

Abstract
Approximately half of the NH2 terminus of inward rectifier (Kir) channels can be deleted without significant change in channel function, but activity is lost when more than ∼30 conserved residues before the first membrane spanning domain (M1) are removed. Systematic replacement of the positive charges in the NH2 terminus of Kir6.2 with alanine reveals several residues that affect channel function when neutralized. Certain mutations (R4A, R5A, R16A, R27A, R39A, K47A, R50A, R54A, K67A) change open probability, whereas an overlapping set of mutants (R16A, R27A, K39A, K47A, R50A, R54A, K67A) change ATP sensitivity. Further analysis of the latter set differentiates mutations that alter ATP sensitivity as a consequence of altered open state stability (R16A, K39A, K67A) from those that may affect ATP binding directly (K47A, R50A, R54A). The data help to define the structural determinants of Kir channel function, and suggest possible structural motifs within the NH2 terminus, as well as the relationship of the NH2 terminus with the extended cytoplasmic COOH terminus of the channel.
Keywords: K+ current, KATP, PIP2, Kir6.2, ATP
INTRODUCTION
KATP channels are normally formed as an octameric complex of four Kir6.2 subunits that comprise the pore of the channel, and four regulatory sulfonylurea receptor (SUR)* subunits (Inagaki et al., 1995a, 1997; Clement et al., 1997; Shyng and Nichols, 1997). This complex confers ATP, sulfonylurea, potassium channel opener, and PIP2 sensitivity to a weak inwardly rectifying K+ channel (Inagaki et al., 1995a; Fan and Makielski, 1997; Shyng et al., 1997a,b; Baukrowitz et al., 1998; Shyng and Nichols, 1998). Several investigations show that KATP channels formed from Kir6.2 expressed without SUR maintain many of the key properties of the hetero-octameric complex: they are still ATP sensitive (Tucker et al., 1997) and they still respond to PIP2 (Baukrowitz et al., 1998; Enkvetchakul et al., 2000), indicating that these properties are inherent to the Kir6.2 subunit.
In the search for where these various modulators are acting, attention has focused on the cytoplasmic domains of Kir6.2. Both NH2 and COOH termini are cytoplasmic, with the NH2 terminus consisting of ∼70 amino acids, and the large cytoplasmic COOH terminus consisting of ∼220–240 residues. Although the ATP binding site on Kir6.2 remains elusive, several residues in the COOH terminus, including K185 (Tucker et al., 1997; Reimann et al., 1999a), I182 (Li et al., 2000), R201 (Shyng et al., 2000), and G334 (Drain et al., 1998), have been proposed to be directly involved in ATP binding. Various studies have also indicated several residues in the COOH termini of Kir channels that may contribute to PIP2 binding (Fan and Makielski, 1997, 1999; Baukrowitz et al., 1998; Shyng and Nichols, 1998; Huang et al., 1998; Liou et al., 1999; Rohacs et al., 1999; Soom et al., 2001). Systematic alanine replacement of all positive residues in the COOH terminus of Kir6.2 identified residues clustering in two main regions (residues 176–222 and 301–314) that caused a dramatic shift in PIP2 activation when mutated (Shyng et al., 2000), and analogous regions of isolated COOH-terminal fragments of Kir2.1 are implicated in PIP2 binding (Soom et al., 2001). The role of the NH2 terminus remains incompletely defined. Although ∼30 residues can be cleaved from the NH2 terminus of Kir6.2 without loss of activity, further truncation abolishes channel activity (Koster et al., 1999b). Tolbutamide sensitivity, conferred by coassembly with SUR1, is altered in these truncated constructs, indicating a role for the NH2 terminus in the coupling of the channel to SUR1 (Koster et al., 1999a; Reimann et al., 1999b). NH2-terminal truncations demonstrate a decreased ATP sensitivity (Babenko et al., 1999; Koster et al., 1999b; Reimann et al., 1999b) and residues I49 and R50 in the NH2 terminus have also been suggested as potential inhibitory ATP binding sites (Tucker et al., 1998; Proks et al., 1999; Reimann et al., 1999b; Tanabe et al., 1999).
Both the NH2 and COOH termini are involved in conferring sensitivity of other Kir channels to various agonists, including G-protein sensitivity of Kir3 channels and pH sensitivity of Kir1.1 channels (Woodward et al., 1997; Schulte et al., 1998). Cyclic-nucleotide gating of CNG channels analogously requires an interaction of the NH2 and COOH termini (Zagotta and Siegelbaum, 1996; Varnum and Zagotta, 1997). The nonadditive effects of combining mutations in the NH2 and COOH termini of Kir6.2 on ATP sensitivity has been taken to indicate cooperativity between the two termini (Proks et al., 1999). Tucker and Ashcroft (1999) further demonstrated interaction and binding of the Kir6.2 NH2 terminus to the COOH termini of Kir6.2, Kir6.1, and Kir2.1 using exogenously produced fusion proteins. They mapped the NH2-terminal interaction region to residues 30–46, i.e., the most proximal part of the conserved segment (Koster et al., 1999b; Reimann et al., 1999b; Tucker and Ashcroft, 1999). Jones et al. (2001) have recently mapped the complimentary interacting regions in the COOH terminus and defined three critical segments (170–204, 214–222, and 279–323) that include the regions identified by Shyng et al. (2000) as being involved in PIP2 interaction. The identification of interacting portions of Kir6.2 NH2 and COOH termini leads to the possibility that residues in both the NH2 and COOH termini are involved in determining the sensitivity of the channel to agonists such as ATP and PIP2. We have recently defined a probable α-helix in Kir channel COOH-terminal domains that is similar to the COOH-terminal α-helix of pleckstrin homology (PH) domains. We hypothesize that Kir channels contain a conserved lipid interaction (KIRLI) domain, that may be related to PH domains, with structural elements defined by Tucker and Ashcroft (1999) and Jones et al. (2001) contributed by both NH2- and COOH-terminal regions (Cukras et al., 2002).
To generate a primary dataset for consideration of NH2-terminal residues in channel function, we have now systematically mutated each positive charge to alanine. We report several residues in the NH2 terminus that affect the apparent ATP sensitivity or the open probability when mutated. Furthermore, we distinguish between residues that might be involved in ATP binding and those that affect the open state stability of the channel, and are thus likely to be involved in PIP2 interaction.
MATERIALS AND METHODS
Molecular Biology
Point mutations were prepared by overlap extension at the junctions of the relevant residues by sequential PCR. Resulting PCR products were subcloned into pCMV6b vector. Before transfection, constructs were sequenced to verify the correct mutations.
Expression in COSm6 Cells
COSm6 cells were plated at a density of ∼2.5 × 105 cells per well (30 mm six-well dishes) and cultured in Dulbecco's Modified Eagle Medium plus 10 mM glucose (DMEM-HG), supplemented with FCS (10%). The next day, cells were transfected by adding FUGENE and 1 μg each of pCMV6b-Kir6.2 or mutant isoforms, pECE-SUR1 cDNA, and pECE-GFP (green fluorescent protein) directly to the media. The cells were replated the next day onto coverslips for patch-clamping. All experiments were performed 24–48 h posttransfection.
Patch-clamp Measurements
Patch-clamp experiments were made at room temperature, in an oil-gate chamber that allowed rapid exchange of bathing solution by moving patches from one channel to another (Lederer and Nichols, 1989). Micropipettes were pulled from thin-walled glass (WPI, Inc.) on a horizontal puller (Sutter Instrument Co.). Electrode resistance was typically 0.5–1 MΩ when filled with K-INT solution (below). Inside-out patches were voltage-clamped at −50 mV with an Axopatch 1B amplifier (Axon Instruments, Inc.). Standard bath and pipette solutions (K-INT) had the following composition: 140 mM KCl, 10 mM K-HEPES, 1 mM K-EGTA, pH 7.3. PIP2 was bath sonicated in ice for 30 min before use. ATP was added as the potassium salt. All currents were measured at a membrane potential of −50 mV, and inward currents at this voltage are shown as upward deflections. Data were filtered at 0.5–3 kHz, digitized at 22 kHz (Neurocorder; Neurodata) and stored on videotape. Experiments were replayed onto a chart recorder, or digitized into a computer using Axotape software (Axon Instruments, Inc.) and analyzed off-line using Microsoft Excel. Wherever possible, data are presented as mean ± SEM. Microsoft Solver was used to fit data by a least-square algorithm.
Interpretation of PIP2 Response Data
Wild-type (WT) KATP (Kir6.2 + SUR1) channels have an intrinsic open probability in the absence of ATP (Po,zero) of ∼0.4 and are half maximally inhibited at an ATP concentration (K1/2,ATP) of ∼10 μM (Enkvetchakul et al., 2000; Inagaki et al., 1995b). Many mutations of Kir6.2 residues, or exposure of channels to cytoplasmic PIP2, cause changes in both Po,zero and ATP sensitivity. In many cases, Po,zero and K1/2,ATP are strongly correlated, and this correlation can be explained by assuming that the action of ATP is on the closed channel, such that both Po,zero and K1/2,ATP are increased when open state stability is increased, for example, by addition of PIP2 (Baukrowitz et al., 1998; Shyng and Nichols, 1998; Enkvetchakul et al., 2000). Mutations could affect the response of the channel to PIP2 by altering either the affinity or availability of a PIP2 binding site, or by altering the coupling of PIP2 binding to open state stability. These possibilities are experimentally unresolvable and therefore, in the present experiments, we interpret reduced intrinsic activity and greater response to PIP2 as indicating that mutations either affect PIP2 binding directly, or the translation of this binding into an effect on channel open state stability.
Data Analysis
Off-line analysis was performed using ClampFit and Microsoft Excel programs. Two approaches were used to estimate Po,zero, the initial open probability (in zero ATP), after excision of isolated membrane patches containing multiple channels. (1, PIP2 method) PIP2 was added to the patch until the current reached a saturating level (IPIP2). This was assumed to represent a maximum Po,zero of ∼0.9 (Enkvetchakul et al., 2000). The fold increase in current was calculated (fold increase = Iinitial/IPIP2, see Fig. 2) and the Po,zero was estimated from the following equation: Po,zero = 0.9/(fold increase). (2, NA method) Additionally, mean Po,zero was estimated from stationary fluctuation analysis of macroscopic currents (Neher and Stevens, 1977; Sigworth, 1980) on short (<1 s) recordings of currents in zero [ATP] and in 5 mM [ATP]. Currents (4 pA < mean current < 4 nA, at −50 mV, corresponding to ∼1–1,000 channels) were filtered at 1 kHz and digitized at 3 kHz with 12-bit resolution. Mean patch current (I), and variance (σ2) in the absence of ATP were obtained by subtraction of the mean current and variance in 5 mM ATP (i.e., assuming all channels closed), respectively. Single channel current (i) was assumed to be −3.75 pA, corresponding to WT single channel conductance of 75 pS. Po,zero was then estimated from the following equation: Po,zero = 1 − (σ2/[i]). Although the Po,zero estimated from noise analysis (NA) was systematically higher than that from PIP2 response, it is apparent in Fig. 4 that both methods gave correlated estimates of Po,zero. Therefore, although absolute Po,zero estimates are unlikely to be accurate, we have reasonable confidence in comparing relative Po,zero between mutants. Since the PIP2 response (lower estimate) will actually report an upper limit for Po,zero, we utilize these values for examination of Po,zero − K1/2ATP relationships (Fig. 2).
Figure 2.

Representative currents recorded from inside-out membrane patches containing Kir6.2 WT, Kir6.2[R16A], or Kir6.2[R27A], coexpressed with SUR1. In this and subsequent figures, the patch was excised at the arrow, and the bars indicate the application of PIP2 (5 μg/ml, unless indicated) or different [ATP] (as shown). Solutions were switched by moving the patch from one lane of the oil-gate chamber to another. The lower dashed line indicates zero current. Iinitial and IPIP2, used to determine Po,zero (see materials and methods), are indicated by additional dashed lines. The inserts in this and Fig. 3 show expanded records from the first 50–100 s after patch excision, from which ATP sensitivity and open probability (NA method) were estimated.
Figure 4.

Initial open probability in zero ATP (Po,zero) obtained from noise analysis (NA method, mean ± SEM, n = 3–8) plotted versus Po,zero obtained from subsequent increase in current following PIP2 addition (PIP2 method, mean ± SEM). The solid line is a least squares fit to the mean data as indicated. The insert shows mean Po,zero (PIP2 method) plotted against Iinitial (from Fig. 1 B) for each mutant.
ATP sensitivity was estimated from least squares fits of the Hill equation to the currents in 0, 0.1, and 5 mM ATP immediately after patch excision (see insets to Figs. 2 and 3) : Irel = 1/(1 + {[ATP]/K1/2}H), where Irel is the mean current in a given ATP concentration ([ATP]) divided by the mean current in zero ATP, K1/2 is the [ATP] causing half-maximal inhibition, and H is the Hill coefficient (fixed at 1.3, Shyng et al., 2000).
Figure 3.

Representative currents recorded from inside-out membrane patch containing Kir6.2[K47A], Kir6.2[R50A], or Kir6.2[R54A] coexpressed with SUR1. The dashed line indicates zero current.
RESULTS
Alanine Scan of NH2 Terminus Identifies Residues That Affect Open Probability (Po,zero)
There are 17 positive charges (R, arginine; K, lysine; H, histidine) in the NH2 terminus of Kir6.2 (Fig. 1 A) any of which could contribute to interaction with either PIP2, ATP, or both. To assess the role of these charged residues in KATP channel function, we mutated each one to alanine and examined channel properties in inside-out membrane patches. Fig. 1 B shows a summary of current density in patches from COSm6 cells expressing each mutant channel. In contrast to the severe detrimental effects of charge neutralization in some residues in the COOH terminus (Shyng et al., 2000), all of the NH2-terminal mutants generated measurable K+ currents. Wide variability of Iinitial is reflective of variability of Po,zero (see below, Fig. 4, inset).
Figure 1.

Alanine scanning mutagenesis of NH2-terminal basic residues of Kir6.2. (A) Each positive charged residue in the NH2 terminus (bold), up to the beginning of the M1 transmembrane domain, was individually mutated to alanine. (B) Initial current (Iinitial) in macropatches isolated from COSm6 cells cotransfected with mutant Kir6.2 subunits (as indicated) + SUR1 (mean ± SEM, n ≥ 3 in each case). See materials and methods for details.
Figs. 2 and 3 show representative recordings for various mutations. From such recordings we estimated initial Po,zero and ATP sensitivity after patch excision. Initial Po,zero was estimated in two ways: (a) by measuring the response to PIP2 and (b) by noise analysis (see materials and methods). WT channels typically have a Po,zero around 0.4 (Fig. 2, top; Shyng and Nichols, 1998; Enkvetchakul et al., 2000; Shyng et al., 2000). After application of PIP2, WT open probability increases and saturates at ∼0.9 (Shyng and Nichols, 1998; Enkvetchakul et al., 2000; Shyng et al., 2000), such that macroscopic currents approximately double. Fig. 4 summarizes the estimates of Po,zero using the two independent methods. Each method clearly gives correlated estimates, even though Po,zero as estimated by noise analysis is systematically higher than estimated by the PIP2 method, probably due to inevitable underestimation of noise due to filtering. Although the Po,zero of many mutations were not significantly different from WT, some (R16A, R27A, Fig. 2) have a much higher Po,zero, whereas others (R4A, R5A, R39A, K47A, R50A, R54A, K67A) have a significantly lower Po,zero. The inset of Fig. 4 demonstrates that the initial current following patch excision is also reasonably well correlated with Po,zero.
The behavior of mutant R34A was somewhat unique in that the channels showed high Po,zero (0.86 ± 0.08, n = 4), as estimated by noise analysis, but ran down very quickly (Fig. 5 A). After PIP2 addition there was no increase in channel current (Fig. 5 B), precluding estimate of Po,zero (see discussion).
Figure 5.

(A) Representative currents recorded from inside-out membrane patches containing Kir6.2[R34A] coexpressed with SUR1. The dashed line indicates zero current. (B) Mean current (in zero ATP) versus time after application of PIP2 for Kir6.2 WT coexpressed with SUR1 and for Kir6.2[R34A] coexpressed with SUR1.
Alanine Scan of NH2 Terminus Identifies Residues That Affect ATP Sensitivity
We evaluated the ATP sensitivity for each mutant (Fig. 6 A), after patch excision, from records such as those in Figs. 2 and 3. In addition to the previously identified R50A (Proks et al., 1999), R16A is considerably (∼10-fold) less sensitive to ATP than WT channels, and R27A, K47A, and R54A mutants are also, though less markedly, reduced in ATP sensitivity (P < 0.05) (Fig. 2).
Figure 6.
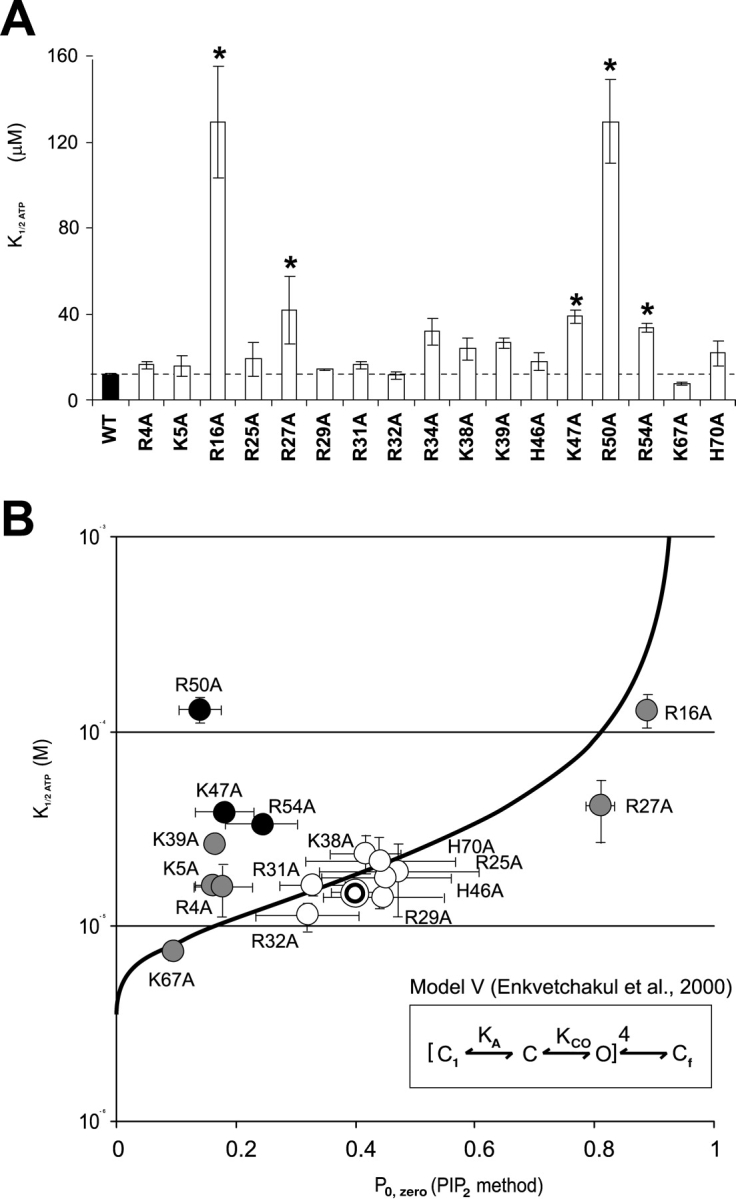
(A) Mean K1/2ATP for each mutant immediately following patch excision (mean ± SEM, n = 3–9, asterisk indicates less ATP-sensitive than WT, P < 0.05). Dose-response curves were obtained from recordings like those in Figs. 2 and 3 and were fit using a Hill equation (relative current = 1/{1 + ([ATP]/K1/2)H}) with K1/2,ATP = [ATP] causing half-maximal inhibition and H = 1.3. (B) K1/2,ATP versus Po,zero for each mutant (mean ± SEM, n = 3–9; for each parameter). The double circle represents mean values for WT. For different Kir6.2 mutant constructs, gray/black indicates Po,zero significantly different than WT (P < 0.05). The heavy line represents predicted Po,zero versus K1/2,ATP relationship calculated from Model V of Enkvetchakul et al. (2000)(see insert), black indicates mutants with K1/2,ATP significantly different than that predicted from the estimated Po,zero (P < 0.05).
A mutation can change ATP sensitivity by directly reducing the affinity of binding, by affecting the binding site allosterically, or by affecting the open state stability of the channel. We have previously proposed a model that quantitatively describes KATP channel behavior over a wide range of conditions and mutations (Model V, see inset Fig. 6) (Enkvetchakul et al., 2000), except for mutations that have extremely high open state stability (e.g., Kir6.2[L164C]; Enkvetchakul et al., 2001). An essential feature of such a model is that ATP predominantly acts to stabilize a closed state and, in consequence, the apparent ATP sensitivity can be reduced either by shifting the equilibrium between C and O states toward the open state or by reduction of the ATP binding. Fig. 6 B plots the measured K1/2,ATP as a function of the estimated Po,zero (from PIP2 response) for each mutant. The bold line indicates the prediction of Model V for varying intrinsic open state stability (i.e., the KCO equilibrium constant). From this analysis it is apparent that many mutations actually cluster very close to WT and most of the remainder are distributed along the predicted curve (mutations indicated in gray are shifted significantly (P < 0.05) along the Po,zero axis from WT). This implies that these mutations act to alter the intrinsic open state stability, and in doing so, ATP sensitivity changes according to the relationship indicated. We further estimated whether the measured mean K1/2,ATP of each mutant was different from that predicted given the mean Po,zero. This further identified three residues (K47A, R50A, and R54A, black in Fig. 6 B) that are significantly (P < 0.05) less sensitive to ATP than predicted from Po,zero. A reduction of ATP sensitivity without increase in Po,zero implicates these residues as potentially contributing to ATP binding itself (see discussion).
DISCUSSION
Distinct Phenotypic Consequences of Charge Neutralization
PIP2 and other negatively charged phospholipids increase the open state stability of Kir channels (Hilgemann and Ball, 1996; Fan and Makielski, 1997). The molecular mechanism by which this occurs is not resolved, but it is likely that electrostatic interactions between a Kir cytoplasmic domain and phospholipids in the membrane stabilizes the open state of the channel (Fan and Makielski, 1997; Huang et al., 1998; Shyng et al., 2000; Soom et al., 2001; Cukras et al., 2002). ATP and PIP2 are negative heterotropic regulators of KATP channels, such that binding of ATP or PIP2 stabilizes the closed channel or the open channel respectively (Shyng and Nichols, 1998; Enkvetchakul et al., 2000). Therefore, treatment of membrane patches with PIP2 leads to an increase of Po,zero and decrease of ATP-sensitivity (Baukrowitz et al., 1998; Shyng and Nichols, 1998; Enkvetchakul et al., 2000; Shyng et al., 2000).
Systematic neutralization of positively charged residues in the COOH terminus of Kir6.2 subunits demonstrated distinct sets of residues that affect PIP2-sensitivity and ATP sensitivity (Shyng et al., 2000). The present data demonstrates that distinct sets of basic residues in the NH2 terminus also contribute to ATP sensitivity and PIP2 sensitivity. In every case, channel activity was present in excised patches (Fig. 1 B), and thus none of the mutations caused global structural changes that made subunits nonfunctional. Of 15 positively charged residues, mutation of seven (R4A, K5A, K39A, K47A, R50A, R54A, K67A) significantly reduced Po,zero (Fig. 4). Two mutations (R16A, R27A) led to increased open probability, and an expected decrease of ATP sensitivity. Thus, these two sets of mutations reflect a decrease, or increase, respectively, in the intrinsic open state stability, which may reflect altered affinity for binding of PIP2 (see below).
Three mutations (K47A, R50A, R54A), which cluster closely together, caused a decrease in ATP sensitivity that was not correlated with increased Po,zero (Fig. 6 B). These mutations thus represent possible ATP binding site mutations. These results support and extend the previous suggestion that R50 is involved in ATP binding (Tucker et al., 1998; Reimann et al., 1999b), either directly by forming part of the binding site, or allosterically (Tucker et al., 1998; Proks et al., 1999) by affecting ATP binding. That the Po,zero of each of these mutants is actually significantly lower than that of WT (Fig. 6) is consistent with these residues also being involved in the interaction of the channel with PIP2. In general, our findings from an alanine scan of the COOH terminus indicate that there are likely to be distinct, yet allosterically coupled and possibly overlapping, binding sites for ATP and PIP2 (Shyng et al., 2000). It is possible, therefore, that while the majority of the residues affect only one or the other sites, a few residues might be involved either in the physical overlap of the two sites or be directly involved in one site while allosterically affecting the other. This is the role/location that we previously proposed for R201 (Shyng et al., 2000) and now propose for K47, R50, and R54.
Finally, the R34A mutation demonstrated a novel phenotype. Channel activity was evident after patch excision, but run-down was rapid, and not prevented by PIP2. This effect is distinct from the effect of COOH-terminal mutations (e.g., R176A) that reduce PIP2 sensitivity, putatively by affecting PIP2 binding. In the case of R176A, mutation results in lower intrinsic Po,zero, but with a dramatic increase in channel activity on exposure to PIP2. In the case of R34A, the intrinsic Po,zero is not obviously reduced, but there is no increase in current after PIP2 exposure. Conceivably, this mutation induces a rapid rundown by some different, unknown, mechanism.
Effects of Truncation Experiments
Previous truncation experiments have shown that deletion of the first 20–30 amino acids of various inward rectifiers may be possible, without loss of channel function (Zhou et al., 1994; Boim et al., 1995; Babenko et al., 1999; Koster et al., 1999b; Reimann et al., 1999b). In the case of Kir6.2, significantly higher K1/2ATP and Po,zero values are observed with NH2-terminal truncations. The deletion of the first 30 amino acids generates channels with K1/2ATP of ∼100–120 μM and a Po,zero of ∼0.9 (Babenko et al., 1999; Koster et al., 1999b; Reimann et al., 1999b), and this shift in nucleotide sensitivity may be sufficient to cause dramatic changes in the physiology of cells in which it is expressed (Koster et al., 2000). These reduced ATP sensitivity, increased Po,zero phenotypes are very similar to that we observe for R16A mutant channels, suggesting that loss of the positive charge at residue 16 may underlie the behavior of the truncated channels.
Possible Structure of the Cytoplasmic Domains
Previous experiments have demonstrated direct interaction of isolated COOH-terminal fragments of Kir channels with PIP2 (Huang et al., 1998; Soom et al., 2001) and that the COOH terminus associates with the cell membrane in a PIP2 regulated manner (Cukras et al., 2002). The data suggest the presence of a common Kir lipid–interacting (KIRLI) domain that may be structurally related to PIP2-interacting PH domains (Harlan et al., 1995; Lemmon et al., 1996; Shaw, 1996; Touhara et al., 1995). We proposed that the KIRLI domain is assembled primarily from amino acids 170–320 of the COOH terminus, but that the first β-strand (β1) may be contributed by the NH2 terminus (Cukras et al., 2002). Fig. 7 shows secondary structure predictions for the NH2 terminus from several prediction algorithms. Two β-strands (residues 34–38, 42–46) are predicted in the beginning of the conserved region of the NH2 terminus. Conceivably one of these may form β1 of the proposed KIRLI domain (Cukras et al., 2002) and this is consistent with the association of isolated NH2-terminal fragments that include this segment with the COOH terminus (Jones et al., 2001). The residues we identify as potentially affecting ATP binding (K47, R50, R54) all cluster in a loop between the second proposed β-strand and a preM1 extended helix. While high-resolution structural information is ultimately required, we may speculate that this loop additionally combines with residues in the COOH terminus to form an ATP binding pocket.
Figure 7.

Computer-predicted secondary structures (E, β-strand; H, helix) of conserved NH2-terminal (residues 1–77) region of Kir6.2 using multiple alignments with other inward rectifiers, generated by various algorithms indicated on left. The vertical line indicates the approximate boundary between essential (i.e., conserved) and nonessential regions. Putative β-strands are boxed, and the loop proposed to contribute to ATP binding is indicated above the sequence.
Acknowledgments
This work was primarily supported by National Institutes of Health grant HL54171 (to C.G. Nichols), as well as by a National Institutes of Health Cardiovascular Training grant (T32 HL07275, fellowship support of C.A. Cukras), and the Washington University NIH DRTC (DK20579).
Footnotes
Abbreviations used in this paper: KIRLI, Kir lipid–interacting; PH, pleckstrin homology; SUR, sulfonylurea receptor.
References
- Babenko, A.P., G. Gonzalez, and J. Bryan. 1999. The N-terminus of KIR6.2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels. Biochem. Biophys. Res. Commun. 255:231–238. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., U. Schulte, D. Oliver, S. Herlitze, T. Krauter, S.J. Tucker, J.P. Ruppersberg, and B. Fakler. 1998. PIP2 and PIP as determinants for ATP inhibition of KATP channels. 282:1059-60. Science. 282:1141–1144. [DOI] [PubMed] [Google Scholar]
- Boim, M.A., K. Ho, M.E. Shuck, M.J. Bienkowski, J.H. Block, J.L. Slightom, Y. Yang, B.M. Brenner, and S.C. Hebert. 1995. ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms. Am. J. Physiol. 268:F1132–F1140. [DOI] [PubMed] [Google Scholar]
- Clement, J.P.T., K. Kunjilwar, G. Gonzalez, M. Schwanstecher, U. Panten, L. Aguilar-Bryan, and J. Bryan. 1997. Association and stoichiometry of K(ATP) channel subunits. Neuron. 18:827–838. [DOI] [PubMed] [Google Scholar]
- Cukras, C.A., I. Jeliazkova, and C.G. Nichols. 2002. Structural and functional determinants of conserved lipid interaction domains of inward rectifying Kir6.2 channels. J. Gen. Physiol. 119:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain, P., L. Li, and J. Wang. 1998. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 95:13953–13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul, D., G. Loussouarn, E. Makhina, and C.G. Nichols. 2001. ATP interaction with the open state of the K(ATP) channel. Biophys. J. 80:719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul, D., G. Loussouarn, E. Makhina, S.L. Shyng, and C.G. Nichols. 2000. The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys. J. 78:2334–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z., and J.C. Makielski. 1997. Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 272:5388–5395. [DOI] [PubMed] [Google Scholar]
- Fan, Z., and J.C. Makielski. 1999. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K(+) channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J. Gen. Physiol. 114:251–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan, J.E., H.S. Yoon, P.J. Hajduk, and S.W. Fesik. 1995. Structural characterization of the interaction between a pleckstrin homology domain and phosphatidylinositol 4,5-bisphosphate. Biochemistry. 34:9859–9864. [DOI] [PubMed] [Google Scholar]
- Hilgemann, D.W., and R. Ball. 1996. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science. 273:956–959. [DOI] [PubMed] [Google Scholar]
- Huang, C.L., S. Feng, and D.W. Hilgemann. 1998. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 391:803–806. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., T. Gonoi, J.P. Clement, N. Namba, J. Inazawa, G. Gonzalez, L. Aguilar-Bryan, S. Seino, and J. Bryan. 1995. a. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 270:1166–1170. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., T. Gonoi, and S. Seino. 1997. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 409:232–236. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., Y. Tsuura, N. Namba, K. Masuda, T. Gonoi, M. Horie, Y. Seino, M. Mizuta, and S. Seino. 1995. b. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J. Biol. Chem. 270:5691–5694. [DOI] [PubMed] [Google Scholar]
- Jones, P.A., S.J. Tucker, and F.M. Ashcroft. 2001. Multiple sites of interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6.2. FEBS Lett. 508:85–89. [DOI] [PubMed] [Google Scholar]
- Koster, J.C., B.A. Marshall, N. Ensor, J.A. Corbett, and C.G. Nichols. 2000. Targeted overactivity of beta cell K(ATP) channels induces profound neonatal diabetes. Cell. 100:645–654. [DOI] [PubMed] [Google Scholar]
- Koster, J.C., Q. Sha, and C.G. Nichols. 1999. a. Sulfonylurea and K(+)-channel opener sensitivity of K(ATP) channels. Functional coupling of Kir6.2 and SUR1 subunits. J. Gen. Physiol. 114:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster, J.C., Q. Sha, S. Shyng, and C.G. Nichols. 1999. b. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J. Physiol. 515:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer, W.J., and C.G. Nichols. 1989. Nucleotide modulation of the activity of rat heart KATP channels in isolated membrane patches. J. Physiol. 419:193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, M.A., K.M. Ferguson, and J. Schlessinger. 1996. PH domains: diverse sequences with a common fold recruit signaling molecules to the cell surface. Cell. 85:621–624. [DOI] [PubMed] [Google Scholar]
- Li, L., J. Wang, and P. Drain. 2000. The I182 region of K(ir)6.2 is closely associated with ligand binding in K(ATP) channel inhibition by ATP. Biophys. J. 79:841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou, H.H., S.S. Zhou, and C.L. Huang. 1999. Regulation of ROMK1 channel by protein kinase A via a phosphatidylinositol 4,5-bisphosphate-dependent mechanism. Proc. Natl. Acad. Sci. USA. 96:5820–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher, E., and C.F. Stevens. 1977. Conductance fluctuations and ionic pores in membranes. Annu. Rev. Biophys. Bioeng. 6:345–381. [DOI] [PubMed] [Google Scholar]
- Proks, P., F.M. Gribble, R. Adhikari, S.J. Tucker, and F.M. Ashcroft. 1999. Involvement of the N-terminus of Kir6.2 in the inhibition of the KATP channel by ATP. J. Physiol. 514:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann, F., T.J. Ryder, S.J. Tucker, and F.M. Ashcroft. 1999. a. The role of lysine 185 in the Kir6.2 subunit of the ATP-sensitive channel in channel inhibition by ATP. J. Physiol. 3:661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann, F., S.J. Tucker, P. Proks, and F.M. Ashcroft. 1999. b. Involvement of the n-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J. Physiol. 518:325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs, T., J. Chen, G.D. Prestwich, and D.E. Logothetis. 1999. Distinct specificities of inwardly rectifying K(+) channels for phosphoinositides. J. Biol. Chem. 274:36065–36072. [DOI] [PubMed] [Google Scholar]
- Schulte, U., H. Hahn, H. Wiesinger, J.P. Ruppersberg, and B. Fakler. 1998. pH-dependent gating of Romk (Kir1.1) channels involves conformational changes in both N and C termini. J. Biol. Chem. 273:34575–34579. [DOI] [PubMed] [Google Scholar]
- Shaw, G. 1996. The pleckstrin homology domain: an intriguing multifunctional protein module. Bioessays. 18:35–46. [DOI] [PubMed] [Google Scholar]
- Shyng, S., T. Ferrigni, and C.G. Nichols. 1997. a. Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J. Gen. Physiol. 110:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S., T. Ferrigni, and C.G. Nichols. 1997. b. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol. 110:643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S., and C.G. Nichols. 1997. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., C.A. Cukras, J. Harwood, and C.G. Nichols. 2000. Structural determinants of PIP(2) regulation of inward rectifier K(ATP) channels. J. Gen. Physiol. 116:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., and C.G. Nichols. 1998. Membrane phospholipid control of nucleotide sensitivity of KATP channels [see comments]. Comment in: Science 1998 Nov 6;282(5391):1059-60. Science. 282:1138–1141. [DOI] [PubMed] [Google Scholar]
- Sigworth, F.J. 1980. The variance of sodium current fluctuations at the node of Ranvier. J. Physiol. 307:97–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soom, M., R. Schonherr, Y. Kubo, C. Kirsch, R. Klinger, and S.H. Heinemann. 2001. Multiple PIP2 binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett. 490:49–53. [DOI] [PubMed] [Google Scholar]
- Tanabe, K., S.J. Tucker, M. Matsuo, P. Proks, F.M. Ashcroft, S. Seino, T. Amachi, and K. Ueda. 1999. Direct photoaffinity labeling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. J. Biol. Chem. 274:3931–3933. [DOI] [PubMed] [Google Scholar]
- Touhara, K., W.J. Koch, B.E. Hawes, and R.J. Lefkowitz. 1995. Mutational analysis of the pleckstrin homology domain of the beta-adrenergic receptor kinase. Differential effects on G beta gamma and phosphatidylinositol 4,5-bisphosphate binding. J. Biol. Chem. 270:17000–17005. [DOI] [PubMed] [Google Scholar]
- Tucker, S.J., and F.M. Ashcroft. 1999. Mapping of the physical interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6.2. J. Biol. Chem. 274:33393–33397. [DOI] [PubMed] [Google Scholar]
- Tucker, S.J., F.M. Gribble, P. Proks, S. Trapp, T.J. Ryder, T. Haug, F. Reimann, and F.M. Ashcroft. 1998. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 17:3290–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, S.J., F.M. Gribble, C. Zhao, S. Trapp, and F.M. Ashcroft. 1997. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 387:179–183. [DOI] [PubMed] [Google Scholar]
- Varnum, M.D., and W.N. Zagotta. 1997. Interdomain interactions underlying activation of cyclic nucleotide-gated channels. Science. 278:110–113. [DOI] [PubMed] [Google Scholar]
- Woodward, R., E.B. Stevens, and R.D. Murrell-Lagnado. 1997. Molecular determinants for assembly of G-protein-activated inwardly rectifying K+ channels. J. Biol. Chem. 272:10823–10830. [DOI] [PubMed] [Google Scholar]
- Zagotta, W.N., and S.A. Siegelbaum. 1996. Structure and function of cyclic nucleotide-gated channels. Annu. Rev. Neurosci. 19:235–263. [DOI] [PubMed] [Google Scholar]
- Zhou, H., S.S. Tate, and L.G. Palmer. 1994. Primary structure and functional properties of an epithelial K channel. Am. J. Physiol. 266:C809–C824. [DOI] [PubMed] [Google Scholar]