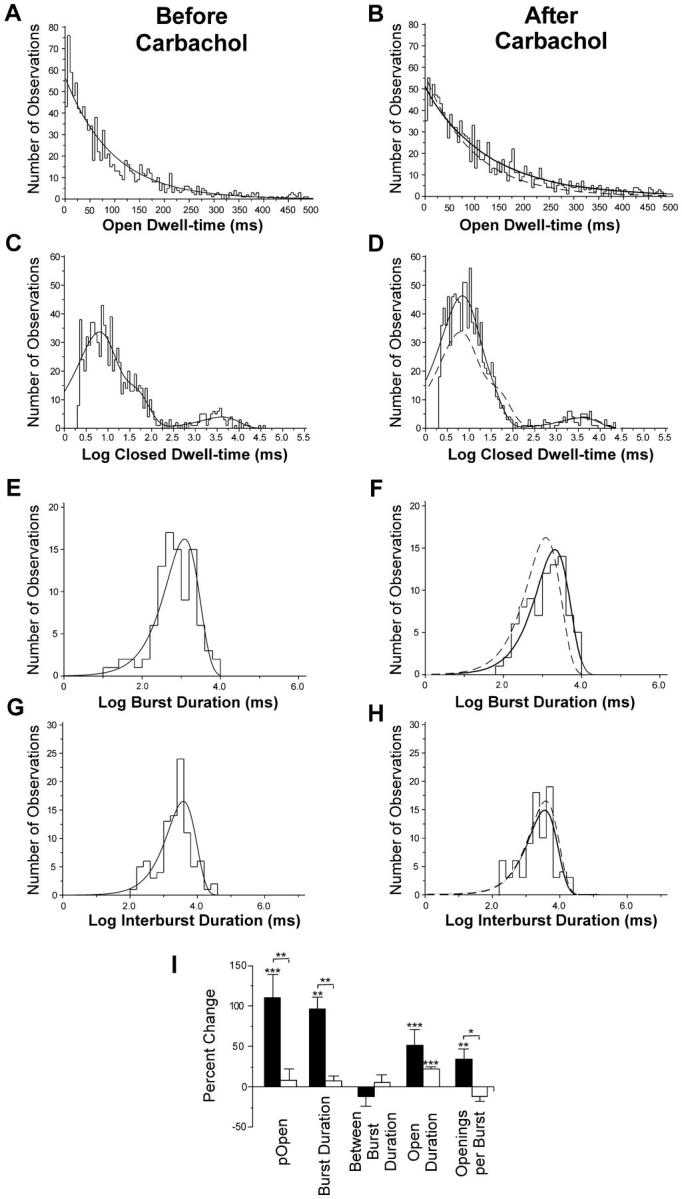
Analysis of GR7.1 single channel parameters. (A–H) Dwell-time histograms from the channel shown in Fig. 3 before (left) and after (right) perfusion of the pipet with carbachol. Maximum likelihood fits to exponential functions are shown for each. All except the closed time histograms were fit to single exponentials. The closed time histograms were fit to sums of three exponentials. The fits of the Before Carbachol data are shown as a dotted line in the After Carbachol histograms for comparison. (I) Mean change in single channel properties after addition of carbachol to the pipet for 5 GR7.1 channels (filled bars) and 4 RB-IRK2 channels (open bars). Stars above bars indicate the significance level of the difference between the properties of each channel before and after carbachol treatment based on a paired t test. Stars above brackets indicate the significance of the difference between the percent changes for each of the two types of channel based on a Kolmogorov-Smirnov test. (***) P > 99%; (**) P > 95%; (*) P > 90%. Error bars represent SEM.
To determine whether wild-type channels display similar changes in single channel activity, we recorded from oocytes injected with cRNAs encoding GIRK1, GIRK4, and the m2 mAChR. Because of the low basal open probability of GIRK1/GIRK4 channels, we could not conclusively determine if our unstimulated patches contained only a single channel and, therefore, could not perform the sort of quantitative analysis applied to GR7.1. However, there is a clear qualitative similarity between the activation of GIRK1/GIRK4 and that of GR7.1. When carbachol is not present in the pipet, recordings from cells expressing GIRK1/GIRK4 and m2 mAChRs are characterized by brief, solitary openings. This can be seen in Fig. 5 A, which shows the current from a patch containing multiple GIRK1/GIRK4 channels in the absence of carbachol. In contrast, when carbachol is present in the pipet, GIRK1/GIRK4 single channel activity is similar to that of GR7.1: bursts of openings are separated by long closings (Fig. 5 B). As with GR7.1, this enhanced tendency to remain in the bursting state appears as a large peak representing short closings within bursts in the closed time distribution observed with carbachol in the pipet (Fig. 5 D). The corresponding region of the closed time distribution for patches containing an unknown number of channels in the absence of carbachol contains very few events (Fig. 5 C).
Figure 5.

Single channel behavior of GIRK1/GIRK4 with and without carbachol in the pipet. (A) A portion of a recording of a patch containing an unknown number of channels at −60 mV without carbachol in the pipet. Bars to the left of traces indicate current level where no channels are open. (B) A portion of a recording from a different patch containing a single GIRK1/GIRK4 channel at −60 mV with 10 μM carbachol in the pipet. (C and E) Closed and open dwell-time histograms from the experiment in A. (D and F) Closed and open dwell-time histograms from the recording in B. The histogram in E is fit to a single exponential function with τ = 2.1 ms. The histogram in F is fit to the sum of two exponential functions with τ1 = 0.7 ms and τ2 = 5.9 ms. The closed dwell-time histograms are shown to illustrate the increased presence of short, intraburst closings. Because the number of channels in each patch is unknown, the durations of longer closings (shown at the right of each histogram) do not have any significance.
DISCUSSION
We have used patch-clamp methodology to study the mechanism of activation of inward rectifier potassium channels by mAChRs. Our results confirm that GIRK1/GIRK4 and chimera GR7.1 channels are both activated by a similar membrane delimited mechanism. Furthermore, at the single channel level, activation appears as an increase in the duration of bursts of channel openings.
There have been a number of studies in which the single channel properties of GIRKs were examined in the presence of various concentrations of neurotransmitters or exogenously applied Gβγ (Ivanova-Nikolova and Breitwieser 1997; Ivanova-Nikolova et al. 1998; Yakubovich et al. 2000). Such studies have revealed that GIRK channels have a variety of gating modes, and it has been proposed that binding to G proteins alters the modal preference such that the channel spends more time in modes with higher open probabilities. A detailed model of the nature of this shift has been difficult to formulate, however, because of the complexity and number of gating modes, as well as the difficulty of obtaining recordings of single, unstimulated GIRK channels.
The simpler single channel behavior and robust unstimulated activity of GR7.1 enable us to investigate the role of the G protein signal in the regulation of single channel kinetics of this model system. If one assumes a simplified model of a bursting channel in which a single open state (O) can be exited either to a short-lived intraburst closing (CS) or a long-lived interburst closing (CL), then both the increased duration of bursts and the increased τo of activated GR7.1 can be explained if the relative probability of exiting an opening to CL rather than CS is decreased upon binding to Gβγ.
This explanation of activation by Gβγ can be extended to account for our qualitative observations of the single channel behavior of GIRK1/GIRK4 channels. In this case, the rare solitary openings observed in the absence of stimulation can be considered as the limiting case of bursts with only a single opening, i.e., the probability of leaving O for CL is much larger than for CS. A decrease in the probability of exiting O for CL might allow the bursts that are observed in the presence of stimulation to occur. Although such an explanation can account for the large-scale differences between GIRK channels in the presence and absence of Gβγ, it cannot account for the observation of Ivanova-Nikolova and Breitwieser that the Po within a burst increases when the concentration of Gβγ is increased (Ivanova-Nikolova and Breitwieser 1997). It may be that binding of G proteins to GIRK affects multiple conformational changes involved in gating, whereas binding to GR7.1 affects only one of these.
Our recordings suggest that the only difference between GR7.1 and IRK2 is in the G protein sensitivity of the transition from the bursting state to the long closed state. Because the sequences of these two channels differ only in their intracellular domains, it is likely that this transition is mediated by conformational changes in this intracellular domain. This would be the case if, as several groups have suggested (Dascal et al. 1995; Kunkel and Peralta 1995; Pessia et al. 1995), the intracellular domain includes an inactivation particle analogous to that of voltage-gated potassium channels (Hoshi et al. 1990). In some other effector systems, Gβγ is thought to function by increasing the affinity of its targets for the membrane (Clapham and Neer 1997). Interestingly, it has been shown that interactions between members of the Kir family and membrane phospholipids are essential for their activity (Huang et al. 1998; Kim and Bang 1999). Taken together, these observations suggest that G protein activation of GIRKs could arise, in part, from an increase in the affinity of an intracellular blocking particle for the membrane, which in turn decreases its availability to occlude the pore.
Acknowledgments
This work is dedicated to the memory of Dr. Ernest G. Peralta (1959–1999).
The authors would like to thank Markus Meister, Guido Guidotti, Gary Yellen, and Jennifer Hill for helpful discussions and comments on the manuscript, and Karl Magleby, Anthony Morielli, and Reed Carroll for invaluable technical advice.
This work was supported by a National Institutes of Health grant GM42843-09 and by a Howard Hughes Medical Institute predoctoral fellowship to J. Bard.
Footnotes
Dr. Bard's current address is Boston Biomedical Research Institute, 64 Grove Street, Watertown, MA 02472. Dr. Kunkel's current address Department of Anatomy and Neurobiology, Washington University Medical School, St. Louis, MO 63110
Abbreviations used in this paper: ACh, acetylcholine; GIRK, G protein–activated inward rectifier potassium channel; IRK, inward rectifier potassium channel; mAChR, muscarinic acetylcholine receptor.
References
- Breitwieser G.E., Szabo G. Uncoupling of cardiac muscarinic and beta-adrenergic receptors from ion channels by a guanine nucleotide analogue. Nature. 1985;317:538–540. doi: 10.1038/317538a0. [DOI] [PubMed] [Google Scholar]
- Clapham D.E., Neer E.J. G protein beta gamma subunits. Annu. Rev. Pharmacol. Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- Dascal N., Doupnik C.A., Ivanina T., Bausch S., Wang W., Lin C., Garvey J., Chavkin C., Lester H.A., Davidson N. Inhibition of function in Xenopus oocytes of the inwardly rectifying G-protein-activated atrial K channel (GIRK1) by overexpression of a membrane-attached form of the C-terminal tail. Proc. Natl. Acad. Sci. USA. 1995;92:6758–6762. doi: 10.1073/pnas.92.15.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik C.A., Davidson N., Lester H.A. The inward rectifier potassium channel family. Curr. Opin. Neurobiol. 1995;5:268–277. doi: 10.1016/0959-4388(95)80038-7. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes 2nd ed 1992. Sinauer Associates, Inc; Sunderland, MA: pp. 607 pp [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Huang C.L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Ivanova-Nikolova T.T., Breitwieser G.E. Effector contributions to Gβγ-mediated signaling as revealed by muscarinic potassium channel gating. J. Gen. Physiol. 1997;109:245–253. doi: 10.1085/jgp.109.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova-Nikolova T.T., Nikolov E.N., Hansen C., Robishaw J.D. Muscarinic K+ channel in the heart. Modal regulation by G protein beta gamma subunits. J. Gen. Physiol. 1998;112:199–210. doi: 10.1085/jgp.112.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Bang H. Modulation of rat atrial G protein-coupled K+ channel function by phospholipids. J. Physiol. 1999;517:59–74. doi: 10.1111/j.1469-7793.1999.0059z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G., Gordon E.A., Wickman K., Velimirovic B., Krapivinsky L., Clapham D.E. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K(+)-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- Kubo Y., Baldwin T.J., Jan Y.N., Jan L.Y. Primary structure and functional expression of a mouse inward rectifier potassium channel Nature 362 1993. 127 133a [DOI] [PubMed] [Google Scholar]
- Kubo Y., Reuveny E., Slesinger P.A., Jan Y.N., Jan L.Y. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel Nature 364 1993. 802 806b [DOI] [PubMed] [Google Scholar]
- Kunkel M.T., Peralta E.G. Identification of domains conferring G protein regulation on inward rectifier potassium channels. Cell. 1995;83:443–449. doi: 10.1016/0092-8674(95)90122-1. [DOI] [PubMed] [Google Scholar]
- Logothetis D.E., Kurachi Y., Galper J., Neer E.J., Clapham D.E. The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- Pessia M., Bond C.T., Kavanaugh M.P., Adelman J.P. Contributions of the C-terminal domain to gating properties of inward rectifier potassium channels. Neuron. 1995;14:1039–1045. doi: 10.1016/0896-6273(95)90342-9. [DOI] [PubMed] [Google Scholar]
- Pfaffinger P.J., Martin J.M., Hunter D.D., Nathanson N.M., Hille B. GTP-binding proteins couple cardiac muscarinic receptors to a K channel. Nature. 1985;317:536–538. doi: 10.1038/317536a0. [DOI] [PubMed] [Google Scholar]
- Soejima M., Noma A. Mode of regulation of the ACh-sensitive K-channel by the muscarinic receptor in rabbit atrial cells. Pflügers Arch. 1984;400:424–431. doi: 10.1007/BF00587544. [DOI] [PubMed] [Google Scholar]
- Tang J.M., Wang J., Eisenberg R.S. Perfusing patch pipettes. Methods Enzymol. 1992;207:176–181. doi: 10.1016/0076-6879(92)07012-d. [DOI] [PubMed] [Google Scholar]
- Trautwein W., Dudel J. Zum mechanismus der membranwirkung des acetylcholin an der herzmuskelfaser. Pflügers Arch. 1958;266:324–334. doi: 10.1007/BF00416781. [DOI] [PubMed] [Google Scholar]
- Yakubovich D., Pastushenko V., Bitler A., Dessauer C.W., Dascal N. Slow modal gating of single G protein-activated K+ channels expressed in Xenopus oocytes. J. Physiol. 2000;524:737–755. doi: 10.1111/j.1469-7793.2000.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]