

Abstract
The primary voltage sensor of the sodium channel is comprised of four positively charged S4 segments that mainly differ in the number of charged residues and are expected to contribute differentially to the gating process. To understand their kinetic and steady-state behavior, the fluorescence signals from the sites proximal to each of the four S4 segments of a rat skeletal muscle sodium channel were monitored simultaneously with either gating or ionic currents. At least one of the kinetic components of fluorescence from every S4 segment correlates with movement of gating charge. The fast kinetic component of fluorescence from sites S216C (S4 domain I), S660C (S4 domain II), and L1115C (S4 domain III) is comparable to the fast component of gating currents. In contrast, the fast component of fluorescence from the site S1436C (S4 domain IV) correlates with the slow component of gating. In all the cases, the slow component of fluorescence does not have any apparent correlation with charge movement. The fluorescence signals from sites reflecting the movement of S4s in the first three domains initiate simultaneously, whereas the fluorescence signals from the site S1436C exhibit a lag phase. These results suggest that the voltage-dependent movement of S4 domain IV is a later step in the activation sequence. Analysis of equilibrium and kinetic properties of fluorescence over activation voltage range indicate that S4 domain III is likely to move at most hyperpolarized potentials, whereas the S4s in domain I and domain II move at more depolarized potentials. The kinetics of fluorescence changes from sites near S4-DIV are slower than the activation time constants, suggesting that the voltage-dependent movement of S4-DIV may not be a prerequisite for channel opening. These experiments allow us to map structural features onto the kinetic landscape of a sodium channel during activation.
Keywords: conformational changes, fluorescence, gating currents, sodium channel
INTRODUCTION
Voltage-gated sodium channels open rapidly on depolarization, leading to a large inward sodium flux followed by a time-dependent inactivation (Hodgkin and Huxley, 1952). According to the Hodgkin and Huxley model, three independent “m” gates control the opening of the channel, and an “h” gate controls the inactivation of the channel. The model also predicts gating currents with kinetic components of the m and the h gates. Subsequent measurements of gating currents led to modification of the original hypothesis (Armstrong and Bezanilla, 1977; Bezanilla and Armstrong, 1977). Gating currents of Na channels in the squid axon were found to have two components: one corresponded to activation but the other was faster than inactivation. The development of inactivation correlated with an increase in immobilization of gating charge and subsequent removal of inactivation relieved charge immobilization. Therefore, in the coupled inactivation model, inactivation develops when the “inactivation particle” binds to the pore, which occurs preferentially during the open state of the channel. Thus, inactivation derives its voltage dependence by coupling to activation. Macroscopic ionic currents, gating current measurements and single channel studies in other preparations also support the coupled inactivation model (Goldman and Schauf, 1972; Hille, 1976; Aldrich et al., 1983; Aldrich and Stevens, 1987).
With the advent of molecular cloning, many structural features of voltage-gated ion channels were clarified (Noda et al., 1986). The α subunit of sodium channel consists of four internally homologous domains, referred to as DI to DIV, connected by cytoplasmic linkers. The fourth transmembrane helix in each domain, also called the S4 segment, has a positively charged residue at every third position and is believed to be the voltage sensor (Stuhmer et al., 1989; Papazian et al., 1991). Direct evidence that charged residues in S4 sensed and moved across the electric field came from studies that measured gating charge per channel after neutralizing S4 charges of the Shaker potassium channel (Aggarwal and MacKinnon, 1996; Seoh et al., 1996). Similar measurements have not been performed for the sodium channel where the nonidentical S4 segments are expected to have variable contributions to the gating process. Kinetic and steady-state analysis of channel opening in charge neutralized mutants of the sodium channel indicate that the S4 charges from all domains are involved in activation, albeit to a variable extent (Chen et al., 1996; Kontis and Goldin, 1997). Interestingly, perturbation of charges in the fourth domain have the largest effects on both inactivation and deactivation with little influence on activation (Chahine et al., 1994; Kontis et al., 1997; Groome et al., 1999). These results suggest that S4 domain IV may be uniquely involved in coupling activation gating to the inactivation process.
In recent years, fluorescence-based methods have been used to probe dynamic changes in the structure since they provide site-specific information in real time (Bezanilla, 2000). In sodium channels, fluorescence studies have demonstrated that the S4 segment of domains III and IV are immobilized by fast inactivation, whereas those of domains I and II are unaffected (Cha et al., 1999). To dissect the functional roles of the four voltage sensors in sodium channel during activation gating, we monitor site-specific fluorescence simultaneously with electrophysiological recordings. Using this technique, we attempt to address a number of important questions. First, how does the conformational change, in each of the domains, correlate with the gating charge movement? Second, do these changes occur in a predetermined sequence in activation or are they random (Bezanilla, 2000). If S4 segments move randomly, one can envision a stochastic sequence determined by steady-state properties whereby the domain opening at most hyperpolarized potentials is likely to move the earliest. Third, does the voltage-dependent movement of S4-DIV track the inactivation process (Sheets and Hanck, 1995). Fourth, and perhaps most intriguing, can we detect ionic currents in the absence of conformational changes in a particular domain, especially domain IV. Such a result would suggest that S4-DIV activation may not be necessary for channel opening (Sheets et al., 1999, 2000). The major finding of our study is that the fluorescence from S4 proximal sites in domains I, II, and III track the fast component of gating currents, whereas the fluorescence from S4 proximal site in domain IV tracks only the slow component. This result is a direct evidence of functional differences between S4 in domain IV and S4s in other domains. A comparison of the rate of channel opening with S4-DIV activation suggests that there exists an open state of sodium channel that does not require domain IV to be activated. These findings allow us to describe the structural changes that take place in a voltage-gated sodium channel during its activation.
MATERIALS AND METHODS
Modified Cut Open Oocyte Epifluorescence Setup
The optical setup used for simultaneous measurement of gating currents and fluorescence signals has been described previously (Cha and Bezanilla, 1998). The oocyte was voltage clamped and gating currents were measured in a cut-open configuration for spatial voltage homogeneity and fast temporal resolution. The cut-open setup was placed on the stage of an upright microscope (BX50WI; Olympus Optical) and the light was focused using a LUMPlanFl 40× water-immersion objective with a numerical aperture of 0.8. The tetramethylrhodamine probe was excited with light from tungsten lamp, which was filtered by a 535DF35 excitation filter, passed through a 570DRLP dichroic mirror that split the emission and the excitation light, and the emitted light was filtered with a 565EFLP filter (Chroma Technologies and Omega Optical). This emitted light was then focused onto a PIN-020A photodiode (UDT Technologies) by a microscope condenser lens at the microscope's epifluorescence port. The photodiode was connected to the headstage of an integrating Axopatch 1B patch clamp amplifier (Axon Instruments, Inc.). The current from the photodiode was offset before being fed onto the patch-clamp amplifier by a circuit of a 45-V battery and a 10-GΩ resistor connected to the input of the patch amplifier headstage to prevent the feedback capacitor from saturating during acquisition. The excitation light from the lamp was interrupted with a TTL triggered VS25 shutter (Vincent Associates) between measurements.
Data Acquisition and Analysis
Gating, ionic, and fluorescence signals were acquired with a PC44 board (Innovative Integration), which interfaced with a Pentium-based computer. The fluorescence and electrophysiology were simultaneously acquired on two 16-bit A/D converters and transferred to two separate channels of the PC-44 board. When the data is sampled at intervals longer than 5 μs (all the traces presented in the paper), the program acquired the data at 5 μs/point, digitally filtered them to the new Nyquist frequency, and decimated them to the new sampling frequency. The acquisition and data analysis programs were developed in the lab and were run in Windows 95.
The external solutions contained 115 mM n-methylglucamine (NMG)-Mes, 20 mM Hepes, and 2 mM Ca(Mes)2 at pH 7.4 and the internal solution contained 115 mM NMG-Mes, 20 mM Hepes, and 2 mM EGTA at pH 7.4. Since the recorded ionic currents were contaminated by gating currents, they had to be corrected by subtraction of the gating currents, which were obtained after TTX block, from the total membrane currents obtained before addition of the toxin. The gating currents used for subtraction were obtained by using P/−4 protocol from a subtracting holding potential of −130 mV. All gating current recordings used for analysis were recorded by using P/4 from a subtracting holding potential of 50 mV. The gating currents were recorded in response to 20-ms test pulses and the traces were baselined after 15 ms.
Molecular Biology, Expression, and Labeling
The rSkM1 cDNA was transferred into a pBSTA vector where the untranslated regions were replaced with Xenopus β-globin untranslated sequences and a Kozak consensus sequence was introduced immediately before the translational start site. The coding region of the Na channel DNA was mutated to generate unique restriction sites for three enzymes BstEII, AvrII, and SpeI to facilitate subcloning. The resulting clone of the rat skeletal muscle sodium channel, z3Na-pBSTA, was used as a template for all the subsequent mutagenesis. Site-directed mutagenesis was performed using the Quikchange site-directed mutagenesis Kit (Stratagene). After the mutation was confirmed by sequencing, a fragment containing the mutation was subcloned back into the wild-type cDNA in order to reduce the possibility of second site mutations.
The Na channel cRNA was transcribed in vitro with T7 polymerase (Ambion) from NotI-linearized cDNA clone. The β subunit was similarly transcribed. 50 nl cRNA were injected into each Xenopus oocyte containing equal volumes of 2 μg/μl of α subunit and 0.4 μg/μl of β subunit. The oocytes were incubated for 4–7 d after injection at 12°C in a incubation solution of 100mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, 10 mM EDTA, and 100 μM DTT. Approximately 14 h before the experiment, the oocytes were treated with 500 μM β-maleimidopropionic acid (Sigma-Aldrich) in oocyte incubation solution without DTT for 45 min to block the native cysteines. After the treatment, the oocytes were washed and incubated in incubation solution containing 100 μM DTT for 14 h at room temperature. The oocytes were labeled by incubation on ice in a depolarizing solution with 10 μM of 5′-tetramethylrhodamine (cat. # T-6027; Molecular Probes, Inc.) for 45 min. This labeling protocol is a modification of the method originally described by Mannuzzu et al. (1996).
Kinetic Simulations
The kinetic models (in Scheme II and Fig. 8 A) were expressed as a series of equations for the time derivatives of the probability of being in each state. Thus, the change in probability of being in each state with time equals the probability of entering that state from all other states and of leaving that state to any other state.
Figure 8.

Limits on the rates to resolve sequential activation of S4 movement. (A) Sequential activation model for a hypothetical channel where the S4 in domain III activates later in the sequence. The state where all the S4s are deactivated is represented as C and the activation of any of the three S4s converts the channel into a state O. The number in the subscript represents the domain whose S4 is activated. α, β, and γ represent the rate constants of transition. (B) The fluorescence signals from S216C, S660C and L1115C were obtained by pulsing to −30 mV. The fluorescence signals of S4 domain I (smooth line) and S4 domain III (broken lines) representing transition of S4 segment from a resting to an activated state were simulated using a kinetic model described in Fig. 8 A. The fluorescence signals of S4 domain III were simulated for three different values of γ with respect to α.
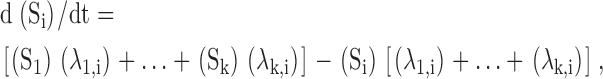 |
for all i, where Si is the probability of being in each state i, k is the total number of states, and λi,k is the rate of going from state i to state k.
For Scheme II, the rate constants were optimized by fitting the predicted ionic currents to the experimental data. The ionic currents were predicted by the probability of being in state O, multiplied with an arbitrary scaling factor N. The ionic current data at different potentials was fitted with five parameters that include the four rate constants x, y, c, and d and scaling factor N. The activation time constants were estimated by fitting the predicted gating current decay to a single exponential. The gating current is the rate of charge movement during a voltage pulse, which is equal to the total sum of all the charge movement occurring at various transitions:
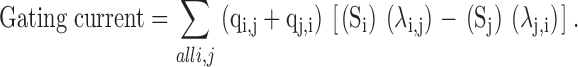 |
qi,j and qj,i is the effective valence for transitions between states i to j. The value of qi,j + qj,i was arbitrarily taken to be 1 for all transitions.
SCHEME II.

For the kinetic model shown in Fig. 8 A, the rates were assumed to be simple exponential function of voltage (Stevens, 1978). The voltage dependence of the transition rate, λi,j, from state i to j under the assumption that the midpoint of transition in all the S4s is at −30 mV, is given by:
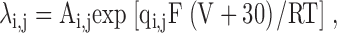 |
where V is the applied voltage, Ai,j is the rate constant from states i to j at −30 mV, qi,j is the effective valence of the i to j transition rate, F is the Faraday constant, R is the gas constant, and T is the absolute temperature. For simplicity, the fluorescence time constants for S4 movement in the three domains can be approximated to 200 μs (comparable to the actual data from sites S216C, S660C, L1115C). Since the time constants of fluorescence, τF, is the inverse of sum of backward and the forward rate constants:
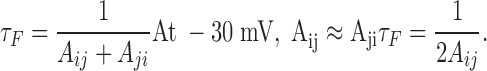 |
Thus, the transition rate constant of each S4 at −30 mV becomes equal to 2.5 ms−1. The effective valence for forward and backward transition was taken as 1. The fluorescence signals from S4 of domain i was predicted as the sum of all states Si,j
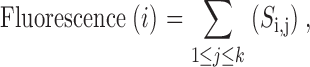 |
where k is total number of states containing activated S4 domain i.
The time-dependent quantities were “sampled” every 2 μs and the data was digitally filtered at 20 KHz by convoluting the discretized signal with a Gaussian kernel (Colquhoun and Sigworth, 1995). Simulations and fitting of the kinetic models were performed on SCoP simulation environment (Simulation Resources).
RESULTS
To characterize the voltage-dependent movement of S4 segments, we introduced single cysteines proximal to each S4 segment in the rat skeletal muscle sodium channel. Earlier efforts to carry out similar studies on sodium channel were hampered by poor signal to noise ratios. To overcome this problem, we first attempted to increase the sodium channel expression. A Kozak sequence in optimal context to the start site of the α subunit of sodium channel was introduced (Kozak, 1991; Starace et al., 1997). Next, sodium channel expression was followed as a function of coinjecting different molar ratios of α to β subunits. The expression of sodium channel with normal inactivation kinetics was highest when the ratio of the α to β subunits was 1:2. Typically, under these conditions, 1010 molecules of wild-type sodium channels were expressed in a single oocyte as determined by measuring total gating charge (Hirschberg et al., 1995). Finally, to reduce the background fluorescence, native cysteines were blocked with β-maleimidopropionic acid, a cysteine-modifying reagent (see materials and methods).
In this report, we characterize four mutants that produce robust fluorescence signals, namely, S216C, S660C, L1115C, and S1436C (Fig. 1) . Since the temperature coefficient of conformational changes is much larger than that of membrane capacitance, we initially attempted our fluorescence measurements at low temperatures to exaggerate the kinetic features. However, in the case S1436C, lowering the temperature slowed the fluorescence kinetics dramatically. As a result, any lag phase due to late activation of the particular S4 segment merged into the slowly activating fluorescence, making it hard to resolve. In addition, we encountered difficulty recording fluorescence signals from S216C mutant at low temperature where it was irreversibly quenched. Therefore, all the experiments described in this work were performed at 20°C. The results described here are supported by fluorescence and electrophysiological data from additional sites located in each of the four domains. All the fluorescence, gating, and ionic current measurements were performed on labeled mutants.
Figure 1.

The S4 sequences from α subunit of the rat skeletal muscle sodium channel were aligned with respect to their charged residues, shown in bold face. The residues (′, *, #) were mutated individually to cysteines and labeled with tetramethylrhodamine. The residues, shown in boxes, correspond to mutants whose fluorescence and electrophysiological properties were characterized in this study. Voltage-dependent fluorescence changes could be measured from additional sites (*) in each S4. The remaining mutants (#) did not show any voltage-dependent fluorescence signals.
Presence of Two Components in the Gating Currents of the Wild-type Skeletal Muscle Sodium Channels
The kinetic characteristics of ON gating currents from the wild-type rat skeletal muscle sodium channel were assessed using the cut-open oocyte technique in order to resolve the fast kinetics of skeletal muscle sodium channels (Stefani and Bezanilla, 1998). After blocking ionic currents with external application of TTX, the gating currents were measured using P/4 protocol with a subtracting holding potential at 50 mV. Alternate measurements of gating currents by subtracting at holding potential of −130 mV resulted in subtraction of charge that moved at hyperpolarized potentials affecting the Q-V relationship. Therefore, in all the experiments described here, the gating currents were recorded by subtracting at a holding potential of 50 mV. A family of representative gating current traces is shown in Fig. 2 A. The gating currents at −30 and −50 mV are characterized by presence of a slow component in the latter part of the trace; arrowheads mark the transitions between fast and slow components. With larger depolarization, the amplitude of the slow component diminishes while its rate increases. To resolve and characterize the slow component of gating from baseline, it is necessary to utilize preparations that yield high channel densities. Typically, the gating currents described here have peak amplitudes between 4 to 8 μA at 0 mV (20°C).
Figure 2.

Gating current characteristics of the wild type rat skeletal muscle sodium channel. (A) Gating currents of the wild type sodium channel were elicited by pulsing to −50, −30, −10, and 30 mV after 400 ms prepulse to −130 mV. The recorded traces were obtained by using a P/4 protocol at a subtracting holding potential of 50 mV. The arrowheads separate the fast and the slow component of the gating currents at potentials −50 and −30 mV. (B) Time constants of gating, activation, and inactivation. The data was obtained by fitting the decay phase of the gating current to the equation: A*exp(−t/τ1) + B*exp(−t/τ2). The slow gating component, τq(s), is shown with squares and the fast gating component, τq(f), with circles. The filled triangles correspond to inactivation time constants, τinact, obtained by fitting ionic currents. The activation time constants, (τact) filled circle, were obtained by fitting the ionic currents using a simplified version of the model proposed by Vandenberg and Bezanilla (1991).
The voltage dependence curves of both slow and fast components of the gating currents are shown in Fig. 2 B and are plotted along with the activation and the inactivation time constants. The time constants of inactivation were obtained by simply fitting the decaying phase of ionic currents with single exponentials. On the other hand, extracting activation time constants from inactivating sodium current data is not straightforward since the activating phase of the sodium current has contribution from the inactivation process. Therefore, the activation time constants of voltage-dependent sodium channel are obtained by fitting the ionic current traces to a kinetic model. Here, we used the sodium channel gating model proposed by Vandenberg and Bezanilla (1991). This kinetic model of sodium channel activation is based on data obtained from macroscopic ionic currents, single channel currents, and gating currents (Scheme I).
SCHEME I.

To fit our data, the model could be simplified to reduce the number of free parameters (Scheme II).
This reduction was achieved by eliminating the parallel pathway leading to inactivation from C4 and making the rates between all the states leading unto the open state equal. Thus, the number of fitted parameters was reduced to four rates (y, z, c, and d) and a scaling factor. These parameters were optimized for ionic current data obtained by pulsing to a particular potential. The optimized parameters were used to generate gating current traces at the test potential (see materials and methods). The main decay phase of the simulated gating current trace was then fitted to a single exponential whose time constant corresponds to the main activation time constant (the reciprocal of the predominant eigenvalue). In this manner, time constants of activation of sodium channel were obtained for potentials ranging from −30 to 30 mV.
The time constants of activation were compared with the fast and the slow components of the recorded gating currents over the activation voltage range. The plot shows that the activation time constants correlate with fast component of gating over most of the voltage range. At less depolarized potentials (−30 mV), however, the activation time constants appear slower than fast component of gating. The slow component of gating does not appear to correlate either with activation time constants or with inactivation time constants. Furthermore, the slow component in the gating currents is consistently faster than the inactivation rates (Fig. 1 C). These results, obtained from the rat skeletal muscle sodium channel, are consistent with previous studies of gating currents from squid sodium channels and other preparations (Armstrong and Bezanilla, 1977; Sheets and Hanck, 1995). In the squid giant axon, this slow component of ON gating was postulated to correspond to the formation of a second open state. The fast component, on the other hand, has been shown to correlate with activation.
Comparison of Fluorescence, Gating, and Ionic Properties of Reporter-labeled Mutants
Fluorescence changes were monitored simultaneously with either gating or ionic currents to determine whether the voltage-induced conformational changes proximal to S4s of various domains are temporally resolved from major charge movement or channel opening. In Fig. 3 , the fluorescence signals (in red), ionic currents (in blue), and time integral of gating currents (in black) in response to a wide range of voltage pulses have been overlaid for each mutant. The time integrals of gating currents represent the position of gating charge with time and were recorded in presence of TTX, which blocked ionic currents. The ionic currents were recorded in 120 mM external NMG-MES with KCl electrode to minimize series resistance errors resulting from large sodium currents. The ionic currents shown here represent outward flux of internal potassium through the sodium channels. These ionic currents were obtained by subtracting the gating currents from the total membrane currents.
Figure 3.

Comparison of fluorescence, ionic currents, and the gating charge position for various cysteine mutants. Ionic currents (in blue), fluorescence (in red), and the gating charge position (time integral of the gating current) (in black) for TMRM labeled S216C (A), S660C (B), L1115C (C), and S1436C (D). Each panel is also marked with a domain name where the cysteine mutation is located. The ionic currents were elicited by pulsing to various test potentials following a 400 ms prepulse to −130 mV. The ionic traces were obtained by using P/−4 protocol with a subtracting holding potential of −130 mV. Fluorescence traces were obtained by making an average of 20 traces at each test potential with an interval of 2 s between pulses. The gating charge position was obtained by integrating the gating currents over the duration of the test pulse. The gating currents were obtained by subtracting at 50 mV using P/4 protocol. The data were scaled so that the peaks of ionic currents, fluorescence, and gating integrals have comparable amplitudes at 30 mV. Typical values of ΔF/F were between 1 and 2%. The time constant of membrane capacitance measured by small hyperpolarizing pulses from −130 mV to −140 mV was between 75 and 90 μs.
On depolarization, the fluorescence signals from sites S216C, S660C, and S1436C are quenched whereas those from L1115C are dequenched. The fluorescence signals from L1115C were inverted and overlaid with the gating and ionic current traces. A quick visual examination reveals that the fluorescence signals from each of the mutants, comprises of two components although the amplitude of the second component is small in S660C and S1436C. Fluorescence from the sites S216C, S660C, L1115C appears to follow the gating currents in the early phase. In contrast, the fluorescence from S1436C is clearly slower than the early phase of gating current. The voltage dependence of a single S4 segment would be predicted to initiate before the ionic currents if the movement of S4s are coupled to channel opening. The fluorescence signals from S216C, S660C, and L1115C initiates before the ionic current rise (−30 and −10 mV). The fluorescence from S1436C initiates along with the ionic currents and are kinetically slower than the ionic currents. Thus, a visual comparison of fluorescence shows that the voltage-dependent conformational changes at S1436C are kinetically different in contrast to those from corresponding sites in other S4 segments.
Steady-state Behavior of Labeled Mutants
The effect of introduced cysteines and labeling on the steady-state properties of the sodium channel was assessed from gating charge-voltage relationships of various mutants. The total gating charge moved at a particular potential was calculated by integrating the gating current trace at that potential and normalizing with respect to maximum charge displaced. The normalized gating charge was then plotted as a function of pulse potential in Fig. 4 A. The data was fit to a single Boltzmann equation with variable parameters, V1/2, the midpoints of activation and, z, the gating valence (Table I) . The differences in the gating valences between the wild-type Na channels and the mutants are insignificant. The midpoints of the Q-V curves for the mutants L1115C (domain III) and S1436C (domain IV) are slightly shifted to hyperpolarized potentials relative to the wild-type. The midpoints of the charge-voltage curves of the mutants S216C (domain I) and S660C (domain II) are similar to the wild-type sodium channel. Overall, the introduction of cysteine mutation and labeling does not significantly perturb equilibrium properties of the Na channel.
Figure 4.
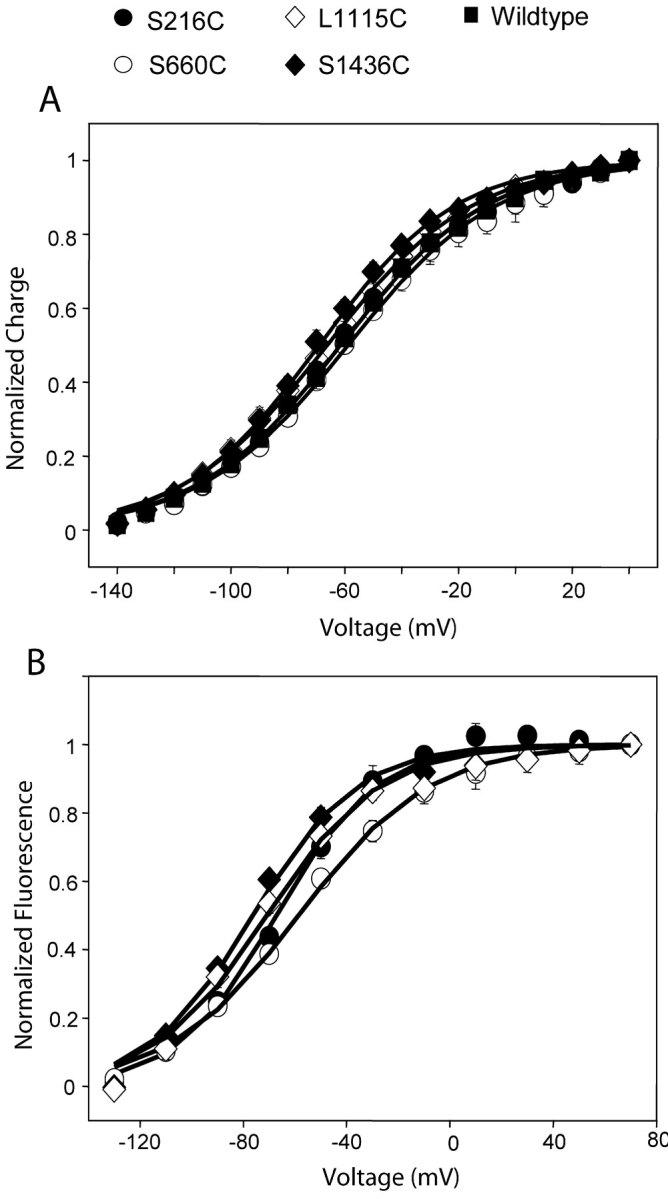
Steady-state fluorescence and gating characteristics of various mutants. (A) Normalized gating charge displacement as a function of pulse potential for S216C, S660C, L1115C, S1436C, and the wild-type Na channel. For each oocyte, the charge displaced was normalized to Qmax and the values were plotted as means ± SEM with n ≥ 3 oocytes. The continuous lines represent best fits of the means to a single Boltzmann. (B) Normalized fluorescence change is plotted as a function pulse potential for S216C, S660C, L1115C, and S1436C. The steady-state fluorescence was determined by measuring the fluorescence change at the end of a 20-ms pulse. The fluorescence change recorded at various test potentials was normalized to the maximum fluorescence change recorded in the particular oocyte. The values were plotted as means ± SEM with n ≥ 3 oocytes and the continuous lines are best fits to a Boltzmann distribution.
TABLE I.
Steady-state Properties of Fluorescence and Charge Movement for Wild-type Channel and Labeled Mutants
| Fluorescence | Charge | |||
|---|---|---|---|---|
| Midpoint | Slope | Midpoint | Slope | |
| mV | e0 | mV | e0 | |
| Wild-type Na Channel | −61.4 ± 2.4 (5) | z = 0.99 | ||
| S216C (domain I) | −67.1 ± 2.5 (3) | z = 1.3 | −60.9 ± 4.4 (4) | z = 1.0 |
| S660C (domain II) | −58.3 ± 2.3 (3) | z = 1.0 | −58.7 ± 1.8 (3) | z = 0.97 |
| L1115C (domain III) | −75.9 ± 1.3 (3) | z = 1.2 | −68.9 ± 2.4 (4) | z = 1.0 |
| S1436C (domain IV) | −70.8 ± 2.3 (4) | z = 1.1 | −66.1 ± 2.4 (4) | z = 0.98 |
The values of midpoints are shown as mean ± SEM (number of oocytes).
The steady-state fluorescence properties of labeled mutants were assessed by plotting normalized fluorescence change, at the end of a 20-ms pulse, with respect to pulse potential (Fig. 4 B). Again, the data was fit to a single Boltzmann function characterized by two parameters V1/2 and z (Table I). The apparent valence ranges from a maximum of 1.3 for domain III to a minimum of 1.0 for domain II. The fluorescence-voltage plots indicate that L1115C activates at the most hyperpolarized potential (V1/2 of −76 mV), followed by S1436C (V1/2 of −71 mV), S216C (V1/2 of −67 mV), and S660C (V1/2 of −58 mV) in that order. Because fluorescence in all the mutants comprises of two components, the use of a single Boltzmann to fit is not optimal. Therefore, we plotted the amplitudes of the fast component of fluorescence with respect to pulse potentials after fitting the fluorescence data to two exponentials. For a single set of data, the midpoints of activation are −60 mV for L1115C, −52.2 mV for S216C, and −41.1 mV for S660C. The data derived in this manner has uncertainties due to introduction of additional errors, particularly, while fitting traces with low signal to noise ratios. Nevertheless, this analysis corroborates the results from the single Boltzmann fits, namely, that the fluorescence changes at L1115C occurs at more hyperpolarized potentials, followed by those at S216C and then by S660C. Although the midpoint of activation of S1436C fluorescence is −71 mV, its fluorescence kinetics are distinctly slower and, therefore, they are likely to contribute most at the later stages of the gating process.
Kinetics of Fluorescence Change from S216C, S660C and L1115C Correlate with Fast Component of Charge Movement
To describe the kinetic properties of individual S4s over the activation voltage range, we first have to determine whether the conformational changes detected by site-specific fluorescence and gating current measurements report the same phenomenon viz. the movement of elementary S4 charges. Unfortunately, to establish this kind of exact correlation is not straightforward in the case of sodium channels. Although, both gating current and fluorescence measurements independently report structural changes, they differ fundamentally in their information content. The gating currents reflect the movement of all gating charges, whereas fluorescence mainly follows local conformational changes. In the sodium channel due to its heterotetrameric nature, the fluorescence is likely to emphasize only one of the multiple components of gating. If we make a simplistic assumption that all the S4 charges in each domain move in a single step with differing rates, then four components corresponding to the movement of each S4 segment would be expected to appear in the gating currents. However, it is highly unlikely that we will be able to resolve more than three components by fitting gating current traces. Analyses of recorded gating currents in the sodium channel reveal the presence of two components of charge movement, which we call the fast and the slow component. The fluorescence data, in all the instances, is best described using a sum of two exponentials. Therefore, as a first approximation, the time constants of fluorescence were compared with time constants of charge movement over the activation voltage range in an effort to establish its correlation with movement of S4 charges.
To obtain the time constants of charge movement, the decay of the gating currents were fit to double exponentials. Although the time integrals of gating currents reflect the movement of charge, the rates obtained by fitting the integral are always slower than those extracted by fitting the gating currents. This is because the gating integral is a sum of exponentials with amplitudes scaled by their time constants. Therefore, all the data pertaining to voltage-dependent charge movement described here was analyzed by fitting the gating currents rather than the gating integrals.
The time constants of fluorescence change at sites S216C, S660C, and L1115C were compared with the time constants of gating currents over a voltage range (Fig. 5 , A–C and Table II) . For S216C and S660C, the fast time constants of fluorescence and gating correlate well over large depolarizations viz. 10–30 mV (Fig. 5, A–C and Table II), whereas for L1115C the correlation is best at less depolarized potentials, viz. −50 to −30 mV (Fig. 5 C and Table II). This result is consistent with the prediction from steady-state fluorescence data. If conformational changes at S216C, S660C, and L1115C occur independent of one another, then the F-V relationship (Fig. 4 B and Table I) suggests that on depolarization, conformational changes would occur first at L1115C followed by those at S216C and then at S660C. If the measured conformational changes reflect the elementary sensor movement then the gating time constants would be expected to have more contribution from the S4 of domain III at less depolarized potentials and from the S4s of domains I and II on increasing depolarization. The slow time constants of fluorescence, on the other hand, from the sites S216, S660, and L1115 do not correlate with the slow component of gating (Fig. 5, A–C and Table II). The slow fluorescence changes are clearly slower than both gating and fast inactivation and may reflect additional conformational changes that have not been explored in sufficient detail here. This slow movement as followed by fluorescence may or may not transport charge, but if it does, it should produce a third component on the gating current. However, as it is very slow it would tend to disappear into the baseline of gating currents and would be difficult to detect. In sum, the data indicates that the voltage-dependent conformational changes at sites S216, S660, and L1115 are comparable to the fast time constants of charge movement.
Figure 5.
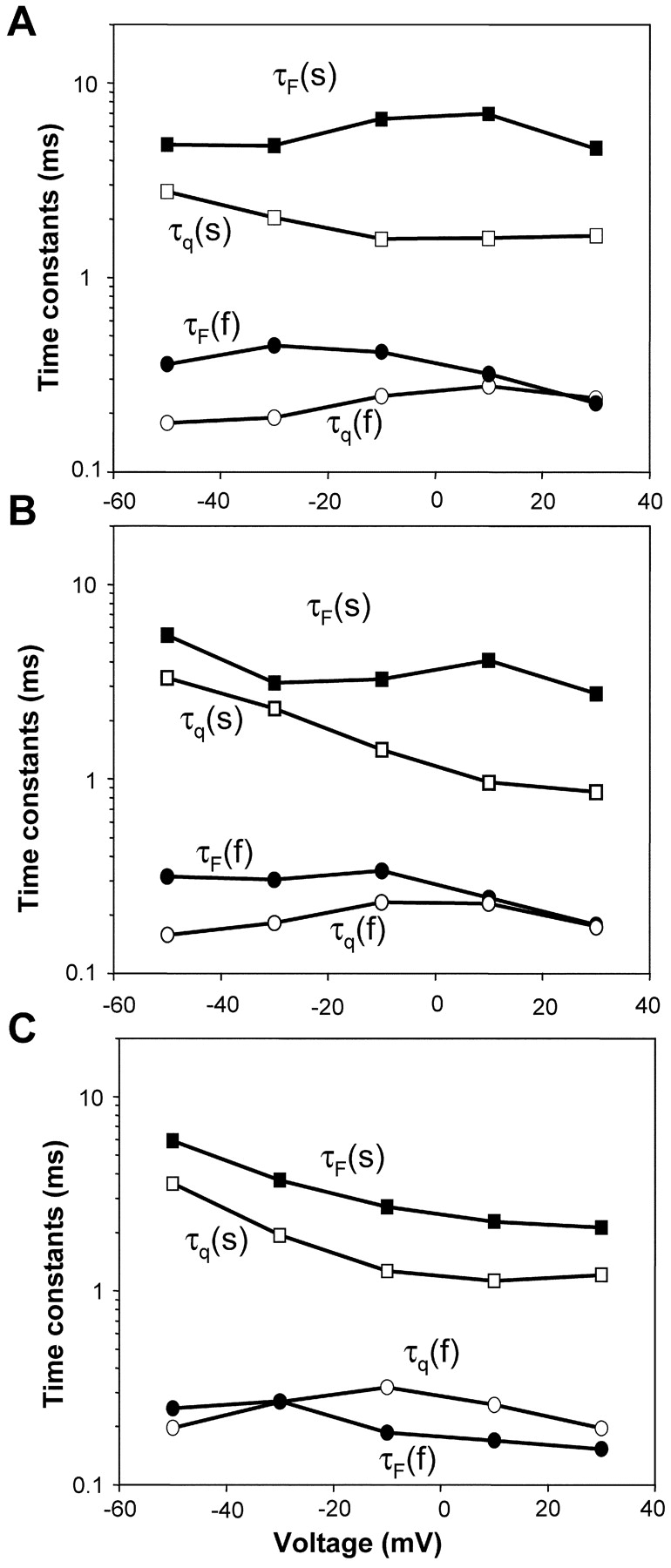
Kinetic characteristics of gating currents and fluorescence change for S216C, S660C, and L1115C. The gating currents and the fluorescence were fitted to a sum of two exponential functions: F(t) or q(t) = a + bexp(−t/τ1) + cexp(−t/τ2), where F(t) and q(t) are the fluorescence rise or the gating current decay as a function of time. The gating currents are shown as unfilled symbols and the fluorescence is shown as filled symbols. (A) Representative time constants of gating currents [slow component, τq(s) and the fast component, τq(f)] and fluorescence [slow component, τF(s) and the fast component, τF(f)] from a S216C-expressing oocyte. (B) Representative time constants of gating and fluorescence [slow component, τF(s) and the fast component, τF(f)] from a S660C-expressing oocyte. (C) Representative time constants of gating and fluorescence [slow component, τF(s) and the fast component, τF(f)] from a L1115C-expressing oocyte.
TABLE II.
Time Constants of Gating and Fluorescence for Various Labeled Mutants
| S216C | S660C | L1115C | S1436C | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
τq
|
τF
|
τq
|
τF
|
τq
|
τF
|
τq
|
τF
|
| n = 4 | n = 3 | n = 3 | n = 3 | n = 4 | n = 3 | n = 4 | n = 4 | ||
| −50 mV | a | 0.27 ± 0.03 | 0.44 ± 0.05 | 0.17 ± 0.06 | 0.47 ± 0.13 | 0.21 ± 0.5 | 0.27 ± 0.03 | 0.17 ± 0.02 | 2.2 ± 0.31 |
| b | 1.79 ± 0.09 | 5.84 ± 0.7 | 2.96 ± 0.25 | 8.32 ± 3.1 | 2.83 ± 0.44 | 4.16 ± 1.25 | 1.7 ± 0.25 | 10.1 ± 2.21 | |
| −30 mV | a | 0.29 ± 0.05 | 0.55 ± 0.07 | 0.20 ± 0.05 | 0.37 ± 0.04 | 0.25 ± 0.11 | 0.23 ± 0.06 | 0.18 ± 0.03 | 1.5 ± 0.20 |
| b | 1.40 ± 0.04 | 5.72 ± 1.24 | 2.2 ± 0.06 | 4.92 ± 2.22 | 1.92 ± 0.26 | 3.29 ± 0.29 | 1.3 ± 0.13 | 6.66 ± 1.39 | |
| −10 mV | a | 0.38 ± 0.1 | 0.48 ± 0.07 | 0.24 ± 0.04 | 0.34 ± 0.04 | 0.27 ± 0.12 | 0.15 ± 0.03 | 0.19 ± 0.03 | 1.17 ± 0.12 |
| b | 1.47 ± 0.16 | 7.3 ± 2.05 | 1.65 ± 0.24 | 5.56 ± 2.5 | 1.5 ± 0.37 | 2.27 ± 0.28 | 1.12 ± 0.13 | 6.51 ± 1.04 | |
| 10 mV | a | 0.39 ± 0.08 | 0.43 ± 0.07 | 0.25 ± 0.04 | 0.26 ± 0.05 | 0.23 ± 0.08 | 0.14 ± 0.04 | 0.16 ± 0.02 | 0.87 ± 0.09 |
| b | 1.89 ± 0.29 | 7.64 ± 0.63 | 1.51 ± 0.4 | 5.26 ± 1.93 | 1.37 ± 0.44 | 2.05 ± 0.17 | 1.08 ± 0.19 | 6.27 ± 0.42 | |
| 30 mV | a | 0.34 ± 0.04 | 0.32 ± 0.06 | 0.19 ± 0.05 | 0.21 ± 0.04 | 0.19 ± 0.04 | 0.16 ± 0.01 | 0.13 ± 0.01 | 0.54 ± 0.02 |
| b | 1.41 ± 0.25 | 5.61 ± 1.07 | 1.4 ± 0.36 | 3.72 ± 1.70 | 1.19 ± 0.25 | 1.74 ± 0.25 | 0.98 ± 0.18 | 4.58 ± 0.4 | |
The time constants are in milliseconds ± SEM (n = number of oocytes). a and b correspond to the fast and the slow time constants, respectively.
Conformational Changes at S1436C Correlate with Slow Component of Gating Current and Precede Inactivation
Next, we compared the fluorescence traces recorded from the site S1436 with gating currents in Fig. 6 A. Superimposed on both fluorescence and gating traces are the double exponential fits to the data. A comparison of time constants of S1436 fluorescence and gating show that the slow component of gating currents correlates with the fast component of fluorescence over most of the voltage range (Fig. 6 B and Table II). Since the slow component of gating current does not correlate with fluorescence from the other domains, this result indicates that the conformational changes at S1436C contribute to most of the slow gating charge movement. Thus, voltage-dependent conformational changes at position S1436C clearly follow charge movement and, hence, reflect movement of the S4 charges in the fourth domain. The slow kinetics of conformational changes at position S1436 is a direct evidence of functional differences between S4 of domain IV and the S4s in other domains.
Figure 6.
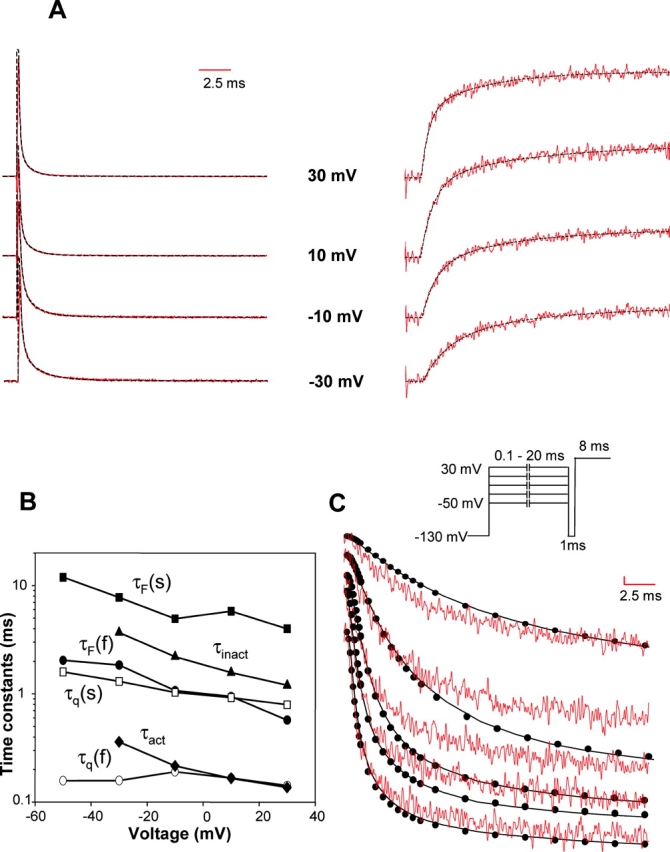
Kinetic characteristics of fluorescence change and gating currents for S1436C. (A) Gating currents traces (left) and the fluorescence traces (right) from a TMRM-labeled S1436C mutant. The superimposed fits (dashed black lines) were generated using a sum of two decaying exponentials. (B) Time constants of fluorescence (slow, τF(s) filled squares and fast, τF(f) circles), charge movement (slow, τq(s), unfilled squares and fast, τq(f), unfilled circles), inactivation (filled triangles), and activation (filled diamonds). The fluorescence and the gating currents were fitted to a sum of two exponentials. Inactivation time constants (τIN) were obtained by fitting the ionic current decay to single exponentials. The activation time constants (τact) were obtained by fitting the ionic current traces with the model shown in Scheme II. (C) Fluorescence decay traces (red) of labeled S1436C channels were compared with development of inactivation (black). The fluorescence was recorded for 20 ms at test potentials varying from −50 to 30 mV after 400 ms prepulse from −130 mV. The development of inactivation was followed by using a two-pulse protocol shown schematically on top of the panel. A conditioning pulse is given to potentials (−50 to 30 mV) where the development of inactivation is to be followed. Each data point (filled circle) represents the peak ionic current at a test potential (50 mV) elicited by varying the length of the conditioning pulse followed by a hyperpolarizing pulse of 1 ms to −130 mV. This 1 ms prepulse to −130 mV resets activation gates without significantly allowing channels to recover from inactivation. The length of the conditioning pulse was incremented by 0.1 ms for pulse lengths up to 1 ms, 0.5 ms for pulse lengths between 1 and 10 ms and 1 ms for pulse lengths between 10 and 20 ms. The fluorescence and the inactivation traces were scaled such that the peak amplitudes were comparable at 30 mV at the end of 20 ms.
Neutralization of charged residues in the S4 of domain IV has been shown to decrease voltage dependence of inactivation kinetics, indicating that S4-DIV may play a unique role in inactivation (Chen et al., 1996). To gain further insight into the role of S4-DIV in inactivation, we compared the time constants of inactivation, obtained by fitting the decay phase of the ionic currents, with the time constants of fluorescence change at S1436 (Fig. 6 B). The plot reveals that inactivation time constants are slower than the slow component of charge movement, as reported previously (Bezanilla and Armstrong, 1975). The protocol for measuring inactivation from the decay of ionic currents follows inactivation in only a subset of channels that are opened by depolarization. Since a significant fraction of sodium channels inactivate without opening, we use a double-pulse protocol to follow the development of inactivation in the total sodium channel population (Gillespie and Meves, 1980; Goldman and Kenyon, 1982). In Fig. 6 C, the time course of voltage-dependent fluorescence changes at S1436 was compared with development of inactivation. The inactivation and fluorescence traces are resolvable at small depolarizations (−50 to −30 mV) with inactivation lagging the S1436C fluorescence. On further depolarization, however, inactivation becomes accelerated and closely correlates with fluorescence changes at S1436. This correlation indicates that, at larger depolarizations, the movement of S4-DIV is the rate-limiting step for fast inactivation. Previous studies have established that the development of inactivation correlates with the time integrals of subtraction gating currents (Sheets and Hanck, 1995). Subtraction gating currents refer to the component of gating currents that disappear on addition of site III toxins and, presumably, reflect the movement of S4 charges in domain IV. However, these comparisons were done at potentials above −20 mV. Our data is in agreement with these results and further reveals that at less depolarized potentials development of inactivation can be kinetically resolved from movement of S4-DIV.
Fluorescence Changes at S1436 Lags in the Sequence of Activation and Is Slower than the Rate of Channel Opening
To establish whether S4s from various domains move in a specific sequence for the Na channel to open, we examined the time course of displacement of individual S4 segments for presence of a lag phase. We hypothesize that the fluorescence change reflects the movement of the S4 where the fluorophore is attached. Thus, the time course of fluorescence can reveal the sequence of activation. If, for instance, the movement of S4 in domain A requires the S4 in domain B to be in an activated position, then the fluorescence signals from S4 domain A should exhibit a lag relative to S4 domain B. On the other hand, if the activation of S4s in the sodium channel were not sequential, then the fluorescence signals from all the S4s would initiate simultaneously. In an attempt to address this issue of sequential activation of S4 segments, we examined the fluorescence signals from the following sites: S216C, S660C, L1115C, and S1436C. These signals, elicited in response to a depolarizing test pulse, were compared over activation voltage range. The comparison shown in Fig. 7 indicates that the fluorescence signals from positions S216C, S660C, and L1115C initiate without any significant delay after the application of a test pulse. This result suggests that the voltage-dependent conformational changes at S216C, S660C, and L1115C initiate in parallel. In contrast, fluorescence changes at S1436 exhibits a clear delay, particularly evident on modest depolarizations to −30 mV. This delay gets shorter on large depolarizations. The delay time is ∼400 μs from the beginning of the depolarizing pulse to −30 mV. More importantly, the delay time is significantly longer than the capacity transient, which is >90% complete by 150 μs (see Fig. 7). The lag in the fluorescence signals at S1436 suggests that the conformational changes at that position occur later in the activation sequence.
Figure 7.
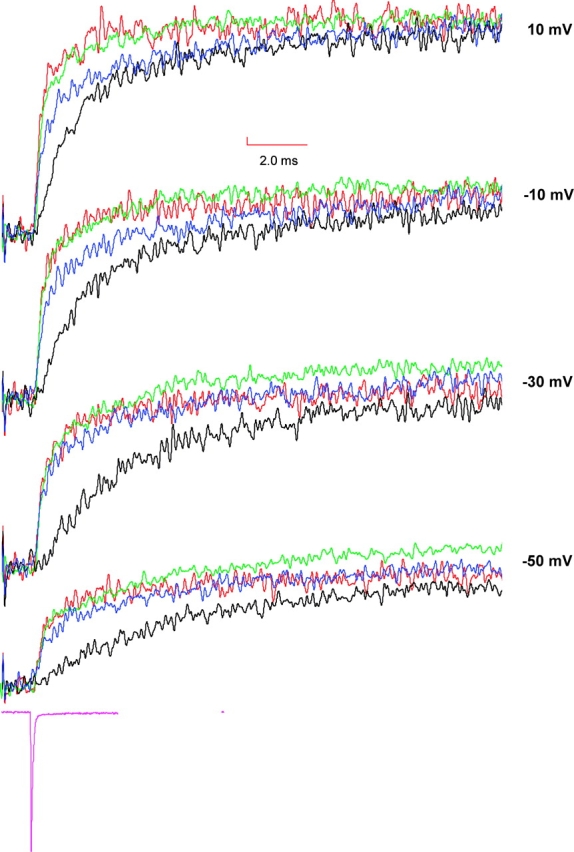
Comparison of fluorescence traces obtained from S216C, S660C, L1115C, and S1436C. Fluorescence signals were recorded by pulsing to test potentials following a prepulse of 400 ms at −130 mV and are color coded in the following manner: S216C in blue, S660C in red, L1115C in green, and S1436C in black. For comparison, the fluorescence traces from various mutants were scaled to have identical steady-state amplitudes at 50 mV. For reference to the clamp speed, a representative capacity transient was obtained by pulsing to −140 mV from −130 mV (in magenta).
Although the fluorescence signals from the sites S216C, S660C, and L1115C initiate simultaneously, the question of whether the conformational changes at those positions initiate simultaneously or sequentially cannot be completely addressed. For instance, we can conceive a kinetic model of a channel where the activation of S4 in domain III requires that the S4s in either domain I or II to be prior activated (see materials and methods, Fig. 8 A). In this channel, the microscopic rate constant of S4 movement in all the three domains is 2.5 ms−1; comparable to the experimentally measured rates at −30 mV for sites S216C, S660C, and L1115C. The simulation shows that, under these circumstances, the delay between fluorescence from S4 domain I and S4 domain II would be clearly resolvable with our data acquisition protocol (Fig. 8 B). Furthermore, the delay is resolvable up until the microscopic rate constant of S4 domain III movement is four times faster than those of S4s in domain I and II. Increasing the rate of S4 domain III movement any further results in merging of the fluorescence signals from S4 domain I with those from S4s in domain I and II. This simulation provides limits within which the sequential activation model can be resolved from a parallel activation model.
The requirement of activation of S4 domain IV in opening of the sodium channel remains highly debated. Gating current recordings in presence of site III toxins reduces the total gating charge displaced by ∼33 percent without affecting activation time constants (Hanck and Sheets, 1995; Sheets and Hanck, 1995). The binding site for these toxins has been localized to the S3-S4 loop of domain IV (Rogers et al., 1996). Together, these results suggest that the activation of S4 charges in domain IV may not be essential for channel opening (Sheets et al., 1999). In contrast, immobilization of S4-DIV using a photocrosslinking reagent has been shown to reduce ionic currents, thereby implying that the voltage-dependent conformational changes of S4-DIV are necessary for channel opening (Horn et al., 2000). Other studies have shown that the neutralization of charged residues in S4-DIV affects the deactivation of a sodium channel (Groome et al., 1999). In Fig. 9 A, we compared the time course of voltage-dependent fluorescence changes at the site S1436 with the time course of channel opening. The rate of channel opening (rising phase of ionic currents) appears faster than the conformational changes at S1436, as monitored by fluorescence. This result is confirmed by comparing the rate constants of activation, extracted from ionic currents, with rate constant of the fast component of fluorescence at S1436 (Fig. 6 B). Over the voltage range, the kinetics of channel opening is faster than the kinetics of fluorescence change at S1436C. This result may be taken as an indication that conformational changes at position S1436 are not essential for channel opening. To further confirm this result, we recorded fluorescence from an additional site near domain IV (Fig. 9 B). The early time courses of fluorescence changes from the position L1439C are identical to S1436C and, therefore, the fluorophore at L1439 does not detect any conformational change that may correspond to the ionic currents. In addition, previous studies at the site equivalent to R1441C (first charge in S4-DIV) in the human skeletal muscle sodium channel have detected only a slow conformational change at that position that may correspond to inactivation (Cha et al., 1999). Thus, unless all the three positions lack a faster component of S4-DIV movement, our data supports the hypothesis that sodium channels do not necessarily require the activation of S4 segment of domain IV to allow ionic current flow.
Figure 9.
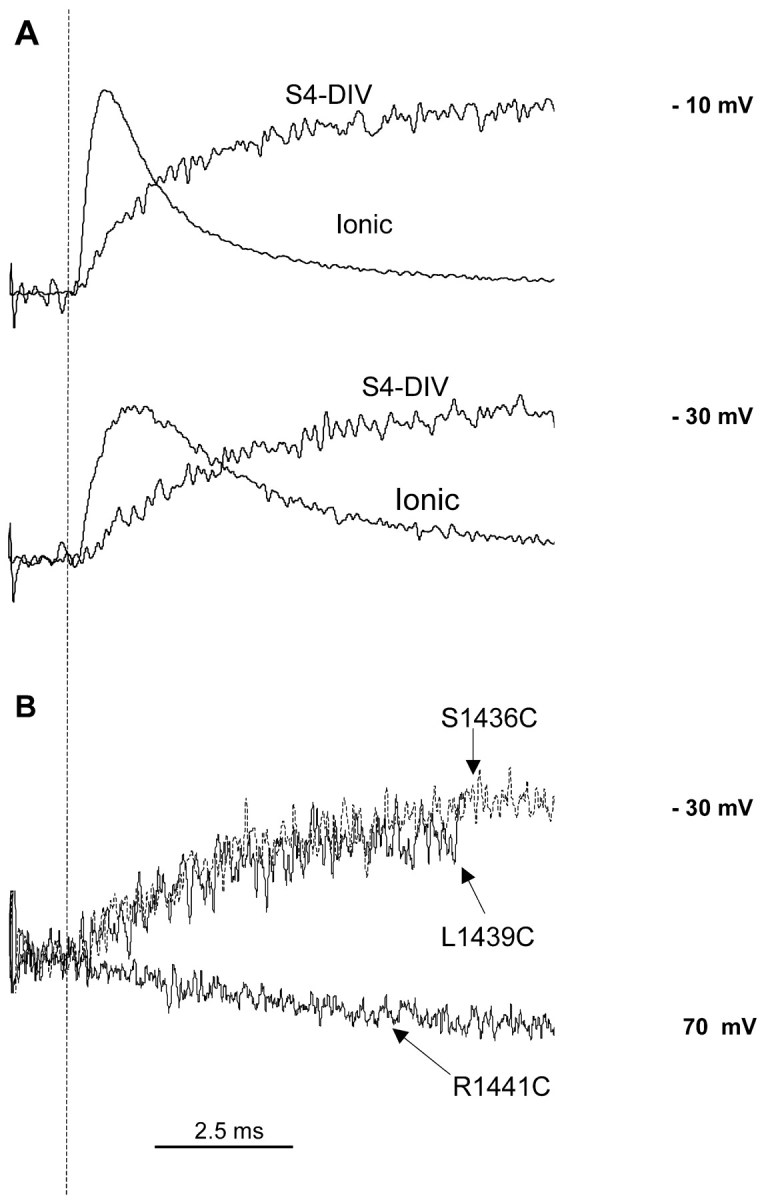
Comparison of ionic currents and fluorescence signals from S1436C. (A) Ionic currents were obtained after subtracting the gating current component. Fluorescence and the ionic traces were scaled to have comparable peak amplitudes at every potential. The beginning of the pulse is marked with a dashed line. (B) Fluorescence traces recorded from three different sites (L1439C, S1436C, and R1441C) on S4 domain IV. The fluorescence data for S1436C and L1439C were recorded by pulsing to −30 mV and that for R1441C was recorded by pulsing to 70 mV.
DISCUSSION
The main purpose of this study was to understand how different S4 segments in the sodium channel contribute to the activation process. Therefore, we characterized the structural changes that take place around the S4 segments of a voltage-gated sodium channel during activation by monitoring local conformational changes using site-specific fluorescence in combination with either gating or ionic current record. Our results indicate that the conformational changes near the various S4 segments of the sodium channel correlates with charge movement. Furthermore, our data provides a direct evidence of the unique kinetic role of S4-DIV during sodium channel activation.
Fluorescence Changes Correlate with Components in the Gating Currents
Do these voltage-dependent conformational changes monitored by fluorescence from specific sites near S4s reflect movement of individual S4 segments in the sodium channel? In an attempt to partially address this question, we first established that the voltage-dependent fluorescence changes from each of the sites have at least one kinetic component that correlates with charge movement. Furthermore, for each S4 segment we measured fluorescence signals from additional sites in the same S4 (Fig. 10) . Our data indicates that at least the fast component of fluorescence appears conserved in different sites within a particular S4 segment (arrows in Fig. 10). It must also be pointed out that many of the positions tested do not show any voltage-dependent fluorescence changes, unlike potassium channels (Fig. 1). In spite of our efforts, it is quite possible that some of the conformational changes in the S4 segment may take place deeper in the S4 segment or are spectroscopically silent. Nonetheless, the correlation between kinetic measurements obtained by two different methods suggests that the voltage dependent fluorescence changes correspond to the movement of a significant fraction of S4 charges in each domain.
Figure 10.

Recordings of voltage-dependent fluorescence changes from additional sites. (Top) Fluorescence change recorded from labeled S216C and I215C by pulsing to 70 mV after a prepulse of 400 ms to −130 mV. (Middle) Fluorescence change recorded from labeled L1115C and S1113C using the same conditions as in A. The fluorescence trace for L1115C was inverted for comparison. (Bottom) Fluorescence change recorded from labeled L1439C and S1436C by pulsing to 70 mV (top) after a prepulse of 400 ms to −130 mV.
Analysis of gating currents from the wild-type skeletal muscle sodium channel reveals that the charge movement can be broadly classified into two components: the fast and the slow charge movement. The faster component of gating charge movement is kinetically comparable to fluorescence changes obtained from sites near voltage sensors of domains I, II, and III. This correlation indicates that the movement of S4 charges in domains I, II, and III contribute to the fast component of gating charge movement. Furthermore, examination of fluorescence-voltage curves suggests that S4 charges in domain III contribute mostly in the early stages of gating followed by S4-DI and by S4-DII. The kinetic data also indicates that S4 charges of domain III contribute in the early stages of charge movement, whereas S4 charges in domain I and II contribute mostly in the later stages. The slow gating charge movement correlates well with the conformational changes at S4-DIV. This suggests that the movement of S4 of domain IV is clearly slower than the S4s in other domains and contributes mostly to the movement of slow gating charge.
Is the Activation of S4 Segments Parallel or Sequential?
Under the assumption that the fluorescence changes follow the movement of individual S4 segments, we have examined the kinetics of activation of sites near the S4s of individual domains with high temporal resolution in order to detect a lag phase. If activation of the domains is sequential, then the domain that activates late will initiate with a lag. The absence of lag phases in the voltage-dependent movement at S4 proximal sites in domains I, II, and III suggests that there is no specific sequence in their activation. Our kinetic simulations indicate that we may be unable to resolve the lag phase even when the S4s activate in a sequential manner if the microscopic rate constants of a late-activating S4 exceeds those of the early-activating S4 by more than four times. Keeping this caveat in mind, we suggest that the S4s of the first three domains do not activate in a specific sequence. The above interpretation of simultaneous activation of S4s in domains I, II, and III is based on the assumption that the voltage-dependent rates measured in this study reflect the microscopic rate constants of voltage-dependent movement. If the movement of the S4s is tightly coupled, then the lag phase in the activation sequence will not be visible. However, under these conditions, the steady-state and the equilibrium properties of activation of S4s measured from different sites should be identical. Since our data indicates otherwise, we suggest that the movement of S4s in the first three domains is not tightly coupled. Therefore, we propose that the activation sequence amongst S4 segments of domains I, II, and III is probabilistic in nature, governed by their rates of activation and their midpoints in the F-V curves. In contrast, the activation of sites proximal to S4 of domain IV shows a clear delay before the onset of fluorescence signals suggesting that the S4 segment of domain IV always moves later in the activation sequence.
Role of Domain IV
Studies in presence of site III toxins suggest that activation of S4 segment in domain IV may not be required for activation (Sheets and Hanck, 1995; Sheets et al., 2000). In contrast, studies involving replacement of charges and immobilization reagents indicate that at least a part of the voltage sensor from domain IV plays an important role in activation (Groome et al., 1999; Horn et al., 2000). According to our data, the kinetics of fluorescence from sites proximal to S4-DIV is slower than the kinetics of activation (determined from ionic currents), suggesting that opening of the channel is not rate limited by activation of S4-DIV. These results may be made compatible with previous reports if we assume that the rat skeletal muscle Na channel has more than one open state and that the activation of S4-DIV may not be required for formation of at least one open state. Since the fluorescence from domain IV correlates with a slow component of gating, we further speculate that domain IV activation may correspond to the formation of a second open state (Armstrong and Bezanilla, 1977).
Inactivation is slower than activation of domain IV at small depolarization and is accelerated on increasing depolarization. However, time constant of fluorescence changes at site S1436C is always faster than the time constant of inactivation. Moreover, the delay in development of inactivation is always larger or equivalent to the delay observed from the fluorescence at S1436C. This data suggests that the inactivation process requires S4-DIV to be activated. However, the activation of S4-DIV is not the only rate-limiting step since at small depolarization, inactivation initiates with a further lag.
It must be pointed out that the fluorescence data from domain IV can also be accounted for with an alternate hypothesis proposed by Horn and his coworkers (Horn et al., 2000). In their model, the S4 of domain IV may move in two steps, where movement of an internal segment is critical for channel opening, whereas movement of an external segment is required for inactivation. As we are only able to monitor the conformational changes on the external side of the S4 of domain IV, we may not detect an activation component contributed by an internal segment, if the segment is not completely rigid. However, the correlation of domain IV fluorescence with the slow component of gating, which accounts for a significant fraction of the total charge, is noteworthy and strongly suggests that the fluorescence signals monitor the movement of most of the S4 segment of domain IV and not a very localized conformational change.
A Kinetic Model of the Na Channel during the Activation and Inactivation Process
Combining the available data, we propose the kinetic model represented schematically in Fig. 11 . This model is similar to the one proposed by Sheets and Hanck (1995). In the first step upon depolarization, S4s in domains I, II, and III activate in parallel. The movement of any one of the S4s in the first three domains to an activated state allows the S4 of domain IV to activate, thereby giving the channel access to six possible states in the next stage. Once S4-DIV is activated, it can initiate inactivation by uncovering a binding site for the inactivation lid, in this manner, activation couples to the inactivation via S4 of domain IV. Therefore, in the third step, the channels can also access three inactivated states. In addition, at this stage at least three of the four domains are in an activated position. If the first three domains are activated, the pore opens and allows ion flux. This represents the first open state (O1). In the fourth step, all four domains are in an activated position and this stabilizes the channel's open state, leading to a second open state (O2). In parallel, at this stage, the channel can access three additional inactivated states. However, in all cases, activation of S4-DIV is a prerequisite for the inactivation lid to bind. Finally, the channel settles into a single inactivated state with the inactivation lid tightly bound and the S4s of all the domains in an activated position. This model can account for the decrease in ionic current that was observed upon immobilization of the S4 DIV in the closed state (Horn et al., 2000). Since inactivation follows activation of S4 DIV, its immobilization will hinder domain IV activation and result in slowing of the inactivation process.
Figure 11.

Proposed kinetic model of sodium channel activation. The S4 segments of each domain are shown as circles that are unfilled in closed state are filled in the activated state. Broadly, the channel exists in two sets of states; those without any inactivation (white bar) are shown in white and those with inactivation particle bound are shown in gray. The opening of the channel is indicated by open circle at the center. The inactivation particle can bind less tightly (slanted bar) or very tightly (horizontal bar). For details, see text.
In this kinetic scheme, we have considered all the possible states with the restrictions imposed by the sequence inferred from the fluorescence data. However, the data also gives us some information on kinetics, namely that the activation is initiated by the fastest moving domain, which is III, followed by II and I. Thus, states where S4-DIV are activated before S4s in DIII, DII, and DI are expected to be rarely populated. This preferred pathway is simpler and is indicated by shading in Fig. 11. It should be emphasized that we cannot completely exclude interaction between domains. Although S4 in DIII appears to be faster than S4s in DII and DI, the measured displacement speeds of these late moving domains may be influenced by the movement of S4 in DIII and the kinetics we measure are the final result of the intrinsic movement of S4s in DII or DI modified by the influence of S4-DIII. This can also be applicable between S4s in DI and DII or even DIV.
It should also be pointed out that this particular sequence applies to experimental conditions described in this paper. The fluorescence data at lower temperature indicates that the temperature coefficient of activation of various S4s differ significantly. For instance, the changes in S4-DI are much slower compared with S4-DII and S4-DIII (unpublished data). Therefore, at low temperatures the S4 segments exhibit a different preferred sequence with S4-DI moving slower than S4-DII. Also, notice that our data does not tell us about the preferred pathway during deactivation, before inactivation takes place. However, once inactivation settles, the deactivation will not occur through any of the pathways indicated in this diagram because S4s in DI and DII can deactivate without waiting for S4s in DIII and DIV to release the inactivating particle (Cha et al., 1999). In that case, S4s in DI and DII will initiate the sequence of deactivation and only after the inactivating particle is released, S4s in DIII and DIV will be able to return to the closed position.
The representation of S4 movement as a single step in this model is probably an oversimplification of the activation process. Fluorescence measurements in the Shaker potassium channel have provided direct evidence of multiple steps involving movement of charges during activation (Sorensen et al., 2000). Measurements from additional sites and at low temperatures may provide evidence for multiple transitions in the movement of S4 of the sodium channel, which will have to be accounted. Nevertheless, this model represents a first step in our attempt to describe the structural features involved in the activation of a voltage-gated sodium channel. In addition, the model makes a number of testable predictions. For instance, perturbations to S4 movement in the first three domains should be clearly manifested in the voltage-dependent S4-DIV kinetics if the S4-DIV follows the activation of S4s in those domains. On the other hand, neutralization of charges on the S4-DIV would be predicted to reduce the slow component of gating charge without affecting the fast component of gating charge movement. Experiments along these lines will extend our understanding of the mechanistic basis of sodium channel activation.
Acknowledgments
We thank Kwame Asamoah, Rikard Blunck, and Dorine Starace for valuable discussion and comments on the manuscript. We would also like to thank Hongyan Guo for technical assistance.
This work was supported by National Institutes of Health grant GM-30376.
References
- Aggarwal, S.K., and R. MacKinnon. 1996. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 16:1169–1177. [DOI] [PubMed] [Google Scholar]
- Aldrich, R.W., D.P. Corey, and C.F. Stevens. 1983. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 306:436–441. [DOI] [PubMed] [Google Scholar]
- Aldrich, R.W., and C.F. Stevens. 1987. Voltage-dependent gating of single sodium channels from mammalian neuroblastoma cells. J. Neurosci. 7:418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and F. Bezanilla. 1977. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70:567–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla, F. 2000. The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80:555–592. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F., and C.M. Armstrong. 1975. Kinetic properties and inactivation of the gating currents of sodium channels in squid axon. Philos. Trans. R. Soc. Lond. B Biol. Sci. 270:449–458. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F., and C.M. Armstrong. 1977. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 70:549–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, A., and F. Bezanilla. 1998. Structural implications of fluorescence quenching in the Shaker K+ channel. J. Gen. Physiol. 112:391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, A., P.C. Ruben, A.L. George, Jr., E. Fujimoto, and F. Bezanilla. 1999. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 22:73–87. [DOI] [PubMed] [Google Scholar]
- Chahine, M., A.L. George, Jr., M. Zhou, S. Ji, W. Sun, R.L. Barchi, and R. Horn. 1994. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 12:281–294. [DOI] [PubMed] [Google Scholar]
- Chen, L.Q., V. Santarelli, R. Horn, and R.G. Kallen. 1996. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J. Gen. Physiol. 108:549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun, D., and F.J. Sigworth. 1995. Fitting and statistical analysis of single channel records. In Single-Channel Recording. B. Sakmann and E. Neher, editors. Plenum Press, New York. 483–587.
- Gillespie, J.I., and H. Meves. 1980. The time course of sodium inactivation in squid giant axons. J. Physiol. 299:289–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, L., and J.L. Kenyon. 1982. Delays in inactivation development and activation kinetics in myxicola giant axons. J. Gen. Physiol. 80:83–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, L., and C.L. Schauf. 1972. Inactivation of the sodium current in Myxicola giant axons. Evidence for coupling to the activation process. J. Gen. Physiol. 59:659–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groome, J.R., E. Fujimoto, A.L. George, and P.C. Ruben. 1999. Differential effects of homologous S4 mutations in human skeletal muscle sodium channels on deactivation gating from open and inactivated states. J. Physiol. 516(Pt 3):687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanck, D.A., and M.F. Sheets. 1995. Modification of inactivation in cardiac sodium channels: ionic current studies with Anthopleurin-A toxin. J. Gen. Physiol. 106:601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 1976. Gating in sodium channels of nerve. Annu. Rev. Physiol. 38:139–152. [DOI] [PubMed] [Google Scholar]
- Hirschberg, B., A. Rovner, M. Lieberman, and J. Patlak. 1995. Transfer of twelve charges is needed to open skeletal muscle Na+ channels. J. Gen. Physiol. 106:1053–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin, A.L., and A.F. Huxley. 1952. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117:500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn, R., S. Ding, and H.J. Gruber. 2000. Immobilizing the moving parts of voltage-gated ion channels. J. Gen. Physiol. 116:461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis, K.J., and A.L. Goldin. 1997. Sodium channel inactivation is altered by substitution of voltage sensor positive charges. J. Gen. Physiol. 110:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis, K.J., A. Rounaghi, and A.L. Goldin. 1997. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J. Gen. Physiol. 110:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak, M. 1991. Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 266:19867–19870. [PubMed] [Google Scholar]
- Noda, M., T. Ikeda, H. Suzuki, H. Takeshima, T. Takahashi, M. Kuno, and S. Numa. 1986. Expression of functional sodium channels from cloned cDNA. Nature. 322:826–828. [DOI] [PubMed] [Google Scholar]
- Papazian, D.M., L.C. Timpe, Y.N. Jan, and L.Y. Jan. 1991. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 349:305–310. [DOI] [PubMed] [Google Scholar]
- Rogers, J.C., Y. Qu, T.N. Tanada, T. Scheuer, and W.A. Catterall. 1996. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 271:15950–15962. [DOI] [PubMed] [Google Scholar]
- Seoh, S.A., D. Sigg, D.M. Papazian, and F. Bezanilla. 1996. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 16:1159–1167. [DOI] [PubMed] [Google Scholar]
- Sheets, M.F., and D.A. Hanck. 1995. Voltage-dependent open-state inactivation of cardiac sodium channels: gating current studies with Anthopleurin-A toxin. J. Gen. Physiol. 106:617–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets, M.F., J.W. Kyle, and D.A. Hanck. 2000. The role of the putative inactivation lid in sodium channel gating current immobilization. J. Gen. Physiol. 115:609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets, M.F., J.W. Kyle, R.G. Kallen, and D.A. Hanck. 1999. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys. J. 77:747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, J.B., A. Cha, R. Latorre, E. Rosenman, and F. Bezanilla. 2000. Deletion of the S3-S4 linker in the Shaker potassium channel reveals two quenching groups near the outside of S4. J. Gen. Physiol. 115:209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace, D.M. E. Stefani, and F. Benzanilla. 1997. Voltage dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron. 19:1219–1327. [DOI] [PubMed] [Google Scholar]
- Stefani, E., and F. Bezanilla. 1998. Cut-open oocyte voltage-clamp technique. Methods Enzymol. 293:300–318. [DOI] [PubMed] [Google Scholar]
- Stevens, C.F. 1978. Interactions between intrinsic membrane protein and electric field. An approach to studying nerve excitability. Biophys. J. 22:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer, W., F. Conti, H. Suzuki, X.D. Wang, M. Noda, N. Yahagi, H. Kubo, and S. Numa. 1989. Structural parts involved in activation and inactivation of the sodium channel. Nature. 339:597–603. [DOI] [PubMed] [Google Scholar]
- Vandenberg, C.A., and F. Bezanilla. 1991. A sodium channel gating model based on single channel, macroscopic ionic, and gating currents in the squid giant axon. Biophys. J. 60:1511–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]