

Abstract
β-Scorpion toxins shift the voltage dependence of activation of sodium channels to more negative membrane potentials, but only after a strong depolarizing prepulse to fully activate the channels. Their receptor site includes the S3–S4 loop at the extracellular end of the S4 voltage sensor in domain II of the α subunit. Here, we probe the role of gating charges in the IIS4 segment in β-scorpion toxin action by mutagenesis and functional analysis of the resulting mutant sodium channels. Neutralization of the positively charged amino acid residues in the IIS4 segment by mutation to glutamine shifts the voltage dependence of channel activation to more positive membrane potentials and reduces the steepness of voltage-dependent gating, which is consistent with the presumed role of these residues as gating charges. Surprisingly, neutralization of the gating charges at the outer end of the IIS4 segment by the mutations R850Q, R850C, R853Q, and R853C markedly enhances β-scorpion toxin action, whereas mutations R856Q, K859Q, and K862Q have no effect. In contrast to wild-type, the β-scorpion toxin Css IV causes a negative shift of the voltage dependence of activation of mutants R853Q and R853C without a depolarizing prepulse at holding potentials from −80 to −140 mV. Reaction of mutant R853C with 2-aminoethyl methanethiosulfonate causes a positive shift of the voltage dependence of activation and restores the requirement for a depolarizing prepulse for Css IV action. Enhancement of sodium channel activation by Css IV causes large tail currents upon repolarization, indicating slowed deactivation of the IIS4 voltage sensor by the bound toxin. Our results are consistent with a voltage-sensor–trapping model in which the β-scorpion toxin traps the IIS4 voltage sensor in its activated position as it moves outward in response to depolarization and holds it there, slowing its inward movement on deactivation and enhancing subsequent channel activation. Evidently, neutralization of R850 and R853 removes kinetic barriers to binding of the IIS4 segment by Css IV, and thereby enhances toxin-induced channel activation.
Keywords: sodium channels, Centruroides suffusus suffusus toxin IV, β-scorpion toxin, voltage sensor, voltage-dependent gating
INTRODUCTION
Voltage-gated sodium channels are responsible for the voltage-dependent increase in sodium permeability and, therefore, play a critical role in the initiation and propagation of action potentials in excitable cells (Hodgkin and Huxley 1952). Sodium channels are transmembrane proteins composed of a pore-forming α subunit of 260 kD associated with one or two smaller auxiliary subunits β1, β2, and β3 (for review see Catterall 2000). The α subunit consists of four homologous domains (I–IV), each containing six transmembrane segments (S1–S6) and one reentrant segment (SS1/SS2) connected by internal and external polypeptide loops (for review see Catterall 2000). Transmembrane segments S5 and S6 and the membrane-reentrant segments SS1 and SS2 form the narrow ion selectivity filter and the walls of the pore (Noda et al. 1989; Terlau et al. 1991; Heinemann et al. 1992; Ragsdale et al. 1994). In response to changes in membrane potential, the S4 segments move outward and act as voltage sensors to initiate activation (Catterall 1986; Guy and Seetharamulu 1986; Stühmer et al. 1989; Yang and Horn 1995; Yang et al. 1996). The intracellular loop connecting domains III and IV forms the inactivation gate, which mediates voltage-dependent inactivation of sodium channels (Vassilev et al. 1988, Vassilev et al. 1989; Stühmer et al. 1989; West et al. 1992). Outward movement of the S4 segments in domains III and IV is likely to couple activation to inactivation (Chahine et al. 1994; Ji et al. 1996; Rogers et al. 1996; Sheets et al. 1999), and these S4 segments are immobilized in their outward positions by fast inactivation (Cha et al. 1999a; Sheets et al. 2000).
Voltage-gated sodium channels are the molecular target of several groups of neurotoxins, which bind to specific receptor sites and strongly alter sodium channel function (for review see Cestèle and Catterall 2000). The voltage-dependent gating of sodium channels is specifically modified by binding of polypeptide neurotoxins to receptor sites 3 and 4. The α-scorpion toxins, sea anemone toxins, and spider toxins bind to receptor site 3 and slow sodium channel inactivation (Catterall 1977, Catterall 1979; Catterall and Beress 1978; Nicholson et al. 1994). Receptor site 4 binds β-scorpion toxins, which shift the voltage dependence of activation to more negative potentials (Cahalan 1975; Jover et al. 1980; Jaimovich et al. 1982; Meves et al. 1982; Wang and Strichartz 1983; Vijverberg et al. 1984; Jonas et al. 1986; Cestèle et al. 1998). The S3–S4 loops at the extracellular ends of the S4 segments of domains IV and II are involved, respectively, in the formation of neurotoxin receptor sites 3 and 4 (Rogers et al. 1996; Cestèle et al. 1998; Sheets et al. 1999). A voltage-sensor trapping mechanism was proposed to account for the effects of these toxins on channel gating (Cestèle et al. 1998). Binding of α-scorpion toxins and sea anemone toxins to receptor site 3 is proposed to slow inactivation by preventing the normal outward movement of the IVS4 transmembrane segment (Rogers et al. 1996; Sheets et al. 1999). β-Scorpion toxins shift the voltage dependence of activation to more negative potentials only after a strong depolarizing prepulse, indicating that interaction of β-scorpion toxins with receptor site 4 is dependent on activation of the channels. Since the IIS4 segment moves outward during activation, the toxins are proposed to bind to newly accessible residues in the IIS3–S4 loop and the extracellular end of the IIS4 segment during the conditioning prepulse, trapping IIS4 in an outward, activated position. Voltage-sensor trapping favors sodium channel activation and causes the negative shift in the voltage dependence of activation characteristic of β-scorpion toxins.
In this paper, we demonstrate that neutralization of the two outermost arginine residues of the IIS4 voltage sensor markedly enhances β-scorpion toxin effects on sodium channels. This effect is proposed to result from an increase in mobility of the IIS4 segment within the membrane. Our results reveal crucial roles of the two first arginines of the IIS4 segment in voltage-dependent activation, β-scorpion toxin action, and stabilization of the IIS4 segment within the membrane.
MATERIALS AND METHODS
Materials
β-Scorpion toxin Css IV was purified from the venom of Centruroides suffusus suffusus (Martin-Eauclaire et al. 1987). 2-Aminoethyl methanethiosulfonate hydrobromide (MTSEA) and 2-(trimethylammonium)ethyl methanethiosulfonate bromide (MTSET) were purchased from Toronto Research Chemicals Inc. Restriction endonucleases and other molecular biology reagents were purchased from New England Biolabs and Boehringer Mannheim. pCDM8 vector and the MC1061 Escherichia coli bacterial strain were purchased from Invitrogen. Human embryonic kidney tsA-201 cells, a simian virus (SV-40) large T-antigen–expressing derivative of HEK-293 cells, were provided by Dr. Robert Dubridge (Cell Genesis, Foster City, CA). cDNA encoding rat Nav1.2a α subunits (Auld et al. 1990; Goldin et al. 2000) in the pCDM8 vector was used for expression.
Site-directed Mutagenesis
Mutations R850Q, R853Q, K859Q, and K862Q were produced using an M13 construct containing a XmaI-SphI fragment (nt 541–1,897) of the Nav1.2a cDNA. Uracil-containing mutagenesis template was prepared from this construct and oligonucleotide-directed mutagenesis was performed using the dut− ung− selection procedure (Kunkel 1985). Mutations made in the M13 construct were confirmed by sequencing, excised by restriction at the sites XmaI-SphI and isolated by low melting point agarose gel electrophoresis and Prep-a-gene (BioRad Inc.). Fragments were then subcloned into the Nav1.2a sodium channel cDNA in pCDM8. The mutations were confirmed in the final constructs by DNA sequencing using the ABI Prism dye terminator cycle sequencing kit (Perkin-Elmer Applied Biosystems). Mutations R850C, R853C, and R856Q were amplified by PCR in an 800-bp cDNA fragment from the XmaI site to the SphI site and were subcloned into the Nav1.2a cDNA in pCDM8. To facilitate the screening of the clones, a silent HinfI site was introduced along with the mutation. All clones were sequenced through the entire PCR fragment.
Transient Expression in tsA-201 Cells
The tsA-201 cells were maintained at 37°C in 10% CO2 in DMEM/Ham's F12 medium (GIBCO BRL and Life Technologies) supplemented with 10% FBS (Gemini Biological Products), 20 μg/ml penicillin, and 10 μg/ml streptomycin (Gemini Biological Products). TsA-201 cells were transiently cotransfected with cDNAs for the channel α subunit and pEBFP-N1 vector encoding the enhanced green fluorescent protein (CLONTECH Laboratories, Inc.) using calcium phosphate precipitation (Jordan et al. 1996). Successfully transfected cells were detected by fluorescence microscopy.
Electrophysiological Recording and Analysis
Whole-cell sodium currents were recorded from tsA-201 cells expressing Nav1.2a wild-type or mutant sodium channel α subunits. The external recording solution consisted of the following (in mM): 150 NaCl, 10 Cs-HEPES, 1 MgCl2, 2 KCl, and 1.5 CaCl2, pH 7.4. The internal recording solution consisted of the following (in mM): 190 N-methyl-d-glutamine, 10 HEPES, 4 MgCl2, 10 NaCl, and 5 EGTA, pH 7.4. Patch electrodes were pulled from 75-μl micropipette glass (VWR Scientific) and were fire-polished before use. Electrode resistances were typically 1.5–2.5 mΩ in the bath. Recordings were obtained using a patch-clamp amplifier (model Axopatch 200B; Axon Instruments, Inc.). Voltage pulses were applied and data were acquired using pClamp6 software (Axon Instruments Inc.). Linear leak and capacitance currents have been subtracted using an online P/−4 subtraction paradigm. Css IV, MTSEA, and MTSET were dissolved in the extracellular solution at the final concentration. Css IV was applied to cells using fast local perfusion of the cell with background perfusion of the chamber; MTSEA and MTSET were added to the extracellular solution in the recording bath. All experiments were performed at room temperature. Conductance-voltage (activation) curves were derived from peak sodium current versus voltage measurements according to: G = I/(V − VR) where I is the peak current, V is the test voltage, and VR is the apparent reversal potential. Normalized conductance-voltage and inactivation curves were fit with a Boltzmann relationship of the form 1/{1 + exp[(V1/2 − V)/k]} or with the sum of two such expressions, where V1/2 is the voltage for half-maximal activation or inactivation, and k is a slope factor with the dimensions of millivolts.
RESULTS
Effects of Mutations of the Gating Charges in Domain II on Activation and Inactivation
Previous studies have demonstrated the important role of the positively charged amino acid residues of the S4 segments in the voltage-dependent gating of sodium channels (Stühmer et al. 1989; Chen et al. 1996; Kontis et al. 1997; Groome et al. 1999). To examine the importance of the gating charges of the IIS4 segment on β-scorpion toxin action, we constructed mutants in which each of the five arginine and lysine residues in IIS4 were mutated to glutamine. In the absence of toxin, analysis of the conductance-voltage relationships for the wild-type and mutant sodium channels showed a positive shift in the activation curve for R850Q, R853Q, R856Q, K859Q, and K862Q of 3 to 16 mV (Fig. 1 and Table ). Thus, each mutation had effects that opposed voltage-dependent channel activation. In addition, substitutions of R850, R853, R856, and K859 by glutamine reduced the steepness of the activation curve, as expected for neutralization of the gating charges of the sodium channel. Therefore, these positively charged residues are likely to move outward through the membrane electric field during activation and contribute to the electrostatic driving force for channel gating.
Figure 1.
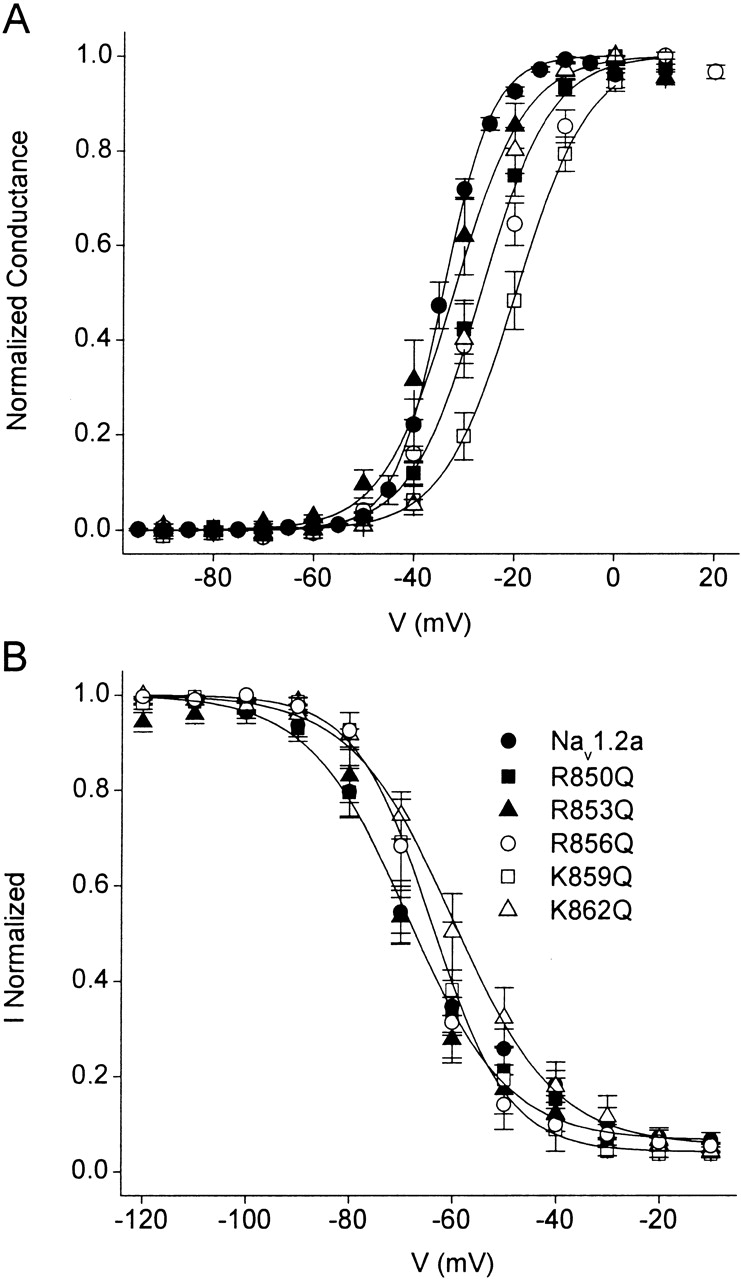
Effects of the neutralization of each of the positive charges of the IIS4 segment of rat brain Nav1.2a sodium channel. Voltage dependence of activation (A) and inactivation (B) for wild-type Nav1.2a, R850Q, R853Q, R856Q, K859Q, and K862Q. Conductance-voltage curves were determined as described in materials and methods . Inactivation curves were determined using 150-ms-long prepulses to the indicated potentials followed by test pulses to 0 mV.
Table 1.
Voltage Dependence of Nav1.2a and Mutants
| Activation | Inactivation | |||||
|---|---|---|---|---|---|---|
| Control V1/2 | Slope | n | Control V1/2 | Slope | n | |
| mV | mV | mV | mV | |||
| Nav1.2a | −36.7 ± 0.6 | 5.8 ± 0.5 | 4 | −68.3 ± 1.0 | −10.2 ± 0.8 | 4 |
| R850Q | −27.5 ± 0.2 | 6.6 ± 0.2 | 4 | −68.8 ± 1.3 | −9.7 ± 0.6 | 4 |
| R850C | −24.1 ± 0.1 | 6.8 ± 0.1 | 11 | −69.3 ± 0.4 | −8.1 ± 0.3 | 10 |
| R853Q | −33.7 ± 0.3 | 7.1 ± 0.3 | 5 | −70.9 ± 0.6 | −7.1 ± 0.4 | 4 |
| R853C | −31.7 ± 0.2 | 7.1 ± 0.1 | 12 | −69.3 ± 0.8 | −7.5 ± 0.7 | 7 |
| R856Q | −25.7 ± 0.3 | 8.5 ± 0.2 | 5 | −66.1 ± 0.2 | −6.0 ± 0.2 | 2 |
| K859Q | −20.8 ± 0.2 | 7.2 ± 0.2 | 3 | −64.0 ± 0.4 | −7.4 ± 0.4 | 3 |
| K862Q | −27.6 ± 0.2 | 5.1 ± 0.2 | 3 | −59.9 ± 0.6 | −9.9 ± 0.5 | 4 |
Measurements of steady-state inactivation for Nav1.2a, R850Q, and R853Q using 150-ms prepulses under control conditions indicate that these mutations do not significantly modify the voltage of half-maximal inactivation when compared with wild-type sodium channels (Fig. 1 B and Table ), despite the difference in voltage dependence of activation. On the other hand, mutations R856Q, K859Q, and K862Q cause small but significant positive shifts in the voltage dependence of steady-state inactivation (Fig. 1 B and Table ). These data agree with previous results implicating the IIS4 segment in activation, rather than inactivation (Chen et al. 1996; Kontis et al. 1997; Cha et al. 1999a), and suggest that outward movement of the IIS4 segment during activation is only weakly coupled to inactivation.
Effects of Css IV on Mutant Sodium Channels with Neutralized Gating Charges in Transmembrane Segment IIS4
As previously described for the wild-type Nav1.2a channels (Cestèle et al. 1998), treatment with 200 nM Css IV initially has no effect on channel activation, as measured by the voltage dependence of the conductance-voltage curve at a holding potential of −100 mV (Fig. 2 A). However, after a 1-ms depolarization to +50 mV, Css IV induced a negative shift in the voltage dependence of activation of a fraction of the sodium channels (Fig. 2 A; Cestèle et al. 1998). Similar results are observed for the mutants R856Q, K859Q, and K862Q (Fig. 2, D–F). In contrast, the neutralization of either of the two outermost positive charges (R850 and R853) of the IIS4 segment strongly altered the effect of Css IV. For R850Q, Css IV caused no shift in activation without a prepulse, but the prepulse induced a much larger shift of the activation curve to more negative membrane potentials, and the voltage dependence of activation of nearly all of the sodium channels was shifted (Fig. 2 B). More remarkably, for the mutant R853Q, Css IV caused a substantial negative shift in the activation of a large fraction of the sodium channels without a depolarizing prepulse and depolarization caused a further shift (Fig. 2 C). The ability of Css IV to induce a negative shift in the voltage dependence of activation without a depolarizing prepulse suggests that toxin binding by itself is sufficient for the IIS4 segment of R853Q channels to adopt an outward, activated position at the resting potential. Evidently, neutralization of the two outermost arginine residues, R850 and R853, favors voltage-sensor trapping by β-scorpion toxins, whereas substitutions of other positively charged residues in IIS4 do not.
Figure 2.

Activity of Css IV on mutant sodium channels with neutralized positive charges in segment IIS4. Conductance-voltage relationships measured as described in materials and methods in control (closed circles) or in the presence of 200 nM Css IV without (open circles) or with (open squares) a +50 mV, 1–ms prepulse that preceded the test pulse by 61 ms for wild-type Nav1.2a (A), R850Q (B), R853Q (C), R856Q (D), K859Q (E), and K862Q (F) sodium channels. The holding potential was −100 mV. The insets show sodium current traces evoked by a test pulse to −65 mV before and after a prepulse to +50 mV in the presence of Css IV.
Effects of Cysteine Substitution and Reaction with Methanethiosulfonate (MTS) Reagents at Positions 850 and 853
To further examine the role of the two outermost arginines of the IIS4 segment in the activation process of sodium channels as well as in the mechanism of action of β-scorpion toxins, we replaced the two outermost arginine residues by cysteine. Those mutants allowed us to use cysteine-modifying MTS reagents to chemically modify the channel at these positions. This method has been extensively used to monitor the movement of the S4 segments during activation and the inactivation gate during inactivation of voltage-gated sodium channels (Yang and Horn 1995; Yang et al. 1996, Yang et al. 1997; Kellenberger et al. 1996; Mitrovic et al. 1998; Vedantham and Cannon 1998).
Neutralizing R850 or R853 by substitution with cysteine reduced the slope of the activation curve and shifted the voltage of half activation by 5–12 mV in the depolarizing direction (Fig. 3 and Table ). Compared with glutamine substitution, neutralization of R850 and R853 by cysteine substitution induces a 2–3-mV additional positive shift in the voltage dependence of activation (Table ). We studied the modification of the mutants R850C and R853C by MTSEA and MTSET. These two reagents covalently modify accessible cysteinyl sulfhydryl groups by the transfer of a positively charged, substituted amino group, but MTSEA is smaller and can reach less accessible sites of reaction. Our experiments were performed by adding MTSET or MTSEA to the extracellular recording solution, allowing modification of the cysteine residues by MTS reagents at the resting membrane potential. Modification of cysteine residues in S4 segments by the MTS reagents is fast and complete under similar conditions (Mitrovic et al. 1998). Treatment with MTS reagents had no effect on wild-type sodium channels (unpublished data). Modification of R850C by MTSEA or MTSET caused a 6.5- or 16.9-mV negative shift in the voltage dependence of activation, respectively, thereby partially or fully reversing the effect of neutralization of this arginine residue (Fig. 3 A). Surprisingly, modification of R853C with MTSEA caused a 15.7-mV positive shift of the conductance-voltage relationship (Fig. 3 B). In contrast, exposure of R853C to MTSET had no effect on the voltage-dependent properties of R853C, presumably because R853C was inaccessible to this relatively large reagent (Fig. 3 B). These data indicate that restoration of the positive charge of R850C by MTSEA or MTSET favors activation of the sodium channels at more negative potentials, and are consistent with restoration of charge providing increased electrostatic driving force for movement of IIS4 to its outward, activated position. On the other hand, modification of R853C by MTSEA shifts activation to more positive potentials and, therefore, favors the resting position of IIS4. The side chain produced when MTSEA reacts with R853C may be too large, have the incorrect shape, or induce an inappropriate conformation to allow the IIS4 segment to move outward easily upon depolarization and, therefore, may inhibit the activation process.
Figure 3.

Modification of R850C and R853C sodium channels by MTSEA and MTSET. Conductance-voltage relationships for R850C (A) and R853C (B) in control and after modification by MTSEA or MTSET. The data were fit with a Boltzmann relationship with V1/2 = −24.1 ± 0.1 mV, k = 6.8 mV for R850C (n = 11); V1/2 = −30.5 ± 0.3 mV, k = 7.8 mV for R850C-MTSEA (n = 9); V1/2 = −39.9 ± 0.5 mV, k = 6.3 mV for R850C-MTSET (n = 18); V1/2 = −31.7 ± 0.2 mV, k = 7.1 mV for R853C (n = 12); V1/2 = −16.1 ± 0.3 mV, k = 7.7 mV, for R853C-MTSEA (n = 11); and V1/2 = −29.6 ± 0.2 mV, k = 7.4 mV for R853C-MTSET (n = 9).
Effects of Css IV on R850C-MTSET and R853C-MTSEA
Because R850C-MTSET and R853C-MTSEA have activation curves that are strongly shifted relative to wild-type Nav1.2a or the unmodified cysteine-containing channels, they represent powerful tools for elucidating the relationship between the voltage dependence of channel gating and the ability of β-scorpion toxins to trap the IIS4 segment in its activated position. Therefore, we analyzed the effects of 200 nM Css IV on R850C-MTSET and R853C-MTSEA (Fig. 4, A–D).
Figure 4.
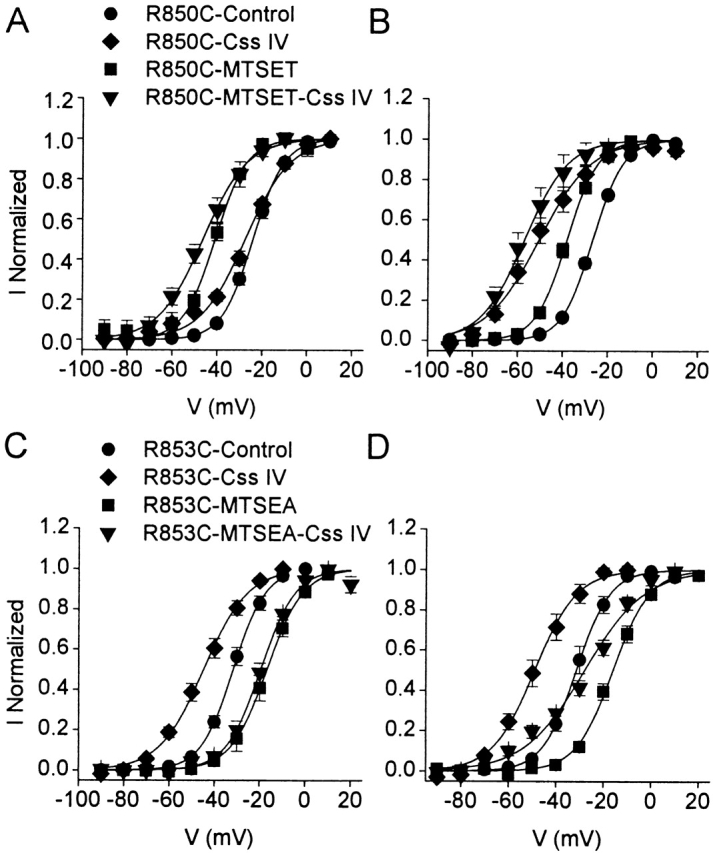
Activity of Css IV on MTS-modified R850C and R853C sodium channels. Conductance-voltage relationships for R850C-MTSET (A and B) or R853C-MTSEA (C and D) in the presence of 200 nM Css IV without (A and C) or with a +50 mV, 1–ms prepulse preceding the test pulse (B and D). Data were fit with Boltzmann relationships with (A) V1/2 = −25.6 ± 0.8 mV, k = 10.2 mV for R850C-Css IV (n = 4) and −46.3 ± 0.6 mV, k = 9.8 mV for R850C-MTSET-Css IV; (B) with V1/2 = −54.3 ± 0.9 mV, k = 12.3 mV for R850C-Css IV (n = 6), and −57.1 ± 0.9 mV, k = 9.6 mV for R850C-MTSET-Css IV (n = 4); (C) V1/2 = −44.8 ± 0.5 mV, k = 9.7 mV for R853C-Css IV (n = 7), −20.1 ± 0.7 mV, k = 7.1 mV for R853C-MTSEA-Css IV (n = 3); (D) V1/2 = −49.0 ± 0.7 mV, k = 8.8 mV for R853C-Css IV (n = 6); −28.0 ± 1.01 mV, k = 12.6 mV for R853C-MTSEA-Css IV (n = 3). The toxin-free curves in A and C are repeated from Fig. 3.
For R850C without MTSET modification, 200 nM Css IV causes a negative shift in the conductance-voltage relationship of a small fraction of sodium channels in the absence of a depolarizing prepulse (Fig. 4 A). Although R850C-MTSET channels have a negative voltage dependence of activation (Fig. 3), treatment with 200 nM Css IV causes an additional negative shift of activation of a similar small fraction of the sodium channels (Fig. 4 A). Comparison of the effects of Css IV on unmodified R850C channels and R850C-MTSET channels shows that they are similar in magnitude, but shifted in absolute voltage dependence by the −17-mV difference in voltage-dependent activation between the two channels. Thus, the negative shifts of activation caused by modification by MTSET and modification by Css IV are additive and independent. Consistent with this, depolarizing prepulses in the presence of Css IV cause a larger negative shift in the activation curves for R850C and R850C-MTSET, and the voltage dependence of a large fraction of the modified channels is shifted (Fig. 4 B). Thus, modification by MTSET causes a negative voltage shift in sodium channel activation, but once channels are fully modified after a depolarizing prepulse, the effects of Css IV are similar before and after treatment with the MTS reagent.
Effects of MTSEA modification of R853C were quite different. For unmodified R853C channels, a prepulse was not required to observe a substantial shift in the activation curve for nearly all of the sodium channels using 200 nM Css IV (Fig. 4 C). However, after modification with MTSEA, a prepulse was required to observe a strongly shifted activation curve (Fig. 4C and Fig. D). This result contrasts with those for unmodified R853Q and R853C sodium channels where the prepulse was not required to observe the complete shift in the activation curve (Fig. 2 C and 4 C). Thus, MTSEA modification of R853C markedly inhibited the effects of Css IV on R853C sodium channels and restored the requirement for a prepulse for Css IV to cause a negative shift in the voltage dependence of activation. Restoration of the positive charge by reaction with MTSEA may stabilize the IIS4 segment in an inward position by restoring ionic interactions.
Voltage Dependence of Wild-Type and Mutant Sodium Channels with IIS4 Segments Trapped in Their Activated Position by Css IV
Although the wild-type and mutant sodium channels studied here had different voltage dependence of activation (V1/2 from −16 to −39 mV; Table and Fig. 3), successful modification by Css IV shifted the voltage dependence of these sodium channels to approximately the same position on the voltage axis (Fig. 5). This is most clearly illustrated for the channels that are strongly shifted to a more negative activation curve after a depolarizing prepulse in the presence of Css IV (i.e., R850Q, R853Q, R850C, and R850C-MTSET; Fig. 5). The activation curves for these toxin-modified channels are all similar, within the experimental error for measurements of the voltage dependence of activation and fits to the sum of two Boltzmann functions (Fig. 5). When only a fraction of the channels are shifted negatively by a prepulse in the presence of toxin (e.g., WT), the activation curves are well-fit with two Boltzmann components: a negative one having approximately the same voltage dependence as the fully shifted activation curves and a positive one resembling unmodified channels (Fig. 5). Thus, approximately the same negative voltage dependence is attained by these toxin-modified channels, whether their activation curves are relatively positive or negative under control conditions and whether their activation curves are fully (R850Q, R853Q, R850C, and R850C-MTSET) or partially (WT) shifted by the prepulse in the absence of toxin (Fig. 5). Exceptions to this rule are presented by toxin-modified R853C and R853C-MTSEA, whose voltage dependence of activation is not as negatively shifted by Css IV. Nevertheless, our results overall indicate that, once wild-type and most mutant sodium channels are successfully modified by toxin binding alone or in response to a depolarization, the channels adopt the same or similar toxin-modified conformation for wild-type, mutants, and MTS-modified mutants, perhaps having the IIS4 segment in its outward, activated position bound to Css IV.
Figure 5.

Voltage dependence of Css IV-trapped sodium channels. Mean conductance-voltage relationships of Nav1.2, R850C/Q, R850C-MTSET, R853C/Q, and R853C-MTSEA in the presence of 200 nM Css IV after a +50 mV, 1 ms–prepulse. The solid lines through the data are fits to a sum of two Boltzmann relationships. The voltage dependence of the positive component was fixed to the half activation and slope values obtained from fits of the control channels to a single Boltzmann as shown in Table . The voltage for half activation (V1/2), slope factors (k), and the fraction of the total represented by the negative, toxin-shifted, component (Aneg) in these fits were the following: for Nav1.2a, V1/2 = −62.9 mV, k = 8.93 mV, Aneg = 0.13; for R850C, V1/2 = −57.4 mV, k = 7.9 mV, Aneg = 0.77; for R850C MTSET, V1/2 = −65.1 mV, k = 6.0 mV, Aneg = 0.64; for R850Q, V1/2 = −62.6 mV, k = 10.88 mV, Aneg = 0.70; and for R853Q, V1/2 = −67.0 mV, k = 6.80 mV, Aneg = 0.76.
Accessibility of the IIS4 Segment in R853C Sodium Channels
The R853C mutant is modified by binding of Css IV at the holding potential of −100 mV, since a strong shift in the voltage dependence of activation is observed immediately on the application of Css IV. To test whether it might be possible to reverse effects of this mutation on Css IV action by using a strongly hyperpolarizing holding potential, we analyzed the effects of Css IV on R853C by measuring the threshold currents elicited by a test pulse at −65 mV when the holding potential was maintained at −100, −120, and −140 mV. In the absence of Css IV, no sodium current is activated at −65 mV in this mutant (Fig. 3 B and 4 C). Transfected cells were exposed to 200 nM Css IV for 2 min at the holding potential. A subsequent test pulse to −65 mV resulted in an inward sodium current that was as large as that normally observed at this potential in the current-voltage curve (Fig. 6, A–C). Thus, R853C renders IIS4 directly accessible for voltage-sensor trapping by the toxin, and hyperpolarizing the membrane potential to −120 or −140 mV is not sufficient to prevent toxin action. This result indicates that neutralization of R853C alters the accessibility of IIS4 to the toxin at all membrane potentials positive to −140 mV. Modification of R853C with MTSEA prevents this change in accessibility of IIS4. When R853C-MTSEA is exposed to 200 nM Css IV using the same holding potential protocol, the test pulse to −65 mV does not result in sodium current, even when the membrane potential is maintained at −100 mV (Fig. 6 D). These data reinforce the hypothesis that modification of R853C by MTSEA stabilizes the IIS4 segment in an inward position, thus, altering dramatically its accessibility to the toxin and preventing voltage-sensor trapping.
Figure 6.
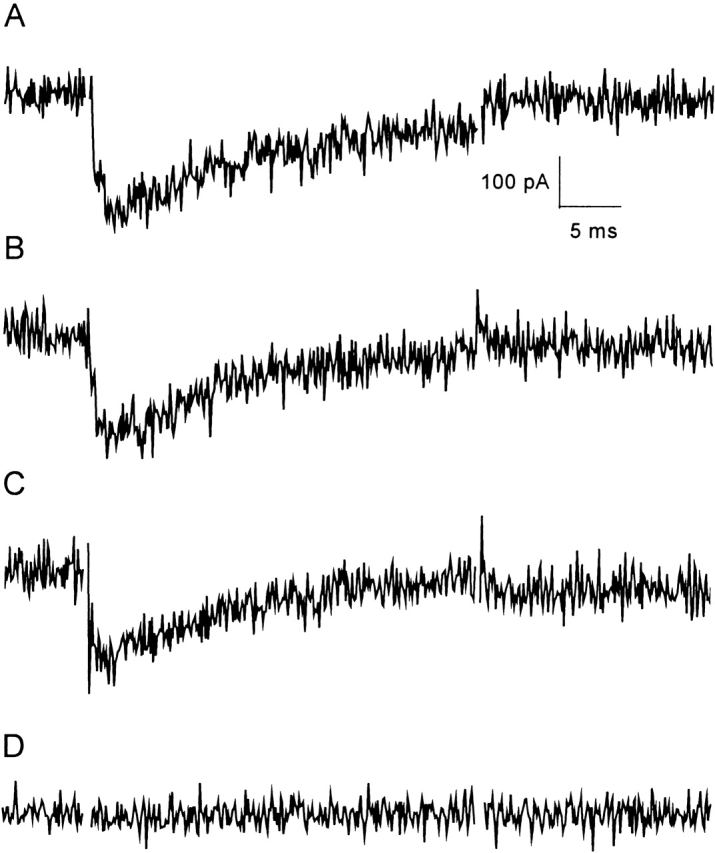
Effects of MTSEA on Css IV-induced threshold sodium currents of mutant R853C. R853C currents elicited by a single test pulse to −65 mV were measured 2 min after introduction of 200 nM Css IV into the bath at a holding potential of −100 (A), −120 (B), or −40 mV (C). (D) R853C-MTSEA currents recorded in the same way with a holding potential of −100 mV.
Toxin-induced Tail Current Reflects Voltage-Sensor Trapping
If the IIS4 segment is trapped in its outward, activated position by binding of Css IV, the toxin would be expected to induce long-lasting tail currents due to slowed deactivation and inward movement of the trapped voltage sensor. In fact, Css venom induces long-lasting tail currents after depolarizing pulses (Cahalan 1975). Tail currents were recorded after a 28-ms test pulse to −20 mV for Nav1.2a, R850Q/C, and R853Q/C in the absence or in the presence of 200 nM Css IV. This long depolarizing pulse allows complete inactivation of the wild-type channels. Therefore, repolarization is not followed by tail currents in the absence of Css IV because one or more of the S4 voltage sensors must deactivate before recovery from inactivation allows current flow (Kuo and Bean 1994). In the presence of 200 nM Css IV, a novel inward tail current is observed upon repolarization for all the channels studied (Fig. 7 A), even though all of the channels are inactivated by the end of the depolarizing pulse. The toxin-induced tail current is larger for the glutamine-substituted channels, R850Q and R853Q (Fig. 7 A). In contrast, the toxin-induced tail currents for R850C and R853C are similar in magnitude to the wild-type Nav1.2a channels (Fig. 7 A). Despite the increased magnitude of the tail currents in the glutamine-substituted mutants, all of the toxin-induced tail currents for Nav1.2a and the Q and C mutants have the same time course, as illustrated by superimposition of the current traces (Fig. 7 B). Analysis of the peak toxin–modified tail current as a function of repolarization potential showed that tail currents are not observed positive to −90 mV and increase steeply with hyperpolarization (Fig. 7 C). This voltage dependence is similar for wild-type and mutant channels, but it differs from tail currents that would be measured in unmodified channels after short depolarizations due to closing of open, noninactivated channels. In that case, tail currents would be observed at all potentials, becoming smaller with depolarization toward the reversal potential, and then increasing again at very positive voltages. The voltage dependence of the toxin-induced tail currents likely reflects inactivation at potentials more positive than −90 mV. Thus, the increase in the amplitude of the toxin-induced tail current with hyperpolarization can be attributed to both an increase in the fraction of conducting channels created by reversal of inactivation as well as to an increase in the driving force for sodium entry. Evidently, trapping the IIS4 segment in its activated position is sufficient to slow sodium channel deactivation and to produce large tail currents through sodium channels that are not inactivated.
Figure 7.
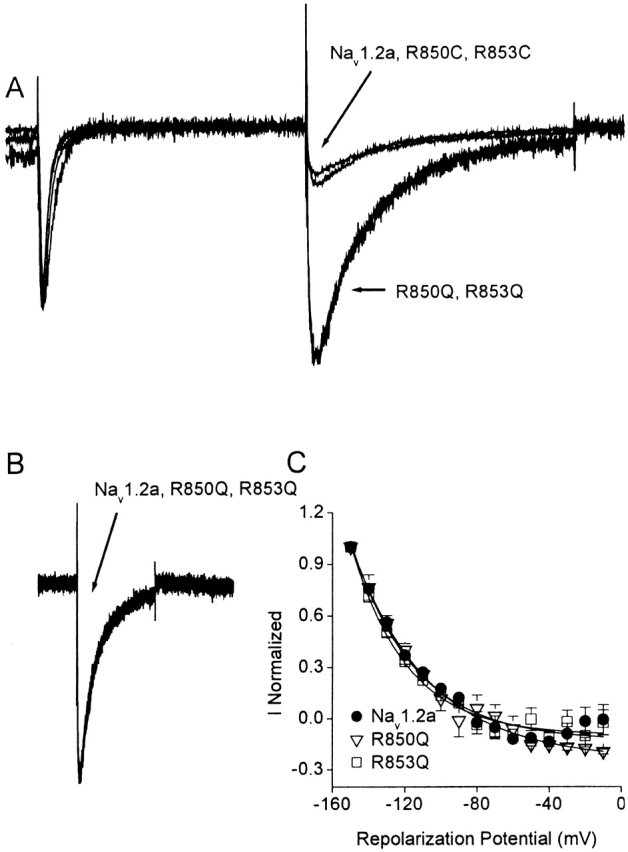
Css IV-induced tail currents. (A) Whole-cell currents elicited by a 28-ms test pulse to −20 mV followed by a repolarization to −150 mV from a holding potential of −100 mV in the presence of 200 nM Css IV for wild-type Nav1.2a, R850Q, R853Q, R850C, and R853C sodium channels. Each trace was normalized to the maximal amplitude of the peak sodium current. Each trace is an average of at least four recordings. (B) Comparison of the time course of the Css IV-induced tail currents for Nav1.2a, R850Q, and R853Q. The data were normalized to the maximal amplitude of the toxin-induced tail current. (C) Amplitude of the toxin-induced tail current as a function of the repolarization potential for Nav1.2a (black circle), R850Q (open triangle), and R853Q (open square) sodium channels in the presence of 200 nM Css IV.
DISCUSSION
Gating Charges in the IIS4 Segment of Sodium Channels
The activation process of sodium channels corresponds to a voltage-driven transition from a resting, closed conformation to an open conformational state, which is accompanied by the translocation of several positive charges outward across the membrane (Hodgkin and Huxley 1952; Armstrong 1981). The S4 segments have been proposed to serve as voltage sensors, moving outward to initiate channel activation (Catterall 1986; Guy and Seetharamulu 1986). Consistent with this idea, mutation of the positive charges in S4 segments reduces the steepness of voltage-dependent activation (Stühmer et al. 1989), and covalent modification experiments show that S4 segments move outward during the activation process (Yang and Horn 1995; Yang et al. 1996, Yang et al. 1997). Recent studies with fluorescently labeled S4 segments suggest that they also undergo a rotation and possible tilt, in response to depolarization (Cha et al. 1999b; Glauner et al. 1999).
Comparison of the voltage dependence of gating of sodium channels having mutations in each of the five positively charged amino acid residues in segment IIS4 indicates that all five mutations affect activation gating. Neutralization of each of the five positive charges shifts the voltage dependence of activation in the positive direction. Moreover, the slope of the activation curve was reduced by neutralization of R850, R853, R856, and K859. Neutralization of K862 did not have an effect on the steepness of the activation curve. These results are consistent with the idea that R850, R853, R856, and K859 all move outward through the membrane electric field as the sodium channel activates. However, these charges are not equally important for activation gating. Neutralization of K859 produced the largest shift in the voltage dependence of activation, whereas neutralization of R856 has the smallest effect (Fig. 1 and Table ; Kontis et al. 1997). These results indicate that neutralization of the positive gating charges in these positions in wild-type sodium channels stabilizes the resting state of the channel relative to the open state. This presumably results from an increase in the electrostatic driving force necessary to push the IIS4 segment outward, but steric effects of these neutralizing mutations cannot be excluded.
Neutralization of the Two Outermost Arginines of the IIS4 Segment Favors Voltage-Sensor Trapping by Css IV
Bound β-scorpion toxins induce only a partial shift of the activation curve of wild-type sodium channels, even after a strong depolarizing prepulse, as indicated by the biphasic activation curves for wild-type channels (Cahalan 1975; Jover et al. 1980; Jaimovich et al. 1982; Meves et al. 1982; Wang and Strichartz 1983; Vijverberg et al. 1984; Jonas et al. 1986; Cestèle et al. 1998; Tsushima et al. 1999). The biphasic curves for activation in the presence of the β-scorpion toxin are proposed to be due to the trapping of the IIS4 segment of a fraction of sodium channels in an outward position by binding the extracellular end of the S4 segment to the previously bound toxin in its receptor site, which includes the IIS3–S4 extracellular loop (Cestèle et al. 1998). In the experiments presented here, analysis of mutations that neutralize each of the five positive charges of the IIS4 voltage sensor indicates that β-toxin action can be strongly enhanced by substitution of the two outermost arginines by glutamine or cysteine. The increase in β-toxin activity is not caused by a general enhancement of activation, since the neutralization of R850 or R853 favors the resting state of the channels by shifting the voltage dependence of activation toward more positive potentials (Stühmer et al. 1989; Kontis et al. 1997; present study). These two sets of results imply that neutralization of R850 and R853 reduces the energy barrier for trapping the IIS4 segment in its outward position by binding of β-scorpion toxin while increasing the electrostatic force required to drive the IIS4 segment outward on depolarization.
Neutralization of R850 and R853 Increases the Mobility of IIS4 Segment within the Membrane
We interpret the enhanced effects of Css IV on activation of R850C/Q and R853C/Q in terms of a role for these two amino acid residues in stabilizing the position of the IIS4 segment with respect to interactions with Css IV. The ability of Css IV to shift the voltage dependence of activation of R853C/Q in the negative direction without a depolarizing prepulse supports the idea that this mutation increases the mobility of the IIS4 segment, allowing the toxin to induce IIS4 movement without depolarization. We consider two mechanisms that may contribute to this stabilizing influence of R850 and R853.
First, it is possible that these neutralizing mutations increase the mobility of the IIS4 segment in the presence of Css IV because the normal positive charges at these positions make unfavorable electrostatic interactions with the amino acid residues in the strongly positively charged toxin. Neutralization of these charges may enhance β-toxin action by removing this unfavorable electrostatic interaction, allowing easier outward movement of the IIS3–S4 loop and the IIS4 segment. This would promote more effective interaction with the bound Css IV toxin and more complete shift of the voltage dependence of activation.
Alternatively, it has been shown for K+ channels that basic amino acid residues in the S4 segment interact with acidic residues in the S2 and S3 segments. These electrostatic interactions are structural constraints that can stabilize the S4 segments and impede their motion (Papazian et al. 1995; Planells-Cases et al. 1995; Tiwari-Woodruff et al. 1997, Tiwari-Woodruff et al. 2000; Li-Smerin et al. 2000). Similarly, R850 and R853 may interact with acidic amino acid residues localized in the S2 and S3 segments of sodium channels. Neutralization is expected to abolish these electrostatic interactions and reduce the kinetic barrier for S4 movement inward or outward, resulting in a greater mobility of the IIS4 segment. An increase in the mobility of the IIS4 segment would contribute to the enhanced effects of β-scorpion toxin on activation that we have observed. With greater mobility, the probability that the IIS3–S4 loop and the IIS4 segment will be exposed at the extracellular surface of the channel by random motions is increased. Voltage-sensor trapping would be facilitated because the IIS3–S4 loop and the extracellular end of the IIS4 segment would be more often available to bind to the toxin, even at negative membrane potentials where the channel is not activated.
Our results obtained with R853Q/C suggest a dominant role of R853 in stabilizing the position of the IIS4 segment within the membrane, because voltage-sensor trapping and the resulting negative shift of activation occur in the absence of a depolarizing prepulse with these mutations. The voltage-sensor trapping mechanism can also account for the marked effect of MTSEA on the activation of R853C. We suggest that reaction of MTSEA with this residue stabilizes the IIS4 segment in its inward position through steric hindrance and/or ion pair formation, and thereby impedes movement of the IIS4 segment. In contrast to the unmodified-R853C channel, R853C-MTSEA requires a depolarizing prepulse to observe a shift in the activation curve due to Css IV application, and the voltage dependence of only a fraction of the sodium channels is shifted. These results indicate that the modification of R853C by MTSEA stabilizes the IIS4 segment within the membrane, opposes depolarization-induced movement of the IIS4 segment toward the extracellular side of the membrane, and inhibits voltage-sensor trapping by Css IV.
Toxin-induced Tail Currents Caused by Slow Movement of the IIS4 Segment
The toxin-induced tail currents observed upon repolarization give direct information about the behavior of the toxin-modified channels. The appearance of this tail current upon repolarization is a direct consequence of the IIS4 voltage-sensor trapping and its slow reversal with time after repolarization. The tail currents of toxin-modified channels are different in waveform than tail currents of unmodified channels. For unmodified sodium channels, voltage pulses long enough to allow complete inactivation are not followed by tail currents because one or more of the S4 voltage sensors deactivate before the inactivation gate opens to allow current flow (Kuo and Bean 1994). In contrast, tail currents of toxin-modified sodium channels rise to a peak followed by a slow decay, even when the sodium channels have inactivated completely during the depolarizing pulse. Most models of ion channel gating indicate that current can flow only when all four S4 segments are in the activated position (Armstrong 1981; Groome et al. 1999). According to these models, the rise of the tail currents of toxin-modified channels must reflect reversal of inactivation of sodium channels whose voltage sensors are all held in the activated position. Because Css IV holds the IIS4 segment in the activated position, we propose that the rapid deactivation of unmodified sodium channels is mediated by the inward movement of IIS4. Measurements of movement of the IIS4 segment fluorescently labeled on a substituted cysteine residue are consistent with this idea (Cha et al. 1999a). When inward movement of IIS4 is prevented by binding of Css IV, the inactivation gate in the intracellular loop between domains III and IV can reopen faster than deactivation, and tail currents are observed upon repolarization and reversal of inactivation. The time course of decay of the toxin-induced tail current was approximately equal for wild-type and mutants. This is expected if another S4 segment (perhaps IVS4, which is responsible for inactivation; Chen et al. 1996; Ji et al. 1996; Cha et al. 1999a) deactivates with normal kinetics and closes the channel with IIS4 still trapped in its outward position. Therefore, the toxin-induced tail currents provide a direct measure of trapping of the IIS4 voltage sensor in its activated position.
Alternative Interpretations of the Results
We have interpreted our results in terms of the voltage-sensor trapping hypothesis presented previously to explain the actions of α- and β-scorpion toxins (Rogers et al. 1996; Cestèle et al. 1998). We believe that this simple model can accommodate all of the results presented here. However, our results do not constitute proof of the voltage-sensor trapping model because the data can also be accommodated by more complex models invoking broader conformational changes in the sodium channel induced by the mutations and chemical reactions. For example, neutralization of gating charges may cause conformational changes that alter voltage-dependent activation and β-scorpion toxin action, and reaction of R853C with MTSEA may inhibit channel activation by altering the conformation of domain II in a way that prevents normal activation. These alternative interpretations of our results seem unlikely because multiple ad hoc assumptions are required to explain the results. Nevertheless, further work will be required to obtain direct proof for the voltage-sensor trapping model for the action of polypeptide neurotoxins.
Acknowledgments
We thank Dr. Marie-France Martin-Eauclaire (CNRS, UMR 6560, Marseille, France) for purifying the β-scorpion toxin, Css IV.
Footnotes
Abbreviations used in this paper: MTS, methanethiosulfonate; MTSEA, 2-aminoethyl methanethiosulfonate hydrobromide; MTSET, 2-(trimethylammonium)ethyl methanethiosulfonate bromide.
References
- Armstrong C.M. Sodium channels and gating currents. Physiol. Rev. 1981;61:644–683. doi: 10.1152/physrev.1981.61.3.644. [DOI] [PubMed] [Google Scholar]
- Auld V.J., Goldin A.L., Krafte D.S., Catterall W.A., Lester H.A., Davidson N., Dunn R.J. A neutral amino acid change in segment IIS4 dramatically alters the gating properties of the voltage-dependent sodium channel. Proc. Natl. Acad. Sci. USA. 1990;87:323–327. doi: 10.1073/pnas.87.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan M.D. Modification of sodium channel gating in frog myelinated nerve fibres by Centruroides sculpturatus scorpion venom. J. Physiol. 1975;244:511–534. doi: 10.1113/jphysiol.1975.sp010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W.A. Membrane potential-dependent binding of scorpion toxin to the action potential Na+ ionophore. Studies with a toxin derivative prepared by lactoperoxidase-catalyzed iodination. J. Biol. Chem. 1977;252:8660–8668. [PubMed] [Google Scholar]
- Catterall W.A. Binding of scorpion toxin to receptor sites associated with sodium channels in frog muscle. Correlation of voltage-dependent binding with activation. J. Gen. Physiol. 1979;74:375–391. doi: 10.1085/jgp.74.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W.A. Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem. 1986;55:953–985. doi: 10.1146/annurev.bi.55.070186.004513. [DOI] [PubMed] [Google Scholar]
- Catterall W.A. From ionic currents to molecular mechanismsthe structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Catterall W.A., Beress L. Sea anemone toxin and scorpion toxin share a common receptor site associated with the action potential sodium ionophore. J. Biol. Chem. 1978;253:7393–7396. [PubMed] [Google Scholar]
- Cestèle S., Catterall W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000;82:883–892. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- Cestèle S., Qu Y., Rogers J.C., Rochat H., Scheuer T., Catterall W.A. Voltage sensor trappingenhanced activation of sodium channels by β-scorpion toxin bound to the S3-S4 loop in domain II. Neuron. 1998;21:919–931. doi: 10.1016/s0896-6273(00)80606-6. [DOI] [PubMed] [Google Scholar]
- Cha A., Ruben P.C., George A.L., Jr., Fujimoto E., Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation Neuron. 22 1999. 73 87a [DOI] [PubMed] [Google Scholar]
- Cha A., Snyder G.E., Selvin P.R., Bezanilla F. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy Nature. 402 1999. 809 813b [DOI] [PubMed] [Google Scholar]
- Chahine M., George A.L., Jr., Zhou M., Ji S., Sun W., Barchi R.L., Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chen L.Q., Santarelli V., Horn R., Kallen R.G. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J. Gen. Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauner K.S., Mannuzzu L.M., Gandhi C.S., Isacoff E.Y. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 1999;402:813–817. doi: 10.1038/45561. [DOI] [PubMed] [Google Scholar]
- Goldin A.L., Barchi R.L., Caldwell J.H., Hofmann F., Howe J.R., Hunter J.C., Kallen R.G., Mandel G., Meisler M.H., Netter Y.B. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Groome J.R., Fujimoto E., George A.L., Ruben P.C. Differential effects of homologous S4 mutations in human skeletal muscle sodium channels on deactivation gating from open and inactivated states. J. Physiol. 1999;516:687–698. doi: 10.1111/j.1469-7793.1999.0687u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy H.R., Seetharamulu P. Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. USA. 1986;83:508–512. doi: 10.1073/pnas.83.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann S.H., Terlau H., Imoto K. Molecular basis for pharmacological differences between brain and cardiac sodium channels. Pflügers Arch. 1992;422:90–92. doi: 10.1007/BF00381519. [DOI] [PubMed] [Google Scholar]
- Hodgkin A.L., Huxley A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaimovich E., Ildefonse M., Barhanin J., Rougier O., Lazdunski M. Centruroides toxin, a selective blocker of surface Na+ channels in skeletal musclevoltage-clamp analysis and biochemical characterization of the receptor. Proc. Natl. Acad. Sci. USA. 1982;79:3896–3900. doi: 10.1073/pnas.79.12.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji S., George A.L., Jr., Horn R., Barchi R.L. Paramyotonia congenita mutations reveal different roles for segments S3 and S4 of domain D4 in hSKM1 sodium channel gating. J. Gen. Physiol. 1996;107:183–194. doi: 10.1085/jgp.107.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P., Vogel W., Arantes E.C., Giglio J.R. Toxin gamma of the scorpion Tityus serrulatus modifies both activation and inactivation of sodium permeability of nerve membrane. Pflügers Arch. 1986;407:92–99. doi: 10.1007/BF00580727. [DOI] [PubMed] [Google Scholar]
- Jordan M., Schallhorn A., Wurm F.M. Transfecting mammalian cellsoptimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jover E., Couraud F., Rochat F. Two types of scorpion neurotoxins characterized by their binding to two separate receptor sites on rat brain synaptosomes. Biochem. Biophys. Res. Commun. 1980;95:1697–1714. doi: 10.1016/s0006-291x(80)80082-9. [DOI] [PubMed] [Google Scholar]
- Kellenberger S., Scheuer T., Catterall W.A. Movement of the Na+ channel inactivation gate during inactivation. J. Biol. Chem. 1996;271:30971–30979. doi: 10.1074/jbc.271.48.30971. [DOI] [PubMed] [Google Scholar]
- Kontis K.J., Rounaghi A., Goldin A.L. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J. Gen. Physiol. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C.C., Bean B.P. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y., Hackos D.H., Swartz K.J. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel. Neuron. 2000;25:411–423. doi: 10.1016/s0896-6273(00)80904-6. [DOI] [PubMed] [Google Scholar]
- Martin-Eauclaire M.F., Garcia y Perez L.G., El Ayeb M., Kopeyan C., Bechis G., Jover E., Rochat H. Purification and chemical and biological characterization of seven toxins from the Mexican scorpion, Centruroides suffusus suffusus . J. Biol. Chem. 1987;262:4452–4459. [PubMed] [Google Scholar]
- Meves H., Rubly N., Watt D.D. Effect of toxins isolated from the venom of the scorpion Centruroides sculpturatus on the Na currents of the node of Ranvier. Pflügers Arch. 1982;393:56–62. doi: 10.1007/BF00582392. [DOI] [PubMed] [Google Scholar]
- Mitrovic N., George A.L., Jr., Horn R. Independent versus coupled inactivation in sodium channels. Role of the domain 2 S4 segment. J. Gen. Physiol. 1998;111:451–462. doi: 10.1085/jgp.111.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson G.M., Willow M., Howden M.E., Narahashi T. Modification of sodium channel gating and kinetics by versutoxin from the Australian funnel-web spider Hadronyche versuta . Pflügers Arch. 1994;428:400–409. doi: 10.1007/BF00724524. [DOI] [PubMed] [Google Scholar]
- Noda M., Suzuki H., Numa S., Stuhmer W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989;259:213–216. doi: 10.1016/0014-5793(89)81531-5. [DOI] [PubMed] [Google Scholar]
- Papazian D.M., Shao X.M., Seoh S.A., Mock A.F., Huang Y., Wainstock D.H. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R., Ferrer-Montiel A.V., Patten C.D., Montal M. Mutation of conserved negatively charged residues in the S2 and S3 transmembrane segments of a mammalian K+ channel selectively modulates channel gating. Proc. Natl. Acad. Sci. USA. 1995;92:9422–9426. doi: 10.1073/pnas.92.20.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale D.S., McPhee J.C., Scheuer T., Catterall W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Rogers J.C., Qu Y., Tanada T.N., Scheuer T., Catterall W.A. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. J. Biol. Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Sheets M.F., Kyle J.W., Kallen R.G., Hanck D.A. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys. J. 1999;77:747–757. doi: 10.1016/S0006-3495(99)76929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets M.F., Kyle J.W., Hanck D.A. The role of the putative inactivation lid in sodium channel gating current immobilization. J. Gen. Physiol. 2000;115:609–620. doi: 10.1085/jgp.115.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stühmer W., Conti F., Suzuki H., Wang X.D., Noda M., Yahagi N., Kubo H., Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Terlau H., Heinemann S.H., Stuhmer W., Pusch M., Conti F., Imoto K., Numa S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991;293:93–96. doi: 10.1016/0014-5793(91)81159-6. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff S.K., Lin M.A., Schulteis C.T., Papazian D.M. Voltage-dependent structural interactions in the Shaker K+ channel. J. Gen. Physiol. 2000;115:123–138. doi: 10.1085/jgp.115.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari-Woodruff S.K., Schulteis C.T., Mock A.F., Papazian D.M. Electrostatic interactions between transmembrane segments mediate folding of Shaker K+ channel subunits. Biophys. J. 1997;72:1489–1500. doi: 10.1016/S0006-3495(97)78797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsushima R.G., Borges A., Backx P.H. Inactivated state dependence of sodium channel modulation by beta- scorpion toxin. Pflügers Arch. 1999;437:661–668. doi: 10.1007/s004240050830. [DOI] [PubMed] [Google Scholar]
- Vassilev P.M., Scheuer T., Catterall W.A. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Vassilev P., Scheuer T., Catterall W.A. Inhibition of inactivation of single sodium channels by a site- directed antibody. Proc. Natl. Acad. Sci. USA. 1989;86:8147–8151. doi: 10.1073/pnas.86.20.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantham V., Cannon S.C. Slow inactivation does not affect movement of the fast inactivation gate in voltage-gated Na+ channels. J. Gen. Physiol. 1998;111:83–93. doi: 10.1085/jgp.111.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijverberg H.P., Pauron D., Lazdunski M. The effect of Tityus serrulatus scorpion toxin gamma on Na channels in neuroblastoma cells. Pflügers Arch. 1984;401:297–303. doi: 10.1007/BF00582600. [DOI] [PubMed] [Google Scholar]
- Wang G.K., Strichartz G.R. Purification and physiological characterization of neurotoxins from venoms of the scorpions Centruroides sculpturatus and Leiurus quinquestriatus . Mol. Pharmacol. 1983;23:519–533. [PubMed] [Google Scholar]
- West J.W., Patton D.E., Scheuer T., Wang Y., Goldin A.L., Catterall W.A. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc. Natl. Acad. Sci. USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., George A.L., Jr., Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Yang N., Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Yang N.B., George A.L., Horn R. Probing the outer vestibule of a sodium channel voltage sensor. Biophys. J. 1997;73:2260–2268. doi: 10.1016/S0006-3495(97)78258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]