

Abstract
To investigate the mechanisms by which low intracellular pH influences calcium signaling, I have injected HCl, and in some experiments CaCl2, into snail neurons while recording intracellular pH (pHi) and calcium concentration ([Ca2+]i) with ion-sensitive microelectrodes. Unlike fluorescent indicators, these do not increase buffering. Slow injections of HCl (changing pHi by 0.1–0.2 pH units min−1) first decreased [Ca2+]i while pHi was still close to normal, but then increased [Ca2+]i when pHi fell below 6.8–7. As pHi recovered after such an injection, [Ca2+]i started to fall but then increased transiently before returning to its preinjection level. Both the acid-induced decrease and the recovery-induced increase in [Ca2+]i were abolished by cyclopiazonic acid, which empties calcium stores. Caffeine with or without ryanodine lowered [Ca2+]i and converted the acid-induced fall in [Ca2+]i to an increase. Injection of ortho-vanadate increased steady-state [Ca2+]i and its response to acidification, which was again blocked by CPA. The normal initial response to 10 mM caffeine, a transient increase in [Ca2+]i, did not occur with pHi below 7.1. When HCl was injected during a series of short CaCl2 injections, the [Ca2+]i transients (recorded as changes in the potential (VCa) of the Ca2+-sensitive microelectrode), were reduced by only 20% for a 1 pH unit acidification, as was the rate of recovery after each injection. Calcium transients induced by brief depolarizations, however, were reduced by 60% by a similar acidification. These results suggest that low pHi has little effect on the plasma membrane calcium pump (PMCA) but important effects on the calcium stores, including blocking their response to caffeine. Acidosis inhibits spontaneous calcium release via the RYR, and leads to increased store content which is unloaded when pHi returns to normal. Spontaneous release is enhanced by the rise in [Ca2+]i caused by inhibiting the PMCA.
Keywords: pH, endoplasmic reticulum, ryanodine, caffeine, Ca2+ pump
INTRODUCTION
It is a truth universally acknowledged that all intracellular reactions are sensitive to pH, and that inside nerve cells pH is well buffered and regulated. Yet local pH can change during acidosis, ischemia, or anoxia. It is also possible that intracellular pH (pHi) will be lowered by a period of hyperactivity since Ca2+ ions entering the cell are extruded in exchange for H+ by the plasma membrane Ca-ATPase (PMCA)* (Ahmed and Connor, 1980; Schwiening et al., 1993; Trapp et al., 1996). Changes in pHi can also result from bicarbonate flux through inhibitory postsynaptic channels (Ballanyi and Kaila, 1998).
Such changes in pHi are likely to have important modulatory effects due to the high pH sensitivity of cellular proteins such as enzymes, ion channels, and ion transporters (Busa, 1986; Deitmer and Rose, 1996; Tombaugh, 1998). In particular, these pH changes may have significant effects on calcium regulation in nerve cells (Benham et al., 1992). Although changes in pHi have been associated with changes in intracellular calcium levels ([Ca2+]i) in a number of cell types, the mechanisms underlying them remain unclear. Increases in [Ca2+]i occur as a consequence of electrical activity that may allow calcium influx through voltage-gated channels, and that in turn may trigger calcium release from intracellular stores (Neering and McBurney, 1984; Friel and Tsien, 1992; Orkand and Thomas, 1995). These increases in [Ca2+]i can be modulated by several different processes. These are: the rate of extrusion from the cell via the PMCA, the rate of uptake into intracellular stores via the sarco-endoplasmic reticulum Ca ATPase (SERCA), intracellular calcium buffering, and mitochondrial uptake.
Previous work on the effects of pHi changes on resting [Ca2+]i in nerve cells has shown that relatively large experimentally imposed shifts in pHi (0.3–0.9 pH unit) can modulate neuronal calcium regulation (Dickens et al., 1989; Ouyang et al., 1994; Willoughby et al., 2001). In these studies, pHi was changed by superfusing weak acids and bases, which may well change pH inside organelles as well as in the cytoplasm. I have, therefore, now investigated the effects of the strong acid HCl, the iontophoretic injection of which should decrease pHi without directly changing pH in other compartments. I have explored the effects of short periods of low pHi on resting [Ca2+]i and on calcium transients generated by caffeine by direct injection of CaCl2 or by depolarization. In contrast to previous work I have simultaneously measured both pHi and [Ca2+]i with ion-sensitive microelectrodes that do not increase intracellular buffering.
I have found that small acidifications (0.1–0.2 pH units) caused a decrease in resting [Ca2+]i, which appeared to be due to a reduction in spontaneous release from the stores via ryanodine receptor channels. Larger intracellular acidosis caused an increase in [Ca2+]i and had little effect on depolarization-induced calcium transients, but did block release from the stores by caffeine. During pHi recovery from large acid injections there was an additional increase in [Ca2+]i that appeared to be due to release from overloaded stores. Some preliminary data on the effect of low pHi on responses to caffeine have been published as an abstract (Thomas, 2002).
MATERIALS AND METHODS
General
All the experiments were done at room temperature (18–24°C) on snail neurons 0.2–0.25 mm in diameter. I voltage clamped a suitable cell at a holding potential of −50 or −60 mV using two microelectrodes. I measured [Ca2+]i and pHi using ion-sensitive microelectrodes and induced changes in pHi by injecting HCl. As far as possible the tips of the microelectrodes were placed equidistantly from each other in the top part of the cell, with the calcium-sensitive microelectrode inserted first at the top center of the cell. This electrode often needed manipulation up and down to obtain a stable potential. Other microelectrodes were inserted at the corners of a square or pentagon centered on the calcium-sensitive one, with tips 50–75 μm apart.
Solutions
The subesophageal ganglia preparation was continuously superfused with one of up to seven different solutions held in bottles mounted on top of a Faraday cage. Flow was controlled by solenoid-operated valves near each bottle. From each valve a silica tube led to a manifold built into the experimental bath. The gravity-driven flow rate was 1.0–1.2 ml/min, and bath volume ∼0.1 ml. The normal snail Ringer contained (mM): 80 NaCl, 4 KCl, 7 CaCl2, 5 MgCl2, 20 HEPES, titrated with NaOH to pH 7.5. Low pH (6.5) Ringer was the same but buffered with 20 mM PIPES. Low calcium Ringer was made as normal except that CaCl2 was omitted, MgCl2 was increased to 12 mM, and 1 mM EGTA was added. Caffeine was added to normal Ringer to a concentration of 10, 20, or 50 mM. Bicarbonate Ringer was made by adding 5 mM NaHCO3 to normal Ringer just before use, the bottle being covered to reduce loss of CO2. Cyclopiazonic acid (CPA) was kept frozen as a 50 mM stock solution in dimethyl sulphoxide and diluted in snail Ringer on the day of use. Ryanodine was dissolved directly in 20 mM caffeine Ringer immediately before use. All these chemicals were from Sigma-Aldrich.
Calcium-sensitive Microelectrodes
These were made by a method refined from those described previously (Thomas, 1994, 2001). Every few days I pulled 8–10 long, sharp micropipettes from unfilamented aluminosilicate glass (1.2 mm diameter) and broke the tips on a glass rod to outer diameters of 1.5–2 μm. I left them in air for ∼30 min, then stacked them horizontally on a metal tray that I put into a borosilicate glass tube (12 mm internal diameter). One end of the tube was connected via a tap to a vacuum pump, the other sealed with a rubber cap. The tube was then evacuated for 30 s, the tap closed, and 3 μl bis(dimethylamino)dimethylsilane injected through the cap. A heating current, set to generate a temperature of ∼450°C around the pipette tips, was then passed through a wire coil round the middle of the tube for 10 min. The tube was then briefly reevacuated, sealed, and left to cool.
Functional microelectrodes were prepared as follows on the day of the experiment. First I backfilled three or four silanized micropipettes with a solution of 10 mM KCl and 0.01 mM CaCl2, using a 10 ml syringe and rubber tube to force air out of the tips. I then sucked a short column (150–250 μm) of sensor cocktail into the tip of each micropipette. The cocktail was a modification of that described by Ammann et al. (1987) with the composition (mg/ml): 12 Ca ionophore ETH 129, 6 sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, 200 2-Nitrophenyloctylether, 34 high molecular weight PVC, and 748 Tetrahydrofuran (all from Fluka). Filled microelectrodes were left to stabilize for at least 1 h before use, and had useful lives of only 12 h or so. During the experiment I applied ∼100 kPa of air pressure to the back of the Ca2+-sensitive microelectrode, since I have found that this maximizes its response. Before use each microelectrode was tested: the bath was superfused with Ca-free Ringer's solution (1 mM EGTA, pCa > 8), and any electrode showing a potential change of less than 150 mV was discarded. The potentials recorded from the electrodes during the experiments were displayed as VCa, and I have not converted them either to pCa or nM free [Ca2+]i. Calibration of Ca2+-sensitive microelectrodes is difficult because, in my hands, their responses change when they are withdrawn from a cell at the end of an experiment. Previous work suggests that their responses are essentially linear down to pCa 8 (10 nM), with a potential change of 28 mV per decade. For a detailed account see Kennedy and Thomas (1996). Experimental data were discarded if the VCa outside the cell had changed by >7 mV at the end of the experiment.
pH-sensitive Microelectrodes
The same method was used as above except that the micropipettes were made from borosilicate glass and the tips broken to ∼1 μm diameter before silanization. The backfill solution was pH 6.5 Ringer, and the pH sensor Hydrogen ionophore II, cocktail A (Fluka), sucked up to give a 200–500 μm column. They were calibrated by superfusing the preparation before and after the experiment with pH 6.5 Ringer. If the potential change in response to this solution was <58 mV, or if the potential in normal Ringer changed during the experiment by >5 mV, the result was discarded.
Other Microelectrodes
Microelectrodes for recording membrane potential (filled with 2 M KCl), and for passing clamp current (filled with 1 M KCl or CsCl) were pulled from filamented borosilicate glass (1.5 mm diameter), and broken on a pin in the bath to a resistance of 7–15 MΩ. For injecting HCl, such micropipettes were unbroken, filled with 1 M HCl, and had resistances of 40–60 MΩ. For injecting CaCl2, micropipettes were filled with 0.1 M CaCl2 and broken to a resistance of ∼10MΩ; for injecting ortho-vanadate, they were filled with 100 mM Na o-vanadate (BDH), titrated to pH 9.5, and similarly broken. If the potential of the membrane potential microelectrode in normal Ringer changed by more than 5 mV during the experiment the result was discarded.
To check that the effects of HCl injection were not related to the Cl− ions injected, and that the Cs+ ions in the clamp electrode had no effect on calcium transients, I tested the effects of large CsCl injections on [Ca2+]i and on the Ca2+ transients induced by caffeine application (unpublished data). Injections that, based on earlier experiments with NaCl injections (Thomas, 1972), I estimate would have increased intracellular Cs+ and Cl− concentrations inside the cell by 30–50 mM, had no obvious effect on [Ca2+]i or the response to caffeine. The clamp current required to depolarize the membrane was, however, reduced by two-thirds, since Cs+ ions block K+ channels.
Snail Brain Preparation
Common garden snails, Helix aspersa, were collected from the wild by local gardeners and kept in the laboratory where they estivated spontaneously. I isolated the subesophageal ganglia as described by Kennedy and Thomas (1995), mounted them on a plastic bath insert, and superfused them with normal snail Ringer. I then tore the inner connective sheath to expose the neurons and inserted pins to ensure that some of the large neurons at the back of the visceral and right pallial ganglia were accessible and well supported. Up to six microelectrodes were arranged over the preparation, five mounted on simple manual micromanipulators (Prior), but the Ca2+-sensitive microelectrode held on a Luigs and Neumann (Ratingen) motorized manipulator that could be controlled from outside the Faraday cage.
Data Collection and Analysis
Potentials from the microelectrodes were led to preamplifiers in the cage. Outputs from these and the clamp amplifier were filtered (8-pole Bessel) and recorded at 5 Hz on a PC via a CED interface and data collection program (Cambridge Electronic Design). For more details of most of the apparatus see Kennedy and Thomas (1995). Rates of recovery were calculated by fitting a single exponential to the data. Means are reported ± SEM. Significance was tested using the t test for two samples assuming unequal variances.
RESULTS
The Effect of HCl Injection on Resting Intracellular Calcium
In preliminary experiments I found that injecting sufficient HCl to lower pHi by more than one unit for more than a few minutes led to irreversible changes in the electrical properties of the neuron. Thus I sought to minimize the time at such low pHi values. Fig. 1 A shows the effects of one period of acidification by HCl injection and of the subsequent bicarbonate-assisted pHi recovery on the resting [Ca2+]i level in a neuron clamped at a potential of −60 mV. During the initial part of the experiment, as the Ca2+-sensitive microelectrode sealed into the cell and VCa decreased to a value below (i.e., more negative than) −120 mV, four brief depolarizations were imposed to allow calcium entry from outside the cell. (VCa values of −100, −120, and −140 mV correspond to [Ca2+]i levels of ∼1.7 μM, 300 nM, and 60 nM, respectively, assuming that the properties of my Ca2+-sensitive microelectrodes have changed little from those used by Kennedy and Thomas (1996). The HCl injection electrode was then inserted and a 20 nA current passed through it for 410 s. The resulting injection of H+ and Cl− ions lowered pHi from 7.2 to 6.3. Within a few seconds of the start of the injection [Ca2+]i began to decrease, reaching a minimum as pHi reached 6.75 (VCa fell from −123 to −132 mV). As the injection continued, [Ca2+]i began to increase, reaching almost the preinjection level by the time the injection current was switched off as pHi reached 6.3. As pHi then was allowed to recover from the injection, assisted by the added bicarbonate, [Ca2+]i at first changed little, then increased, reaching a peak during an additional pHi increase just after bicarbonate was removed. (This additional pHi increase was caused by intracellular bicarbonate ions taking up intracellular H+ ions and leaving the cell as CO2.) Fig. 1 B shows the relationship between VCa and pHi during the part of the experiment outlined by the box in Fig. 1 A. In a total of 13 similar tests of the effect of HCl injection on the resting calcium level there was, during the acidification, always an initial decrease in [Ca2+]i, with an average change in VCa of 4.7 ± 0.8 mV, followed by an increase as pHi continued to fall during each injection. During the pHi recovery there was always a transient increase in [Ca2+]i, although often on a changing baseline that made it hard to quantify. A second example of these effects of HCl injections and the subsequent pHi recovery is shown in the first part of Fig. 2 A. In this experiment VCa first decreased by 4 mV then increased to a value 11 mV above the preinjection level as pHi reached 6.4. In the 13 similar tests of the effect of large HCl injections, the average increase in VCa by the end of each injection was 9 mV, with VCa changes ranging from a 6 mV decrease to a 25 mV increase. The average minimum pHi reached in these experiments was 6.5.
Figure 1.


The effect of HCl injection on pHi and steady-state [Ca2+]i in a voltage-clamped snail neuron. (A) Record of membrane potential, clamp and HCl injection current, pHi and VCa, during a short experiment. VCa values of −100, −120, and 140 mV correspond, respectively, to [Ca2+]i values of 1.7 μM, 300 nM, and 60 nM. The Ca2+-sensitive microelectrode was inserted first, then the membrane potential and clamp electrodes (both filled with KCl, the latter pushed against the cell membrane before being zapped in by switching on the clamp) and then the pH-sensitive microelectrode. The membrane potential was clamped at −60 mV except for one brief hyperpolarization and six brief depolarizations. After the first four depolarizations the HCl electrode was inserted (arrow) and used to lower pHi to ∼6.3. The injection current, shown as a downward deflection between a and b on the current trace, was then switched off and pHi allowed to start recovering. 3 min after the injection current was switched off a solution with 5 mM bicarbonate was superfused (shown as bar under pHi record) for 7 min to accelerate pHi recovery. (B) Graph of VCa versus pHi for the period (shown as within dashed box on Fig. 1 A) from the start of the HCl injection to the end of the bicarbonate superfusion. Arrows show direction in which time passed.
Figure 2.

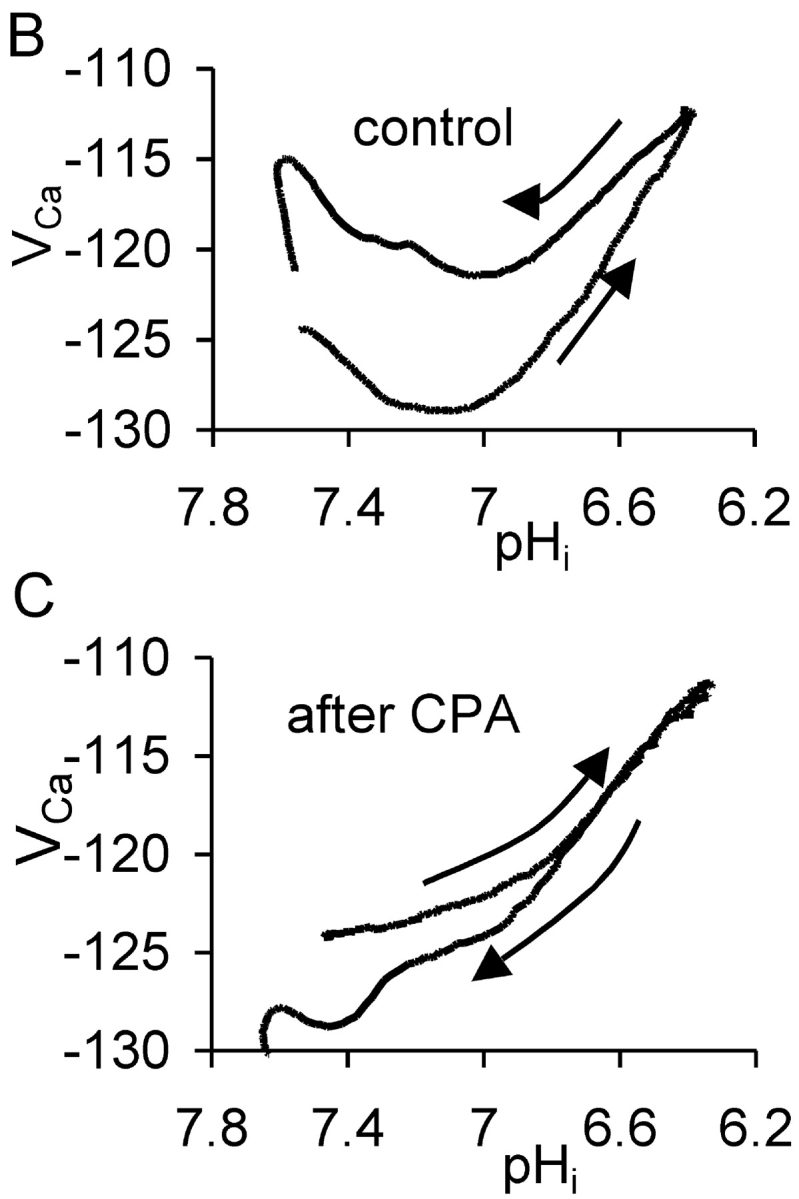
The effect of cyclopiazonic acid on the VCa response to acid injection. (A) Experiment showing two periods of acid injection (downward deflections of the current trace) before and after the start of application of 50 μM CPA. The membrane potential was held at −60 mV throughout the period displayed. (B) Plots of VCa versus pHi for the first period during which acid was injected and then pHi allowed to recover to the point where CPA was added. Arrows show direction of time. (C) Graph of VCa versus pHi for the second period, after the start of CPA application, during which acid was injected and pHi recovered. Passage of time again shown by arrows.
Willoughby et al. (2001) have shown that a pHi increase (induced by a weak base) caused a rise in [Ca2+]i in snail neurons via release from the stores. I therefore investigated the effect on the pHi recovery–induced transient [Ca2+] increase of blocking the store-loading pump with cyclopiazonic acid (CPA). Fig. 2 A shows an experiment much like that of Fig. 1 A, except that after pHi had recovered from the first HCl injection I superfused the preparation with Ringer to which CPA had been added shortly before. Many previous studies (e.g., Seidler et al., 1989; Orkand and Thomas, 1995) have shown that CPA blocks the SERCA pump. This leads to a slow emptying of the stores, since they seem to be inherently leaky. (In the experiment shown in Fig. 2 A, CPA application caused an unexpected, immediate, but transient fall in [Ca2+]i. In a total of seven similar experiments in which CPA was applied after a period of acid injection the transient fall in VCa ranged from 1 to 10 mV.)
After the transient fall in [Ca2+]i, CPA caused the expected slow rise in calcium as the stores emptied, followed by recovery as the PMCA restored the normal resting level. At this point the HCl injection was repeated, lowering pHi from 7.4 to 6.35 over 12 min. This time there was no fall in [Ca2+]i before the rise. In a total of seven similar experiments in which HCl was injected before and after CPA, the average fall in VCa before CPA was 7.7 ± 2.4 mV. After CPA, HCl injection failed to decrease [Ca2+]i in any of the experiments. Fig. 2 B shows the control relationship between VCa and pHi from the first acid injection and recovery, whereas Fig. 2 C shows the relationship after CPA. Inhibition of the SERCA pump clearly removed most of the hysteresis in the VCa/pHi relationship.
Next I investigated the possible roles of the PMCA and the steady-state calcium level on the response to acidification. I injected ortho-vanadate, a potent ATPase inhibitor (Cantley et al., 1977), which should block the PMCA, the sodium pump and possibly also the SERCA pump. Fig. 3 shows one of three similar experiments in which o-vanadate was injected iontophoretically, in this case after two periods of HCl injection. The first injection lowered pHi from 7.3 to 7.0, and VCa from −129 to −133 mV. The second, larger, injection lowered pHi from 7.35 to 6.8, and VCa from −134 mV to −137 mV. Once pHi and VCa had stabilized after recovering from the second injection, a microelectrode filled with Na-o-vanadate was inserted and a 5 nA current passed to inject o-vanadate ions. The resulting inhibition of the PMCA caused [Ca2+]i to start increasing immediately. Three depolarizations imposed an extra calcium load. When VCa had reached −110 mV (from −137 mV, equivalent to almost a tenfold increase in [Ca2+]i), I injected HCl for 210 s, lowering pHi from 7.35 to 7.15. This time VCa fell rapidly to −121 mV, and rose again as rapidly when the HCl injection was stopped. After superfusion with CPA, the acid injection was repeated. It had no immediate effect on intracellular calcium, although as pHi recovered there was a small increase. These effects of vanadate and CPA were seen in a total of three similar experiments: vanadate injection to block the PMCA and raise [Ca2+]i made the VCa response to HCl injection larger, whereas CPA, which inhibits the SERCA pump, abolished it.
Figure 3.

The effect of Ca-ATPase inhibitors on the response of VCa to acid injections. Records of membrane potential, clamp, and injection current, pHi and VCa, during a complete experiment. The membrane potential was clamped at −50 mV except for one brief hyperpolarization and many brief depolarizations to test homeostatic mechanisms. After the first five depolarizations the HCl electrode was inserted and current passed through it to lower pHi four times, as shown by the downward current deflections (a–d). When pHi had recovered from the second HCl injection, o-vanadate was injected between points x and z to inhibit the PMCA. In this experiment pHi was allowed to recover after each injection without bicarbonate superfusion.
The Effect of Caffeine and Ryanodine on the Response to HCl Injection
Caffeine-sensitive calcium stores seem to be universal in nerve cells, although their function remains unclear (Bootman et al., 2001). Calcium is released physiologically from the stores via the RYR calcium-release channels by a local increase in [Ca2+]i in the process known as calcium-induced calcium release (CICR). High levels of caffeine lower the threshold for this process so that it occurs at normal levels of [Ca2+]i, leading to store emptying. The subsequent undershoot in [Ca2+]i when caffeine is removed has been ascribed to local depletion of calcium by the hyperactive SERCA pump (Collins and Thomas, 2001). Since the RYR calcium-release channels of the sarcoplasmic reticulum of muscle cells are known to be inhibited by low pH (Meissner and Henderson, 1987; Rousseau and Pinkos, 1990; Meissner, 1994), I have tested the effect of caffeine and ryanodine on the VCa response to small acid injections. Fig. 4 A shows an experiment in which caffeine was applied after two short HCl injections, both of which caused a decrease in [Ca2+]i. Superfusion with 20 mM caffeine then caused a large transient increase in [Ca2+]i as CICR was induced, followed by a prolonged undershoot. Then, 5 min after beginning the caffeine application, during the undershoot, I made a third HCl injection. This caused no change in [Ca2+]i, although it was followed by a small release of residual calcium just before I ended the caffeine superfusion. A fourth HCl injection made 22 min later caused a small decrease in [Ca2+]i. Similar results were obtained in two other experiments in which HCl was injected during a long exposure to 20 mM caffeine.
Figure 4.


The effect of caffeine alone and with ryanodine on the response of VCa to HCl injection. (A) The cell was clamped at −60 mV except at the start of the section shown, when five depolarizations to −20 mV for 10 s were applied. Then HCl was injected for two short periods, and when pHi had recovered, caffeine was superfused for 12 min. During caffeine superfusion a third HCl injection was made, and near the end of the experiment, a fourth. The two missing parts of the VCa trace after caffeine washout are due to removal of noise from the clamp electrode. (B) Another cell, which was clamped at −60 mV throughout. After two acid injections, a freshly-made solution of ryanodine and caffeine was superfused for 3 min. Then another HCl injection was made. A second application of ryanodine and caffeine showed that the first had not been completely effective. A second postryanodine injection again caused no decrease in [Ca2+]i.
While caffeine opens RYR and thus empties the stores, ryanodine blocks the RYR. Unfortunately, it rapidly blocks only open RYR in snail neurons, so I applied it together with 20 mM caffeine to maximize its effectiveness. Fig. 4 B shows an example result. After two periods of HCl injection, each of which caused a fall in [Ca2+]i, the preparation was superfused with ryanodine and caffeine for 210 s. This caused a large transient increase in [Ca2+]i followed by a long-lasting undershoot. Both of the two subsequent HCl injections caused an increase in [Ca2+]i. In a total of six similar experiments, before ryanodine the mean decrease in VCa caused by a 0.1 pH unit acidification was −1.5 ± 0.7 mV. After ryanodine, the same acidification caused a mean increase in VCa of 0.9 ± 0.4 mV. Ryanodine and caffeine thus converted the effect of small HCl injections from a decrease in [Ca2+]i to no change or an increase.
The application of ryanodine and caffeine together led to a prolonged decrease in steady-state VCa that was maintained long after the return to normal snail Ringer, unlike with the application of caffeine alone. The average VCa before ryanodine application was −132 ± 3 mV, while 10 min after removing ryanodine it was −145 ± 2 mV. This is equivalent to a decrease of [Ca2+]i from 122 to 42 nM.
The Effect of HCl Injection on the Response to Caffeine
At normal pHi values 10 mM caffeine always causes a large calcium release from snail neurone calcium stores within 1 min as long as the stores have had time (4–6 min) to refill after any previous caffeine application. To test the effects of low pHi on the effectiveness of caffeine in initiating CICR, I have observed the responses to 10 mM caffeine, applied for 2 min, as shown in Fig. 5 . Seven applications of caffeine were made at different pHi levels as changed by HCl injection or bicarbonate superfusion. At pHi levels below seven there was no clear response to caffeine, while at higher pHi levels caffeine consistently caused a transient increase in calcium.
Figure 5.

The effect of low pHi on the response of VCa to the application of 10 mM caffeine. The first half of a long experiment is shown, including the period during which the calcium-sensitive microelectrode was pushed right through the cell and then withdrawn back into the cell. After two applications of 10 mM caffeine, an HCl microelectrode was inserted through which currents were passed to decrease pHi, shown as downward deflections on the current record. Caffeine was applied as shown by the bars below the VCa record, and bicarbonate (5 mM) was applied for two periods as indicated.
Fig. 6 shows results from a total of 25 caffeine applications from three experiments in which pHi was manipulated as in Fig.5. As shown in Fig. 6 A all the caffeine applications made when pHi was above 7.1 produced a fall in VCa of at least 6 mV; but the six application made when pHi was lower than 7.1 caused a change in VCa of, at most, less than 3 mV. Fig. 6 B shows the values of VCa and pHi at which the caffeine was applied. Although low pHi did tend to increase the steady-state calcium level, this cannot explain the failure of the caffeine response. When caffeine was applied at 50 mM (unpublished data) it caused a large release (ΔVCa over 10 mV) at pHi values down to 6.75, but only a small release at lower values.
Figure 6.
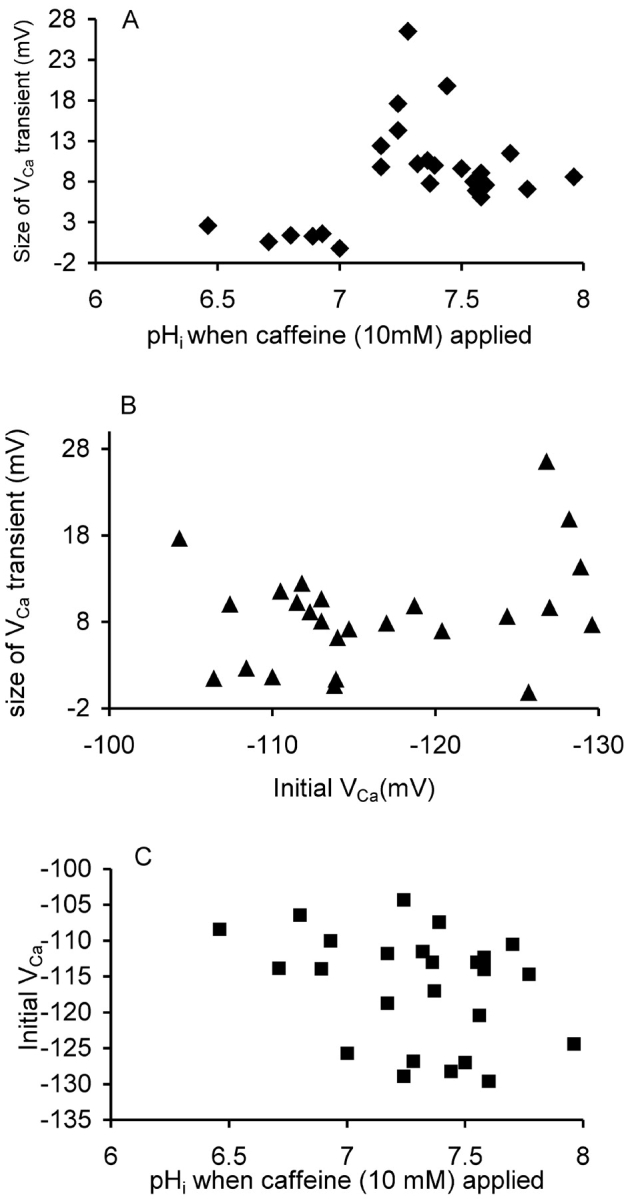
Caffeine-induced calcium release at different pHi. (A) The size of the transient change in VCa occurring within 60 s of the start of 25 applications of 10 mM caffeine onto three different cells is plotted against the pHi at the time of application. (B) The size of the transient change in VCa plotted against the VCa at the time of application. (C) The initial VCa and pHi when the caffeine applications were made. Data from three experiments in which caffeine was applied a total of 25 times.
The Effects of Acid Injections on Calcium Transients.
Since the calcium pump (PMCA) that extrudes Ca2+ ions from the cell is known to carry H+ ions into the cell, it seems probable that it would be inhibited by low pHi, as it is by high extracellular pH (Benham et al., 1992; Schwiening et al., 1993). I therefore examined the effects of HCl injection on calcium transients. I increased [Ca2+]i by two methods: by direct iontophoretic injection of Ca2+ (and Cl−) ions or by brief depolarizations. The number of ions injected for a given charge, using the same procedure, has been shown not to change in vitro when [Ca2+] was varied over the normal [Ca2+]i range of 10–100 nM (Schwiening and Thomas, 1996). The effect of varying pH in vitro on Ca2+ iontophoresis has not been studied, but there is no reason to believe it would be sensitive to pHi, at least in the range used here. The voltage-sensitive calcium channels are, however, likely to be inhibited by low pHi, so that calcium entry will decrease as HCl is injected (see Tombaugh and Somjen, 1998). Fig. 7 shows an experiment in which I tested the effect of low pHi on both types of calcium transient. Calcium injections were made with currents of 10 or 20 nA for 10 s. The depolarizations were by 40 mV for 10 s. After inserting the HCl electrode I passed an H+ injection current which was increased as necessary to lower pH by ∼1 U over a period of 18 min; Ca2+ ions were injected at ∼2 min intervals. Once the pHi was below 6.5 I switched off the acid injection and superfused the preparation for a short time with bicarbonate Ringer to allow the cell to extrude the injected acid more rapidly. After allowing some time for pHi and VCa to stabilize, I began a series of 10 s depolarizations, and repeated the acid injection protocol. The depolarizations caused rather smaller changes in VCa than the injections, although the pHi changes, presumably generated by the PMCA, associated with the calcium extrusions at normal pHi were similar: 0.009 for the injections and 0.015 for the depolarizations. (The scale used in Fig. 7 makes this hard to detect, but I made the comparison by displaying the original data at high gain.) This suggests that the calcium load generated by the depolarizations was larger than that generated by the injections, assuming in both cases that the complete calcium load was extruded by the PMCA. The location of the Ca2+-sensitive microelectrode some distance from the cell membrane presumably accounts for the smaller recorded size of the depolarization-induced transients.
Figure 7.

The effect of HCl injection on pHi and on VCa transients generated either by injection of CaCl2 or by depolarizations to −20 mV for 10 s. The membrane potential was clamped at −60 mV except for brief depolarizations. After the first two depolarizations the CaCl2 electrode was inserted and used to make several injections. Finally the HCl electrode was inserted and used to reduce pHi. Downward offsets on the current trace indicate the periods during which Ca2+ or H+ and Cl− ions were injected iontophoretically, and upward deflections indicate depolarizations. After the two periods when pHi was reduced by HCl injection, recovery was assisted by superfusion of Ringer containing 5 mM added bicarbonate. The large vertical excursions of the pH and calcium traces were caused by electrical pickup by the amplifier inputs.
The size of the VCa transients caused by the injections presumably reflects a combination of local buffering and diffusion from the injection site to the measurement point. The large acidification appears to have had little overall effect. On the other hand, the transients caused by depolarizations were greatly reduced by acidification. Fig. 8 shows another experiment in which transients resulting from injections and depolarizations were compared. In this case the depolarizations were to 0 mV, which as pHi fell caused the opening of outward H+ (Thomas and Meech, 1982) as well as Ca2+ channels. Thus, pHi tended to increase briefly during the depolarizations at low pHi. As before, the VCa transients generated by Ca2+ influx from outside the cell were more sensitive to low pHi than were those arising from the iontophoretic injections, although both seemed more sensitive to low pHi here than in the experiment shown in Fig. 7. The first pHi decrease, from 7.28 to 6.24, reduced the VCa transient generated by CaCl2 injection by almost 50%, from 15.7 to 8 mV. The second pHi decrease, from 7.24 to 6.49, reduced the VCa transient generated by depolarization by 90%, from 17.3 to 1.8 mV depolarization.
Figure 8.

The effect of HCl injection on pHi and on VCa transients generated either by injection (downward deflections on the current record) of CaCl2 or by depolarizations to 0 mV for 10 s (upward deflections on the current record). After the first three injections, the position of the calcium-sensitive microelectrode was adjusted; this increased the recorded VCa transients. After the two periods when pHi was reduced by HCl injection, recovery was assisted by superfusion of Ringer containing 5 mM added NaHCO3. The vertical excursions of the pH and calcium traces were caused by electrical pickup by the amplifier inputs.
Data on normalized VCa transients from five experiments are collected in Fig. 9 . The series of transients recorded just before and during each period of HCl injection were normalized to the largest response of that series, then collected in 0.2 pH unit bins. The injection-induced calcium transients were only ∼20% smaller at pH 6.5 than in the physiological range of 7.2 to 7.4, although only the point at pHi 6.4 was significantly different (P < 0.05) from that at pHi 7.4. On the other hand, the depolarization-induced transients were reduced by ∼60% by the same acidification. Only the points at pH 6.8 and 6.6 were significantly different from that at 7.3, that at pH 6.6 at the 0.0003 level.
Figure 9.

Effect of low pHi on the size of the transient increases in VCa induced by either iontophoretic injections (♦) or 10 s depolarizations (▪). The calcium transients from each period of acid injection were normalized relative to the largest transient in that series, then collected in 0.2 pH unit bins (the pH at the start of each injection). The mean transient size and SEM for each bin was plotted against the average starting pHi for the transients in each bin. Data from five different experiments, with a total of 26 injections and 25 depolarizations. Lines are least squares best fits to the original normalized data of the following relationship: pHst = pK + log (ΔCa/1 − ΔCa), where pHst is the pHi at the start of a VCa transient, pK the pH when ΔCa = 0.5, and ΔCa the relative size of the calcium transient. The pKs giving the two lines were 6.05 for the injections and 6.51 for the depolarizations.
Since calcium extrusion by the PMCA involves H+ transport into the cell, low pHi will increase the work required to extrude Ca2+ ions, and may slow the rate of recovery after a calcium injection. Fig. 10 shows the recovery time constants averaged in 0.2 pH unit bins for 25 injection-induced VCa transients. Acidification did not slow the rates of recovery; none of the low pHi points were significantly different from that at pHi 7.45.
Figure 10.

The effect of low pHi on the rate of recovery of [Ca2+]i (expressed as VCa) for calcium injections from three experiments, those of Figs. 6 and 7 and another. Transients were grouped into 0.2 pH unit bins (pHi at the midpoint of the recovery), and averaged with respect to both pHi and time constant. The bars show the standard errors of the mean time constants.
DISCUSSION
In this study I have shown that changes in pHi induced by HCl injection can have marked effects on neuronal [Ca2+]i. When resting calcium was recorded while pHi was lowered, [Ca2+]i initially fell with modest acidifications but then rose as pHi was further reduced. When pHi was allowed to recover, [Ca2+]i at first also began to recover (if it had been increased above baseline by the acidification), but then transiently increased again. Both the acid-induced fall in [Ca2+]i and the recovery-induced increase were abolished by CPA. Ortho-vanadate increased the steady-state [Ca2+]i and magnified the acid-induced fall [Ca2+]i. The decrease in [Ca2+]i caused by small HCl injections was abolished by caffeine and ryanodine. Calcium release from stores by 10 mM caffeine did not occur when the pHi was below 7.1. While injections sufficient to lower pHi by one unit had little effect on calcium transients generated by CaCl2 injection, such acidifications greatly reduced transients caused by opening voltage-gated calcium channels.
The Effect of HCl Injection on Resting [Ca2+]i
Resting [Ca2+]i in snail neurons is influenced by several processes. These are (a) influx from outside the cell, mainly through channels, (b) extrusion from the cell by the PMCA (Kennedy and Thomas [1995] showed that snail neurons have no Na/Ca exchanger), (c) uptake into stores by the SERCA, (d) release from the stores via the RYR and inositol 1,4,5-trisphosphate channels, and (e) uptake and release by mitochondria. Since there is no evidence for any mitochondrial involvement at the low levels of [Ca2+]i in a cell clamped at −50 or −60 mV I will not discuss this further.
My results show that modest reductions in pHi caused by HCl injection consistently reduced resting [Ca2+]i. This effect of acidification is similar to that reported by Willoughby et al. (2001). They used propionate superfusion to lower pHi by 0.2–0.3 pH units and recorded a small fall in [Ca2+]i using both fura-2 and a Ca2+-sensitive microelectrode. They did not investigate the effects of SERCA inhibitors on the response to low pHi, although they showed that CPA blocked a large fraction of the rise in [Ca2+]i seen with pHi increases induced with weak bases. My findings that CPA blocked both the fall in [Ca2+]i induced by modest acidifications and the rise in [Ca2+]i induced by the pHi recovery after acidification show that both phenomena reflect changes in the net calcium uptake or release of calcium by the calcium stores.
What effect of pHi on the stores caused the fall in [Ca2+]i seen with acidification? Calcium uptake into the stores is driven by the SERCA pump, whereas spontaneous release is believed to occur through RYR channels. Thus, a fall in [Ca2+]i may result from either stimulation of the pump or inhibition of a continuous release process. In the experiments in which I saw clear falls in [Ca2+]i with HCl injection the membrane potential was clamped near the resting potential, so that [Ca2+]i was presumably well below the normal threshold for calcium-induced calcium release. Perhaps, nevertheless, continuous release of calcium from the stores was occurring, until inhibited by the fall in pHi. The finding that caffeine and ryanodine block, indeed reverse, the fall in [Ca2+]i caused by acid injection supports this suggestion. There is also considerable evidence from other preparations that low pHi inhibits release by the stores. In intact cardiac myocytes both uptake and release of Ca2+ were inhibited by acidosis (Hulme and Orchard, 1998) and the frequency of spontaneous calcium sparks, caused by release from the stores, was reduced by low pHi (Balnave and Vaughan-Jones, 2000). There is also clear evidence that calcium release by isolated sarcoplasmic reticulum RYR receptors is pHi sensitive (Meissner and Henderson, 1987; Rousseau and Pinkos, 1990; Meissner, 1994), although recent work with RYR receptors from rabbit skeletal muscle incorporated in lipid bilayers suggests low pH on the cytoplasmic side may be less inhibitory than reported previously (Laver et al., 2000).
Alternatively or additionally, the reduction in [Ca2+]i might indicate that low pHi was stimulating the SERCA pump. However, there is no evidence for this in other preparations. On the contrary, a recent study of the effect of acidification on sarcoplasmic reticulum Ca-ATPase activity found that it was ∼60% less at pH 6.5 than 7.4. (Stienen et al., 1999). Thus, the processes controlling both calcium store uptake and release are inhibited by acidosis in muscle.
Together, the inhibitory effects of caffeine, ryanodine, and CPA show that the initial effects of HCl injection on [Ca2+]i must be due to inhibition of release via RYR. As pHi is reduced calcium release is inhibited, causing [Ca2+]i to fall. Further acidification, and/or the passage of time, caused [Ca2+]i to rise, often to levels higher than the baseline. This increase was not blocked by CPA, suggesting that it was due to low pH effects on either the PMCA or mitochondria. It was unlikely to be due to an effect on the plasma membrane calcium channels, since they are inhibited by low pHi.
The long-term fall in [Ca2+]i produced by ryanodine, as well as the effects of small HCl injections, lead to the conclusion that resting [Ca2+]i, at least as recorded by a Ca2+-sensitive microelectrode, may be supplemented by a continuous leak from the stores. It is difficult to understand how such a steady leak could contribute in the long term to resting [Ca2+]i, unless the release and uptake sites are in different parts of the endoplasmic reticulum. If they are colocalized, as usually assumed, and the rate of uptake depends on the store content, there is presumably a balance at rest between local release and uptake. Reducing release by HCl injection would increase the store content and thus reduce uptake. Any reduction in [Ca2+]i would be only transient. Perhaps, as suggested by Verkhratsky and Petersen (2002) the ER acts as a conduit, in this case carrying calcium from the plasma membrane into the cell interior. There would then be a cytoplasmic gradient from the release sites to the plasma membrane. Blocking release would lead to a long-term fall in resting [Ca2+]i in the cell interior.
The transient decrease in [Ca2+]i sometimes seen on the application of CPA, as in Fig. 2 A, has not been observed before. In my experiments the calcium stores were often likely to have been overloaded when CPA was applied. Perhaps the sudden blockage of the SERCA pump also briefly reduces the leakage by some unknown mechanism. If the SERCA pump is electrogenic, there might be a change in the potential across the ER membrane.
Further evidence for a continuous resting leak is that inhibition of the SERCA pump by CPA does lead to store emptying, which has always been presumed to occur via leakage. On the other hand, CPA has not been reported to cause a significant long-term reduction in [Ca2+]i as might be expected when leakage ceases when the stores are exhausted, while ryanodine and caffeine do cause such a reduction. In nonexcitable cells, of course, emptying the stores greatly increases calcium influx from outside the cell via store-operated calcium entry. Perhaps at least a modest increase occurs in neurons too when the stores are emptied by CPA, sufficient to offset the effects on [Ca2+]i when there is no further leakage from stores. The stores are not emptied by ryanodine, so the same increase in influx would not occur.
If the extent of the fall in [Ca2+]i with acidification reflects a fall in the rate of leakage from the stores, then my results with o-vanadate suggest that blocking the PMCA and thus raising [Ca2+]i greatly increases leakage from the stores. Indeed this is further evidence that CICR operates at near-normal [Ca2+]i levels. Alternative mechanisms that could explain both the large fall in [Ca2+]i with acidification and the blocking effect of CPA are difficult to conceive.
The Effect of the Postacidification pHi Recovery on Resting [Ca2+]i
The transient rise in [Ca2+]i, seen when pHi was allowed to recover from HCl injection, is similar to that reported recently by Willoughby et al. (2001) for a pHi increase from its resting level induced by application of a weak base. Thus, it seems that a relative pHi increase magnifies spontaneous calcium release. The stores in my experiments behave as if the period in low pHi has overloaded them, suggesting that the uptake pump was much less inhibited by low pHi than was the release process.
The pHi sensitivity of Ca2+ release via RYR implies that pHi shifts seen during normal neuronal activity might influence [Ca2+]i also (Chesler and Kaila, 1992). A study of rat hippocampal neurons showed a train of action potentials (for 1 min at 0.5 Hz) was accompanied by a fall in pHi of 0.18 pH unit (Trapp et al., 1996). Similarly, in Helix neurons a burst of depolarizations to 0 mV (50 times at 5 Hz) produced an acidification of 0.06 pH unit (Willoughby et al., 1999). In both studies pHi returned rapidly to resting values. Such activity-induced acidifications will lead to a build-up of calcium in the stores, so that, as pHi recovers, the stores will be hypersensitive to a subsequent influx of calcium. This could be a mechanism contributing to long-term changes in neuronal properties.
Of all the processes I have tested which are involved in influencing or controlling [Ca2+]i, the RYR calcium release channels seem to be in practice the most sensitive to pHi. Modest acidifications inhibit release much more than they inhibit the SERCA pump, so acidified stores increase their calcium content. Conversely, alkalinizations or simple recovery of normal pHi from acidifications sensitize the stores and enhance CICR.
Inhibition by Low pHi of the Response to Caffeine
It is well established that the calcium stores in a wide variety of preparations are sensitive to pHi (see Willoughby et al., 2001) but complete inhibition of the response to caffeine by low pHi has not been reported before. It is unlikely that the stores were empty, or that access of caffeine was restricted in any way by the low pHi. Although the calcium release channel is inhibited by low pH in various sarcoplasmic vesicle preparations, the inhibition seen at pHi 7.0 in the recent careful study of Laver et al. (2000) was <20%.
Lack of Effect of Acidification on Injection-induced Calcium Transients
The repeated iontophoretic injections of CaCl2 caused transient increases in [Ca2+]i at the tip of the Ca2+-sensitive microelectrode that was some tens of micrometers distant from the injection site. Assuming that calcium stores were so well-loaded that they played no significant role in shaping the transients, the recovery after the end of the injections presumably reflects Ca2+ extrusion by the PMCA (Schwiening et al., 1993). The modest slowing of the rate of VCa recovery caused by large HCl injections correlates well with the relatively low sensitivity of the PMCA of rat sensory and other neurons to high extracellular pH (Benham et al., 1992). Presumably the PMCA has a very low affinity for intracellular H+ ions. Similarly, the size of the injection-induced VCa transients was little reduced by low pHi, presumably because calcium buffering is not very sensitive to pHi. Much of the size reduction with acidification may be due to the rise in [Ca2+]i that accompanied the HCl injection at low pHi levels. Schwiening and Thomas (1996) found that the calcium transients (recorded with fura-2) generated by iontophoretic injection were reduced while [Ca2+]i was increased by changing the clamp potential. The greater sensitivity of the depolarization-induced calcium transients found here was presumably due to the pH sensitivity of the calcium channels. The identity of the channels likely to operate in snail neurons is unknown, but all such channels are likely to be inhibited by low pHi (Tombaugh and Somjen, 1998).
In summary, the mechanism by which modest intracellular acidosis is most likely to influence [Ca2+]i in nerve cells is by inhibiting spontaneous or calcium-induced calcium release from the endoplasmic reticulum. All the other processes which raise or lower [Ca2+]i in the cytoplasm appear to be far less sensitive to changes in pH.
Acknowledgments
I am grateful to the Medical Research Council for financial support, to Christof Schwiening for much help with data analysis and many critical remarks, to Debbie Willoughby, Ann Silver, and Mike Mason for comments on the draft MS, to local gardeners for collecting snails, and to Andrea Nistri for his hospitality while I revised the manuscript in Italy.
Footnotes
Abbreviations used in this paper: CPA, cyclopiazonic acid; pHi intracellular pH; PMCA, plasma membrane calcium ATPase; SERCA, sarcoplasmic/endoplasmic reticulum calcium ATPase.
References
- Ahmed, Z., and J.A. Connor. 1980. Intracellular pH changes induced by calcium influx during electrical activity in molluscan neurons. J. Gen. Physiol. 75:403–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammann, D., T. Bührer, U. Schefer, M. Müller, and W. Simon. 1987. Intracellular neutral carrier-based Ca2+ microelectrode with subnanomolar detection limit. Pflugers Arch. 409:223–228. [DOI] [PubMed] [Google Scholar]
- Ballanyi, K., and K. Kaila. 1998. Activity-evoked changes in intracellular pH. pH and Brain Function. K. Kaila and B.R. Ransom, editors. Wiley-Liss, New York. 291–308.
- Balnave, C.D., and R.D. Vaughn-Jones. 2000. Effect of intracellular pH on spontaneous Ca2+ sparks in rat ventricular myocytes. J. Physiol. 528:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham, C.D., M.L. Evans, and C.J. McBain. 1992. Ca2+ efflux mechanisms following depolarization evoked calcium transients in cultured rat sensory neurones. J. Physiol. 455:567–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman, M.D., P. Lipp, and M.J. Berridge. 2001. The organisation and functions of local Ca2+ signals. J. Cell Sci. 114:2213–2222. [DOI] [PubMed] [Google Scholar]
- Busa, W.B. 1986. Mechanisms and consequences of pH-mediated cell regulation. Annu. Rev. Physiol. 48:389–402. [DOI] [PubMed] [Google Scholar]
- Cantley, L.C., L. Josephson, R. Warner, M. Yanagisawa, C. Lechene, and G. Guidotti. 1977. Vanadate is a potent (Na,K)-ATPase inhibitor found in ATP derived from muscle. J. Biol. Chem. 252:7421–7423. [PubMed] [Google Scholar]
- Chesler, M., and K. Kaila. 1992. Modulation of pH by neuronal activity. Trends Neurosci. 15:396–402. [DOI] [PubMed] [Google Scholar]
- Collins, R., and R.C. Thomas. 2001. The effects of calcium pump inhibitors on the response of intracellular calcium to caffeine in snail neurones. Cell Calcium. 30:41–48. [DOI] [PubMed] [Google Scholar]
- Deitmer, J.W., and C.R. Rose. 1996. pH regulation and proton signaling by glial-cells. Prog. Neurobiol. 48:73–103. [DOI] [PubMed] [Google Scholar]
- Dickens, C.J., J.I. Gillespie, and J.R. Greenwell. 1989. Interactions between intracellular pH and calcium in single mouse neuroblastoma (N2A) and rat pheochromocytoma cells (PC12). Q. J. Exp. Physiol. 74:671–679. [DOI] [PubMed] [Google Scholar]
- Friel, D.D., and R.W. Tsien. 1992. A caffeine- and ryanodine-sensitive Ca2+ store in bullfrog sympathetic neurones modulates effects of Ca2+ entry on [Ca2+]i. J. Physiol. 450:217–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme, J.T., and C.H. Orchard. 1998. Effect of acidosis on Ca2+ uptake and release by sarcoplasmic reticulum of intact rat ventricular myocytes. Am. J. Physiol. Heart. 44:H977–H987. [DOI] [PubMed] [Google Scholar]
- Kennedy, H.J., and R.C. Thomas. 1995. Intracellular calcium and its sodium independent regulation in voltage-clamped snail neurones. J. Physiol. 484:533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, H.J., and R.C. Thomas. 1996. Effects of injecting calcium-buffer solutions on [Ca2+]i in voltage-clamped snail neurons. Biophys. J. 70:2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver, D.R., K.R. Eager, L. Taoube, and G.D. Lamb. 2000. Effects of cytoplasmic and luminal pH on Ca2+ release channels from rabbit skeletal muscle. Biophys. J. 78:1835–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neering, I.R., and R.N. McBurney. 1984. Role for microsomal Ca storage in mammalian neurones? Nature. 309:158–160. [DOI] [PubMed] [Google Scholar]
- Meissner, G. 1994. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu. Rev. Physiol. 56:485–508. [DOI] [PubMed] [Google Scholar]
- Meissner, G., and J.S. Henderson. 1987. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J. Biol. Chem. 262:3065–3073. [PubMed] [Google Scholar]
- Orkand, R.K., and R.C. Thomas. 1995. Effects of low doses of caffeine on [Ca2+]i in voltage-clamped snail (Helix aspersa) neurones. J. Physiol. 489:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang, Y.B., P. Mellergard, T. Kristian, V. Kristianova, and B.K. Siesjo. 1994. Influence of acid-base changes on the intracellular calcium concentration of neurons in primary culture. Exp. Brain Res. 101:265–271. [DOI] [PubMed] [Google Scholar]
- Rousseau, E., and J. Pinkos. 1990. pH modulates conducting and gating behaviour of single calcium release channels. Pflugers Arch. 415:645–647. [DOI] [PubMed] [Google Scholar]
- Schwiening, C.J., and R.C. Thomas. 1996. Relationship between intracellular calcium and its muffling measured by calcium iontophoresis in snail neurones. J. Physiol. 491:621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening, C.J., H.J. Kennedy, and R.C. Thomas. 1993. Calcium-hydrogen exchange by the plasma membrane Ca-ATPase of voltage-clamped snail neurones. Proc. R. Soc. Lond. B Biol. Sci. 253:285–289. [DOI] [PubMed] [Google Scholar]
- Seidler, N.W., I. Jona, M. Vegh, and A. Martonosi. 1989. Cyclopiazonic acid is a specif inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 264:17816–17823. [PubMed] [Google Scholar]
- Stienen, G.J.M., Z. Papp, and R. Zaremba. 1999. Influence of inorganic phosphate and pH on sarcoplasmic reticular ATPase in skinned muscle fibres of Xenopus laevis. J. Physiol. 518:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, R.C. 1972. Intracellular sodium activity and the sodium pump in snail neurones. J. Physiol. 220:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, R.C. 1994. Current construction protocol for calcium-sensitive intracellular microelectrodes. J. Physiol. 476:9P. [Google Scholar]
- Thomas, R.C. 2001. Electrophysiological measurements using Ca2+-sensitive microelectrodes. : Measuring Calcium and Calmodulin Inside and Outside Cells. O.H. Petersen, editor. Springer, Berlin. 91–102.
- Thomas, R.C. 2002. The effect of HCl injection-induced acidosis on caffeine-induced calcium release in snail neurones. J. Physiol. 539P:96P–97P. [Google Scholar]
- Thomas, R.C., and R.W. Meech. 1982. Hydrogen ion currents and intracellular pH in depolarised voltage-clamped snail neurones. Nature. 299:826–828. [DOI] [PubMed] [Google Scholar]
- Tombaugh, G.C. 1998. Intracellular pH buffering shapes activity-dependent Ca2+ dynamics in dendrites of CA1 interneurons. J. Neurophysiol. 80:1702–1712. [DOI] [PubMed] [Google Scholar]
- Tombaugh, G.C., and G.C. Somjen. 1998. pH modulation of voltage-gated ion channels. pH and Brain Function. K. Kaila and B.R. Ransom, editors. Wiley-Liss, New York. 395–416.
- Trapp, S., M. Luckermann, K. Kaila, and K. Ballanyi. 1996. Acidosis of hippocampal-neurons mediated by a plasmalemmal Ca2+/H+ pump. Neuroreport. 7:2000–2004. [DOI] [PubMed] [Google Scholar]
- Verkhratsky, A., and O.H. Petersen. 2002. The endoplasmic reticulum as an integrating signalling organelle: from neuronal signalling to neuronal death. Eur. J. Pharmacol. 447:141–154. [DOI] [PubMed] [Google Scholar]
- Willoughby, D., R.C. Thomas, and C.J. Schwiening. 1999. A role for Na/H exchange in pH regulation in Helix neurones. Pflugers Arch. 438:741–749. [DOI] [PubMed] [Google Scholar]
- Willoughby, D., R.C. Thomas, and C.J. Schwiening. 2001. The effects of intracellular pH changes on resting cytosolic calcium in voltage-clamped snail neurones. J. Physiol. 530:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]