

Abstract
By defining functional defects in a congenital myasthenic syndrome (CMS), we show that two mutant residues, located in a binding site region of the acetylcholine receptor (AChR) epsilon subunit, exert opposite effects on ACh binding and suppress channel gating. Single channel kinetic analysis reveals that the first mutation, ɛN182Y, increases ACh affinity for receptors in the resting closed state, which promotes sequential occupancy of the binding sites and discloses rate constants for ACh occupancy of the nonmutant αδ site. Studies of the analogous mutation in the δ subunit, δN187Y, disclose rate constants for ACh occupancy of the nonmutant αɛ site. The second CMS mutation, ɛD175N, reduces ACh affinity for receptors in the resting closed state; occupancy of the mutant site still promotes gating because a large difference in affinity is maintained between closed and open states. ɛD175N impairs overall gating, however, through an effect independent of ACh occupancy. When mapped on a structural model of the AChR binding site, ɛN182Y localizes to the interface with the α subunit, and ɛD175 to the entrance of the ACh binding cavity. Both ɛN182Y and ɛD175 show state specificity in affecting closed relative to desensitized state affinities, suggesting that the protein chain harboring ɛN182 and ɛD175 rearranges in the course of receptor desensitization. The overall results show that key residues at the ACh binding site differentially stabilize the agonist bound to closed, open and desensitized states, and provide a set point for gating of the channel.
Keywords: congenital myasthenic syndrome, single channel kinetics, agonist binding, channel gating, mutation analysis
INTRODUCTION
Nicotinic acetylcholine receptors (AChRs)* and their relatives mediate synaptic transmission in the peripheral and central nervous systems. They are pentamers in which each subunit contributes to a large extracellular domain as well as to an assembly of transmembrane domains. AChRs belong to the ionotropic class of receptors and function as allosteric proteins that bind agonist and transduce binding free energy into opening and closing of the receptor ion channel (Changeux, 1990). Over the past decade studies of site-directed as well as naturally occurring mutations have related key residues in the AChR to their elementary functions.
The initial step in receptor function, ACh binding, is accomplished by specialized residues at interfaces between α-epsilon and α-delta subunits of the adult muscle AChR (Prince and Sine, 1998). Located in the large extracellular domain, the interface residues provide receptors in the resting state with sufficient affinity to bind synaptically released ACh, and endow the active receptor with increased affinity necessary to promote high activity (Jackson, 1989). Our current picture of the ACh binding site views these key residues purely as sensors for ACh, promoting channel gating through the large difference in affinity between resting and active states. However, because the binding site also serves as a trigger for opening the channel, key residues at the binding site may stabilize resting relative to active states independently of ACh occupancy. Previous work showed that mutations of residues at the binding site affect both agonist binding and channel gating steps (Chen et al., 1995, Ohno et al., 1996; Akk et al., 1999; Akk, 2001), but effects on gating independent of agonist binding have not been documented.
Congenital myasthenic syndromes (CMSs) are presynaptic, synaptic, or postsynaptic in origin (Engel et al., 1998). The postsynaptic CMS reported to date are caused by mutations in subunits of the AChR that reduce expression or alter the kinetic properties of the receptor. Two major types of kinetic mutations and associated diseases have emerged: slow-channel syndrome mutations that enhance agonist affinity or gating efficiency, and fast-channel syndrome mutations that reduce agonist affinity for the open channel or impair gating. Investigations of the molecular basis of these syndromes have uncovered novel structures key to AChR function (Engel et al., 1998).
The present study arose from analysis of a fast-channel CMS defined by clinical, electrophysiological and electron microscopic criteria, which prompted us to search for mutations in AChR subunits and delineate their mechanistic consequences. We trace the cause of the CMS to two heteroallelic mutations in a region of the epsilon subunit that contributes to the ACh binding site, and show that the CMS results from an attenuated postsynaptic response. Kinetic analysis of engineered mutant receptors reveals that the two mutations exert opposing effects on ACh binding, but each mutation impairs gating of the ion channel. The overall results suggest that key residues in the ACh binding site contribute locally to stabilize bound ACh and globally to stabilize the closed relative to the open state of the channel.
MATERIALS AND METHODS
Endplate Studies
Intercostal muscle specimens were obtained intact from origin to insertion from the patient and from control subjects without muscle disease undergoing thoracic surgery. All human studies were in accord with the guidelines of the Institutional Review Board of the Mayo Clinic. Endplates (EPs) were localized for electron microscopy by established methods (Engel, 1994). α-Bungarotoxin (α-Bgt) labeled with peroxidase was used for the ultrastructural localization of AChR (Engel et al., 1977). The number of AChRs per EP was measured with 125I-labeled α-bgt, as described previously (Engel et al., 1993).
Miniature EP potential (MEPP), miniature EP current (MEPC), and EP potential recordings, and estimates of the number of transmitter quanta released by nerve impulse were performed as described previously (Engel et al., 1993; Uchitel et al., 1993). Patch-clamp recordings from EP AChRs were performed in cell-attached mode by a slight modification of the previously described method (Milone et al., 1994). For all patches, the membrane potential was set to −80 mV. Whenever possible, recordings were also obtained at −40, −120, and −160 mV. These membrane potentials were achieved by applying a corresponding potential to the patch pipette and assuming a resting potential of 0 mV. A null resting potential was confirmed by the absence of detectable channel events with 0 mV applied to the pipette. Channel currents were recorded using an Axopatch 200A amplifier (Axon Instruments, Inc.) and analyzed at a final bandwidth of 8 kHz (patient EPs) and 12 kHz (control EPs) using the programs PClamp 6 (Axon Instruments, Inc.). Burst durations were determined by grouping openings separated by a specified closed time that misclassifies equal proportions of long closed times within bursts and short intervals between bursts (Colquhoun and Sakmann, 1985). Dwell time histograms were plotted on a logarithmic abscissa and fitted to the sum of exponentials by maximum likelihood (Sigworth and Sine, 1987).
Mutation Analysis
We directly sequenced the AChR α, β, δ, and ɛ subunit genes using genomic DNA isolated from blood (Ohno et al., 1995). To investigate family members, we traced ɛD175N with allele-specific PCR and ɛN182Y by RsaI restriction analysis of PCR products. ɛN182Y results in gain of an RsaI site. To prove that the patient's two mutations are heteroallelic, we selectively amplified the alleles with and without ɛD175N using mutant and wild-type allele-specific PCR primers, respectively, and digested the products with RsaI that recognizes only the ɛN182Y allele. To exclude that ɛD175N and ɛN182Y represent polymorphisms, we screened for them in 200 normal alleles using allele-specific PCR. To check paternity, we determined the numbers of tri- or tetranucleotide repeats at D17S969, D17S918, and D17S2196 by direct sequencing. The paternity index was calculated using a Bayesian approach (Sozer et al., 2001).
Construction and Expression of Wild-Type and Mutant AChRs
Human AChR α, β, δ, and ɛ subunit cDNA were cloned into the CMV-based expression vector pRBG4 as described elsewhere (Ohno et al., 1996). ɛD175N and ɛN182Y were introduced into the ɛ subunit cDNA in pRBG4 by the QuickChange site-directed mutagenesis kit (Stratagene). The presence of the desired mutations and absence of unwanted artifacts were confirmed by sequencing the entire insert. HEK 293 cells were transfected with AChR subunit cDNAs using calcium phosphate precipitation (Sine et al., 1995; Ohno et al., 1996). In some experiments the BOSC cell line was used, which is a variant of the 293 HEK cell line (Wang et al., 2000).
Patch-clamp Recordings from AChRs Expressed in HEK Cells
Recordings were obtained in the cell-attached configuration at a membrane potential of −70 mV and at 22°C. The bath and pipette solutions contained (mM): KCl 142, NaCl 5.4, CaCl2 1.8, MgCl2 1.7, HEPES 10, pH 7.4. Single channel currents were recorded using an Axopatch 200A amplifier at a bandwidth of 50 kHz, digitized at 5-μs intervals using an ITC-16 AD interface (Instrutech), recorded to hard disk using the program Acquire, and detected by the half-amplitude threshold criterion using the program Tac (Bruxton Corporation) at a final bandwidth of 10 kHz (Wang et al., 1999). To estimate rate constants in Scheme I (see text and Table III) , clusters of events corresponding to a single channel were identified as a series of closely spaced openings preceded and followed by closed intervals greater than a specified critical time; this time was taken as the point of intersection of the predominant closed time component and the succeeding closed time component. Each recording was examined for homogeneity by determining the mean open probability and open duration for each cluster, and accepting clusters with parameters within two standard deviations of the means (Ohno et al., 1996; Wang et al., 1997). These selections included >90% of the total clusters. The resulting table of open and closed intervals were transferred to a Dell Precision 610 work station, and analyzed according to Scheme I using the program MIL (Qin et al., 1996), which uses an interval-based maximum likelihood method that corrects for missed events. A uniform filter bandwidth of 10 kHz and a dead time of 23 μs were imposed for all recordings. For each type of receptor, single channel dwell times, obtained at several concentrations of ACh, were fitted simultaneously. For wild-type AChR, data were obtained at 3, 5, 10, 30, 50, 100, and 300 μM ACh. For ɛN182Y, ɛN182F, and ɛN182D data were obtained at 1, 3, 10, 30, 100, and 300 μM ACh. For ɛD175N data were obtained at 10, 30, 100, 300, and 1,000 μM ACh. An average of ∼6,000 dwell times were analyzed for each ACh concentration, with the range from 2,300 to 17,000. After fitting, error estimates were determined for each parameter (Qin et al., 1996). Theoretical open and closed duration histograms were calculated from the fitted rate constants and superimposed on the experimental dwell time histograms. To check the final set of fitted rate constants, we used the method of Clay and DeFelice (1983) to simulate open and closed intervals according to Scheme I modified to include short, intermediate, and long desensitized states to mimic real data. The desensitized states were connected to the doubly liganded closed state with forward rate constants of 200 s−1 and backward rate constants of 200, 20, and 2 s−1 to approximate the three desensitized states present in the data. After defining the critical closed time as described above, clusters were identified, events briefer than the experimental dead time were removed, and the resulting dwell times were fitted with the program MIL. In all cases the original rate constants were recovered to within 10% of the original rate constants.
TABLE III .
Fits of Scheme I to Dwell Times from Wild-Type and Mutant AChRs
| Receptor | k+1 | k−1 | K1 | k+2 | k−2 | K2 | β1 | α1 | θ1 | β2 | α2 | θ2 | k+b | k−b | KB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
μM |
μM |
μM |
μM |
μM |
μM |
|
|
|
|
|
|
mM |
mM |
mM |
| Wild-type | 197 ± 14 | 5,180 ± 450 | 26 | 120 ± 4 | 16,600 ± 290 | 138 | 99 ± 10 | 4,070 ± 470 | 0.024 | 47,000 ± 1,200 | 1,800 ± 45 | 26 | 36 ± 5 | 139,000 ± 6,000 | 3.9 |
| Wild-type constraint | 200 | 7,400 | 37 | 150 ± 3 | 16,600 ± 260 | 111 | 155 ± 9 | 3,750 ± 360 | 0.041 | 43,700 ± 1,060 | 1,630 ± 36 | 27 | 28 ± 4 | 128,000 ± 7,200 | 4.6 |
| εN182Y | 267 ± 17 | 529 ± 40 | 2.0 | 202 ± 3 | 8,080 ± 110 | 40 | 23 ± 2 | 9,380 ± 670 | 0.0025 | 8,340 ± 100 | 3,110 ± 17 | 2.7 | 17 ± 5.8 | 130,000 ± 1,400 | 7.6 |
| εN182F | 144 ± 12 | 443 ± 50 | 3.0 | 143 ± 4 | 7,130 ± 140 | 50 | 53 ± 4 | 7,880 ± 520 | 0.0067 | 6,860 ± 100 | 3,350 ± 23 | 2.1 | 18 ± 9 | 123,000 ± 2,600 | 6.8 |
| εN182D | 187 ± 6 | 1,400 ± 70 | 7.5 | 263 ± 7 | 7,250 ± 110 | 28 | 124 ± 9 | 1,370 ± 110 | 0.091 | 46,600 ± 660 | 1,180 ± 19 | 40 | 48 ± 7 | 167,000 ± 5,900 | 3.5 |
| δN187Y | 251 ± 16 | 3,100 ± 240 | 12 | 174 ± 4 | 15,260 ± 240 | 88 | 44 ± 3 | 5,760 ± 410 | 0.0076 | 15,240 ± 240 | 2,590 ± 20 | 5.9 | 11 ± 1 | 81,500 ± 5,600 | 7.4 |
| εD175N | 50.7 ± 8 | 5,920 ± 1,100 | 117 | 35 ± 4 | 27,200 ± 1,700 | 777 | 90 ± 10 | 7,400 ± 300 | 0.012 | 2,930 ± 160 | 2,170 ± 31 | 1.4 | 22 ± 1.3 | 99,400 ± 2,500 | 4.5 |
| εD175N constraint | 200 | 7,400 | 37 | 16 ± 1 | 31,700 ± 1500 | 1,980 | 30 ± 1 | 9,600 ± 420 | 0.0031 | 5,010 ± 290 | 2,390 ± 26 | 2.1 | 21 ± 1 | 98,600 ± 2,600 | 4.7 |
Receptor indicates the mutant subunit expressed with complementary wild-type subunits. Rate constants are as defined in Scheme I, in units of μM−1 s−1 for association rate constants, and s−1 for all others. Values are results of global fits to data obtained over a range of ACh concentrations, with standard errors indicated by ± (see materials and methods). The channel gating equilibrium constants, θ, are ratios of the corresponding opening to closing rate constants, β/α. Constraint indicates k+1 and k−1 were constrained to the average of values obtained for the low affinity site in ɛN182Y, ɛN182F, and ɛN182D receptors.
α-Bgt and ACh Binding Measurements
The total number of 125I α-bgt sites on the cell surface of transfected HEK cells and ACh competition against the initial rate of 125I α-bgt were determined as previously described (Ohno et al., 1996). ACh competition measurements were analyzed according to either the Hill equation (Eq. 1) or the two binding site equation (Eq. 2):
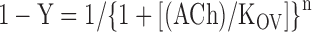 |
(1) |
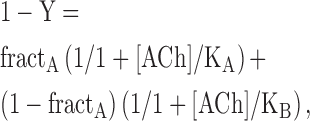 |
(2) |
where Y is fractional occupancy by ACh, n is the Hill coefficient, KOV the overall dissociation constant, KA the dissociation constant for one site, and KB the dissociation constant for the second site.
RESULTS
Characteristics of the CMS Patient
A 5-yr-old male had decreased movements in utero, eyelid ptosis since early infancy, and delayed motor development. He fatigued rapidly on mild exertion and could not keep up with his peers in physical activities. He had a decremental electromyographic response on stimulation of motor nerves, negative tests for anti-AChR antibodies, and responded partially to cholinesterase inhibitors. He has a high-arched palate, mild scoliosis, slight limitation of the ocular ductions, and fluctuating eyelid ptosis. He can extend his arm horizontally for only 30 s. The parents and a younger sibling are unaffected.
Endplate Studies
The configuration of the EPs, evaluated from the cytochemical reaction for acetylcholinesterase on 40 teased single muscle fibers, was normal on 33 fibers and consisted of 2–3 closely spaced small EP regions on 7 fibers. The number of 125I-α-bgt sites per EP was normal for the patient's age (Table I) . The reaction for AChR at the EPs, detected in cryostat sections with rhodamine-labeled α-bgt, was normal. Electron microscope studies of 45 EP regions at 20 EPs revealed that the structural integrity of the pre and postsynaptic regions was preserved, but 5 EP regions had few or no secondary clefts and junctional folds. The reaction for AChR on the junctional folds, detected with peroxidase-labeled α-bgt, was normal.
TABLE I .
Endplate Studies
| Patient | Controls | |
|---|---|---|
| 125I-α-bgt binding sites/EP | 5.5 E6 | 12.82 ± 0.79 E6 (13 adults) 4.7 E6 (3-yr-old control) |
| EPP quantal content (1 Hz)a | 34 ± 4 (11) | 31 ± 1 (190) |
| MEPP amplitude (mV)b | 0.32 ± 0.069 (15) | 1.00 ± 0.025 (164) |
| MEPC amplitude (nA) | 1.60 ± 0.086 (14) | 3.95 ± 0.10 (79) |
| τMEPC | 0.85 ± 0.057 (14) | 3.23 ± 0.06 |
Values represent mean ± SE. Numbers in parentheses indicate number of endplates, except for α-bgt binding sites where they indicate number of adult controls. T = 29 ± 0.5°C for EPP and MEPP recordings, and 22 ± 0.5°C for MEPC studies.
Quantal content of EP potential (EPP) at 1 Hz stimulation corrected for resting membrane potential of −80 mV, nonlinear summation, and non-Poisson release.
Corrected for resting membrane potential of −80 mV and a mean muscle fiber diameter of 55 μm.
Quantal release by nerve impulse was normal. Although the density and distribution of AChR on the junctional folds appeared normal, the amplitude of the MEPPs was reduced to 32%, and that of the MEPCs to 40% of normal. The MEPCs decayed abnormally fast, so that τMEPC was only 26% of normal (Table I).
To confirm the kinetic defect of AChR predicted by the rapidly decaying MEPCs, we compared single channel currents recorded from control and patient EPs. In contrast to control EPs, only few channel openings could be recorded from patient EPs with ACh concentrations lower than 5 μM, suggesting a decreased probability of channel opening. At patient EPs, 96% of all channel openings had a normal conductance of 60 pS, but they showed abnormally brief open durations; by contrast, 4% (∼1% at three EPs and 16% at one EP) of channel openings had a conductance of ∼44 pS and four- to fivefold longer than normal open durations, typical of fetal receptors that harbor the γ instead of the ɛ subunit. Analysis of the 60 pS channel events at patient and control EPs resolved two components both in bursts of openings and single openings (τ1 and τ2), but in the patient the briefer component, τ1, was dominant, whereas at control EPs the longer component, τ2, was dominant (Table II and Fig. 1) . To summarize, EP studies revealed no AChR deficiency, rapidly decaying MEPCs, abnormally brief channel openings, and a small population of fetal AChRs.
TABLE II .
Open Intervals at Seven Control and Four Patient EPs
| Open intervals | Bursts | |||
|---|---|---|---|---|
| Controls | Patient | Controls | Patient | |
| τ1 (ms) | 0.07 ± 0.02 | 0.17 ± 0.021 | 0.09 ± 0.03 | 0.17 ± 0.025 |
| area | 0.14 ± 0.04 | 0.65 ± 0.19 | 0.13 ± 0.02 | 0.61 ± 0.061 |
| τ2 (ms) | 1.13 ± 0.10 | 0.51 ± 0.037 | 2.99 ± 0.26 | 0.59 ± 0.061 |
| area | 0.86 ± 0.04 | 0.34 ± 0.096 | 0.87 ± 0.02 | 0.39 ± 0.061 |
Mean ± SE. ACh = 1 μM for controls, 5 μM for patient. Bandwidth = 12 kHz for controls, 8 kHz for patient. Membrane potential = −80 mV, T = 22 ± 0.5°C.
Figure 1.

AChR channel currents recorded from control and patient EPs. Left column shows AChR channel currents elicited by 1 μM ACh from control EP and by 5 μM ACh elicited from patient EP, with openings as upward deflections. Channel opening events are briefer at patient than at control EP. Right column shows the corresponding burst duration histograms with dotted lines indicating burst components. Time constants, τn, and fractional areas, an, for each component of bursts: Control: τ1 = 0.04 ms, a1 = 0.09, τ2 = 2.38 ms; a2 = 0.91, total events, 1,315. Patient: τ1 = 0.185, a1 = 0.67, τ2 = 0.66 ms, a2 = 0.33; total events, 4,025. Bandwidth = 12 kHz for control, 8 kHz for patient; membrane potential = −80 mV, T = 22°C ± 0.5.
Mutation Analysis
To determine the basis of the observed kinetic abnormalities, we proceeded with mutation analysis. Direct sequencing of all exons and flanking regions of the AChR α, β, δ, and ɛ subunit genes revealed two heterozygous mutations in the extracellular domain of the AChR ɛ subunit: a G-to-A substitution at nucleotide 523 in ɛ exon 6, predicting an aspartate-to-asparagine substitution at codon 175 (ɛD175N), and an A-to-T substitution at nucleotide 544 in ɛ exon 7, predicting an asparagine-to-tyrosine substitution at codon 182 (ɛN182Y). Aspartate at ɛ codon 175 is conserved in the human α, δ, and γ subunits, and in ɛ subunits of all species. Asparagine at ɛ codon 182 is conserved in the human β, δ, and γ subunits, and in ɛ subunits of all species (Fig. 2 C). Neither ɛD175N nor ɛN182Y was found in 200 normal alleles. Analysis of family members showed that the asymptomatic mother and brother are heterozygous for ɛN182Y, but no other family member carries ɛD175N (Fig. 2 A). Combined allele-specific PCR and RsaI restriction analysis revealed that ɛD175N and ɛN182Y are heteroallelic in the patient (Fig. 2 B). Although the father does not carry ɛD175N, analysis of the D17S969, D17S918, and D17S2196 markers in the parents revealed 90.6% probability of paternity (unpublished data). Thus, ɛD175N either arose in the paternal germline or was a spontaneous mutation.
Figure 2.
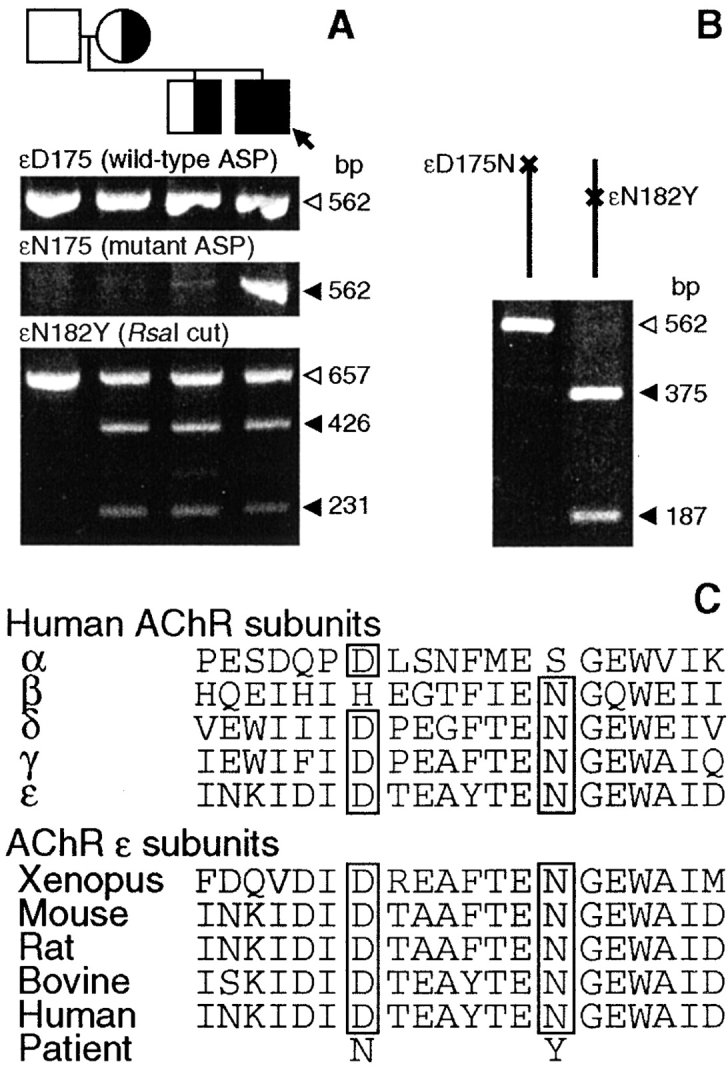
(A) Allele-specific PCR (ASP) for ɛD175N and RsaI restriction analysis for ɛN182Y of genomic DNA from patient (arrow) and family members. The patient carries ɛD175N and ɛN182Y; patient, mother, and brother harbor ɛN182Y. Open and closed arrowheads indicate wild-type and mutant fragments, respectively. (B) The ɛD175N and ɛN182Y mutations are heteroallelic. Allele-specific PCR selectively amplifies the ɛD175N allele (left), and the ɛD175N allele (right). PCR product amplified from each allele is digested with RsaI that only cuts the ɛN182Y allele. That only allele “b” is cut by RsaI indicates that ɛN182Y is on allele “b”. (C) Aspartate at ɛ codon 175 (D175) is conserved in human α, δ, and γ subunits, and in ɛ subunits of all species. Asparagine at ɛ codon 182 (N182) is conserved in human β, δ, and γ subunits, and in ɛ subunits of all species.
Activation of Wild-Type, ɛD175N, and ɛN182Y Receptors in Transfected Cells
To establish that ɛN182Y and ɛD175N cause the fast-channel CMS, we recorded single channel currents from 293 human embryonic kidney (293 HEK) cells transfected with wild-type α, β, and δ plus either wild-type or mutant ɛ subunit cDNAs. For each wild-type or mutant receptor, we applied a range of high concentrations of ACh (1–1000 μM) to elicit clustering of the currents into epochs during which only one channel is active. Recording individual clusters of channel activity allows subsequent removal of intercluster closed periods due to desensitization, allowing analysis of receptor activation independent of desensitization (Sine et al., 1990). Inspection of current pulses within clusters reveals briefer openings and prolonged closings for both ɛN182Y and ɛD175N receptors compared with wild-type receptors (Fig. 3) . The briefer openings of the two mutant receptors mirror the briefer single channel currents and faster decays of MEPCs recorded at patient EPs. Thus, ɛN182Yand ɛD175N together cause the fast-channel CMS.
Figure 3.

Kinetics of activation of wild-type and mutant receptors. Left column displays individual clusters of single channel currents recorded at the indicated ACh concentrations from cells expressing adult human AChRs containing the indicated mutant ɛ subunits. Currents are displayed at a bandwidth of 10 kHz, with channel openings shown as upward deflections. Center and right columns display the corresponding closed and open duration histograms with the fits for Scheme I superimposed. Fitted rate constants are given in Table III.
Elementary Kinetic Steps Altered by ɛD175N and ɛN182Y
To uncover the mechanistic consequences of ɛN182Y and ɛD175N, we constructed dwell time histograms from open and closed intervals within individual clusters of single channel activity (see materials and methods). Histograms of open dwell times show a major peak, or exponential component at all ACh concentrations (Fig. 3). A second brief component, visible as a rise in the distribution to the left of the major component, is present at low but not high ACh concentrations. The second component is readily discerned in the data for wild-type and ɛD175N receptors selected for display. However, because the ɛN182Y mutation increases agonist affinity, as described below, the second component is present only at ACh concentrations <10 μM (unpublished data). Histograms of closed dwell times show two predominant components, the longer of which moves to briefer time as the ACh concentration increases. These concentration dependencies of open and closed dwell times are observed for wild-type and each mutant AChR, and are characteristic of an activation process driven by ACh concentration.
To identify elementary kinetic steps altered by ɛN182Y and ɛD175N, we analyzed the closed and open dwell times according to the following standard description of receptor activation,
Figure.

where A is the agonist, R is the closed receptor, R* is the open receptor, k+1 and k+2 are association rate constants, and k−1 and k−2 are dissociation rate constants. Singly occupied receptors AR open with rate constant β1 and close with rate constant α1, whereas doubly occupied receptors A2R open with rate constant β2 and close with rate constant α2. A blocked state A2RB is included to account for brief channel closures elicited by high ACh concentrations.
We estimated rate constants for each state transition in Scheme I by computing the likelihood of the experimental series of dwell times, given a set of trial rate constants, and then changing the rate constants to maximize the likelihood (see materials and methods). For wild-type and each mutant receptor, we fitted Scheme I simultaneously to open and closed dwell times obtained for the entire range of ACh concentrations. Use of a range of ACh concentrations optimizes the contribution of dwells in each receptor state to the cumulative likelihood. The fitted rate constants yield probability density functions that superimpose upon the open and closed duration histograms (Fig. 3), showing that Scheme I describes activation of wild-type, ɛN182Y, and ɛD175N receptors.
Functional Consequences of the ɛN182Y Mutation
The fitted rate constants show that ɛN182Y markedly increases ACh affinity for one of the two binding sites in the resting closed state of the receptor, but does not affect ACh affinity for the second binding site (Table III). The dissociation constant for the altered site falls from 138 μM for the wild-type receptor to 2 μM for the mutant, owing to a 30-fold slowing of the rate constant for ACh dissociation and a twofold increase of the rate constant for ACh association. Because the mutation is present in the ɛ subunit, the binding site with increased ACh affinity corresponds to the αɛ site. The increased affinity conferred by ɛN182Y defines the sequence in which ACh occupies the binding sites of the mutant receptor in the course of opening the channel; the αɛ site is occupied first, whereas the αδ site is occupied second.
The ɛN182Y mutation not only alters ACh binding, but it also alters gating of the channel. The gating equilibrium constants for singly occupied (θ1) and doubly occupied (θ2) receptors are reduced by an order of magnitude. Reduction of θ2 likely follows directly from the reduction in θ1 because ACh occupancy of the second binding site begins with a singly occupied receptor less prone to opening. Thus, although the second occupancy step in the mutant receptor promotes channel gating as well as the second occupancy step in wild-type (θ2/θ1 = 1,080 for the mutant and θ2/θ1 = 1,083 for wild-type; see Table III), gating of the doubly occupied mutant receptor is reduced by an order of magnitude.
Mechanistic underpinnings for the reduced θ1 can be understood by inspection of the following expanded form of Scheme I (Monod et al., 1965):
Figure.

where K1* and K2* are dissociation constants for ACh binding to the open states and θ0 is the gating equilibrium constant in the absence of agonist. According to Scheme II, θ1 could decrease due to (a) a decrease of K1 relative to K1* or (b) a decrease of θ0 without a change in K1 relative to K1*. Because neither K1* nor θ0 are defined by the data, both alternatives are formal possibilities.
Comparison of ACh binding steps for wild-type and ɛN182Y mutant receptors shows that the second binding site in the ɛN182Y receptor is similar to the first binding site in the wild-type receptor; both the association and dissociation rate constants are similar between the two types of receptors (Table III). A common unaltered site is also observed for the site-directed mutations ɛN182F and ɛN182D described below. The most likely explanation for a common site is ɛN182Y affects the αɛ binding site without altering the αδ binding site. Thus, mutations at ɛN182 not only reveal a new determinant of ACh binding affinity, but also provide independent estimates of the rate constants underlying ACh occupancy of the wild-type αδ site.
Structural Basis of the Effects of the ɛN182Y Mutation
To determine the structural basis for the effect of ɛN182Y on receptor activation, we constructed mutations with electron-rich side chains, ɛN182F and ɛN182D, and examined single channel kinetics of the resulting mutant receptors (Fig. 4) . Mutation to phenylalanine mimics the effects of the original tyrosine mutation, increasing ACh affinity for one of the two binding sites in the resting closed state and impairing gating of singly and doubly occupied receptors (Table III). Mutation to aspartic acid increases ACh affinity of the mutant binding site similar to the original tyrosine mutation, but enhances rather than impairs channel gating (Table III). For receptors containing either ɛN182F or ɛN182D, rate constants underlying ACh binding to the nonmutant α–δ site are similar to those obtained for the nonmutant site in the ɛN182Y receptor, and to one of the two sites in wild-type AChR. Thus, as observed for ɛN182Y, both ɛN182F and ɛN182D promote sequential occupancy by ACh because they decrease K1 relative to K2 (Table III).
Figure 4.
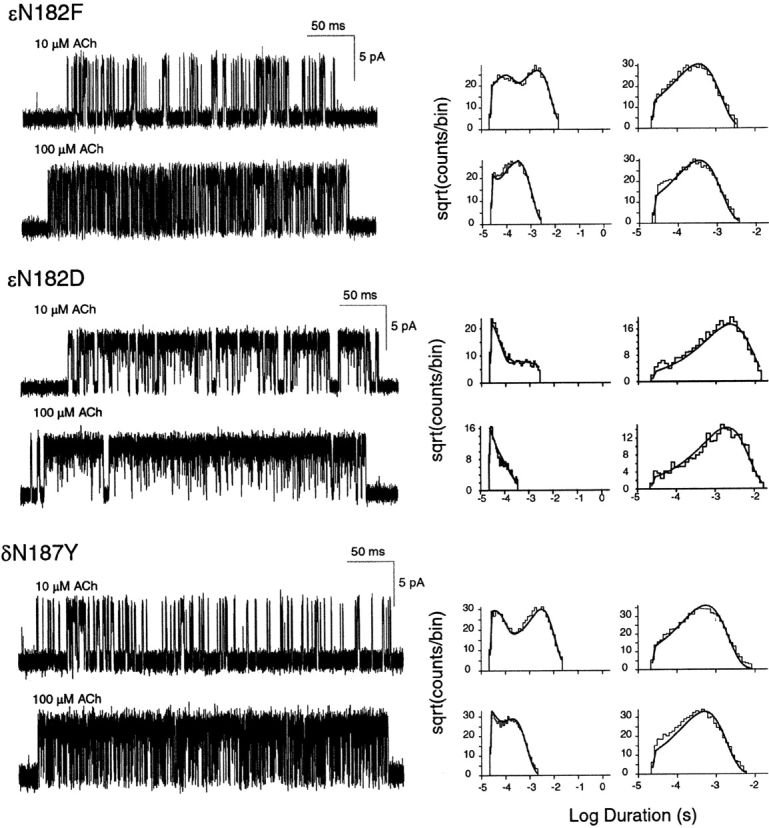
Kinetics of activation of receptors with site-directed mutations. Left column displays individual clusters of single channel currents recorded at the indicated ACh concentrations from cells expressing adult human AChRs containing the indicated mutant subunits, as in Fig. 3.
Unlike the tyrosine and phenylalanine mutations, the aspartic acid mutation increases rather than decreases channel opening equilibrium constants for singly and doubly occupied receptors (Table III). The increased θ1 may owe to enhanced binding of ACh to the open relative to the closed state of the mutant α–ɛ site, or to an effect of ɛN182D on gating of the channel in the absence of ACh (see Scheme II above). However, the increase of θ2 is likely a direct consequence of the increase of θ1 because ACh occupancy of the second binding site begins with a singly occupied receptor more prone to opening. The overall results of side chain substitutions indicate that the consequences of the original mutation ɛN182Y owe to substitution of an aromatic ring, and that electron density at this position affects ACh affinity for the resting closed state of the receptor. Altogether, analyses of the three mutations of ɛN182 provide independent estimates of rate constants underlying ACh binding to the nonmutant α–δ site.
Studies of the Analogous δN187Y Mutation
We constructed the analogous mutation in the δ subunit, δN187Y, to determine whether it affects ACh binding to one of the two binding sites, as observed for ɛN182Y, and to independently estimate rate constants underlying ACh occupancy of the nonmutant αɛ site. Single channel currents and the corresponding fitted rate constants show that the δN187Y mutation enhances ACh affinity for one binding site in the resting closed state, but does not affect ACh affinity for the second site (Fig. 4; Table III). The rate constant for ACh association accelerates 1.3-fold, and the rate constant for ACh dissociation slows 1.7-fold; the resulting dissociation constant for the altered site falls from 26 μM for wild-type AChR to 12 μM for the mutant. Because the mutation is present in the δ subunit, the binding site with increased agonist affinity corresponds to the αδ site. Moreover, rate constants underlying ACh occupancy of the nonmutant αɛ site are very similar to those of the low affinity binding site in the wild-type receptor (Table III). Thus, the ɛN182Y and δN187Y mutations locally affect ACh affinity of the corresponding αɛ or αδ sites, providing independent estimates of rate constants underlying ACh binding to nonmutant αδ and αɛ sites. In the wild-type human AChR, the αδ subunit interface forms the binding site with high affinity for ACh, whereas αɛ interface forms the site with low affinity.
Functional Consequences of ɛD175N
Single channel kinetic analysis of currents through receptors containing ɛD175N suggests that ACh binding is impaired at both binding sites (Table III). However, because the mutation is present in the ɛ subunit, we tested the idea that ɛD175N affects the affinity of only one binding site. A mutational effect restricted to the αɛ site is likely because four mutations, ɛN182Y, ɛN182F, ɛN182D, and δN187Y, had local effects at the corresponding α–ɛ or α–δ sites. Furthermore, the protein chain harboring ɛN182 and ɛD175 is not part of a secondary structural element and lodges in a crevice on the periphery of the subunit (Sine et al., 2002). Thus, we constrained rate constants for one binding site to the average of those obtained for the nonmutant α–δ site from analyses of the three mutations at ɛN182, and allowed rate constants for the other binding site to vary. The resulting fits provided well-defined rate constants for both wild-type and ɛD175N receptors (Table III); probability density functions computed from the fitted rate constants superimpose upon the dwell time histograms for both types of receptors (Fig. 3), and were indistinguishable from those obtained from the unconstrained fitting (not shown). Moreover, the resulting fitted parameters reveal that ɛD175N slows the rate constant for ACh association ∼10-fold, and accelerates the rate constant for ACh dissociation twofold; the mutation increases the dissociation constant for ACh binding to the αɛ site from 111 to 1,980 μM (Table III).
The fitting analysis also reveals that ɛD175N reduces gating of singly and doubly occupied receptors by about an order of magnitude (Table III). The gating equilibrium constant θ1 is particularly well discerned in the data where a predominant brief component of openings, present at low ACh concentrations, is replaced by a longer-lived component at high concentrations (Fig. 3). The decrease of θ2 is likely a direct consequence of the decrease of θ1 because binding of the second ACh starts with a singly occupied receptor less prone to opening. Thus, although ACh occupancy of the mutant binding site promotes channel gating nearly as well as occupancy of the second binding site in wild-type (θ2/θ1 = 677 for ɛD175N and θ2/θ1 = 1,083 for wild-type), gating of the doubly occupied mutant receptor is reduced by an order of magnitude.
The possibility that ɛD175N preferentially affects ACh affinity for the open relative to the closed state of the receptor was assessed by applying detailed balancing to the following cycle taken from Scheme II:
Figure.

Here K2* is the dissociation constant for agonist binding to the open state of the receptor, which equals K2θ1/θ2. For the wild-type receptor, K2* calculated from the present data is 168 nM, whereas K2* for the ɛD175N mutant is 2,920 nM. Thus, ɛD175N increases K2* by 17-fold, which is close to its 18-fold increase of K2, indicating that the mutation does not preferentially affect ACh affinity for the open relative to the closed state of the receptor.
If the ɛD175N mutation maintains a large K2/K2* ratio, providing a normal increase of θ2 relative to θ1, how does ɛD175N impair overall gating of the channel? The earliest discernable effect in Scheme I is reduction of θ1, but this observation leads to the following paradox: how does a mutation at the αɛ site affect the gating equilibrium constant associated with ACh occupancy of the nonmutant αδ site? A likely explanation is that residues at the binding site not only mediate recognition of ACh, but also set the trigger point for opening the channel. Thus, the results suggest that residues at the binding site may contribute to stability of closed relative to open states of the channel even when agonist is not bound.
Mutational Effects on Channel Open Probability
To illustrate the overall consequences of ɛD175N, ɛN182Y, and ɛN182D, and to confirm our estimated rate constants, we determined the fraction of receptors activated as a function of ACh concentration, and compared it with the dose–response relationship calculated from the fitted rate constants in Table III. Our measure of the fraction of receptors activated is the mean open probability (Popen) within identified clusters due to single AChRs (see traces in Fig. 3). The ɛN182Y mutation reduces the maximum channel open probability, but does not significantly shift the curve along the concentration axis (Fig. 5) . By contrast the engineered mutation, ɛN182D, shifts the curve to the left and increases the maximum channel open probability due to enhanced gating and increased affinity of one of the two binding sites. The ɛD175N mutation shifts the dose–response curve far to the right and decreases the maximum channel open probability due to impaired gating and reduced affinity of one of the two binding sites. The kinetically derived dose–response curves superimpose upon the Popen measurements, thus supporting the rate constant estimates.
Figure 5.

Dependence of channel open probability (Popen) on ACh concentration for receptors containing the indicated mutant ɛ subunits plus complementary α, β, and δ subunits. The mean fraction of time the channel was open during a cluster (Popen) was determined at the indicated concentrations of ACh. The theoretical Popen was calculated from Scheme I using the fitted rate constants in Table III, and superimposed on the Popen measurements. Error bars indicate standard deviations of Popen for the population of clusters at the specified ACh concentration.
Effects of ɛD175N and ɛN182Y on ACh Binding to Desensitized AChRs
Our single channel kinetic analysis reveals that ɛN182Y enhances, whereas ɛD175N reduces, ACh affinity for receptors in the resting closed state. To determine whether these mutations affect ACh affinity for receptors in the desensitized state, we converted receptors to the desensitized state using the local anesthetic proadifen (Sine and Taylor, 1982), and then measured binding of ACh (Ohno et al., 1996). We incubated intact 293 HEK cells expressing recombinant receptors with specified concentrations of ACh, with or without proadifen, and then measured binding of ACh by competition against the initial rate of 125I-α-bgt binding. For the wild-type receptor, the presence of proadifen increases ACh affinity ∼100-fold due to high affinity of the desensitized state (Fig. 6) . Binding of ACh to the desensitized wild-type receptor is described by a two-site model in which the fitted dissociation constants differ by three- to fivefold, indicating slightly different ACh affinities of the two binding sites in the desensitized state.
Figure 6.

ACh binding to receptors in the presence of the desensitizing agent proadifen. (A) Comparison of ACh binding to wild-type and ɛN182Y receptors. (B) Comparison of ACh binding to wild-type and ɛD175N receptors. Binding of ACh was determined by competition against the initial rate of 125I-α-bgt binding. Intact HEK cells expressing the indicated receptor type were incubated in the presence of ACh, with or without 100 μM proadifen (pro), 30 min before measuring the initial rate of α-bgt binding. Smooth curves are fits to the Hill equation (measurements in the absence of proadifen) or to an equation describing the sum of two binding sites (measurements in the presence of proadifen). Fitted parameters (A): wild-type, Kov = 7.9 × 10−7, n = 1.4; wild-type plus proadifen, KA = 3.3 × 10−8, KB = 2.1 × 10−9; ɛN182Y, Kov = 1.4 × 10−7, n = 0.9; ɛN182Y plus proadifen, KA = 2.3 × 10−8, KB = 1.2 × 10−9. Fitted parameters (B): wild-type, Kov = 1.5 × 10−6, n = 1.1; ɛD175N, Kov = 1.2 × 10−5, n = 1.1; wild-type plus proadifen, KA = 2.9 × 10−8, KB = 8.9 × 10−9; ɛD175N plus proadifen, KA = 2.7 × 10−8, KB = 1.7 × 10−6.
For receptors containing the ɛN182Y mutation, proadifen also increases ACh affinity, but the competition curve closely approaches that of the wild-type receptor in the presence of proadifen (Fig. 6 A); the desensitized state affinity for the ɛN182Y receptor is only 1.6-fold greater than that of the wild-type receptor. Thus, although ɛN182Y markedly increases agonist affinity for receptors in the resting closed state, it only slightly increases agonist affinity for receptors in the desensitized state.
For receptors containing the ɛD175N mutation, proadifen again increases ACh affinity, but the binding profile shows two clear components (Fig. 6 B). The dissociation constant for the high affinity component of the ɛD175N receptor (27 nM) coincides with that for the low affinity component of the wild-type receptor (range: 29–33 nM; see Fig. 6, legend), indicating a common unaltered site in the desensitized state, which corresponds to the αδ site. On the other hand, the low affinity component (1,700 nM) for the ɛD175N receptor is shifted some 800-fold to the right of the high affinity component for the wild-type receptor (range: 2–9 nM; see Fig. 6, legend), indicating a large decrease in desensitized state affinity at the αɛ site. Thus, both ɛN182Y and ɛD175N are state-specific in affecting ACh affinity; ɛN182Y increases closed state affinity 70-fold but only increases desensitized state affinity 1.6-fold, whereas ɛD175N decreases closed state affinity 17-fold but decreases desensitized state affinity 800-fold.
DISCUSSION
The present work begins by defining a CMS by clinical, morphological, and in vitro electrophysiological criteria, and then traces its cause to two heteroallelic mutations, N182Y and D175N, in the AChR ɛ subunit. Mechanistic analyses of AChR activation reveal that each mutation reduces the magnitude and hastens the decay of the postsynaptic response, classifying the CMS as a fast-channel syndrome. The ɛD175N mutation reduces ACh sensitivity of the resting closed state of the receptor by slowing ACh association and speeding ACh dissociation, whereas ɛN182Y enhances ACh sensitivity by speeding ACh association and slowing ACh dissociation. For both mutations, the effects at the binding site are accompanied by impaired gating of the channel through a mechanism independent of agonist occupancy of the mutant binding sites. The overall results show that key residues at the ACh binding site contribute locally to stabilize ACh bound to closed and open states, as well as contribute globally to provide a set point for triggering gating of the channel. Our results also demonstrate state-selective effects of the mutations on resting relative to desensitized states. When mapped on a structural model of the muscle AChR binding site (Sine et al., 2002), ɛN182Y localizes to the subunit interface with the α subunit and ɛD175N to the entrance of the ACh binding site cavity.
The ɛD175N and ɛN182Y mutations compromise neuromuscular transmission by reducing the amplitude of the postsynaptic response and accelerating its decay. Because the density and distribution of AChR on the postsynaptic membrane was normal, we attribute the reduced MEPP and MEPC amplitudes to the decreased probability that the fully occupied receptor will open. Based on the rate constants in Table III, the probability that a doubly liganded AChR opens in response to an instantaneous transmitter pulse, β2/(β2 + k−2), is 0.74 for the wild-type receptor, but only 0.14 for the ɛD175N receptor, and 0.51 for the ɛN182Y receptor.
The rate constants in Table III also predict burst open durations, (1 + β2/k−2)/α2, of 2.13 ms for wild-type AChR, but only 0.49 for the ɛD175N receptor and 0.65 ms for the ɛ182Y receptor. The predicted burst open durations for either mutant receptor are in excellent agreement with the 0.59 ms burst open duration recorded from patient EPs (see Table II) that reflects the approximate average of burst durations generated by the two mutant species of EP AChRs. The burst open duration observed in single channel patch-clamp recordings from the EP is also an approximate measure of the decay time constant of the MEPC. Concordant with the observed burst open duration, the τMEPC was 0.85 ms at patient EPs (Table I).
The mutations identified here alter rate constants underlying both ACh binding and gating of the channel. A natural way to interpret these findings would be to identify a mechanism that links these two effects. However, for the ɛD175N mutation, the effects at the binding site in the resting closed state, slowing ACh association and speeding ACh dissociation, do not impair the ability of ACh occupancy to promote gating of the channel. Instead, channel gating itself is impaired, which is seen as reduced gating following ACh occupancy of the nonmutant αδ site. The possibility that the ɛD175N mutation allosterically affects the nonmutant αδ site seems unlikely because several mutations in the local protein chain affect rate constants underlying ACh occupancy at only one of the two binding sites. The consequences of ɛD175N are best explained by the idea that residues at the binding site not only mediate recognition of ACh, but also set the trigger point for opening the channel. Thus, both the structure of the ACh binding site and the structure of the ion channel contribute to stability of closed relative to open states of the receptor. A further test of this idea might be to compare channel opening in the absence of agonist for wild-type and the ɛD175N mutation, but this would be difficult in practice because spontaneous opening is rare and difficult to quantify.
Similar to the ɛD175N mutation, ɛN182Y affects rate constants underlying both ACh binding and channel gating. Because ɛN182Y enhances ACh affinity and increases the rate constant for ACh binding to the closed state of the receptor, occupancy of the αɛ site becomes the first step in activating the mutant receptor. This promotion of sequential occupancy means that only two out of four equilibrium constants in the first cycle of Scheme II can be estimated for the mutant receptor. Rate constants underlying ACh binding to the closed state and opening of mono-liganded receptors are well defined by the measurements, but those for the coupled steps, binding of the first ACh to the open state and opening of unliganded receptors, remain unknown. Thus, we find that monoliganded gating decreases following ACh occupancy of the mutant site, but this could be due to either a change in closed relative to open state affinity for ACh, or to a change in spontaneous gating of the receptor.
Previous mutagenesis studies of the ACh binding site revealed decreases in both ACh affinity for receptors in the resting closed state and impaired gating of the channel. The traditional explanation of such dual changes is state-specificity of the mutations; the mutations alter closed relative to open state affinities for ACh, which in turn impair channel gating (see Scheme II). For example, the CMS mutation ɛP121L decreased ACh affinity for the open state, had little effect on affinity for the resting closed state and markedly impaired gating of the channel (Ohno et al., 1996). However, similar to the present observations, the affinity changes were accompanied by a decrease in the monoliganded gating equilibrium constant associated with occupancy of the nonmutant αδ site. Furthermore, the binding site mutations αY190F, αY198F, αW149F, and αY93F all reduced affinity of ACh for closed states of the receptor and impaired gating of the channel. For each of these mutations, the changes in channel gating rate constants were greater than changes in rate constants underlying ACh binding to the resting closed state (Chen et al., 1995; Akk et al., 1999; Akk, 2001). These studies did not distinguish monoliganded from diliganded gating, and therefore could not separate effects on agonist binding from those on channel gating, contrary to our present analysis. Thus, the conclusion that the binding site residues studied here independently affect gating of the receptor channel may be generalized to other key residues at the ACh binding site of the receptor.
Both ɛN182Y and ɛD175N are state specific in affecting affinities of closed relative to desensitized states. ɛD175N preferentially affects the desensitized state because it decreases closed state affinity for ACh by 17-fold, but decreases desensitized state affinity by 800-fold. ɛN182Y, on the other hand, preferentially affects the resting closed state because it increases closed state affinity by 31-fold but increases desensitized state affinity only 1.6-fold. Thus, the state specificity of ɛD175N and ɛN182Y indicates that these residues, and possibly nearby structures, contribute differently to ACh binding in one functional state compared with the other.
Studies over the past decade showed that ACh can distinguish between the two ligand binding sites of an individual receptor in the resting closed state, but that the extent of the distinction depends on the species of the receptor. For the Torpedo receptor, ACh binds with about a 100-fold difference in affinity for the two sites in the resting closed state (Sine et al., 1990), for the fetal mouse receptor the difference is 32-fold (Zhang et al., 1995), for the adult human receptor the difference is sixfold (Ohno et al., 1996; Wang et al., 1999), and for the adult mouse receptor the two binding sites are indistinguishable (Akk and Auerbach, 1996; Wang et al., 1997; Salamone et al., 1999). When affinities of the two binding sites are very different, a sequential description of ACh occupancy (Scheme I) is expected to fully account for the kinetics of receptor activation at the single channel level, as observed for receptors containing the mutations ɛN182Y and ɛD175N.
On the other hand, for our reference wild-type human receptor, ACh occupancy should not be strictly sequential because the binding sites differ by only fivefold and the underlying rate constants differ by approximately twofold at the two binding sites. In such cases, the standard alternative description of receptor activation incorporates independent binding of ACh at the two binding sites. Occupancy is still sequential, but there are two possible pathways for achieving double occupancy of the resting, closed state of the receptor. A second monoliganded state is introduced, which adds four more rate constants for ACh binding and two more for the second monoliganded gating step. The independent occupancy model was used to analyze single channel dwell times from the adult mouse receptor, which contains indistinguishable binding sites (Salamone et al., 1999). However, even in this relatively straightforward case, constraints in fitting were required in order to obtain well-defined estimates of the rate constants; the diagonal binding steps were constrained to be equal, as were the two monoliganded gating steps, in harmony with the observation of indistinguishable binding sites. For the adult human receptor, the binding sites are measurably different and the two monoliganded gating steps are not identical (Milone et al., 1997), so constraining these rate constants could not be justified. With all rate constants free to vary, our attempts to fit the independent model to single channel dwell times from the wild-type human receptor did not yield well-defined estimates of the rate constants. Nevertheless, by combining mutagenesis of both binding sites with kinetic analyses, we estimated ACh association and dissociation rate constants for both αδ and αɛ sites. Our results from analyses of mutations at ɛN182 and the equivalent δN187 clearly show that the site from which ACh dissociates slowly is the αδ site, whereas the site from which ACh dissociates rapidly is the αɛ site.
Studies of the ɛD175N mutation in the adult mouse receptor also revealed markedly impaired gating efficiency when two agonists are bound; the rate constant for channel opening was slowed and the rate constant for channel closing was increased (Akk et al., 1999). The rate constant for ACh association was slowed, as we observe, but the rate constant for ACh dissociation was also slowed, contrary to our observed increase in the rate of dissociation. However, contrary to our analysis, the analysis of the mutant mouse AChR kinetics assumed identical binding sites, which likely spread the effects of the mutation in the ɛ subunit over both the αδ and αɛ binding steps.
The AChR ligand binding site is formed by pairs of subunits: α–ɛ and α–δ. Evidence that the binding sites are formed at subunit interfaces comes from mutagenesis and site-directed labeling studies over the past decade (Prince and Sine, 1998), and recently from structural determination of an acetylcholine binding protein homologous to the major extracellular domain of the AChR (Brejc et al., 2001). The mutant residues identified here are found in binding site loop G of the ɛ subunit, which contributes to the non-α subunit face of the binding site and encompasses residues 161–182 (Prince and Sine, 1998). That ɛD175 contributes to the binding site interface was first demonstrated in studies using a site-directed cross-linking reagent (Czajkowski et al., 1993), whereas the present work is the first demonstration that ɛN182 contributes to the binding site. According to a structural model of the AChR binding site, based on lysine-scanning mutagenesis and homology modeling (Sine et al., 2002), loop G forms an irregular coil on the outer surface of the ɛ subunit, lodging in a crevice formed by underlying β-strands. Loop G approaches the α subunit face of the binding site, with ɛD175 just outside the center of the binding site, which is defined by αW149 and ɛW55 (Fig. 7) . ɛN182 is located farther down loop G near the bottom of the subunit, with the amide group of ɛN182 closely approaching the main chain of the α subunit at αC128, which forms the signature cys-loop found in all AChR subunits. Thus, ɛD175 is poised at the periphery of the binding site interface where it can enhance entry and suppress egress of ACh. ɛN182 is located below the center of the binding site where it closely approaches the subunit interface with the neighboring α subunit; it stabilizes ACh bound to the resting closed state, most likely through effects propagated from the subunit interface to residues in loop G proximal to the binding site. Our observation that the mutations are state-specific in altering ACh affinity for resting relative to desensitized states suggests that loop G harboring ɛN182 and ɛD175 rearranges in the course of receptor desensitization.
Figure 7.
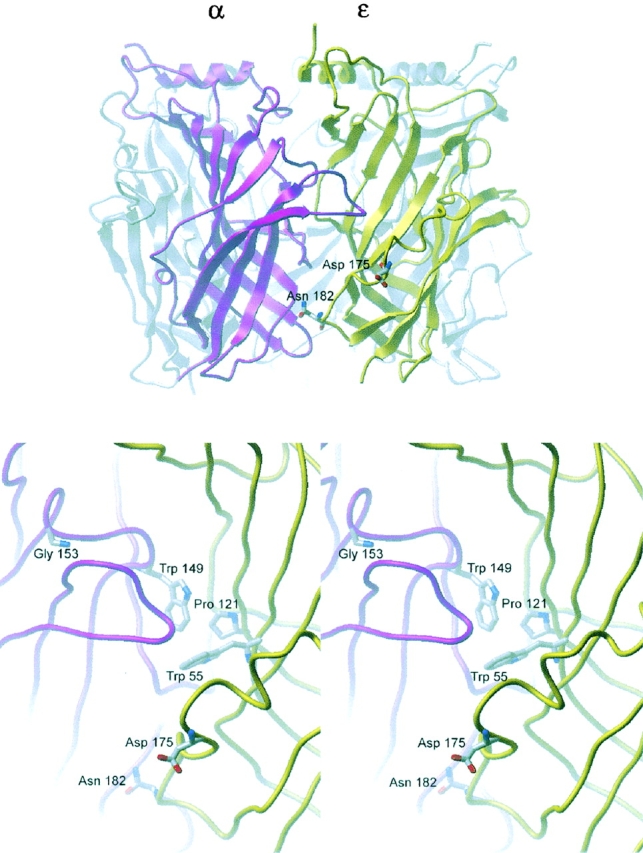
Location of residues ɛD175 and ɛN182 in a structural model of the binding site interface formed by α and ɛ subunits of the human AChR (Sine et al., 2002). Top panel shows the major extracellular domain of the α subunit in magenta, that of the ɛ subunit in yellow and the other subunits in gray. View is from the periphery of the receptor normal to the central vestibule. Bottom panel shows a stereo view of the ligand binding site. Residue side chains are shown in stick representation, with residues at the center of the binding site (αW149 and ɛW55) and other residues mutated in CMS (ɛP121, αG153; see text) shown for reference.
Acknowledgments
This work was supported by National Institutes of Health grants to S.M. Sine (NS31744) and A.G. Engel (NS6277) and by an MDA research grant to AGE.
Footnotes
Abbreviations used in this paper: AChR, acetylcholine receptor; CMS, congenital myasthenic syndrome; EP, endplate; MEPC, miniature EP current; MEPP, miniature EP potential.
References
- Akk, G., and A. Auerbach. 1996. Inorganic, monovalent cations compete with agonists for the transmitter binding site of nicotinic acetylcholine receptors. Biophys. J. 70:2652–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk, G., M. Zhou, and A. Auerbach. 1999. A mutational analysis of the acetylcholine receptor channel transmitter binding site. Biophys. J. 76:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk, G. 2001. Aromatics at the murine nicotinic receptor agonist binding site: mutational analysis of the αY93 and αW149 residues. J. Physiol. 535:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc, K., W. van Dijk, R. Klaassen, M. Schuurmans, J. van der Oost, A. Smit, and T. Sixma. 2001. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 411:269–276. [DOI] [PubMed] [Google Scholar]
- Changeux, J.P. 1990. Functional architecture and dynamics of the nicotinic acetylcholine receptor, an allosteric ligand-gated ion channel. Fida Research Foundation. Neuroscience Award Lectures. 4:21–168. [Google Scholar]
- Chen, J., Y. Zhang, G. Akk, S. Sine, and A. Auerbach. 1995. Activation kinetics of recombinant mouse nicotinic acetylcholine receptors with mutations at α subunit residue tyrosine 190. Biophys. J. 69:849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay, J., and L. DeFelice. 1983. Relationship between membrane excitability and single channel open-close kinetics. Biophys. J. 42:151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun, D., and B. Sakmann. 1985. Fast events in single channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J. Physiol. 369:501–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajkowski, C., C. Kaufmann, and A. Karlin. 1993. Negatively charged amino acid residues in the nicotinic receptor delta subunit that contribute to the binding of acetylcholine. Proc. Natl. Acad. Sci. USA. 90:6285–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, A.G., J.M. Lindstrom, E.H. Lambert, and V.A. Lennon. 1977. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and in its experimental autoimmune model. Neurology. 27:307–315. [DOI] [PubMed] [Google Scholar]
- Engel, A.G., A. Nagel, T.J. Walls, C.M. Harper, and H.A. Waisburg. 1993. Congenital myasthenic syndromes. I. Deficiency and short open- time of the acetylcholine receptor. Muscle Nerve. 16:1284–1292. [DOI] [PubMed] [Google Scholar]
- Engel, A.G. 1994. The muscle biopsy. Myology. A.G. Engel and C. Franzini-Armstrong, eds. McGraw-Hill, New York. 822–831.
- Engel, A.G., K. Ohno, H.-L. Wang, M. Milone, and S.M. Sine. 1998. Molecular basis of congenital myasthenic syndromes: Mutations in the acetylcholine receptor. Neuroscientist. 4:185–194. [Google Scholar]
- Jackson, M. 1989. Perfection of a synaptic receptor: Kinetics and energetics of the acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 86:2199–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone, M., D.O. Hutchinson, and A.G. Engel. 1994. Patch-clamp analysis of the properties of acetylcholine receptor channels at the normal human endplate. Muscle Nerve. 17:1364–1369. [DOI] [PubMed] [Google Scholar]
- Milone, M., H.-L. Wang, K. Ohno, T. Fukudome, N. Pruitt, N. Bren, S. Sine, and A. Engel. 1997. Slow channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity due to mutation in the M2 domain of the acetylcholine receptor α subunit. J. Neurosci. 17:5651–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod, J., J. Wyman, and J.P. Changeux. 1965. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 3:318–356. [DOI] [PubMed] [Google Scholar]
- Ohno, K., D.O. Hutchinson, M. Milone, J.M. Brengman, C. Bouzat, S.M. Sine, and A.G. Engel. 1995. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the ɛ subunit. Proc. Natl. Acad. Sci. USA. 92:758–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno, K., H.-L. Wang, M. Milone, N. Bren, J.M. Brengman, S. Nakano, P. Quiram, J.N. Pruitt, S.M. Sine, and E.G. Engel. 1996. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor ɛ subunit. Neuron. 17:157–170. [DOI] [PubMed] [Google Scholar]
- Prince, R.J., and S.M. Sine. 1998. The ligand binding domains of the nicotinic acetylcholine receptor. The Nicotinic Receptor: Current Views and Future Trends. F.J. Barrantes, editor. Landes Bioscience/Springer Verlag, TX. 31–59
- Qin, F., A. Auerbach, and F. Sachs. 1996. Estimating single-channel kinetic parameters from idealized patch clamp data containing missed events. Biophys. J. 70:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone, F., M. Zhou, and A. Auerbach. 1999. A re-examination of adult mouse nicotinic acetylcholine receptor channel activation kinetics. J. Physiol. 516:315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth, F.J., and S.M. Sine. 1987. Data transformation for improved display and fitting of single-channel dwell time histograms. Biophys. J. 52:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine, S.M., and P. Taylor. 1982. Local anesthetics and histrionicotoxin are allosteric inhibitors of the acetylcholine receptor. J. Biol. Chem. 257:8106–8114. [PubMed] [Google Scholar]
- Sine, S.M., T. Claudio, and F.J. Sigworth. 1990. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts: Single channel current kinetics reveal distinct agonist binding affinities. J. Gen. Physiol. 96:395–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine, S.M., K. Ohno, C. Bouzat, A. Auerbach, M. Milone, J.N. Pruitt, and A.G. Engel. 1995. Mutation of the acetylcholine receptor α-subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 15:229–239. [DOI] [PubMed] [Google Scholar]
- Sine, S.M., H.-L. Wang, and N. Bren. 2002. Lysine scanning mutagenesis delineates structural model of the nicotinic receptor ligand binding domain. J. Biol. Chem. 277:29210–29223. [DOI] [PubMed] [Google Scholar]
- Sozer, A.C., C.M. Kelly, and D.B. Demers. 2001. Molecular analysis of paternity. Current Protocols in Human Genetics. N.C. Dracopoli, editor. Wiley and Sons, New York. [DOI] [PubMed]
- Uchitel, O., A.G. Engel, T.J. Walls, A. Nagel, and V. Bril. 1993. Congenital myasthenic syndrome attributed to an abnormal interaction of acetylcholine with its receptor. Ann. NY Acad. Sci. 681:487–495. [DOI] [PubMed] [Google Scholar]
- Wang, H.-L., M. Milone, K. Ohno, X.-M. Shen, A. Tsujuno, A. Paola, P. Tonali, J. Brengman, A.G. Engel, and S.M. Sine. 1999. Acetylcholine receptor M3 domain: stereochemical and volume contributions to channel gating. Nat. Neurosci. 2:226–233. [DOI] [PubMed] [Google Scholar]
- Wang, H.-L., A. Auerbach, N. Bren, K. Ohno, A.G. Engel, and S.M. Sine. 1997. Mutation in the M1 domain of the acetylcholine receptor α subunit decreases the rate of agonist dissociation. J. Gen. Physiol. 109:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H.-L., K. Ohno, M. Milone, J. Brengman, A. Evoli, A.P. Batocchi, L. Middleton, K. Christodoulou, A.G. Engel, and S.M. Sine. 2000. Fundamental gating mechanism of nicotinic receptor channel gating revealed by mutation causing a congenital myasthenic syndrome. J. Gen. Physiol. 116:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., J. Chen, and A. Auerbach. 1995. Activation of recombinant mouse acetylcholine receptors by acetylcholine, carbamylcholine and tetramethylammonium. J. Physiol. 486:189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]