

Abstract
Hyperpolarization-activated cation (HCN) channels regulate pacemaking activity in cardiac cells and neurons. Like the related depolarization-activated K+ channels (Kv channels), HCN channels use an intracellular activation gate to regulate access to an inner cavity, lined by the S6 transmembrane regions, which leads to the selectivity filter near the extracellular surface. Here we describe two types of metal interactions with substituted cysteines in the S6, which alter the voltage-controlled movements of the gate. At one position (L466), substitution of cysteine in all four subunits allows Cd2+ ions at nanomolar concentration to stabilize the open state (a “lock-open” effect). This effect depends on native histidines at a nearby position (H462); the lock-open effect can be abolished by changing the histidines to tyrosines, or enhanced by changing them to cysteines. Unlike a similar effect in Kv channels, this effect depends on a Cd2+ bridge between 462 and 466 in the same subunit. Cysteine substitution at another position (Q468) produces two effects of Cd2+: both a lock-open effect and a dramatic slowing of channel activation—a “lock-closed” effect. The two effects can be separated, because the lock-open effect depends on the histidine at position 462. The novel lock-closed effect results from stabilization of the closed state by the binding of up to four Cd2+ ions. During the opening conformational change, the S6 apparently moves from one position in which the 468C cysteines can bind four Cd2+ ions, possibly as a cluster of cysteines and cadmium ions near the central axis of the pore, to another position (or flexible range of positions) where either 466C or 468C can bind Cd2+ in association with the histidine at 462.
Keywords: SPIH, gating, Cd2+, cysteine mutagenesis
INTRODUCTION
Hyperpolarization-activated cation currents modulate rhythmic activity in the heart and brain (Brown et al., 1979; Brown and DiFrancesco, 1980; Pape and McCormick, 1989; for reviews see DiFrancesco, 1993; Pape, 1996). These currents are produced by hyperpolarization-activated cation (HCN) channels (Santoro et al., 1998; Gauss et al., 1998; Ludwig et al., 1998), which are relatives of depolarization activated K+ channels (KV channels) and cyclic nucleotide–gated (CNG) channels. HCN channels contain six putative transmembrane regions and an intracellular cyclic nucleotide binding domain that accounts for the direct modulation of native pacemaker current by intracellular cAMP (DiFrancesco and Tortora, 1991).
Despite the inverted voltage dependence of HCN gating, the HCN channel's voltage-controlled gate and pore are similar to those of KV channels like Shaker (Shin et al., 2001; Rothberg et al., 2002). Most notably, it has been possible to engineer a cysteine-substituted spHCN channel (T464C) in which cysteines from each of the four subunits form a single high-affinity site that binds Cd2+ irreversibly, similar to the Shaker mutant V474C (Liu et al., 1997). As with Shaker V474C, Cd2+ access to the spHCN 464C site from the cytoplasmic side can be prevented by holding the channel closed; once bound, Cd2+ can be trapped inside the closed 464C channel (Rothberg et al., 2002). These results provide strong evidence for a voltage-controlled gate at the intracellular entrance to the pore of HCN channels. However, we do not know what part of the channel forms the gate itself, or how the gate moves between the open and closed position.
This study further investigates movements in the S6 region that underlie the gating of HCN channels. We have found that replacing residues L466 or Q468 with cysteines results in a high-affinity effect of Cd2+. In the case of 466C, Cd2+ produces a strong stabilization of the open state (“lock-open”), with tight binding that involves both 466C and the native histidine at position 462. The Q468C mutant has two opposing effects of Cd2+, both lock-open and lock-closed, which can be cleanly separated by mutation of the histidine at 462. We exploited both L466C and Q468C mutant spHCN channels to gain insight into the workings of the channel's gate.
MATERIALS AND METHODS
Expression of Recombinant spHCN Channels
spHCN (SPIH) channels (containing the M349I mutation to increase functional expression levels) were transiently expressed in human embryonic kidney 293 cells (HEK 293; American Type Culture Collection) as described previously (Shin et al., 2001). Cells were cotransfected with the πH3-CD8 plasmid (Seed and Aruffo, 1987), which expresses the α subunit of the human CD8 lymphocyte antigen. Cells expressing the CD8 antigen were identified by decoration with antibody-coated beads (Jurman et al., 1994).
Construction of Tandem Dimers and Site-directed Mutagenesis
Point mutations were introduced by PCR and confirmed by sequencing. The tandem dimer construct was generated by eliminating the stop codon of the “A” protomer and inserting the “B” protomer cDNA using an introduced KpnI site in the pcDNA4 expression vector, as described previously (Rothberg et al., 2002).
Solutions and Electrophysiological Recordings
All experiments were performed with excised inside-out patches (Hamill et al., 1981) from identified transfected cells 1–2 d after transfection. Experiments were done at room temperature (22–24°C). Currents were low-pass filtered at 1–2 kHz and digitized at 5–50 kHz. Solutions bathing both sides of the membrane contained (in mM) 160 KCl, 0.5 MgCl2, and 10 HEPES (pH 7.4). The solution at the extracellular face of the patch additionally contained 1 mM EGTA. Solutions at the intracellular face of the patch all contained 100 μM cAMP to prevent channel inactivation. Cd2+ solutions with <130 nM Cd2+ additionally contained 10 mM nitrilo triacetic acid (NTA) and sufficient amounts of CdCl2 and MgCl2 to bring the free [Cd2+] and [Mg2+] to the indicated levels. Buffer calculations were based on equilibrium constants for binding of H+, Cd2+, and Mg2+ to NTA and to chloride ion (Martell and Smith, 1998). Note that throughout this paper we report free [Cd2+] based on these buffer calculations; in previous papers from this lab using [Cd2+], we have reported total added [Cd2+]. In the presence of 160 mM chloride ion, the calculated ratio of free [Cd2+] to total [Cd2+] is 0.13. The control solution contained 20 μM EGTA and no added Cd2+.
Kinetic Modeling
Kinetic parameters for Scheme I were estimated using a Q-matrix method, implemented in MATLAB, to simulate currents and to fit them by minimizing the sum of squared residuals.
SCHEME I.

RESULTS
Cd2+ Locks the L466C Mutant in the Open State
We observed in a previous study of the spHCN channel S6 region that in the presence of 2.6 μM free Cd2+ (applied to the cytoplasmic side), the mutant L466C could not be closed upon depolarization, despite gating that was similar to wild-type in the absence of Cd2+ (Rothberg et al., 2002). Fig. 1 A illustrates that in the L466C mutant, 130 nM Cd2+ dramatically slowed the tail current upon depolarization to 50 mV. Because a slower closing rate is explained intuitively by stabilization of the open state, we refer to this gating phenotype in the presence of Cd2+ as “lock-open”.
Figure 1.

Intracellular Cd2+ traps L466C channels in the open state. (A) In the absence of Cd2+ (thin black trace), L466C opened in response to a hyperpolarized voltage step at −120 mV. The channels securely closed at depolarized voltage at +50 mV. In the presence of 130 nM Cd2+ (thick gray trace), L466C channels hardly closed even at +50 mV. (B) Replacing the neighboring native histidine (L466C H462Y) almost abolished the lock-open effect of Cd2+ (thick gray trace). There is some time-dependent blockade of the inward current by Cd2+. In longer activation pulses, this blockade reaches a steady-state value of ∼30% reduction in current, with a time constant of ∼200 ms. (C) Aligned amino acid sequences of the S6 regions of spHCN, Shaker, KcsA, and MthK channels. The dots indicate spHCN positions 462, 466, and 468.
High-affinity binding sites for Cd2+ tend to require the close proximity of multiple cysteine or histidine sidechains. We first looked at H462 as a potential partner with 466C to form a Cd2+ binding site in the open state; this residue was substituted with tyrosine to generate the double mutant H462Y-L466C. The gating behavior of the double mutant in the absence of Cd2+ was similar to that of L466C, but the lock-open effect was essentially abolished in the H462Y-L466C double mutant (Fig. 1 B). As for the original L466C mutant, there is some reduction in the inward current, which may be caused by binding of the divalent Cd2+ ions near the conduction pathway.
This is consistent with a contribution of H462 in coordinating Cd2+ in the open state, but it remains possible that the mutation of the histidine to tyrosine simply had some nonspecific effect on lock-open by Cd2+. We therefore mutated H462 to cysteine, another amino acid capable of strong direct interaction with Cd2+ ions. In the absence of Cd2+, the H462C-L466C mutant opened and closed normally. In the presence of 130 nM Cd2+, however, channel closing was essentially eliminated (Fig. 2 A). Perhaps even more remarkably, the lock-open effect in the double cysteine mutant was nearly irreversible; in contrast to the rapid reversal (<10 s) seen for the H462-L466C combination, in the double mutant, even after several minutes at +10 mV there was only ∼50% recovery of normal gating. The H462C single mutant was unaffected by 130 nM Cd2+ (Fig. 2 B). The gain of function seen specifically when both positions 462 and 466 are mutated to cysteine argues that these two side chains are close to each other in the open state.
Figure 2.

High-affinity Cd2+ binding in L466C H462C double mutant channels. (A) In the absence of Cd2+, L466C H462C channels opened normally in response to the hyperpolarization step (black trace). In the presence of 130 nM Cd2+ (gray trace), channel closing was essentially eliminated. Note the large instantaneous currents upon hyperpolarization and the persistent current at +50 mV. These effects required several minutes for partial reversal when Cd2+ was washed out. (B) 462C alone in the absence of 466C was not affected by 130 nM Cd2+.
We next asked whether Cd2+ is coordinated by a 466C from one subunit and a 462C from a neighboring subunit (intersubunit), or by a 466C and a 462C from the same subunit (intrasubunit). To test for an intersubunit interaction, we constructed a tandem-dimer channel with H462C in one subunit and both H462Y and L466C in the adjacent subunit (Fig. 3 A). The resulting channels would contain H462C and L466C in adjacent subunits, but never in the same subunit. These channels were not locked open in the presence of 130 nM Cd2+, suggesting that interactions between 462C and 466C in different subunits contribute little to the Cd2+ lock-open effect.
Figure 3.

Cd2+ binding is coordinated by 466C and 462C of the same subunit. (A) A tandem dimer construct with 466C and 462C in different subunits (protomer A: L466 H462C, protomer B: L466C H462Y) did not show any Cd2+ effect. (B) In a tandem dimer construct with 466C and 462C in the same subunit (protomer A, L466 H462Y; protomer B, L466C H462C), 130 nM Cd2+ produced a strong lock-open effect.
To test for an intrasubunit interaction, we constructed a tandem-dimer channel with both H462C and L466C in one subunit, and H462Y in the adjacent subunit (Fig. 3 B). These channels were locked open in the presence of 130 nM Cd2+, suggesting that Cd2+ forms a bridge between 462C and 466C in the same subunit in the open state.
Cd2+ Modifies the Gating of the Q468C Mutant
In our previous study of the spHCN channel S6 region, we observed that Q468C is strongly inhibited in the presence of 2.6 μM free Cd2+ (Rothberg et al., 2002). However, despite this strong inhibition of ∼90% of the current, the channels displayed a very rapid and complete recovery from inhibition. This was in sharp contrast to Cd2+ inhibition of the nearby T464C mutant, which does not recover from Cd2+ inhibition except in the presence of a dithiol like dimercaptopropanesulfonate.
Upon closer inspection of the 468C currents, we found that the channels were in fact opening in the presence of Cd2+, but at a greatly slowed rate. This is illustrated in Fig. 4 . This effect was apparent at [Cd2+] as low as 25 nM, indicating a very high affinity. Because a slower opening rate is explained intuitively by stabilization of the closed state, we refer to this effect in the presence of Cd2+ as “lock-closed”.
Figure 4.

Cd2+ has two high-affinity effects on gating of Q468C channels. Cd2+ was applied to the intracellular side of the patch. At 2.6 μM Cd2+ (left), channel opening was slowed, and maximal current and tail current were strongly inhibited. At 130 nM Cd2+ (right), currents were less inhibited, and an additional strong effect of Cd2+ on channel closing is revealed. Effects of Cd2+ were rapidly reversible (<10 s).
Interestingly, when we reduced the [Cd2+] to 130 nM or less, we found that the Q468C mutant also displayed a lock-open effect, similar to the effect seen with Cd2+ in the L466C mutant. We sought to determine the mechanisms of these two effects in the Q468C mutant to gain further insight into the gating process.
Cd2+ may Form a Bridge between H462 and 468C to Stabilize the Open State
As with L466C, we first looked at the nearby H462 as a potential partner with 468C to form a Cd2+ binding site; this residue was substituted with tyrosine to generate the double mutant H462Y-Q468C. The activation gating of the double mutant in the absence of Cd2+ was similar to that of Q468C; the deactivation gating was slower and clearly multiexponential (Table I) .
TABLE I.
Effects of Cd2+ on Deactivation Kinetics in HCN Channel Mutantsa
| Control | 100 nM Cd2+ | |||||
|---|---|---|---|---|---|---|
| Mutant | τfast | τslow | %fast | τfast | τslow | %fast |
| ms | ms | ms | ||||
| Q468C | 14 ± 0.8 | 240 ± 56 | 81 ± 3.2 | 30 ± 6.5 | 500 ± 67 | 38 ± 6.5 |
| H462Y-Q468C | 37 ± 2.4 | 220 ± 23 | 60 ± 3.8 | 32 ± 6.0 | 210 ± 19 | 56 ± 3.7 |
Kinetic parameters were estimated by fitting tail currents at +50 mV to a double-exponential function (n = 3 for each mutant). τfast and τslow are the fast and slow time constants, and %fast is the fractional amplitude of the component described by the fast time constant. All parameters are expressed as mean ± SEM.
In the presence of 100 nM Cd2+, H462 seemed to be critical to the Q468C lock-open effect. Fig. 5 shows that the lock-open effect was greatly reduced in the H462Y-Q468C double mutant compared with the 468C single mutant in 100 nM Cd2+. 100 nM Cd2+ had little effect on the tail current kinetics of the double mutant, compared with the greatly slowed tail current in the Q468C single mutant (Fig. 5 and Table I). The decreased lock-open effect in the double mutant was similar at 50 and 130 nM Cd2+.
Figure 5.

Cd2+ slows closing of 468C channels through an interaction with H462. Current from inside-out patches containing 468C channels (left) and the H462Y-Q468C double mutant (right). Channels were held closed at +50 mV, stepped to −100 mV for 3 s, then stepped back to +50 mV. 100 nM Cd2+ slows both opening and closing when H462 is present (left). The H462Y mutation eliminates the slowed closing (right). The schematic insets indicate the amino acids at positions 462 and 468 in each of the four subunits. Lower traces show the tail currents on a larger scale (control = black traces, Cd2+ = gray traces).
In contrast, the lock-closed effect was preserved even without a histidine at 462. Because the activation kinetics for both the single mutant and double mutant are multiexponential, we quantified activation by taking a weighted average of the activation time constants (<τ>), determined by a two-exponential fit. Using this index, we found that <τ> of both the double mutant and 468C single mutant are slowed to nearly the same extent at 100 nM Cd2+ (19-fold slower <τ> for double mutant vs. 16-fold slower <τ> for 468C; <τ>s at 0 and 100 nM Cd2+ were 52 ± 5.3 ms and 970 ± 210 ms, double mutant, n = 3 each; 56 ± 14 ms and 870 ± 350 ms, single mutant, n = 3 each). Slowing of double mutant activation kinetics was similar to that observed in the single mutant at 50 and 130 nM Cd2+ as well.
We reasoned that as for 466C, the Cd2+ might lock 468C channels open by forming a metal bridge between H462 and 468C in the open state. Unfortunately, we were unable to try the gain of function experiment with both residues mutated to cysteine, because the 462C-468C mutant showed no functional expression. Nevertheless, because of the high apparent affinity of the Cd2+ effect and the absence of good alternative partners (see discussion), we suspect that this lock-open effect also involves a direct interaction of both partners. We therefore asked whether the putative bridge is formed between an H462 and 468C residue in the same subunit (as in the L466C mutant), or between H462 in one subunit and 468C in the adjacent subunit. To answer this, we again constructed two tandem-dimer channels: one with the H462 and 468C present in the same subunit (with an adjacent subunit having neither H nor C; “YQ-HC”), and the other with an H462 and 468C present in adjacent subunits (but never in the same subunit; “HQ-YC”).
The high-affinity lock-open effect occurs preferentially in the YQ-HC dimer channels, suggesting the formation of an intrasubunit bridge (Fig. 6 A). At the same free [Cd2+] of 50 nM, there was no effect on the closure of the HQ-YC dimer (Fig. 6 B). At higher [Cd2+] of 130 nM or 2.6 μM, both dimers exhibited slower deactivation (for YQ-HC, τ at 2.6 μM Cd2+ = 78 ± 1.2 ms, τ control = 22 ± 2.1 ms; for HQ-YC, τ at 2.6 μM Cd2+ = 86 ± 8.4 ms, τ control = 22 ± 1.4 ms). Although there was a stronger effect of Cd2+ when H462 and 468C are in the same subunit, we cannot rule out an alternative intersubunit interaction that is somewhat weaker.
Figure 6.
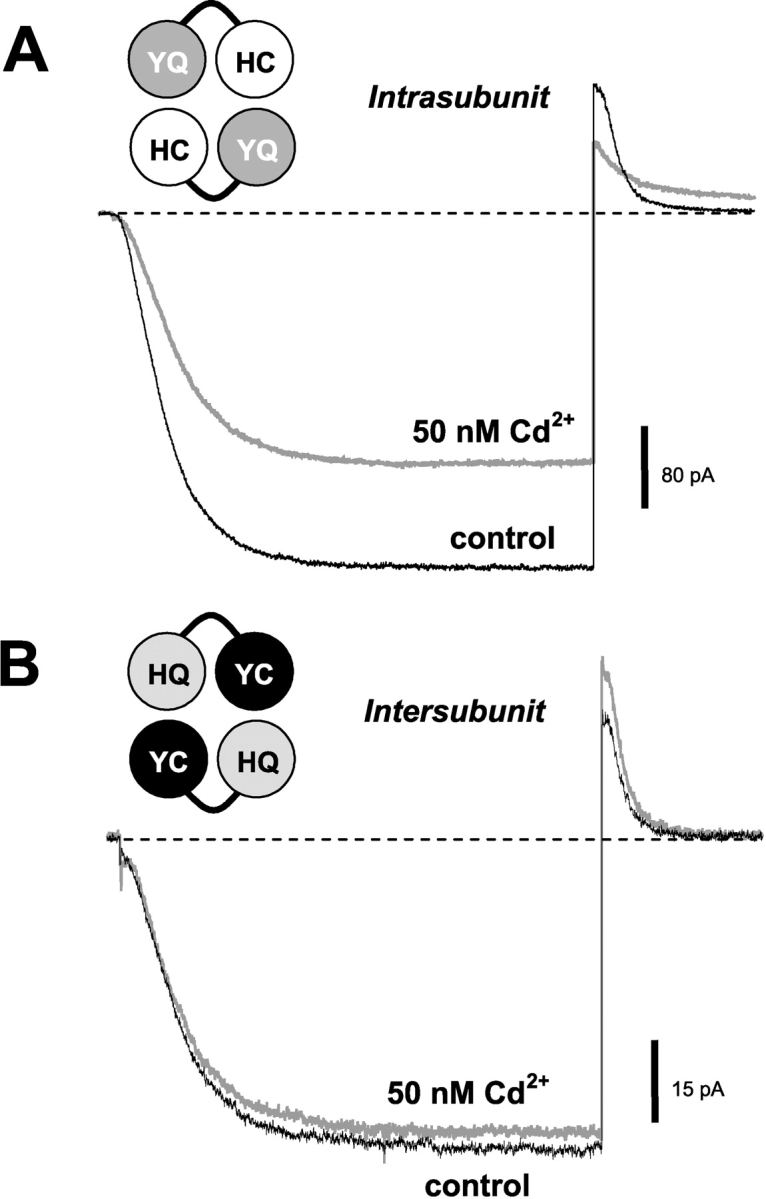
H462 and 468C need to be on the same subunit for the high-affinity Cd2+ lock-open effect. (A) In the YQ-HC tandem dimer, channel closing is slowed and maximal currents are inhibited in the presence of 50 nM Cd2+. (B) In the HQ-YC tandem dimer, channel closing is not slowed and there is little apparent inhibition. Voltage protocols for A and B are as in Fig. 2.
Cd2+ may Form a Bridge among 468C Residues to Stabilize the Closed State
The H462Y-Q468C double mutant provides a clear separation between the lock-open and lock-closed effects of Cd2+ on Q468C: the lock-open effect disappeared in the double mutant, but the lock-closed effect remained (Fig. 5). This allowed us to explore the nature of the lock-closed effect in isolation.
Given the high affinity of the lock-closed effect, we thought it was likely that position 468 faces the central axis of the pore in the closed state, so that two or more 468C sidechains coordinate a Cd2+ among themselves to stabilize this conformation and lock the channel closed. We tested this possibility using a tandem-dimer construct in which each subunit contained the H462Y mutation (to eliminate the high-affinity Cd2+ lock-open effect), but only half of the subunits contained the Q468C mutation. Thus, the resulting channels (called YQ-YC) only contained two 468C residues, presumably in nonadjacent subunits.
The YQ-YC channels displayed a greatly reduced lock-closed effect at 50 nM Cd2+ (Fig. 7) , suggesting that two 468C residues in diagonal subunits are not sufficient to produce the high-affinity Cd2+ lock-closed phenotype; the high-affinity lock-closed effect was also absent in the tandem construct containing the same subunits in reverse order (YC-YQ channels; not depicted). The high-affinity lock-closed effect was also apparently absent in the YQ-HC channels (Fig. 6 B). Thus, it appears that 468C residues must be present in adjacent subunits, or in more than two subunits, to produce the high-affinity lock-closed phenotype.
Figure 7.

Two diagonally opposed cysteines at 468 are not sufficient for the high-affinity Cd2+ lock-closed effect. Channels were held closed at +50 mV, stepped to −100 mV for 1.5 s, then stepped back to +50 mV. Opening of the H462Y-Q468C double mutant is slowed in the presence of 50 nM Cd2+ (left), but the effect is greatly reduced in channels made from YQ-YC tandem subunits, in which 468C residues are present in diagonal subunits and not in adjacent subunits.
The Lock-closed Effect has a Steep Dependence on Cd2+ Concentration
We also investigated the concentration dependence of the Cd2+-induced lock-closed effect (Fig. 8) . The simplest picture of a single Cd2+ binding to a site formed by four 468 cysteines facing the central axis predicts a first-order concentration dependence for the effect. We were therefore surprised to see a much steeper concentration dependence. Over a narrow (∼5-fold) range of Cd2+ concentration, the effect varied from a relatively subtle slowing and ∼5% inhibition to a dramatic slowing and >80% inhibition. Using the nearly steady-state current at the end of a 3-s activation step as an estimator of the Cd2+ effect, we found a steep concentration dependence consistent with the coordinated action of ∼4 Cd2+ ions (Fig. 8 B).
Figure 8.

Current through the H462Y-Q468C double mutant is steeply inhibited by Cd2+. (A) Channels were held closed at +50 mV, stepped to −100 mV for 3 s, then stepped back to +50 mV, in the presence of the free [Cd2+] indicated to the right of the current. The dotted lines are fits to the data traces using the model in Scheme I, with the following parameters: β1/α1 = β2/α2 = 220 nM, β3/α3 = 1.4 μM, β4/α4 = 125 pM, β4 = 0.86 s−1, kO = 12.1 s−1, kC = 1.2 s−1, and β1–3 are all fast (>100 s−1). (B) Current amplitudes were measured at the end of a 3 s pulse to −100 mV and normalized to 0 Cd2+ controls; these values were plotted as a function of [Cd2+] on semilogarithmic coordinates (filled circles). The solid line shows a fit with the Hill equation with IC50 = 72 nM and nH = 4, consistent with four Cd2+ ions underlying the inhibitory effect. The dashed line shows a fit with nH = 1, for comparison.
A Semiquantitative Description of the Lock-closed Effect
If Cd2+ binds with high affinity only to the closed state of the H462Y-Q468C double mutant, then it should be possible to describe the major features of the lock-closed effect using a simple kinetic model. Such a model is depicted by Scheme I. In this model, up to four Cd2+ ions bind to the closed state (C). The rate of opening upon hyperpolarization is slowed in the presence of Cd2+ because the channel cannot open until Cd2+ is released. Although Scheme I clearly oversimplifies the gating of HCN channels, we used this model to explore the minimal requirements for a model to describe the lock-closed effect.
Fig. 8 shows that a model of this type can describe the major features of the lock-closed effect, including the reduced current and slowed opening kinetics observed with increasing [Cd2+]. The parameters for this model were estimated using global fitting of currents obtained at 0, 25, 50, 100, and 130 nM Cd2+. Although the properties of the individual binding sites are not well-determined by the model, the four binding sites and the overall high cooperativity of the model (with the binding of the final Cd2+ much stronger than that of the initial Cd2+) are required to explain the high Hill coefficient.
Aside from the requirement of multiple Cd2+ binding steps, another essential property of the model is the relation between Cd2+ binding and voltage-controlled gating. It is clear that voltage activation works against the Cd2+-bound closed state. Scheme I, which captures the major features of the lock-closed effect, represents a simple two-state channel in which voltage drives the channel toward the open state in a single step. But even more complex models containing multiple activation steps can reproduce this behavior, as long as the Cd2+-bound closed state precedes at least one voltage-dependent step in the activation pathway. Also, note that the voltage-dependent activation does not completely relieve the inhibition of the channels by Cd2+, particularly at higher [Cd2+]. We suspect that this is because even with a maximal voltage stimulus, the closed-open equilibrium is not sufficiently biased toward the open state to overcome the strong binding of Cd2+.
DISCUSSION
What the Cadmium Lock-open Effects Reveal about the Open State
We have identified several positions in the S6 region of an HCN channel where introducing a cysteine permits Cd2+ binding to inhibit the channel closing process. Three positions in a row exhibit strong effects at 130 nM free Cd2+ or below: 466, 467, and 468. The I467C mutant shows a strong effect (Rothberg et al., 2002), but because of generally poor expression we did not investigate it further.
We have the clearest picture for the lock-open effect of Cd2+ on the L466C mutant. In this case we have clear evidence that the native histidine at position 462 is involved in binding Cd2+, together with the introduced cysteine. Eliminating the histidine by mutation to tyrosine abolishes the effect, whereas mutating the histidine to cysteine makes an improved binding site for Cd2+, which takes minutes to release the bound ion (Figs. 1 and 2). The lock-open effect is nearly as strong when only two of the four subunits have a cysteine at 466C, but it requires the coordinated action of a cysteine at 462 in the same subunit; having the 462 and 466 cysteines in neighboring subunits does not produce the effect (Fig. 3).
Based on our previous work showing that position 464 faces the pore (with three or more cysteines at 464 binding a single Cd2+ ion; Rothberg et al., 2002), it is reasonable to expect that positions 462 and 466 both face away from the pore, and lie on the same face of an α helix. Metal coordination at (i,i + 4) cysteine side chains is compatible with a helical conformation (Cline et al., 2003); in this situation, metal binding might favor the open state by inducing a conformational preference for one of two different positions of a bent helix (del Camino et al., 2000; del Camino and Yellen, 2001). Alternatively, it might interfere with the close approach of another protein segment, such as the neighboring S5.
The lock-open effect of Cd2+ on the Q468C mutant is less clear. This effect is also abolished by mutating the 462 histidine to tyrosine. In this case we were unable to assess the ability of a 462 cysteine to produce an improved binding site. Although again there is a preference for an intrasubunit arrangement of the cysteine side chain with the 462 histidine, this preference is not as clear, and the relationship between the two positions is more ambiguous. On the other hand, the 462 histidine is the only apparent partner for the high affinity effect: elimination of two S5 cysteines at positions 369 and 373 by mutation to F and G, respectively (substitution with the corresponding residues in mHCN1), did not prevent lock-open effects at 468C (or at 466C; unpublished data). (A conserved histidine at 380 and cysteine at 384 in S5 seem by sequence alignment to be located much further toward the extracellular side, but we cannot rule out their involvement.)
If the 462H is indeed an intrasubunit Cd2+ binding partner for both 466C and 468C, it is hard to imagine a fixed helix-like structure that would accommodate both pairings. Perhaps the open state is flexible enough to accommodate either pair, while preserving the openness of the gate in either configuration.
A similar Cd2+-induced lock-open effect is seen for a specific cysteine-histidine pairing in the S6 region of Shaker Kv channels (Holmgren et al., 1998). The cysteine mutant is at Shaker position 476, which is homologous either to spHCN 462 (by sequence alignment) or 466 (by alignment of the functionally homologous Shaker 474C and spHCN 464C; Rothberg et al., 2002). The histidine partner is at Shaker position 486, and interaction between partners involves an intersubunit bridge. In contrast to the variety of lock-open mutations in spHCN, the Shaker pairing seems very stringent; attempts to move the histidine partner to other positions in the lower S6 (482–489) were unsuccessful (unpublished data).
The relative ease of producing lock-open effects in spHCN could indicate a greater flexibility in the open state of these channels than in the Shaker channels. Alternatively, it may be easier to lock these channels open due to weaker coupling between their voltage sensors and gate. In other words, even at limiting positive voltages, HCN channels have an open probability of a few percent (unpublished data), so that even a 10-fold increase in this equilibrium constant would be quite noticeable. Voltage-gated K+ channels are closed much more securely (<10−8 at negative voltages; Islas and Sigworth, 1999), so a much stronger interaction is needed to produce a lock-open effect.
In the Q468C Mutant, How Might Cd2+ Lock the Channel Closed?
Again, based on the previous result that T464C faces the pore (Rothberg et al., 2002), we suppose that Q468 is also likely to face the central axis of the pore. If the S6 region of spHCN is primarily helical, as with its bacterial K channel relatives (Doyle et al., 1998; Jiang et al., 2002), then the i + 4 position of 468 would be placed on nearly the same face of the helix as 464, and probably lie near the central axis.
Our tandem-dimer experiments suggest that in the closed state, 468C residues from each of the four subunits participate in the coordination of Cd2+. But the dose dependence of the kinetics and steady-state current further suggests that four Cd2+ ions participate in locking the channel closed—which is inconsistent with a single, central Cd2+ binding site (as postulated for 464C). Either there are four separate high affinity binding sites for Cd2+, involving each of the four 468C residues (perhaps with another partner to permit high affinity binding), or the four cysteines somehow collaborate to make a site capable of binding multiple Cd2+ ions (a “cluster” of metal ions and ligands; Lippard and Berg, 1994). Based on our results, we prefer the cluster hypothesis. Because the lock-closed effect occurs with submicromolar affinity, it seems likely that each Cd2+ is bound to multiple ligands, rather than to a single isolated cysteine. The only obvious partner ligands are the other 468C residues; the effect does not depend on the histidine at 462 (Fig. 5), nor is it altered with mutation of the two cysteines at positions 369 and 373, in the lower S5 (not depicted). Based on sequence alignment with the KcsA channel, these results exclude all of the other potential nitrogen and sulfur candidates in the vicinity of 468.
The plausibility of the cluster hypothesis, in which the four Q468C cysteines coordinate four Cd2+ ions, is supported by a comparison of K+ channel structure with the structure of a known cysteine-cadmium cluster in metallothionein (Furey et al., 1986). Metallothionein proteins contain two metal-cysteine clusters. One of the two clusters contains four Cd2+ ions coordinated by multiple cysteines, some of which bridge two Cd2+s, whereas others contact only one Cd2+. In this cluster, it is possible to identify a ring containing four cysteines alternating with four Cd2+ ions, with the structure shown in the lower right of Fig. 9 . By comparison, placing four cysteines onto the KcsA channel backbone (Doyle et al., 1998) in the position homologous to spHCN 468 could produce an alternating cysteine-cadmium cluster with similar distances (Fig. 9, right). Such a cluster, located below the bundle crossing that closes the channel, would be ideally positioned to hold the channel in the closed position.
Figure 9.

A possible mechanism for the lock-closed effect by four Cd2+ ions at spHCN Q468C. (Left) A key diagram showing the KcsA backbone structure with the equivalent positions for spHCN 462 (as histidine), 466 (as cysteine), and 468 (as cysteine). (Top right) A scale view of the inner helices in the KcsA structure from the intracellular side, with the homologous position V115 mutated to cysteine in all four subunits. Carbon atoms are white, sulfur atoms are gray, and the hypothetical four Cd2+ ions are shown in black. (Bottom right) A known example of four cysteines bridging four Cd2+ ions in metallothionein (PDB identifier 4MT2; Furey et al., 1986), shown at the same scale. Not shown are six cysteines making single contacts with the Cd2+s, and one more bridge between the top and bottom Cd2+ ions. For reference, the Cβ–Sγ distance in the cysteine side chains is 1.80 Å, the Cd2+–Sγ distances range from 2.45–2.62 Å, and the Cd2+ coordination is approximately tetrahedral.
In any case, the Q468C mutant makes it possible for four Cd2+ ions to bind to the lower S6 of spHCN channels, and when they are bound, opening of the channel is prohibited. Once the channel opens, Cd2+ can bind to the 468C side chains in their new position, possibly in the vicinity of H462, and this different interaction holds the channel in the open state.
Acknowledgments
We thank Dr. U. Benjamin Kaupp for the spHCN clone. We are grateful to Tatiana Abramson for expert technical assistance and to members of the Yellen laboratory for helpful discussions.
This work was supported by grants to G.Y. from the National Institutes of Health (HL70320) and the McKnight Endowment Fund for Neuroscience.
Olaf S. Andersen served as editor.
Brad S. Rothberg and Ki Soon Shin contributed equally to this work.
Brad S. Rothberg's present address is Department of Physiology, University of Texas Health Science Center at San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229.
Ki Soon Shin's present address is Department of Anatomy and Neurobiology, College of Medicine, Kyunghee University, Dongdaemun-Gu, Hoegi-Dong 1, Seoul 130-701, Korea.
Abbreviations used in this paper: CNG, cyclic nucleotide–gated channel; HCN, hyperpolarization-activated cation; mHCN, mammalian HCN; NTA, nitrilotriacetic acid; spHCN, sea urchin HCN.
References
- Brown, H., and D. DiFrancesco. 1980. Voltage-clamp investigations of membrane currents underlying pace-maker activity in rabbit sinoatrial node. J. Physiol. 308:331–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, H.F., D. DiFrancesco, and S.J. Noble. 1979. How does adrenaline accelerate the heart? Nature. 280:235–236. [DOI] [PubMed] [Google Scholar]
- Cline, D.J., C. Thorpe, and J.P. Schneider. 2003. Effects of As(III) binding on alpha-helical structure. J. Am. Chem. Soc. 125:2923–2929. [DOI] [PubMed] [Google Scholar]
- del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- del Camino, D., and G. Yellen. 2001. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron. 32:649–656. [DOI] [PubMed] [Google Scholar]
- DiFrancesco, D. 1993. Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 55:455–472. [DOI] [PubMed] [Google Scholar]
- DiFrancesco, D., and P. Tortora. 1991. Direct activation of cardiac pacemaker channels by intracellular cAMP. Nature. 351:145–147. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J. Morais-Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of potassium conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Furey, W.F., A.H. Robbins, L.L. Clancy, D.R. Winge, B.C. Wang, and C.D. Stout. 1986. Crystal structure of Cd,Zn metallothionein. Science. 231:704–710. [DOI] [PubMed] [Google Scholar]
- Gauss, R., R. Seifert, and U.B. Kaupp. 1998. Molecular identification of a hyperpolarization-activated cation channel in sea urchin sperm. Nature. 393:583–587. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Holmgren, M., K.S. Shin, and G. Yellen. 1998. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 21:617–621. [DOI] [PubMed] [Google Scholar]
- Islas, L.D., and F.J. Sigworth. 1999. Voltage sensitivity and gating charge in Shaker and Shab family potassium channels. J. Gen. Physiol. 114:723–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jurman, M.E., L.M. Boland, Y. Liu, and G. Yellen. 1994. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 17:876–881. [PubMed] [Google Scholar]
- Lippard, S.J., and J.M. Berg. 1994. Principles of bioinorganic chemistry. Mill Valley, University Science Books.
- Liu, Y., M. Holmgren, M.E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- Ludwig, A., X. Zong, M. Jeglitsch, F. Hofmann, and M. Biel. 1998. A family of hyperpolarization-activated mammalian cation channels. Nature. 393:587–591. [DOI] [PubMed] [Google Scholar]
- Martell, A.E., and R.M. Smith. 1998. Critically selected stability constants of metal complexes. NIST Standard Reference Database 46, version 5.0.
- Pape, H.C. 1996. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu. Rev. Physiol. 58:299–327. [DOI] [PubMed] [Google Scholar]
- Pape, H.C., and D.A. McCormick. 1989. Noradrenaline and serotonin selectively modulate thalamic burst firing by enhancing a hyperpolarization-activated cation current. Nature. 340:715–718. [DOI] [PubMed] [Google Scholar]
- Rothberg, B.S., K.S. Shin, P. Phale, and G. Yellen. 2002. Voltage-controlled gating at the intracellular entrance to a hyperpolarization-activated cation channel. J. Gen. Physiol. 119:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, B., D.T. Liu, H. Yao, D. Bartsch, E.R. Kandel, S.A. Siegelbaum, and G.R. Tibbs. 1998. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 93:717–729. [DOI] [PubMed] [Google Scholar]
- Seed, B., and A. Aruffo. 1987. Molecular cloning of the CD2 antigen, the T-cell erythrocyte receptor, by a rapid immunoselection procedure. Proc. Natl. Acad. Sci. USA. 84:3365–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, K.S., B.S. Rothberg, and G. Yellen. 2001. Blocker state dependence and trapping in hyperpolarization-activated cation channels: evidence for an intracellular activation gate. J. Gen. Physiol. 117:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]