

Abstract
Voltage-dependent K+ channel gating is influenced by the permeating ions. Extracellular K+ determines the occupation of sites in the channels where the cation interferes with the motion of the gates. When external [K+] decreases, some K+ channels open too briefly to allow the conduction of measurable current. Given that extracellular K+ is normally low, we have studied if negatively charged amino acids in the extracellular loops of Shaker K+ channels contribute to increase the local [K+]. Surprisingly, neutralization of the charge of most acidic residues has minor effects on gating. However, a glutamate residue (E418) located at the external end of the membrane spanning segment S5 is absolutely required for keeping channels active at the normal external [K+]. E418 is conserved in all families of voltage-dependent K+ channels. Although the channel mutant E418Q has kinetic properties resembling those produced by removal of K+ from the pore, it seems that E418 is not simply concentrating cations near the channel mouth, but has a direct and critical role in gating. Our data suggest that E418 contributes to stabilize the S4 voltage sensor in the depolarized position, thus permitting maintenance of the channel open conformation.
Keywords: K+-channel gating, extracellular K+, acidic residues, open-state stabilization, glutamate mutation
INTRODUCTION
Voltage-dependent K+ channels of the plasmalemma participate in fundamental cellular electrical events such as the genesis of the resting membrane potential, action potential repolarization, and repetitive firing (Hille 1992). The transmembrane efflux of K+ ions through the open channels is driven by the chemical gradient existing between the cell's interior, where [K+] is high (≈140 mM), and the extracellular milieu with lower [K+] (≈2.5 mM in most mammalian tissues). However, the low external [K+] imposes a major challenge to many K+ channels since they do not gate properly, or even become nonfunctional if extracellular K+ is too scant. Although the channels were classically viewed as pores with gates that move independently of the permeating ions, there are several reports indicating that occupation of the channels by the permeating ions modulates their gating properties (for review, see Yellen 1997). For example, the rise of extracellular [K+] slows the rates of channel closing (Stanfield et al. 1981; Swenson and Armstrong 1981; Demo and Yellen 1992; Molina et al. 1998) and C-type inactivation (López-Barneo et al. 1993; Marom and Levitan 1994; Baukrowitz and Yellen 1995; Kiss and Korn 1998). These observations indicate that the concentration of external permeant cations determines the occupation of sites in the pore, where they interfere with the motion of the inactivation or closing gates. Some K+ channels have a particularly strong dependence on external K+ because they can easily loose the resident K+ ions. In these cases, removal of the cation from the external solution results in immediate loss of the K+ conductance (Pardo et al. 1992; López-Barneo et al. 1993; Jäger et al. 1998). Given the competition between C-type inactivation and the ions occupying the pore, the channels with fast C-type inactivation rate (such as Kv1.4, HERG, or Shaker mutants T449K and D447E) are, in general, those more exquisitely modulated by the changes in extracellular [K+] (López-Barneo et al. 1993; Schönherr and Heinemann 1996; Smith et al. 1996; Jäger et al. 1998). Even K+ channels with slow C-type inactivation kinetics resistant to brief removal of external K+ become nonfunctional after perfusion of the two sides of the membrane with K+-free media. This last phenomenon, first observed in squid K+ channels (Almers and Armstrong 1980), is a manifestation of the complete depletion of K+ ions from inside the channels, which results in collapse of the pore and stabilization of the channels in a nonconducting or “defunct” state (Gómez-Lagunas 1997; Melishchuk et al. 1998).
In K+ channels, both the intra- and extracellular entryways contain negatively charged amino acids that presumably contribute to increase the local concentration of cations while lowering the concentration of anions (see Doyle et al. 1998). Some of the extracellular acidic residues are probably critical for ensuring channel occupancy by K+ ions because, in contrast with the internal vestibule, which is wide and easily occupied by the highly concentrated K+ ions, the external channel mouth is relatively shallow and in contact with a solution poor in K+. For example, a dicarboxylic residue (E422) located in the region between S5 and the pore helix (turret in the terminology of Doyle et al. 1998) of the Shaker K+ channel is known to favor the electrostatic attraction of positively charged blocking toxins (MacKinnon and Miller 1989; Goldstein et al. 1994). Based on these presumptions, we studied whether neutralization of charges in acidic residues of the extracellular loops of Shaker K+ channels alters their gating properties and modulation by extracellular K+. Here, we show that, surprisingly, substitution of several acidic residues by neutral amino acids has no major effects on channel activity. However, a glutamate in position 418 of Shaker, located at the base of the turret and conserved in voltage-dependent K+ channels, is absolutely required for channel gating in the normal external [K+]. E418 appears to stabilize the open state of the channels, thus preventing the collapse of the K+ conductance. These results give new insight into the structural determinants of the interaction of channel permeation and activation and inactivation gating.
MATERIALS AND METHODS
Molecular Biology and Expression of K+ Channels in Chinese Hamster Ovary Cells
We used as wild-type construct the Shaker BΔ6-46 (Shaker BΔ) channel, which has a deletion in the amino terminus that completely removes N-type inactivation (Hoshi et al. 1991). Point mutations were made on this channel using mismatched oligonucleotides and the Altered Site II kit (Promega). We have studied K+ channel mutants with replacement of one or several amino acids. Glutamate and aspartate were replaced for either glutamine or asparagine. Mutation of glutamate in position 418 (E418Q) was studied in normal Shaker BΔ channels and in combination with mutation T449V. These mutants were done as described previously (López-Barneo et al. 1993; Molina et al. 1997). The cDNAs encoding the various mutant channels were subcloned in plasmid p513, a derivative of pSG5 (Stratagene). For all the mutations, the entire DNA region was sequenced to check for the mutation and ensure against mistakes of the polymerase. For channel expression, we used Chinese hamster ovary cells transiently transfected with 2–10 μg of the cDNAs by electroporation (Gene Pulser; Bio-Rad Laboratories). For transfection, cells were resuspended in a sucrose-phosphate buffer composed of 1 mM sucrose, 2 mM MgCl2, 1 mM KH2PO4, and 1mM K2HPO4 and placed in cuvettes at room temperature (≈22–25°C). Electroporation parameters (350 V and 125 μF) yielded typical time constant values of 24–26 ms. cDNA of green fluorescent protein included in plasmid pRK5 was cotransfected with the K+ channel α subunits to detect by fluorescence those cells expressing K+ currents. After electroporation, the cells were resuspended in culture medium (McCoy's 5A with supplements), plated on slivers of glass coverslips, and maintained in an incubator at 37°C until use.
Electrophysiology and Data Analysis
Electrical recordings were performed on cells 24–72 h after plating. For the experiments, a coverslip was transferred to a recording chamber of ≈0.2 ml with continuous flow of solution that could be completely replaced in <40 s. Potassium currents were recorded using the whole-cell configuration of the patch-clamp technique as adapted to our laboratory (Hamill et al. 1981; Castellano and López-Barneo 1991). We used low resistance electrodes (1–3 MΩ), capacity compensation and subtraction of linear leakage, and capacitive currents. Series resistance compensation was between 40 and 50%. Macroscopic current recordings were low-pass filtered with the cutoff frequency between 3 and 10 kHz. Single-channel recordings were done in excised outside-out membrane patches using pipettes of 5–7 MΩ. In these experiments, subtraction of leakage and capacity currents was done off-line using the average current generated in the same patch by 20 consecutive hyperpolarizing pulses of 10 mV and scaled to the appropriate value. Single-channel records were low-pass filtered at 500 or 700 Hz. For the calculation of the mean cluster duration, cluster criterion were set at 20 and 5 ms for the E418E and E418Q channels, respectively. The deactivation time course of macroscopic K+ tail currents and the C-type inactivation rate were estimated by fitting the appropriate current traces with a single exponential function. The composition of the standard solutions was (mM): external: 140 NaCl, 2.7 KCl, 2.5 CaCl2, 4 MgCl2, 10 HEPES, pH 7.4; solution in the pipette and inside the cell: 30 KCl, 80 K-glutamate, 20 K-fluoride, 2 Mg-Cl2, 4 ATP-Mg, 10 HEPES, 10 EGTA, pH 7.2. For experiments in which we used external solutions with a different [K+], NaCl was replaced for the same amount of KCl. To study C-type inactivation of inward K+ currents, all the K+ of the internal solution was replaced for NMDG. Unless otherwise noted, the holding potential in the electrophysiological experiments was −80 mV. Although the junction potential of the solutions was not compensated, we estimated the changes of junction potential produced by the use of solution of variable [K+]. Switching from the control (2.7 mM K+) external solution to solutions with 30, 70, and 140 mM K+ produced changes of junction potential of approximately −0.6, −2, and −4.5 mV, respectively. Experiments were performed at room temperature (≈22–25°C).
RESULTS
Differential Influences of Extracellular Dicarboxylic Residues on K+ Channel Gating
The dicarboxylic amino acids mutated in this study are indicated in Fig. 1 A on a scheme of the proposed transmembrane topology of the α subunit of voltage-gated K+ channels. Glutamate and aspartate residues located near the transmembrane segments in loops S1-S2 (E251 and D277), S3-S4 (E333, E334, D335, and E336), and S5-S6 (E418 and E422) of the Shaker channel were replaced by glutamine or asparagine. Mutations in loop S3-S4 were all done simultaneously so the channel studied (EEDE333-336QQQQ) had four negative charges neutralized in each α subunit (channel 4Q). On this channel construct, we replaced residues in positions 422 and 418 to obtain the channels EEDE333-336QQQQ, E422Q (5Q), and EEDE333-336QQQQ, E422Q, E418Q (6Q). Representative current traces obtained during depolarizing pulses to +20 mV in cells transfected with either wild-type Shaker BΔ channels or any of the five mutants are shown in Fig. 1 B. Because, in most of the mutants (except channel 6Q), inactivation rates were similar to, or even slightly slower than, the inactivation rate of Shaker BΔ channels, current decay was not appreciable during the short-lasting depolarizing pulses of the figure. Mean values of inactivation time constant and of other voltage-dependent parameters of the various types of K+ currents recorded are summarized in Table . Inactivation kinetics of the 5Q channels was seen to change from one experiment to another, being in some cases two or three times faster than in the wild-type channel. This variability, which could not be attributed to any special modification of the experimental protocol, is illustrated in Fig. 1 B with examples of fast- and slow-inactivating current traces (** and*, respectively). Macroscopic currents resulting from the expression of the 6Q channels had normal activation kinetics, but showed an almost 150-fold acceleration of inactivation. Another notable characteristic of the recordings obtained from cells expressing 6Q channels was a >10-fold reduction of peak current amplitude (Table ). In cells where the efficiency of the 6Q cDNA transfection was not too high, the amplitude of the current in the normal (2.7 mM K+) extracellular solution was almost negligible (see below).
Figure 1.
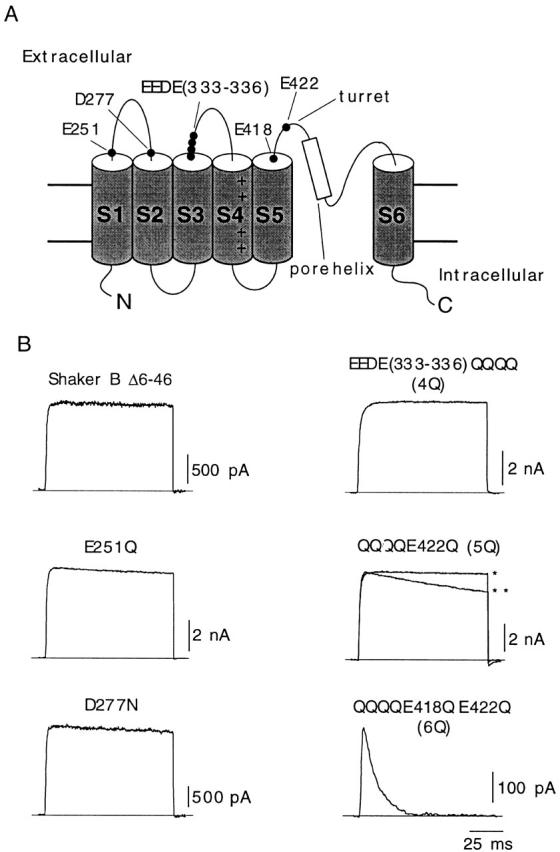
Differential effects of neutralization of charges in extracellular dicarboxylic residues of Shaker K+ channels. (A) Scheme of the transmembrane topology of the α subunit of the K+ channel with indication of the residues mutated in this study. (B) Representative recordings of outward K+ currents from six different types of channels studied. In all cases, the membrane was depolarized from −80 to +20 mV during 100-ms depolarizations, and external K+ was 2.7 mM. Two different recordings corresponding to channel 5Q are superimposed to illustrate the variability of C-type inactivation in this mutant (*time constant, 1,970 ms; **time constant, 542 ms). Current amplitude is scaled to match the value of the current in the trace indicated with the single asterisk.
Table 1.
Gating Parameters of the K+ Channel Mutants Studied
| Mutants | Imax | Rise time | τi | τc | ||||
|---|---|---|---|---|---|---|---|---|
| nA | n | ms | n | ms | n | ms | n | |
| WT | 3.3 ± 2.3 | 9 | 1.44 ± 0.5 | 9 | 2190 ± 250 | 6 | 1.53 ± 0.45 | 8 |
| E251Q | 3.8 ± 2.5 | 8 | 0.93 ± 0.3 | 4 | 1980 ± 140 | 4 | 1.10 ± 0.30 | 3 |
| D277N | 2.7 ± 1.5 | 10 | 0.96 ± 0.3 | 6 | 1770 ± 750 | 5 | 0.97 ± 0.25 | 3 |
| 4Q | 3.4 ± 1.7 | 12 | 1.84 ± 0.4 | 7 | 2400 ± 640 | 8 | — | |
| 5Q | 4.2 ± 3.1 | 23 | 1.84 ± 0.6 | 14 | 1490 ± 950 | 9 | 2.38 ± 0.36 | 3 |
| 6Q | 0.29 ± 0.21 | 16* | 1.27 ± 0.41 | 8 | 11.2 ± 2.70 | 7* | — | |
| E418Q | 0.45 ± 0.49 | 15* | 1.12 ± 0.2 | 7 | 9.22 ± 2.40 | 6* | 0.35 ± 0.06 | 4* |
| E418D | 4.9 ± 3.1 | 17 | 1.36 ± 0.5 | 10 | 2600 ± 120 | 7 | 2.31 ± 0.51 | 3 |
Mean ± SD; n, number of experiments. *Parameters are significantly different (P < 0.001, nonparametric Mann-Whitney U test) from the values in the wild-type channel. Peak current amplitude (Imax), rise time (time between 20 and 80% of current amplitude), and inactivation time constant (τi) were measured at +20 mV. Closing time constant (τc) was measured at −80 mV in 30 mM external K+.
Because it has been shown in previous studies that dependence of C-type inactivation on external K+ is higher after abolishment of outward current (Baukrowitz and Yellen 1995, Baukrowitz and Yellen 1996), we also measured the C-type inactivation rate of the various channel mutants expressed in cells dialyzed with K+-free internal solution. Traces of inward K+ currents of wild type and the channel mutants studied are illustrated in Fig. 2. Values for the C-type inactivation time constant at various external [K+] are given in Table . As described previously (Baukrowitz and Yellen 1995), inactivation of wild-type inward K+ currents recorded with 2.7 mM external [K+] was about three times faster than inactivation of outward K+ currents recorded in the standard experimental conditions (Table and Table ). For unknown reasons (perhaps due to differences in animal species or experimental protocols), inactivation rates of Shaker BΔ outward and inward K+ currents were slower in transfected Chinese hamster ovary cells (this study) than in patches excised from Xenopus oocytes (López-Barneo et al. 1993; Baukrowitz and Yellen 1995). Inactivation rates of the inward K+ currents in the mutant channels (except in channel E418Q, described below) were within the range of the inactivation rate of wild-type channels, and in all cases they were slowed in the same proportion upon raising extracellular [K+] (Fig. 2 and Table ). In solutions with relatively low external [K+] (2.7 and 5 mM), inactivation of mutants D277N and 5Q appeared to be somewhat faster than inactivation of the wild-type channel. These effects, although manifested repeatedly in different experiments, were small and not appreciable at higher external [K+].
Figure 2.

C-type inactivation time course of inward K+ currents mediated by the various channel mutants studied. Representative current traces recorded from cells with the indicated channel types. Pulses applied from −80 to −10 mV. In all cases, we used K+-free internal solution. The external solution contained 5 mM K+ in all cases except in experiments with E418Q channels, which were done with 30 mM K+.
Table 2.
Inactivation Time Constant (ms) of the Channel Mutants Studied with Inward Currents in Various External [K+]
| Mutants | External [K+] | |||||
|---|---|---|---|---|---|---|
| 2.7 mM | n | 5 mM | n | 30 mM | n | |
| WT | 770 ± 71 | 3 | 927 ± 227 | 10 | 1527 ± 85 | 4 |
| E251Q | 657 ± 79 | 2 | 923 ± 138 | 5 | 1492 ± 280 | 4 |
| D277N | 544 ± 146 | 3 | 874 ± 133 | 4 | 1794 ± 237 | 3 |
| 4Q | 722 ± 85 | 3 | 1113 ± 254 | 7 | 2231 ± 606 | 7 |
| 5Q | 587 ± 194 | 3 | 757 ± 191 | 7 | 1450 ± 71 | 2 |
| E418Q | — | — | 16.4 ± 4 | 3* | ||
Mean ± SD; n, number of experiments. *Parameter is significantly different (P < 0.001, nonparametric Mann-Whitney U test) from the value in the wild-type channel. Inactivation time constant measured at −10 mV. The internal solution contained 0 mM K+.
A Conserved Extracellular Glutamate Determines K+ Current Amplitude and Kinetics
Given the pronounced differences in inactivation kinetics between outward currents of channels 5Q and 6Q (Fig. 1), presumably ascribable to the mutation of the residue in position 418, we studied the single mutant E418Q. Macroscopic currents recorded from cells expressing this channel type were of small amplitude and exhibited the fast inactivation time course characteristic of channel 6Q (Fig. 3 A; Table ). Inactivation rate of mutant E418Q was also very fast in cells dialyzed with K+-free internal solutions and, in this condition, measurable currents were recorded only when external [K+] was higher than 5 mM (Fig. 2 and Table II; see below). The action of glutamate 418 appeared to reside mainly on the negatively charged carboxyl group since its replacement for aspartate, with shorter side chain but maintaining the negative charge (mutant E418D), had almost no effect on channel kinetics and current amplitude (Fig. 3 A, Table ). Inactivation and closing kinetics of channel E418D were even slightly slower than in the wild-type Shaker BΔ channel (Table ), but the differences were small and were not studied in further detail. Thus, whereas neutralization of the charge of several dicarboxylic amino acids in the extracellular loops of Shaker K+ channels has little affect on channel activity and kinetics, the maintenance of a negatively charged residue in position 418 is absolutely necessary for the normal function of the channels. Fig. 3 B illustrates that, although the length and number of negatively charged residues of the turret varies among the different types of K+ channels, the glutamate at position 418 of Shaker is conserved in all the families of delayed rectifier voltage-dependent channels of both Drosophila and mammals. This residue is also maintained in silent, regulatory K+ channel α subunits, such as Kv2.3 (see Castellano et al. 1997), and, although absent in most of the K+ channels with two membrane-spanning segments (Kubo et al. 1993), it is present in the KcsA channel from Streptomyces lividans (Doyle et al. 1998).
Figure 3.

A negatively charged residue at position 418 is necessary for normal channel gating. (A) Currents recorded in cells transfected with the indicated channel types. In all cases, 100-ms pulses were applied from −80 to +20 mV. (B) Sequence alignment of the region between the carboxyl end of membrane-spanning segment S5 and the pore helix in several types of K+ channels. In a box is glutamate 418 of Shaker, conserved at the same position in all the channels indicated.
The relatively small size of the K+ current observed in cells transfected with mutants 6Q or E418Q (Table ) was not due to a decrease in the level of channel expression, but to a reduction in the number of open channels, which was strongly dependent on extracellular [K+] (Fig. 4). Superfusion of E418Q-transfected cells with a K+-free solution elicited an immediate abolishment of the voltage-dependent K+ current, which was reversed by reintroduction of K+ in the chamber (Fig. 4 A). In contrast, exposure to high (30 mM) external K+ produced, despite the reduction of the K+ driving force, an increase in current amplitude (Fig. 4 B). K+ currents recorded from a representative E418Q-transfected cell exposed to various external [K+] are shown in Fig. 4 C, and Fig. 4 D summarizes the relationship between peak current amplitude and extracellular [K+] with average data obtained from several cells. The dependence of peak current amplitude on external [K+] had a bell-shaped curve. Rising [K+] evoked a gradual increase in current amplitude, with a maximal value obtained with 70 mM. Current amplitude decreased with higher concentrations due to reduction of the K+ driving force. Since the increase of external [K+] had little effect on E418Q single-channel current amplitude (data not shown), these data indicate that the augmentation of the macroscopic E418Q currents induced by extracellular K+ was due to the increase in the number of channels that were simultaneously open during the depolarizing pulses.
Figure 4.

Modulation by extracellular K+ of E418Q channels. (A and B) Reversible modification of the amplitude of K+ currents recorded in control conditions (2.7 mM K+) and during exposure to low (0 mM) or high (30 mM) extracellular [K+]. Internal [K+] was 130 mM. In all cases, 100-ms pulses were applied to +20 mV. (C, left) K+ currents recorded from a cell with a pulse to +20 mV during exposure to various extracellular [K+]. (Right) Changes of peak K+ current amplitude at +20 mV (ordinate) as a function of extracellular [K+] (abscissa). Data points are the mean and the vertical bars are the standard deviation of measurements done in eight cells.
The Effects of Mutation E418Q Are Similar to Those Produced by Channel Deprivation of K+ Ions
The modulatory effect of extracellular K+ on E418Q current amplitude resembles the effect of the cation on the currents mediated by Kv1.4 channels (Pardo et al. 1992; Jäger et al. 1998) and the Shaker pore mutants at positions 449 and 447 with fast C-type inactivation kinetics (López-Barneo et al. 1993; Baukrowitz and Yellen 1995; Molina et al. 1997). External K+ competes with C-type inactivation (from either open or closed states), thus increasing the number of channels that are simultaneously open during the depolarizing pulse (López-Barneo et al. 1993; Jäger et al. 1998). Therefore, it seems that neutralization of charge at position 418 produces an effect equivalent to depletion of the pore by K+ ions. Besides increasing C-type inactivation rate, channel deprivation of K+ ions also results in acceleration of closing kinetics and reduction of single channel conductance (López-Barneo et al. 1993; Molina et al. 1998). Fig. 5 shows that the increase of inactivation rate in mutant E418Q was partially compensated by elevation of extracellular K+ (Fig. 5 A) and that this mutation also induced four- to fivefold acceleration of closing time course (Table ) observed over a broad range of membrane potentials (Fig. 5 B). As for C-type inactivation, the closing of channel E418Q was also slowed by raising external K+ (deactivation time constant at −80 mV was 0.35 ± 0.06 ms, n = 4, in 30 mM K+, but 0.47 ± 0.02 ms, n = 3, in 70 mM K+).
Figure 5.
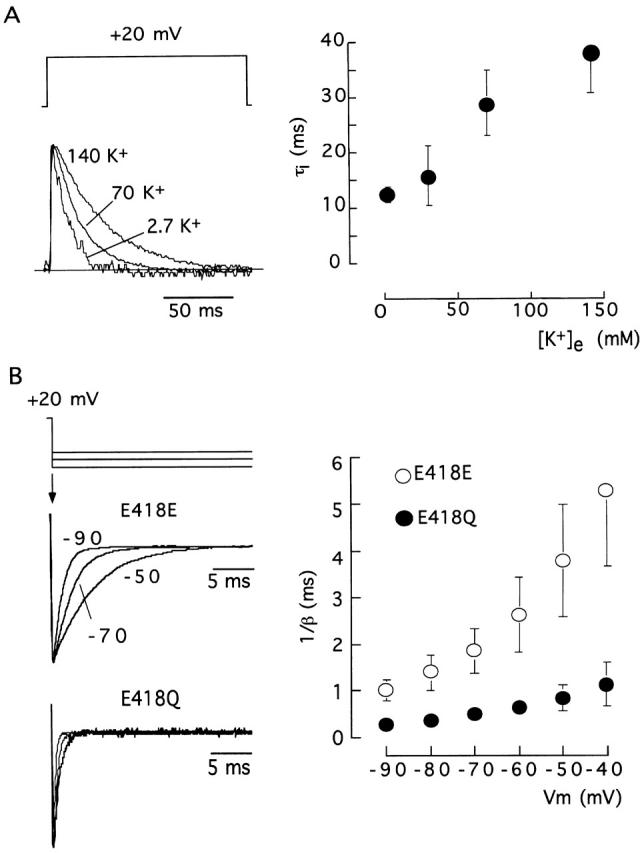
Modulation by extracellular K+ of C-type inactivation and closing time courses in E418Q channels. (A, left) Scaled current traces generated by pulses to +20 mV; (right) inactivation time constant (τi, ordinate) as a function of extracellular [K+] (abscissa). Data points are the mean and the vertical bars are the standard deviation of measurements done in six cells. (B, left) Scaled recordings of tail currents illustrating the deactivation time course at the indicated membrane potentials (−50, −70, and −90 mV) of wild-type (E418E) and E418Q channels with 30 mM external K+. (Right) Closing time constant (expressed in the ordinate as the inverse of the deactivation rate constant) of the two channel types as a function of the membrane potential (abscissa). Data points are the mean and the vertical bars are the standard deviation of measurements done in eight (E418E) and four (E418Q) cells.
Comparison of single-channel currents recorded with the standard asymmetrical [K+] indicated that, as expected, E418Q channels have smaller single-channel current amplitude and conductance than wild-type, E418E, channels (Fig. 6). We estimated the average duration of well-defined clusters of openings recorded from excised patches that appeared to contain a single E418E or E418Q channel. Mean burst duration was voltage independent in the range between +20 and +50 mV, averaging 270 ± 188 ms (mean ± SD, n = 135 in five cells) in Shaker BΔ channels (E418E), but only 10.5 ± 5.5 ms (n = 29 in four cells) in E418Q channels. We also observed clear differences between the two channel types in the number of clusters during each depolarizing pulse, a parameter that at positive membrane potentials indicates the reversibility of the C-type inactivated state. In response to a large depolarization, E418Q channels produced normally only a single cluster of openings (1.18 ± 0.39, mean ± SD, n = 38 clusters in seven cells), whereas two or three clusters were observed in Shaker BΔ channels with pulses of 0.5 or 1 s, even though they lasted less than the time required for inactivation to be complete. The reduction of cluster duration and the number of clusters per trace produced by mutation E418Q is in accord with the acceleration of the rate of macroscopic C-type inactivation described above, and further suggests that neutralization of charge at position 418 destabilizes the open conformation of the channels and favors their transition to the C-inactivated state.
Figure 6.
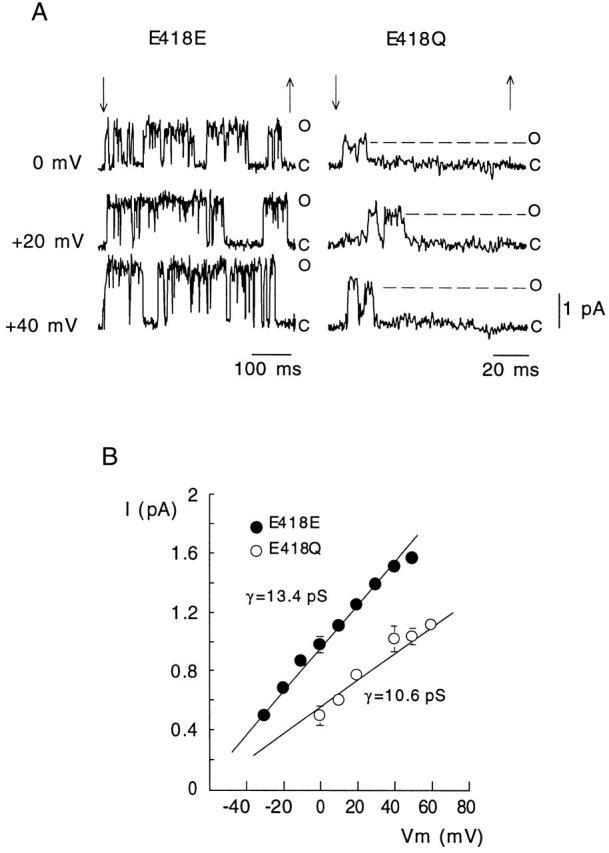
Comparison of single-channel currents recorded in outside-out patches that seemed to contain only one wild-type (E418E) or mutated (E418Q) channel. (A) Examples of current records during application of depolarizing pulses (between the arrows) to the indicated membrane potentials. The current levels indicating the closed (C) and open (O) states of the channels are shown. Extra- and intracellular [K+] were 2.7 and 130 mM, respectively. (B) Plot of single-channel current amplitude as a function of the membrane potential in the two channel types exposed to the indicated asymmetrical [K+]. Data points are the mean and the vertical bars are the standard deviation of measurements done in three (E418E) and six (E418Q) cells. The straight lines are linear regression fits to the data points with slopes giving values of the average single-channel conductance (γ).
Neutralization of residue E418 had effects opposed to those produced by pore mutations, such as T449V, which abolish modulation by extracellular K+ and result in channels with ultraslow C-type inactivation (López-Barneo et al. 1993; Baukrowitz and Yellen 1995; Jäger et al. 1998; Melishchuk et al. 1998). This is illustrated in Fig. 7, with superimposed scaled current records of Shaker BΔ channels, the single mutant T449V, and the double mutant T449V, E418Q. The marked slowing of inactivation time course produced by mutation T449V (∼18-fold reduction of inactivation rate) was partially compensated when the mutation E418Q was done on the T449V channel (channel E418Q, T449V). These data indicate that mutation E418Q alters channel gating in a way similar to deprivation of the channels of K+ and that the effects of this mutation can be compensated by external K+ or mutations in the pore (such as T449V), with slow C-type inactivation.
Figure 7.

Comparison of C-type inactivation time courses of Shaker BΔ channels and of mutants at position 449 alone (T449V) or at positions 449 and 418 (E418Q, T449V). Pulses of 60 s applied from −80 to +20 mV. Mean ± SD (number of experiments is shown in parentheses) of inactivation time constants in the three channel types are also indicated in the figure.
DISCUSSION
The major finding in this study is the identification of a molecular determinant for the stabilization of conductiving versus closed and of conductiving vs. C-inactivated conformations of voltage-dependent K+ channels. Among the various dicarboxylic residues in the extracellular loops of the K+ channels, E418 of Shaker, conserved in most families of voltage-dependent K+ channels, is absolutely necessary for the channels to be functional at the normal extracellular [K+]. The role of glutamate 418 appears to depend on its negatively charged side chain since a mutation conserving the charge (E418D) has no major functional consequences, whereas neutralization of the charge (mutation E418Q) results in almost complete loss of conductance. In a previous work on toxin binding to Shaker K+ channels, the mutant E418K was reported to be unable to express currents in Xenopus oocytes (Goldstein et al. 1994). We also show here that the function of E418 is highly specific since neutralization of up to 16 negative charges of residues located in similar position with respect to the S1, S2, or S3 transmembrane segments (E251, D277, and the group from E333 to E336, respectively) produces little changes of channel activity. Although with subtle effects, residues D277 and E422 might contribute to concentrate cations near the channel mouth. Glutamate 422 is located near the pore helix and is known to have a strong influence on the electrostatic binding of positively charged toxins to the K+ channels (MacKinnon and Miller 1989; Goldstein et al. 1994). However, neutralization of the negative charges at positions 277 and 422 results in minimal gating alterations as compared with the drastic changes in current amplitude and kinetics observed in channel E418Q.
Mutation E418Q causes a decrease of single-channel conductance and reduction of the time that the channels stay open during a maintained depolarization. In parallel with these changes, macroscopic currents mediated by the mutated channels exhibit a marked acceleration of C-type inactivation and closing kinetics. Due to these biophysical alterations, macroscopic K+ currents in E418Q-transfected cells were barely appreciable, although the number of channels expressed was normal. In these same experiments, vigorous K+ currents were recorded when extracellular [K+] was elevated to 30 or 70 mM. The effects of mutation E418Q are qualitatively similar to the changes produced in Shaker K+ channels by removal of extracellular K+ or by mutations reducing the occupation of the pore by permeating ions (López-Barneo et al. 1993; Baukrowitz and Yellen 1996; Jäger et al. 1998; Molina et al. 1998). The resemblance is particularly evident with channel D447E, mutated in a residue adjacent to the selectivity filter, which has also fast closing and C-type inactivation kinetics strongly dependent on extracellular K+ (Molina et al. 1997, Molina et al. 1998). In addition, we have shown that the effects of mutation E418Q are compensated by a mutation in the pore (T449V) that produces ultraslow C-type inactivation and makes the channels resistant to removal of extracellular K+.
Despite the similarity between the gating modifications produced by mutation E418Q and by depletion of the pore of permeating ions, the location of residue 418 makes it unlikely that its primary role is to increase the local [K+] in the external mouth of the channels. Glutamate 418 is in the transition between the membrane-spanning segment S5 and the base of the turret, and therefore far from the entryway of the channel pore (Doyle et al. 1998). Nevertheless, although not exerting a direct electrostatic attraction on potassium ions, residue E418 might indirectly alter occupation of the pore by cations since it is located inside the protein in the same plane and only few angstroms away from residues D447 and D449 (Doyle et al. 1998), which, as indicated above, critically determine closing and C-type inactivation time courses. Neutralization of the charge in position 418 could allosterically produce a modification of the entryway of the pore sufficient to cause the observed changes in conductance and kinetics. Independently of these effects, glutamate 418 could also have a more direct participation in channel activation, being possibly involved in coupling the structural rearrangements of the pore and the voltage sensor during C-type inactivation.
This last role of glutamate 418 is described diagrammatically in Fig. 8 with a scheme of a sectioned K+ channel inspired by the model of Melishchuk et al. 1998 that incorporates the structural data of Doyle et al. 1998 and recent spectroscopy results on the movement of the S4 segment during activation (Cha et al. 1999; Glauner et al. 1999). The P region is shown in white with the negative charges of the carbonyl dipoles lining the selectivity filter and the negative charge of residue 418 inside a circle. Two of the four S4 helices, with seven positive charges each, are shown. The helices are connected to gates that open and permit the outflux of K+ ions when the S4 segments move and rotate by depolarization (Cha et al. 1999; Glauner et al. 1999). Once rotated, positively charged side chains in S4 are attracted by the negative charge of E418, thus stabilizing the voltage sensor in the “depolarized” conformation and making difficult the transition from the open state to either the C-inactivated or closed states. C-type inactivation, initiated by the exit of K+ from the pore, results in closer apposition of the four P regions (Liu et al. 1996), which displaces E418 and weakens its interaction with S4. This is consistent with previous work using fluorophores attached to specific residues, suggesting rearrangement of the S4 segment by either occupation of the pore by permeant ions (Cha and Bezanilla 1998) or during C-type inactivation (Loots and Isacoff 1998). In consequence, both stabilization of the S4 sensor in the “depolarized” position by E418 and occupation of the pore by extracellular K+ are opposed to the conformational change producing C-type inactivation. Therefore, any of the two alterations, removal of external K+ or mutation E418Q, destabilize the conductiving state and favor C-type inactivation. Once C-type inactivation is completed and S4 displaced with respect to the P region, the voltage sensor may adopt a new stable conformation. The lack of electrostatic attraction of the S4 sensor in channel E418Q favors the return of the voltage sensor from the “depolarized” to the “resting” position, thus explaining the acceleration of channel closing on repolarization. Extracellular K+ can retard C-type inactivation and closing, and thus compensate partially the effects of mutation E418Q; however, in the physiological conditions most K+ channels are exposed to, low external K+ and therefore glutamate 418 is necessary to prevent the collapse of the K+ conductance.
Figure 8.

Summary and model of the role of glutamate 418 to stabilize the channel voltage sensor in the conductiving conformation. A section of a K+ channel is shown with the pore region in white containing the negative charges of the carbonyl dipoles lining the selectivity filter. The negatively charged side chain of residue 418 located at the base of the turret is represented inside a circle. Two of the four S4 helices with seven positive charges, connected to the activation gates, are shown. The channel is shown at the closed, open, and C-inactivated states. The open state resulting from the movement of the S4 voltage sensors is stabilized by the charge in residue 418. Transition from the open to the C-inactivated state involves reorganization of the pore and rearrangement of the S4 helices, which is favored by either removal of extracellular K+ or neutralization of the charge at position 418. Destabilization of the open state in channel E418Q accelerates the closing of the channels. See text for further details.
Good candidates for having a part in the electrostatic interaction between S4 and E418 are the positively charged arginines (R362 and R365) located in the external side of the voltage sensor. Naturally, the effect of neutralization of these arginines must be tested in future experimental work; however, it is quite interesting that some of the properties of channel mutants R362Q and R365Q, studied by Seoh et al. 1996 in a different context, fit perfectly well with the expectations indicated above. For example, in channel mutant R362Q, opening probability and the amount of gating charge displaced by depolarization are shifted to positive membrane potentials, suggesting that higher energy is necessary for complete movement of the voltage sensor. As channel E418Q, the mutant R365Q has a conductance smaller than normal and displacement of the open-inactivate equilibrium toward the C-type inactivated state.
The loss of conductance observed at low external K+ in E418Q channels might be related to the defunct state caused by removal of K+ from the two sides of the membrane (Gómez-Lagunas 1997; Melishchuk, et al. 1998). Several mutations that remove C-type inactivation also make the channels resistant to becoming defunct and both C-type inactivation and the defunct state are favored by moderate depolarizations (Gómez-Lagunas 1997; Melishchuk et al. 1998). However, whereas C-type inactivation is readily reversible, recovery from the defunct state is slow and requires repolarization followed by a long depolarization. Therefore, it is possible that a reversible C-type inactivated state precedes the irreversible defunct state that, as suggested by Melishchuk et al. 1998, could be caused by extensive deformation of the P region in the absence of external and internal K+, forcing S4 towards the “resting” (or repolarized) position.
Given the fundamental role of glutamate 418 in gating, its conservation or replacement might have contributed to the evolution of K+ channels with kinetic properties adapted to specific functional roles. As indicated before, a glutamate residue in the position equivalent to 418 of Shaker is conserved in all the voltage-dependent K+ channels where stabilization of the S4 sensor in the “depolarized” state is probably necessary to ensure channel opening. Interestingly, glutamate 418 is replaced by neutral amino acids in most K+ channels with two membrane-spanning segments lacking the S4 voltage sensor (Kubo et al. 1993), in cyclic nucleotide–gated channels, which have an S4 segment but are voltage independent (Chen et al. 1993), and in cardiac HERG channels (Warmke and Ganetzky 1994), with characteristic ultrafast C-type inactivation that converts the channels into functional inward rectifiers (Schönherr and Heinemann 1996; Smith et al. 1996).
Acknowledgments
This study was supported by grants from the Spanish Ministry of Education, Andalusian Government, and Fundaciones La Caixa and R. Areces. P. Ortega-Sáenz and R. Pardal were recipients of fellowships of the Programa de Formación del Personal Investigador of the Spanish Ministry of Education and Fondo de Investigación Sanitaria.
References
- Almers W., Armstrong C.M. Survival of K+ permeability and gating currents in squid axons perfused with K+-free media. J. Gen. Physiol. 1980;75:61–78. doi: 10.1085/jgp.75.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz Y., Yellen G. Modulation of K+ currents by frequency and external [K+]a tale of two inactivating mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Baukrowitz Y., Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Castellano A., López-Barneo J. Sodium and calcium currents in dispersed septal neurons. J. Gen. Physiol. 1991;97:303–319. doi: 10.1085/jgp.97.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano A., Chiara M.D., Mellstrom B., Molina A., Monje F., Naranjo J.R., López-Barneo J. Identification and functional characterization of a K+ channel α-subunit with regulatory properties specific of brain. J. Neurosci. 1997;17:4652–4661. doi: 10.1523/JNEUROSCI.17-12-04652.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A., Bezanilla F. Structural implications of fluorescence quenching in the Shaker K+ channel. J. Gen. Physiol. 1998;112:391–408. doi: 10.1085/jgp.112.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A., Snyder G.E., Selvin P.R., Bezanilla F. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature. 1999;402:809–813. doi: 10.1038/45552. [DOI] [PubMed] [Google Scholar]
- Chen T.Y., Peng Y.W., Dhallan R.S., Ahamed B., Reed R.R., Yau K.W. A new subunit of the cyclic nucleotide-gated cation channel in retinal rods. Nature. 1993;362:764–767. doi: 10.1038/362764a0. [DOI] [PubMed] [Google Scholar]
- Demo S., Yellen G. Ion effects on gating of the Ca2+-activated K+ channels correlate with occupancy of the pore. Biophys. J. 1992;61:639–649. doi: 10.1016/S0006-3495(92)81869-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle D.A., Morais Cabral J.H., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Glauner K.S., Mannuzzu L.M., Gandhi C.S., Isacoff E.Y. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 1999;402:813–817. doi: 10.1038/45561. [DOI] [PubMed] [Google Scholar]
- Goldstein S.A.N., Pheasant D.J., Miller C. The charybdotoxin receptor of a Shaker K+ channelpeptide and channel residues mediating molecular recognition. Neuron. 1994;12:1377–1388. doi: 10.1016/0896-6273(94)90452-9. [DOI] [PubMed] [Google Scholar]
- Gómez-Lagunas F. Shaker B K+ conductance in Na+ solutionsa remarkably stable non-conducting state produced by membrane depolarizations. J. Physiol. 1997;499:3–15. doi: 10.1113/jphysiol.1997.sp021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes 2nd ed 1992. Sinauer Associates, Inc; Sunderland, MA: pp. 607 [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. Two types of inactivation in Shaker K+ channelseffects of alterations in the carboxyl terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Jäger H., Rauer H., Nguen A.N., Aiyar J., Chandy K.G., Grissmer S. Regulation of mammalian Shaker-related K+ channelsevidence of non-conducting closed and non-conducting inactivated states. J. Physiol. 1998;506:291–301. doi: 10.1111/j.1469-7793.1998.291bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss L., Korn S.J. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys. J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y., Reuveny E., Slesinger P.A., Jan Y.N., Jan L. Primary structure and functional expression of the rat G-protein coupled muscarinic K+ channel. Nature. 1993;364:802–806. doi: 10.1038/364802a0. [DOI] [PubMed] [Google Scholar]
- Liu Y., Jurman E., Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- López-Barneo J., Hoshi T., S.H.Heinemann, Aldrich R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Loots E., Isacoff E.Y. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. J. Gen. Physiol. 1998;112:377–389. doi: 10.1085/jgp.112.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marom S., Levitan I. State-dependent inactivation in Kv3 potassium channel. Biophys. J. 1994;67:579–589. doi: 10.1016/S0006-3495(94)80517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R., Miller C. Mutant potassium channels with altered binding of charybdotoxin, a pore-blocking peptide inhibitor. Science. 1989;245:1382–1385. doi: 10.1126/science.2476850. [DOI] [PubMed] [Google Scholar]
- Melishchuk A., Loboda A., Armstrong C.M. Loss of Shaker K channel conductance in 0 K+ solutionsrole of the voltage sensor. Biophys. J. 1998;75:1828–1835. doi: 10.1016/S0006-3495(98)77624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina A., Castellano A.G., López-Barneo J. Pore mutations in Shaker K+ channels distinguish between the sites of TEA blockade and C-type inactivation. J. Physiol. 1997;499:361–367. doi: 10.1113/jphysiol.1997.sp021933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina A., Ortega-Sáenz P., López-Barneo J. Pore mutations alter closing and opening kinetics in Shaker K+ channels. J. Physiol. 1998;509:327–337. doi: 10.1111/j.1469-7793.1998.327bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo L.A., Heinemann S.H., Terlau H., Ludewig U., Lorra C., Pongs O., Stühmer W. Extracellular K+ specifically modulates a rat brain K+ channel. Proc. Natl. Acad. Sci. USA. 1992;89:2466–2470. doi: 10.1073/pnas.89.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr R., Heinemann S.H. Molecular determinants for activation and inactivation of HERG a human inward rectifier potassium channel. J. Physiol. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoh S.-A., Sigg D., Papazian D., Bezanilla F. Voltage-sensing residues in the S2 and S4 segment of the Shaker K+ channels. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- Smith P.L., Baukrowitz Y., Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Science. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Stanfield P.R., Ashcroft F.M., Plant T.D. Gating of a muscle K+ channel and its dependence on the permeating ion species. Nature. 1981;289:509–511. doi: 10.1038/289509a0. [DOI] [PubMed] [Google Scholar]
- Swenson R.P., Armstrong C.M. K+ channels close more slowly in the presence of external K+ and Rb+ . Nature. 1981;291:427–429. doi: 10.1038/291427a0. [DOI] [PubMed] [Google Scholar]
- Warmke J.W., Ganetzky B. A family of K+ channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. USA. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. Single channel seeks permeant ion for brief but intimate relationship. J. Gen. Physiol. 1997;110:83–85. doi: 10.1085/jgp.110.2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]