

Abstract
The objective of this study was to investigate the mechanism of uridine 5′-triphosphate (UTP)-dependent inhibition of Na+ absorption in porcine endometrial epithelial cells. Acute stimulation with UTP (5 μM) produced inhibition of sodium absorption and stimulation of chloride secretion. Experiments using basolateral membrane–permeabilized cell monolayers demonstrated a reduction in benzamil-sensitive Na+ conductance in the apical membrane after UTP stimulation. The UTP-dependent inhibition of sodium transport could be mimicked by PMA (1 μM). Several PKC inhibitors, including GF109203X and Gö6983 (both nonselective PKC inhibitors) and rottlerin (a PKCδ selective inhibitor), were shown to prevent the UTP-dependent decrease in benzamil-sensitive current. The PKCα-selective inhibitors, Gö6976 and PKC inhibitor 20–28, produced a partial inhibition of the UTP effect on benzamil-sensitive Isc. Inhibition of the benzamil-sensitive Isc by UTP was observed in the presence of BAPTA-AM (50 μM), confirming that activation of PKCs, and not increases in [Ca2+]i, were directly responsible for the inhibition of apical Na+ channels and transepithelial Na+ absorption.
Keywords: P2Y receptor, ENaC, benzamil, chloride secretion, calcium-activated chloride channel
INTRODUCTION
In porcine endometrium, surface epithelial cells exhibited a basal Na+ absorption that was inhibited by amiloride and regulated by PGF2α and cAMP (Vetter et al., 1997). More recent studies using glandular endometrial epithelial cells in culture showed distinct basal transport phenotypes depending on the growth factor used to maintain the cells (Deachapunya et al., 1999; Deachapunya and O'Grady, 2001). Basal benzamil–sensitive sodium absorption was characteristic of glandular endometrial cell monolayers grown under serum-free conditions, or in the presence of insulin or insulin-like growth factor (IGF-1).* Acute stimulation with insulin or IGF-1 enhanced sodium absorption by increasing Na+-K+ ATPase transport activity and basolateral membrane K+ conductance, without increasing the apical membrane benzamil-sensitive Na+ conductance. The enhanced Na+-K+ ATPase activity was due to activation of phosphatidylinositol 3-kinase and subsequent stimulation of a protein phosphatase. Long-term treatment with insulin additionally enhanced sodium absorption with a further increase in Na+-K+ ATPase activity and benzamil-sensitive Na+ conductance.
P2Y receptors have been identified on the apical membrane of both mouse and porcine endometrial cells (Chan et al., 1997; Palmer-Densmore et al., 2000). Most P2Y receptor subtypes are G-protein receptors that couple to Gq and activate PLC upon agonist stimulation. PLC activation subsequently initiates both diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) production, resulting in activation of PKC and increases in intracellular calcium concentrations ([Ca2+]i). Previous studies have shown that ATP or uridine 5′-triphosphate (UTP)-dependent increases in [Ca2+]i stimulate chloride secretion in both porcine and mouse endometrial epithelial cells (unpublished data; Chan et al., 2000). In these cells, a DIDS-sensitive, Ca2+-activated anion channel has been identified in the apical membrane and is an essential component of Ca2+-dependent anion secretion.
P2Y receptor activation has been found to decrease sodium absorption in mouse endometrial epithelial cells (Wang and Chan, 2000). In addition, UTP was found to inhibit sodium absorption in wild-type and mutant CFTR–expressing human bronchial epithelia, as well as nasal epithelia from normal and cystic fibrosis patients (Devor and Pilewski, 1999; Mall et al., 2000). However, the mechanism for UTP-dependent inhibition of sodium absorption is not well understood. In the present study, we investigated the effects of UTP on the Na+ transport properties of primary endometrial epithelial cells grown in the absence of serum or presence of insulin. UTP was found to produce a dramatic inhibition of Na+ transport that involved a decrease in apical membrane Na+ conductance. We demonstrated that inhibition of benzamil-sensitive Na+ channels was dependent on PKC activation and was not directly dependent on increases in [Ca2+]i.
MATERIALS AND METHODS
Materials
UTP, 1,2-bis (2-aminophenoxy) ethane–N, N, N′, N′-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM), insulin, indomethacin, nonessential amino acid, and high purity grade salts were purchased from Sigma-Aldrich. 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) was purchased from Research Biochemical International and benzamil was from Molecular Probes. Gö6983, Gö6976 and rottlerin were obtained from Biomol. PKC inhibitor 20–28, cell permeable, myristoylated peptide was obtained from Calbiochem). Dulbecco's modified Eagle's medium (DMEM), Dulbecco's phosphate buffer saline (DPBS), FBS, collagenase type 1, kanamycin, penicillin-streptomycin, and fungizone were purchased from GIBCO BRL.
Cell Isolation and Culture
The epithelial glands were isolated from pig uterus as described previously (Deachapunya and O'Grady, 1998). The isolated glands were suspended in DMEM supplemented with 3.7 g/L sodium bicarbonate, 10% FBS, 5 μg/ml insulin, 1% nonessential amino acid, 5 μg/ml fungizone, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 μg/ml kanamycin (standard media). They were plated onto cell culture dishes and incubated at 37°C in a humidified atmosphere of 5% CO2 in air. Culture medium was changed after 24 h and then every 2–3 d. After 80% confluency (within 2–3 d), the stromal cells were easily removed by trypsinization for 5 min and the epithelial cells were further trypsinized and subcultured onto 24 mm (4.5 cm2) transparent permeable membrane filters (CoStar). After plating on filters, monolayers were fed every 2 d and maintained in standard media for 7 d. The standard media was subsequently replaced with DMEM (serum-free media) or DMEM supplemented with 850 nM insulin for 3 d.
Measurement of Monolayer Electrical Properties
Transepithelial resistance of the cell monolayers was measured using the EVOM epithelial voltohmmeter coupled to Ag/AgCl “chopstick” electrodes (World Precision Instruments). High resistance monolayers (∼3000 Ωcm2) were mounted in Ussing Chambers, bathed on both sides with standard porcine saline solution containing (in mM): 130 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 20 NaHCO3, 0.3 Na H2PO4, 1.3 Na2HPO4, pH 7.4, which was maintained at 37°C and bubbled with 95% O2/5% CO2. Transepithelial potential difference, monolayer conductance and short circuit current (Isc) were measured with the use of voltage-clamp circuitry from JWT Engineering. The data from the voltage clamp experiments was digitized, stored, and analyzed using Workbench data acquisition software (Kent Scientific Corporation) and recorded with a Pentium microcomputer. All cells were pretreated with indomethacin (10 μM) added to both apical and basolateral solutions at least 10 min before the beginning of the experiment to eliminate endogenous prostaglandin production. PGE2 is endogenously produced by porcine endometrial epithelial cells and has been shown to regulate Cl− secretion (Deachapunya and O'Grady, 1998).
For experiments involving measurement of membrane conductance, amphotericin B (10 μM) was used to permeabilize the apical or basolateral membranes of the monolayers mounted in Ussing chambers. The permeabilized membranes were bathed with KMeSO4 saline solution containing (in mM): 120 KMeSO4, 5 NaCl, 3 calcium gluconate, 1 MgSO4, 20 KHCO3, 0.3 KH2PO4, 1.3 K2HPO4, 30 mannitol, pH 7.4, whereas the intact membranes were bathed with standard porcine saline solution. A World Precision Instrument epithelial voltage clamp was used to voltage clamp the monolayers. The data was recorded using a Pentium microcomputer with an Axon Instruments, Inc. TL-1 interface. Voltage step commands and the resultant currents were generated and recorded using pCLAMP 8 software from Axon Instruments, Inc. Current-voltage relationships were obtained by a series of voltage step commands described in the figure legends. The compound-sensitive current components were obtained by subtracting currents before and after addition of UTP or blocker compounds.
Measurement of Intracellular Calcium
Cells were seeded at low density on glass coverslips for 48 h. After adhesion to the coverslips, the cells were washed with Hanks balanced salt solution (HBSS) containing 10 mM glucose, 10 mM HEPES, pH 7.4. The cells were then loaded with 10 μM fura-2-AM (Molecular Probes) for 45 min at 37°C, washed in HBSS, mounted onto a Plexiglas chamber (150 μl volume; Warner Instruments), and transferred to the stage of a Nikon Diaphot inverted microscope with an epifluorescence attachment. The chamber was perfused with HBSS at 2–3 ml/min at room temperature. Fluorescence in single cells was visualized using a Nikon UV-fluor 40× oil-immersion objective. The fluorescence excitation, image acquisition, and real-time data analyses were controlled by the Image-1 Metamorph software (Universal Imaging) running on a Pentium microcomputer. [Ca2+]i was measured as the ratio of fluorescence emitted at 510 nm when the cells are alternately excited at 340 and 380 nm [F340/F380].
Western Blot Analysis
Cell monolayers were solubilized with lysis buffer (50 mM Tris-HCl, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 1 μg/ml aprotinin, and 1 mM NaF, pH 7.4) at 37°C for 30 min. A protein assay was performed using a bicinchoninic acid (BCA) protein assay kit by Pierce Chemical Co. Proteins were separated by PAGE (8%). Electroblotting was done using Immobilon-P from Millipore Corporation. The electroblot assembly was placed into the electroblotting apparatus, the Trans-Blot Cell, from Bio-Rad Laboratories, and blotting was performed at 16 V overnight on ice. After the blots were removed, they were washed twice, and then blocked in freshly prepared 1X TBS-tween containing 3% nonfat dry milk (MLK) for 1 h at 20–25°C with constant agitation. After washing, blots were reacted overnight in primary antibody, 15 ml freshly prepared 1× TBS-tween containing 3% MLK with appropriate dilution of the primary antibody, rabbit anti-human PKCα, β11, β2, δ, ζ, ɛ, θ, μ, η polyclonal antibody from Santa Cruz Biotechnology, Inc. The next day, blots were washed and reacted with secondary antibody, alkaline phosphatase–labeled goat anti–rabbit from Santa Cruz Biotechnology, Inc. Secondary antibody was diluted 1:3,000 in 1X TBS-tween containing 3% MLK and was reacted for 1 h. After washing, alkaline phophatase color reagent was added to 100 ml 1× alkaline phosphatase color development buffer from Bio-Rad Laboratories at room temperature. Blots were incubated in development buffer until bands were clearly developed.
Statistics
All values are presented as means ± SEM, n is the number of monolayers and N is the number of animals in each experiment. The differences between control and treatment means were analyzed using a t test for paired and unpaired means where appropriate. A value of P < 0.05 was considered statistically significant.
RESULTS
Acute Effects of UTP on Sodium Absorption and Chloride Secretion
The basal electrical properties of cultured porcine endometrial epithelial cells have been previously described (Deachapunya and O'Grady, 1998, 2001; Deachapunya et al., 1999). To maximize basal sodium absorption, cells were cultured under serum-free conditions in the presence of insulin for 3 d. To determine the acute effects of UTP on basal sodium absorption and chloride secretion, cell monolayers were mounted in Ussing chambers and bathed on both sides with standard porcine saline solution. In Fig. 1 A, the basal short circuit current (Isc) was predominantly benzamil-sensitive, and the Cl− channel inhibitor, NPPB, blocked the remaining Isc. After the addition of UTP (5 μM), the new steady-state Isc was predominantly NPPB sensitive (Fig. 1 B), whereas the benzamil-sensitive Isc was nearly abolished after stimulation with UTP. Pretreatment with benzamil (5 μM) did not prevent the increase in NPPB-sensitive Isc produced by UTP (Fig. 1 C).
Figure 1.
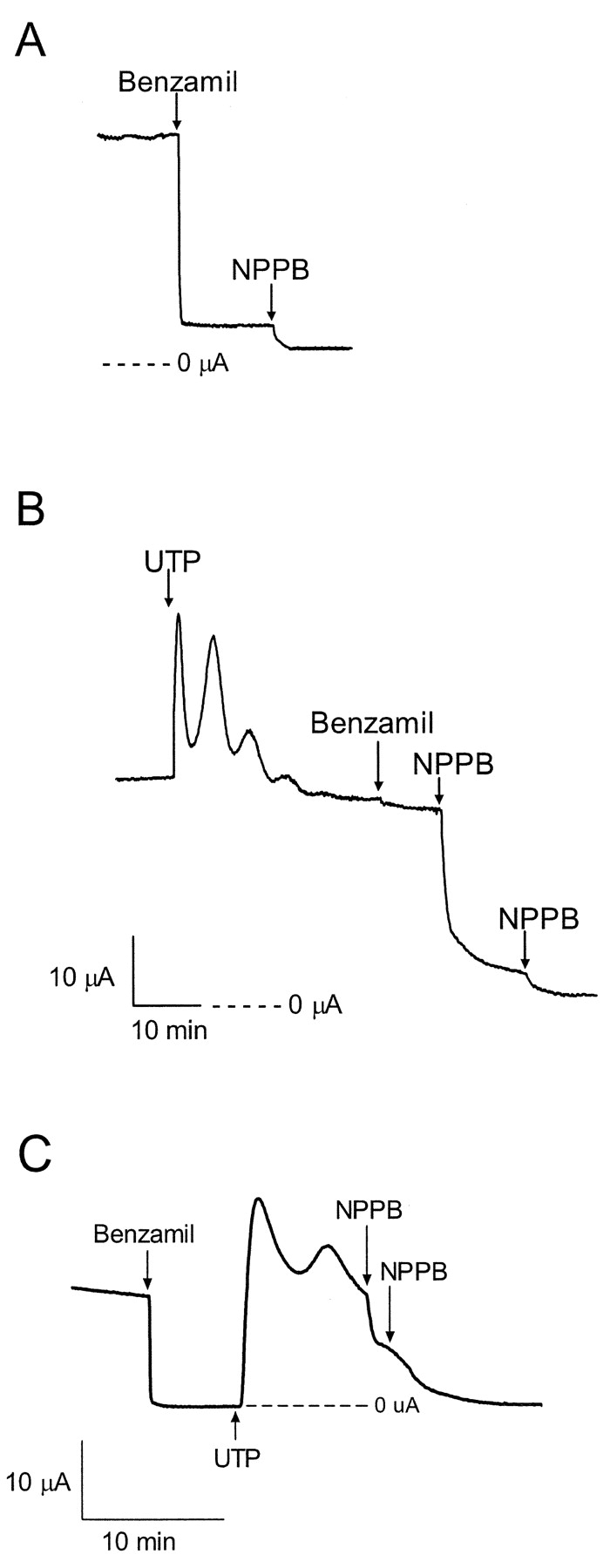
Effect of UTP on basal sodium transport. (A) Representative trace showing that addition of 5 μM benzamil to the apical solution blocked most of the basal Isc in monolayers maintained under serum free conditions, (n = 9, N = 4). (B) Apical addition of UTP (1 μM) caused a rapid increase in Isc followed by a slow decrease back to the basal Isc. Subsequent addition of benzamil had little inhibitory effect, but addition of NPPB (100 μM at each arrow) blocked all of the remaining Isc, (n = 15, N = 4). The scale bar applies to both Fig. 1, A and B. (C) After pretreatment with benzamil (5 μM), apical addition of UTP (5 μM) caused a rapid increase in Isc, similar to what is shown Fig. 1 B. Addition of NPPB (100 μM at each arrow) blocked all of the remaining Isc, (n = 6). Statistical analysis is provided in Fig. 6.
PMA Mimics the Effects of UTP on Inhibition of Sodium Absorption
To illustrate further the inhibition of sodium absorption by UTP, cells were maintained under serum-free conditions and acutely stimulated with insulin (850 nM). Previous studies have characterized the acute insulin response as an increase in benzamil-sensitive sodium absorption resulting from enhanced Na+-K+-ATPase activity and an increase in basolateral membrane K+ conductance (Deachapunya et al., 1999). As shown in Fig. 2 A, addition of UTP (1 μM) inhibited the insulin-stimulated Isc and part of the basal Isc (basal Isc = 19 ± 2, insulin-stimulated Isc = 43 ± 5 and remaining Isc after UTP = 13 ± 1, n = 4). This effect was mimicked by PMA (1 μM), an activator of PKC, (Fig. 2 B; basal Isc = 21 ± 2, insulin-stimulated Isc = 44 ± 4, and remaining Isc after UTP = 7 ± 2, n = 4). To determine whether increases in intracellular calcium were responsible for PMA-mediated inhibition of sodium absorption, calcium-imaging experiments with fura 2–loaded primary endometrial cells were conducted. Addition of PMA (1 μM) failed to show a detectable increase in intracellular calcium, whereas a concentration-dependent increase in [Ca2+]i was observed after stimulation with 1 and 5 μM UTP (Fig. 2 C).
Figure 2.
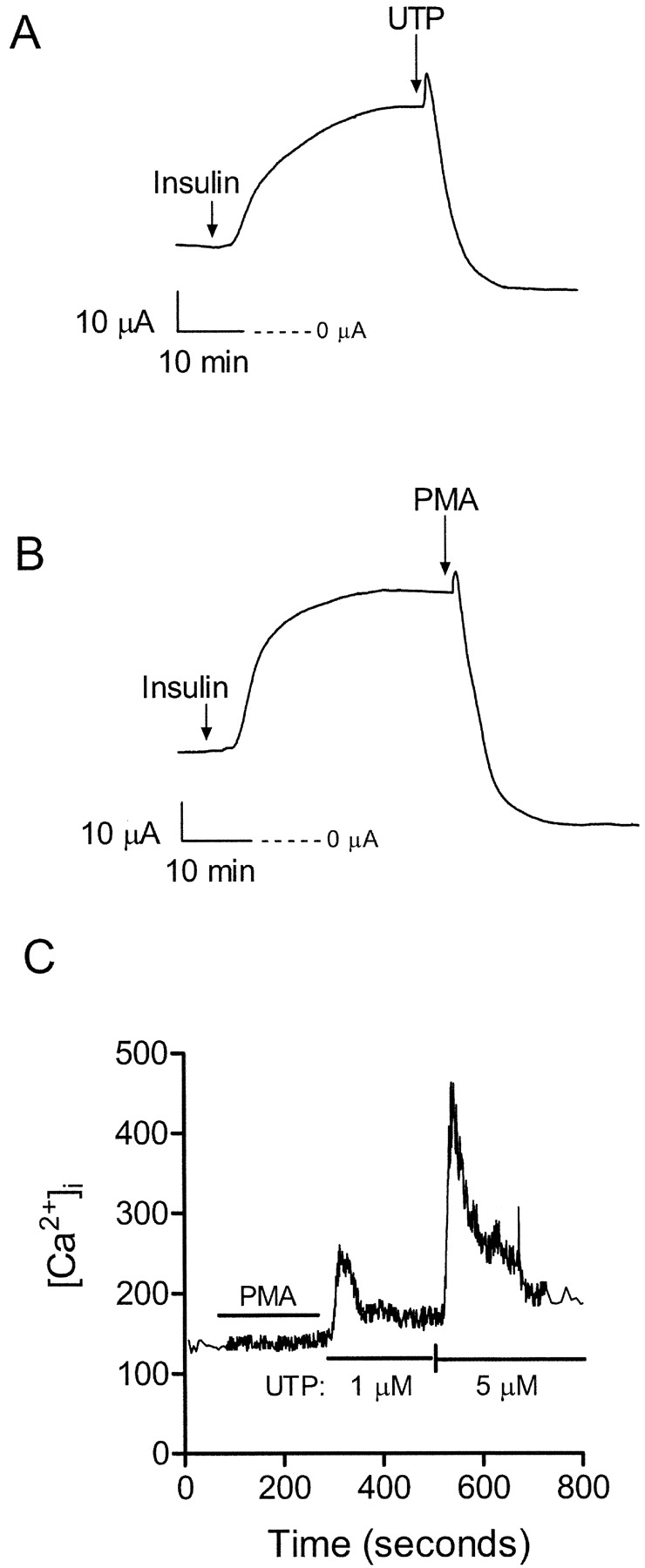
Effects of UTP and PMA on insulin-stimulated Na+ transport. (A) Representative trace showing the time-dependent increase in Isc stimulated by 850 nM insulin added to the basolateral solution. Addition of 1 μM UTP to the apical solution inhibited both the insulin-stimulated and basal Isc. See results for mean ± SEM data for basal Isc, insulin-stimulated Isc and residual Isc after UTP. (B) Addition of 1 μM phorbol 12-myristate 13-acetate (PMA) produced a similar decrease in insulin-stimulated and basal current as observed with UTP. See results for mean ± SEM data for basal Isc, insulin-stimulated Isc, and residual Isc after PMA. (C) Temporal changes in [Ca2+]i in response to PMA and UTP were determined using fura 2-AM as described in materials and methods. Transient elevations in [Ca2+]i were observed after 1 and 5 μM UTP in Ca2+-containing HBSS. No elevation in [Ca2+]i was detected after 1 μM PMA. This data represents the mean trace calculated using calcium measurements from 20 different cells.
Effects of UTP on Sodium Transport Across the Apical Membrane
To investigate the effects of UTP on apical membrane Na+ conductance, benzamil-sensitive difference currents were determined from basolateral membrane–permeabilized monolayers. Apical membrane currents were elicited using a voltage step protocol from −100 to 95 mV in 15-mV increments at a holding potential of 0 mV. Benzamil (5 μM) was added to the apical solution in the absence (control) or presence of 5 μM UTP. The representative traces in Fig. 3 A show the benzamil-sensitive difference current without UTP (top trace) and in the presence of apical UTP (bottom trace). Fig. 3 B represents the benzamil-sensitive current-voltage relationship before and after UTP (1 μM), where a decrease in apical membrane conductance was apparent after comparing the UTP-stimulated I-V relationship to unstimulated controls. Mean reversal potentials for benzamil-sensitive currents were 66.1 ± 4.2 mV (n = 5, N = 3) for control and 57.9 ± 6.3 mV (n = 6, N = 3) after UTP, and were not significantly different.
Figure 3.
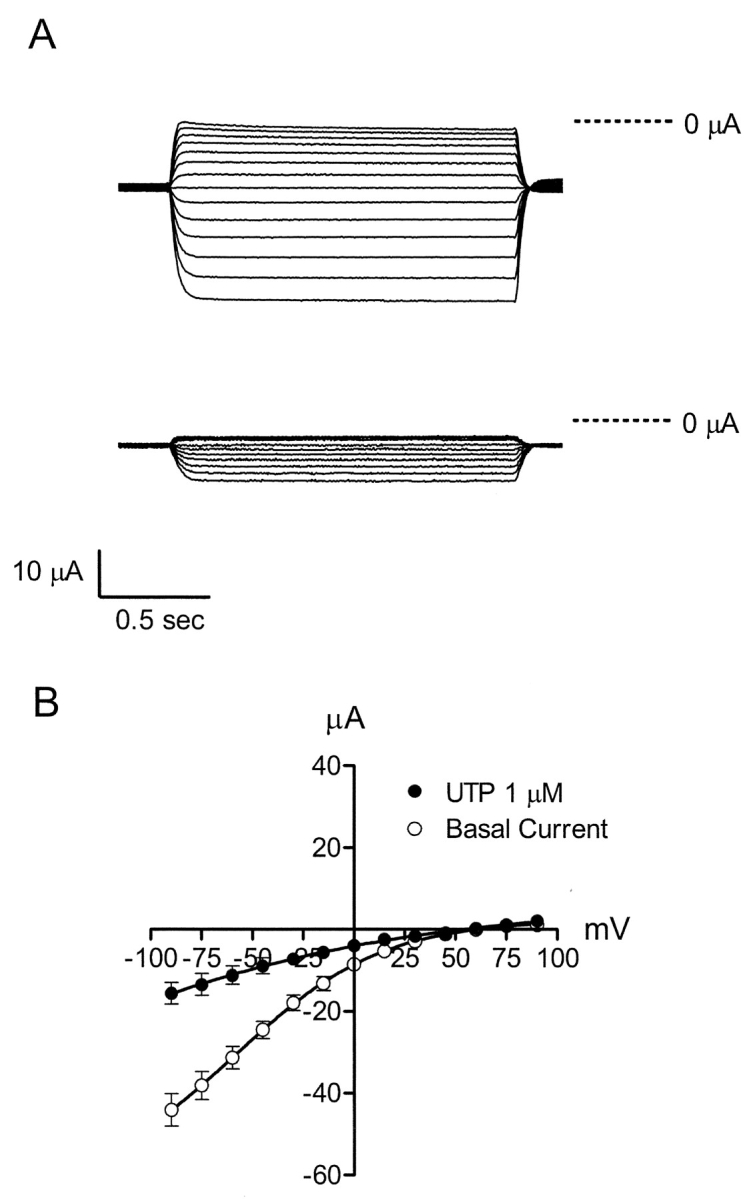
Current-voltage relationships for the benzamil-sensitive pathway in the apical membrane. (A) Experiments were performed using monolayers cultured in serum-free media with insulin for 3 d. Benzamil-sensitive difference currents were obtained using a voltage step protocol that ranged from −100 to 95 mV in 15-mV increments from a holding potential of 0 mV. Benzamil (5 μM) was added apically to either permeabilized control cell monolayers (top trace) or to permeabilized monolayers activated with 5 μM UTP (bottom trace). (B) Benzamil-sensitive current-voltage relationships were obtained in response to a voltage step protocol from −90 to 90 mV in 15-mV increments from a holding potential of 0 mV. Benzamil (5 μM) was added to the apical solution in the absence or presence of UTP (1 μM).
Chloride-dependent Effects of UTP
Cell monolayers were placed in Ussing chambers and bathed on both sides with either chloride-containing or chloride-free saline solutions. A reduction in peak UTP-activated current was evident when comparing monolayers bathed in chloride-free solution to control cell monolayers (Fig. 4 A). In addition, a large reduction in NPPB-sensitive current also occurred, whereas no change in the benzamil-sensitive current was detected (Fig. 4 B). The effect of UTP on apical chloride conductance is shown in Fig. 5 . UTP (1 μM) produced a rapid rise and gradual fall in apical membrane current (Fig. 5 A). The UTP-activated current was outwardly rectifying and chloride dependent (Fig. 5 B). The voltage protocol used to obtain the UTP-activated current-voltage relationship ranged from −90 to 90 mV in 15-mV increments from a holding potential of 0 mV. The mean reversal potential was −28.4 ± 1.3 mV (n = 6, N = 4).
Figure 4.

Effect of UTP on Isc in the presence or absence of chloride. (A) Representative trace showing chloride dependence of the UTP (5 μM) response. Monolayers were bathed in standard porcine saline solution or chloride-free solution, replacing chloride with methane sulfonate. The peak UTP-activated current was reduced under chloride-free conditions (n = 6). (B) Bar graph showing the benzamil (5 μM) and NPPB (100 μM) -sensitive currents in the absence or presence of chloride. The NPPB-sensitive current was reduced under chloride-free conditions, whereas the benzamil-sensitive current remained the same, (n = 6 for each). Asterisk indicates significant difference between the NPPB-sensitive Isc under control and Cl−-free conditions.
Figure 5.
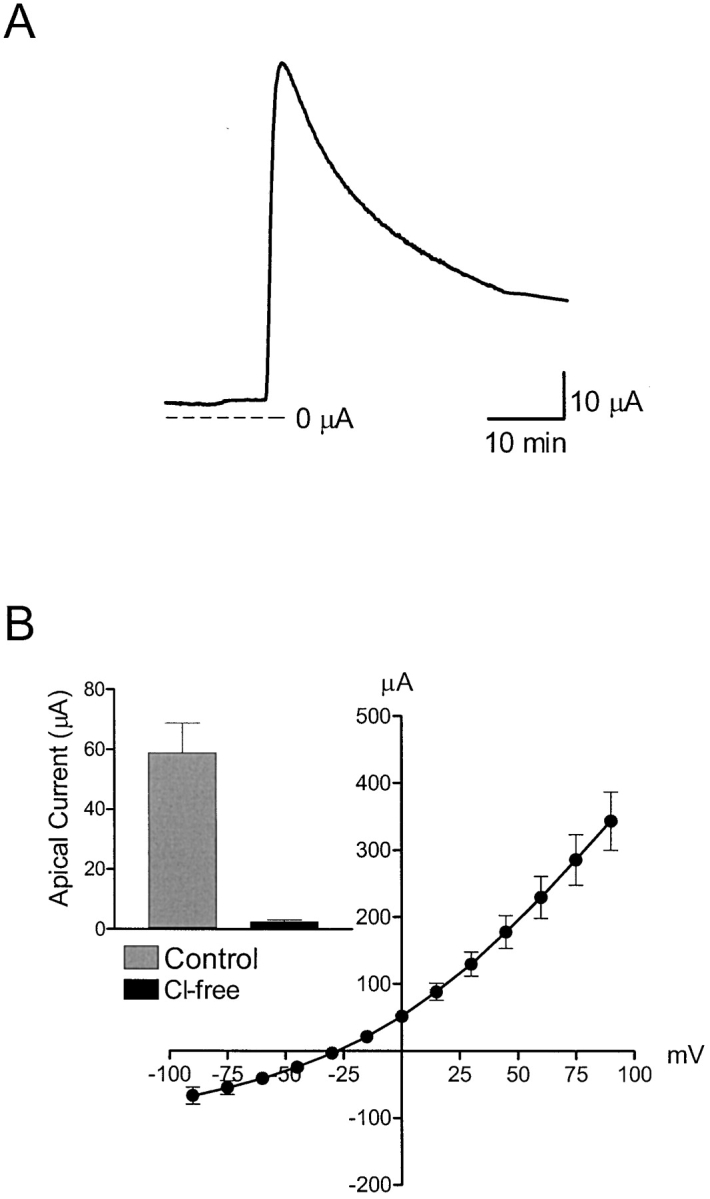
Effect of UTP on apical membrane Cl- conductance. (A) Representative trace showing the transient change in apical membrane current after stimulation with 1 μM UTP using monolayers where the basolateral membrane was permeabilized with amphotericin B (10 μM). The basolateral membrane was bathed with KMeSO4 saline solution, while the apical membrane was bathed with standard porcine saline solution. (B) Current-voltage relationship showing the UTP-activated current obtained in response to voltage steps from −90 to 90 mV in 15-mV increments from a holding potential of 0 mV.
PKC Regulation of Sodium Absorption
To study the possible role of PKC in UTP regulation of sodium absorption, a variety of PKC inhibitors were used in an attempt to block UTP-dependent inhibition of the benzamil-sensitive current. Fig. 6, A and B , illustrate preservation of the benzamil-sensitive current after pretreatment with four different PKC inhibitors. Gö6983, a nonselective PKC inhibitor, and rottlerin, a selective inhibitor of PKCδ, significantly decreased the effects of UTP on benzamil-sensitive Isc, (n = 9 and 5, respectively, N = 2). In addition, 100 nM and 10 μM Gö6976, a PKCα selective inhibitor, partially blocked the effects of UTP on the benzamil-sensitive current, (n = 6 and 4 respectively, N = 2). A similar result was obtained using a cell-permeable, myristoylated PKCα and β inhibitory peptide 20–28 (10 μM; Calbiochem) that also produced partial block of UTP-dependent inhibition of the benzamil-sensitive Isc (n = 4). PMA (0.5 μM) mimicked UTP inhibition of the benzamil-sensitive current as shown previously in Fig. 2, (n = 4 and 14, respectively, N =3). The effects of various PKC blockers on UTP-dependent inhibition of the benzamil-sensitive apical Na+ conductance are shown in Fig. 7 . These experiments were performed on monolayers that were permeabilized with amphotericin B added to the basolateral surface of the cells. The UTP effect was abolished when monolayers were pretreated with the nonselective PKC inhibitor, GF109203X (1 μM, Fig. 7 A). However, pretreatment with the PKCδ selective inhibitor, rottlerin, significantly attenuated the effects of UTP, but did not completely abolish the effect. In addition, pretreatment with the PKCα selective inhibitor, Gö6976, also produced partial block of the UTP effect on apical membrane current. These effects were consistent with the results obtained from Isc measurements, indicating that apical Na+ channels are specific sites of PKC action.
Figure 6.

PKC inhibitors prevent decrease of benzamil-sensitive Isc by UTP. (A) Representative trace showing the benzamil-sensitive current in the absence or presence UTP (5 μM), and after pretreatment with both Gö6983 (10 μM) and UTP, (n = 8, 14, and 9, respectively, N = 3). (B) Bar graph illustrating the benzamil-sensitive current after benzamil only (5 μM, n = 8), after UTP (5 μM, n = 14), after PMA (0.5 μM, n = 4), after UTP in the presence of Gö6983 (10 μM, n = 9), after UTP in the presence of rottlerin (2.5 μM, n = 5), after UTP in the presence of Gö6976 (100 nM, n = 6 and 10 μM, n = 4, respectively), or after UTP in the presence of the cell permeable, myristoylated PKC inhibitor 20–28 (10 μM, n = 4). Asterisk indicates significant differences compared with the benzamil-sensitive current after UTP. The benzamil-sensitive currents following pretreatment with rottlerin and Gö6983 (a nonselective PKC inhibitor) were not significantly different from benzamil treatment alone.
Figure 7.

PKC inhibitors prevent inhibition of the apical membrane benzamil-sensitive current by UTP. (A) Current-voltage relationship comparing the benzamil-sensitive current in monolayers pretreated with GF109203X in the presence and absence of UTP (n = 4). (B) Bar graph showing the fraction of remaining current after UTP stimulation of monolayers pretreated with either GF109203X (n = 4), rottlerin (n = 5) or Gö6976 (n = 4) at −90 mV. Monolayers pretreated with GF109203X did not exhibit significant inhibition of the benzamil-sensitive current following UTP stimulation.
In Fig. 8 , identification of PKC isoforms in whole cell lysates of endometrial epithelial cells was accomplished by Western blot analysis of monolayers maintained under serum-free conditions for 3 d. A total of nine PKC isoform-specific antibodies were tested, representing classical, novel, and atypical PKC isoforms. The results show that the calcium-dependent α isoform and two calcium-independent isoforms (δ and ζ) are expressed in these cells (the θ isoform was not detected; unpublished data).
Figure 8.

Western blot analysis of PKC isoforms in monolayers maintained under serum free conditions. Antibodies used for these Western blots were obtained from Santa Cruz Biotechnology, Inc., and were rabbit anti–human antibodies to various PKC isoforms as indicated in materials and methods. Antibody labeling observed in whole-cell lysates were from epithelial cells maintained under serum-free conditions for 76 h.
BAPTA-AM Does Not Effect UTP-dependent Inhibition of Sodium Absorption
Calcium imaging experiments show that mobilization of intracellular calcium by UTP (5 μM) was dramatically reduced after pretreatment with BAPTA-AM (50 μM) (Fig. 9) . Initially, BAPTA-AM reduced the basal level of intracellular calcium by 80 nM, and upon stimulation with UTP, [Ca2+]i increased by only 20 nM. In contrast, [Ca2+]i increased threefold from 75 to 230 nM after UTP treatment in control monolayers that were not pretreated with BAPTA-AM. Isc results correspond with calcium imaging data showing that peak UTP-activated currents were reduced by 7 ± 0.6 μA (n = 6) in the presence of BAPTA-AM. However, UTP-dependent inhibition of the benzamil-sensitive current was unaffected by treatment with BAPTA-AM (Fig. 10) .
Figure 9.

Effect of BAPTA-AM on changes in [Ca2 +]i in response to UTP. (A) Temporal changes in [Ca2+]i in response to UTP were determined using fura 2-AM as described in materials and methods. Upon stimulation with 5 μM UTP in Ca2+-containing HBSS, control cells initially exhibited a transient elevation in [Ca2+]i, followed by a gradual decline to a new sustained elevated [Ca2+]i. Removing UTP from the bathing solution returned the intracellular calcium concentrations back to baseline levels. (B) BAPTA-AM reduced the basal level of intracellular calcium by 80 nM, and upon stimulation with UTP, [Ca2+]i increased by only 20 nM. For this figure, measurements were obtained from 20 cells in each of three separate experiments.
Figure 10.
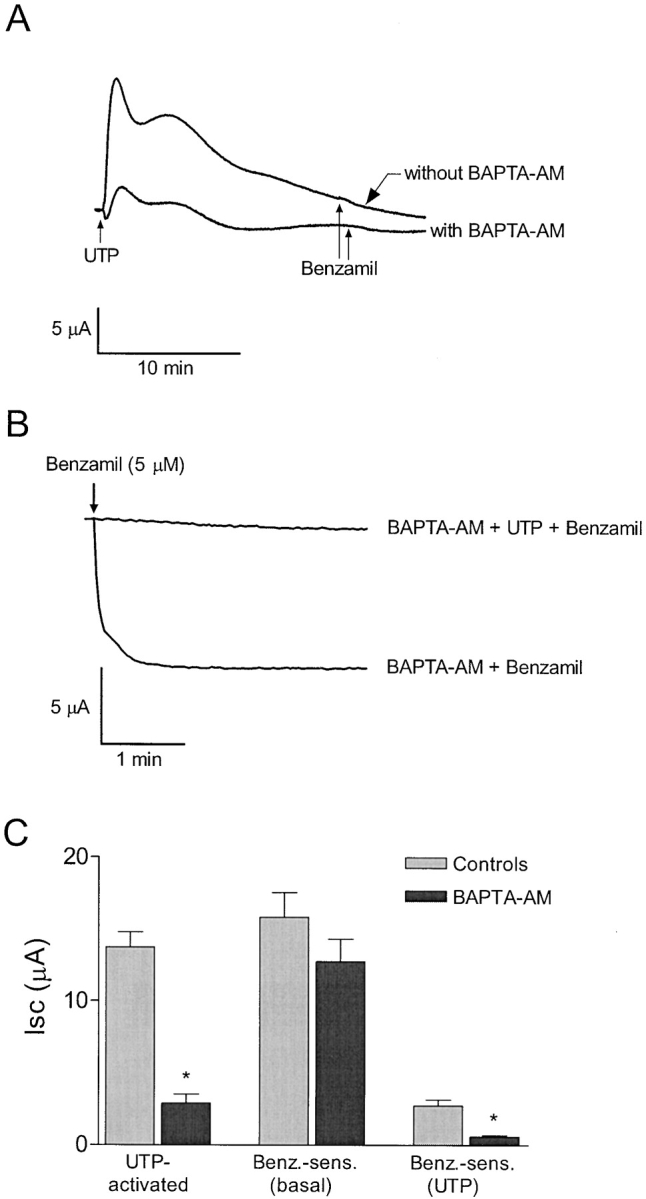
BAPTA-AM does not effect the UTP-mediated inhibition of sodium absorption. (A) Representative trace showing that the peak UTP-activated Isc response is diminished after pretreatment with 50 μM BAPTA-AM. (B) Representative trace showing that benzamil is not effective in reducing Isc following treatment with UTP (5 μM) in monolayers pretreated with BAPTA-AM. (C) Bar graph illustrating that pretreatment with BAPTA-AM diminished the UTP-activated current, but failed to affect inhibition of the benzamil-sensitive current by UTP, (n = 6 for each). Asterisk represents significant difference compared with cells that were not pretreated with BAPTA-AM.
DISCUSSION
CFTR has been shown previously to regulate cAMP-dependent gating of epithelial sodium channels, resulting in increased Na+ absorption observed in CF patients. More recently, UTP has been identified as a potentially useful therapeutic agent for patients with cystic fibrosis as a result of several studies demonstrating that UTP stimulates Cl− secretion and inhibits amiloride-sensitive Na+ absorption in normal and CF airway epithelia (Mason et al., 1991; Knowles et al., 1995; Mall et al., 2000). Therapeutic procedures for CF have used amiloride to inhibit the apical membrane sodium conductance in CF patients (Knowles et al., 1995). An advantage for using UTP in place of amiloride is that UTP can simultaneously stimulate Cl− secretion and inhibit Na+ absorption across the airway epithelium, and thus provide a means for compensating for the defect in CFTR expression in CF airways.
Since Mason et al. (1991) first demonstrated regulation of ion transport by purinergic receptors in normal and cystic fibrosis airway epithelium, the effects of ATP and UTP on sodium absorption have been studied in a variety of epithelial cell systems. Two examples include mouse inner medullary collecting duct (mIMCD) by McCoy et al. (1999) and mouse endometrial epithelium by Wang and Chan (2000). In these studies, most of the basal Isc was amiloride sensitive (∼90%), consistent with Na+ absorption (Garty and Palmer, 1997). However, only modest reductions (∼10%) in amiloride-sensitive current were observed after treatment with ATP (10–100 μM) or UTP (100 μM). These relatively small decreases in amiloride-sensitive Isc may reflect changes in driving force for Na+ influx rather than direct effects on apical Na+ conductance. A greater effect of ATP (100 μM) on amiloride-sensitive Isc was shown in mouse cortical–collecting duct by Thomas et al. (2001). The ATP-dependent reduction in basal Isc was approximately one-third of the total amiloride-sensitive current. In contrast, more complete inhibition (40–75%) of amiloride-sensitive Na+ absorption after UTP was shown in porcine thyroid epithelial cells and airway epithelia (Bourke et al., 1999; Devor and Pilewski, 1999; Ramminger et al., 1999). In these studies, different hypotheses were proposed to account for inhibition of sodium absorption based on experiments where the actions of ATP or UTP on intracellular calcium or PKC activity were investigated. Direct inhibition of sodium absorption mediated by calcium after stimulation with ATP or UTP was proposed in CFTR-expressing human bronchial epithelia, nasal airway epithelium, and distal bronchi epithelial cells (Devor and Pilewski, 1999; Inglis et al., 1999; Mall et al., 2000). However, in rabbit-connecting tubule and CCD cells, ATP or UTP stimulation of PKC was proposed as the mechanism responsible for regulation of sodium absorption (Koster et al., 1996). In contrast, the effects of UTP in mouse cortical–collecting duct (CCD) cells appeared to be mediated by a mechanism independent of either [Ca2+]i or PKC activation (Thomas et al., 2001). It is worth noting that in all of the studies cited above, conclusions regarding the effects of UTP on Na+ absorption were based on measurements of Isc. The specific effects of ATP or UTP on Na+ channel function were not directly measured. Thus, it is possible that decreases in Isc produced by P2Y receptor activation may be a consequence of inhibiting pathways other than apical Na+ channels. In the present study, however, the significant reduction (80%) of the benzamil-sensitive Isc after UTP stimulation was shown to be the result of a decrease in apical membrane Na+ conductance. PKC inhibitors blocked the effect of UTP on both Isc and apical membrane Na+ current. Additionally, effects of the PKC activator, PMA, were identical to the inhibitory effects of UTP on sodium transport. UTP-dependent inhibition of benzamil-sensitive Isc was also observed in the presence of BAPTA-AM (50 μM), leading to the conclusion that increases in [Ca2+]i were not responsible for inhibiting sodium absorption in this system.
Previous studies have demonstrated that activation of PKC directly phosphorylates apical Na+ channels (Shimkets et al., 1998). PKC-mediated inhibition of Na+ channels was shown in rabbit cortical–collecting tubules (Ling et al., 1992), and in an amphibian renal cell line (A6) after acute PGE2 stimulation (Kokko et al., 1994). In these studies, PGE2 was shown to inhibit highly selective apical membrane Na+ channels by increasing Ca2+-dependent PKC activity, resulting in a decrease in channel open probability. In endometrial epithelial cells, inhibition of sodium absorption appears to be dependent on activation of both Ca2+-dependent and Ca2+-independent isoforms of PKC. Results obtained with the PKCδ selective inhibitor, rottlerin, indicate that this calcium-independent PKC isoform plays a role in UTP regulation of benzamil-sensitive sodium channels. The data from experiments with 100 nM Go6976 and 10 μM myristoylated PKCα inhibitory peptide (PKC inhibitor 20–28) suggest that the calcium-dependent isoform, PKCα, may also contribute to regulation of the apical sodium conductance after UTP stimulation. However, our observation that lowering intracellular [Ca2+] with BAPTA-AM does not attenuate UTP-dependent inhibition of the benzamil-sensitive Isc would suggest that activation of PKCα is not required to produce inhibition of apical Na+ channels. Thus, the role of PKCα in UTP regulation of Na+ transport is not certain given that the PKCα-selective inhibitors may have had nonspecific effects on other PKC isoforms, such as PKCδ. In spite of this limitation, we conclude that activation of PKC and not increases in intracellular calcium concentration are responsible for regulation of apical sodium channels by UTP in this epithelium.
PKC regulation of epithelial Na+ channel function has also been demonstrated by Awayda et al. (1996) and Awayda (2000) using purified Na+ channel proteins reconstituted in planar lipid bilayers and cloned ENaC subunits expressed in Xenopus oocytes. Activation of PKC, by PMA and direct injection of purified PKC, inhibited whole-cell currents in ENaC-expressing oocytes. In planar lipid bilayers, addition of PKC, diacyl-glycerol, and Mg-ATP decreased ENaC open probability. More recent studies by Awayda (2000) further describe the activation of PKC by PMA and subsequent inhibition of ENaC activity in oocytes, which included nonspecific effects on membrane capacitance.
Previous studies by Ishikawa et al. (1998) showed a biphasic inhibition of whole-cell Na+ currents in ENaC-expressing MDCK cells when [Ca2+]i was increased to 1 μM. This biphasic effect was due to an initial inhibition of Na+ current within the first 5 min, followed by a secondary decrease in ENaC activity that occurred between 100 and 160 min after raising [Ca2+]i. Stockand et al. (2000) subsequently demonstrated that treatment of A6 cells with PMA decreased the expression levels of β and γ, but not αENaC. The time constant for the decline in γ and βENaC expression was ∼4 and 14 h, respectively, consistent with long-term inhibition of ENaC. Furthermore, direct evidence for PMA-dependent phosphorylation of the carboxyl termini of β and γENaC subunits, but not the αENaC subunit, was shown in a stably transfected MDCK cell line (Shimkets et al., 1998). Thus, PKC activation after UTP stimulation may produce long-term inhibition of Na+ absorption by decreasing the expression of β and γ subunits.
Studies in airway epithelia suggest that increases in [Ca2+]i are responsible for inhibition of Na+ absorption (Devor and Pilewski, 1999). Previous studies have also shown Ca2+ block of ENaC activity in planar lipid bilayers (Ismailov et al., 1997). It is worth noting that open probability and single-channel conductance of αENaC remained stable when [Ca2+]i was varied within a physiologically relevant range (between 10 nM and 1 μM). However, raising the [Ca2+]i beyond 2 μM produced a dose-dependent decrease in single-channel open probability (KD ∼20 μM), or single-channel conductance (KD ∼6 μM) if monomeric actin was present (Berdiev et al., 2001). Studies by Devor and Pilewski (1999) and Mall et al. (2000) suggest a role for Ca2+ in inhibiting Na+ absorption, but the effect of UTP on [Ca2+]i was not reported. Perhaps UTP-dependent increases in [Ca2+]i were of sufficient magnitude to produce inhibition of ENaC activity. Calcium-imaging experiments in the present study show [Ca2+]i increases after UTP stimulation were well below the [Ca2+]i required to influence αENaC activity in planar lipid bilayers. This result is consistent with our conclusion that calcium is not directly responsible for Na+ channel inhibition in porcine endometrial epithelial cells.
The inhibitory effects of UTP on transepithelial Na+ absorption have been shown to differ significantly in magnitude and mechanism of regulation in a variety of epithelial cells types. One explanation for these differences is that PKC isoforms expressed in a given epithelial cell type are variable, and that inhibition of sodium absorption by PKC is isoform specific. Alternatively, variability in α, β, and γENaC subunit expression in different epithelial cell types may also account for the varying levels of UTP effects on sodium absorption. In the present study, UTP was shown to inhibit benzamil-sensitive sodium absorption by 80% in porcine endometrial epithelial cells. This effect was due to direct inhibition of apical membrane sodium conductance and was dependent on activation of PKC. These results are consistent with previous studies of PKC inhibition of ENaC activity and completely account for the decrease in Na+ absorption observed in porcine endometrial cells. The endometrial epithelium therefore represents a useful mammalian model cell system for further studies of P2Y receptor–mediated regulation and PKC-dependent regulation of epithelial Na+ channel function.
Acknowledgments
The authors wish to thank So Yeong Lee for her invaluable comments on the manuscript.
This work was supported in part by a postdoctoral grant to C. Deachapunya from the Thailand Research Fund (PDF/60/2543).
Footnotes
Abbreviations used in this paper: CF, cystic fibrosis; DIDS, 4,4′-diisothiocyanato-stilbene-2,2′-disulfonic acid; ENaC, epithelial Na+ channel; IGF-1, insulin-like growth factor; Isc, short circuit current, MDCK, Madin-Darby canine kidney; PGF2α, prostaglandin F2α; UTP, uridine 5′-triphosphate.
References
- Awayda, M.S., I.I. Ismailov, B.K. Berdiev, C.M. Fuller, and D.J. Benos. 1996. Protein kinase regulation of a cloned epithelial Na+ channel. J. Gen. Physiol. 108:49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awayda, M.S. 2000. Specific and nonspecific effects of protein kinase C on the epithelial Na+ channel. J. Gen. Physiol. 115:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdiev, B.K., R. Latorre, D.J. Benos, and I.I. Ismailov. 2001. Actin modifies Ca2+ block of epithelial Na+ channels in planar lipid bilayers. Biophys. J. 80:2176–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke, J., K. Abel, G. Huxham, V. Cooper, and S. Manley. 1999. UTP-preferring P2 receptor mediates inhibition of sodium transport in porcine thyroid epithelial cells. Br. J. Pharmacol. 127:1787–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, H.C., C.Q. Liu, S.K. Fong, S.H. Law, L.J. Wu, E. So, Y.W. Chung, W.H. Ko, and P.Y. Wong. 1997. Regulation of Cl− secretion by extracellular ATP in cultured mouse endometrial epithelium. J. Membr. Biol. 156:45–52. [DOI] [PubMed] [Google Scholar]
- Chan, L.N., X.F. Wang, L.L. Tsang, and H.C. Chan. 2000. Pyrimidinoceptors-mediated activation of Ca2+-dependent Cl− conductance in mouse endometrial epithelial cells. Biochim. Biophys. Acta. 1497:261–270. [DOI] [PubMed] [Google Scholar]
- Deachapunya, C., M. Palmer-Densmore, and S.M. O'Grady. 1999. Insulin stimulates transepithelial sodium transport by activation of a protein phosphatase that increases Na+-K+ ATPase activity in endometrial epithelial cells. J. Gen. Physiol. 114:561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deachapunya, C., and S.M. O'Grady. 1998. Regulation of chloride secretion across porcine endometrial epithelial cells by prostaglandin E2. J. Physiol. 508:31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deachapunya, C., and S.M. O'Grady. 2001. Epidermal growth factor regulates the transition from basal sodium absorption to anion secretion in cultured endometrial epithelial cells. J. Cell. Physiol. 186:243–250. [DOI] [PubMed] [Google Scholar]
- Devor, D.C., and J.M. Pilewski. 1999. UTP inhibits Na+ absorption in wild-type and ΔF508 CFTR-expressing human bronchial epithelia. Am. J. Physiol. 276:C827–C837. [DOI] [PubMed] [Google Scholar]
- Garty, H., and L.G. Palmer. 1997. Epithelial sodium channels: function, structure, and regulation. Physiol. Rev. 77:359–396. [DOI] [PubMed] [Google Scholar]
- Inglis, S.K., A. Collett, H.L. McAlroy, S.M. Wilson, and R.E. Olver. 1999. Effect of luminal nucleotides on Cl- secretion and Na+ absorption in distal bronchi. Pflugers Arch. 438:621–627. [DOI] [PubMed] [Google Scholar]
- Ishikawa, T., Y. Marunaka, and D. Rotin. 1998. Electrophysiological characterization of the rat epithelial Na+ channel (rENaC) expressed in MDCK cells; effects of Na+ and Ca2+. J. Gen. Physiol. 111:825–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismailov, I.I., B.K. Berdiev, V.G. Shlyonsky, and D.J. Benos. 1997. Mechanosensitivity of an epithelial Na+ channel in planar lipid bilayers: release from Ca2+ block. Biophys. J. 72:1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, M.R., K.N. Olivier, K.W. Hohneker, J. Robinson, W.D. Bennett, and R.C. Boucher. 1995. Pharmacologic treatment of abnormal ion transport in the airway epithelium in cystic fibrosis. Chest. 107:71S–76S. [DOI] [PubMed] [Google Scholar]
- Kokko, K.E., P.S. Matsumoto, B.N. Ling, and D.C. Eaton. 1994. Effects of prostaglandin E2 on amiloride-blockable Na+ channels in a distal nephron cell line (A6). Am. J. Physiol. 267:C1414–C1425. [DOI] [PubMed] [Google Scholar]
- Koster, H.P., A. Hartog, C.H. van Os, and R.J. Bindels. 1996. Inhibition of Na+ and Ca2+ reabsorption by P2U purinoceptors requires PKC but not Ca2+ signaling. Am. J. Physiol. 270:F53–F60. [DOI] [PubMed] [Google Scholar]
- Ling, B.N., K.E. Kokko, and D.C. Eaton. 1992. Inhibition of apical Na+ channels in rabbit cortical collecting tubules by basolateral prostaglandin E2 is modulated by protein kinase C. J. Clin. Invest. 90:1328–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall, M., A. Wissner, T. Gonska, D. Calenborn, J. Kuehr, M. Brandis, and K. Kunzelmann. 2000. Inhibition of amiloride-sensitive epithelial Na+ absorption by extracellular nucleotides in human normal and cystic fibrosis airways. Am. J. Respir. Cell Mol. Biol. 23:755–761. [DOI] [PubMed] [Google Scholar]
- Mason, S.J., A.M. Paradiso, and R.C. Boucher. 1991. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. Br. J. Pharmacol. 103:1649–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy, D.E., A.L. Taylor, B.A. Kudlow, K. Karlson, M.J. Slattery, L.M. Schwiebert, E.M. Schwiebert, and B.A. Stanton. 1999. Nucleotides regulate NaCl transport in mIMCD-K2 cells via P2X and P2Y purinergic receptors. Am. J. Physiol. 277:F552–F559. [DOI] [PubMed] [Google Scholar]
- Palmer-Densmore, M.L., C. Deachapunya, and S.M. O'Grady. 2000. Purinergic receptor mediated regulation of anion secretion in endometrial epithelial cells: Effects of adenosine and UTP. FASEB J. 14:595. [Google Scholar]
- Ramminger, S.J., A. Collett, D.L. Baines, H. Murphie, H.L. McAlroy, R.E. Olver, S.K. Inglis, and S.M. Wilson. 1999. P2Y2 receptor-mediated inhibition of ion transport in distal lung epithelial cells. Br. J. Pharmacol. 128:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimkets, R.A., R. Lifton, and C.M. Canessa. 1998. In vivo phosphorylation of the epithelial sodium channel. Proc. Natl. Acad. Sci. USA. 95:3301–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockand, J.D., H.-F. Bao, J. Schenck, B. Malik, P. Middleton, L.E. Schlanger, and D.C. Eaton. 2000. Differential effects of protein kinase C on the levels of epithelial Na+ channel subunit proteins. J. Biol. Chem. 275:25760–25765. [DOI] [PubMed] [Google Scholar]
- Thomas, J., P. Deetjen, W.H. Ko, C. Jacobi, and J. Leipziger. 2001. P2Y2 receptor-mediated inhibition of amiloride-sensitive short circuit current in M-1 mouse cortical collecting duct cells. J. Membr. Biol. 183:115–124. [DOI] [PubMed] [Google Scholar]
- Vetter, A.E., C. Deachapunya, and S.M. O'Grady. 1997. Na+ absorption across endometrial epithelial cells is stimulated by cAMP-dependent activation of an inwardly rectifying K+ channel. J. Membr. Biol. 160:119–126. [DOI] [PubMed] [Google Scholar]
- Wang, X.F., and H.C. Chan. 2000. Adenosine triphosphate induces inhibition of Na+ absorption in mouse endometrial epithelium: a Ca2+-dependent mechanism. Biol. Reprod. 63:1918–1924. [DOI] [PubMed] [Google Scholar]