

Abstract
When excised inside-out membrane patches are bathed in symmetrical Cl−-rich solutions, the current-voltage (I-V) relationship of macroscopic cystic fibrosis transmembrane conductance regulator (CFTR) Cl− currents inwardly rectifies at large positive voltages. To investigate the mechanism of inward rectification, we studied CFTR Cl− channels in excised inside-out membrane patches from cells expressing wild-type human and murine CFTR using voltage-ramp and -step protocols. Using a voltage-ramp protocol, the magnitude of human CFTR Cl− current at +100 mV was 74 ± 2% (n = 10) of that at −100 mV. This rectification of macroscopic CFTR Cl− current was reproduced in full by ensemble currents generated by averaging single-channel currents elicited by an identical voltage-ramp protocol. However, using a voltage-step protocol the single-channel current amplitude (i) of human CFTR at +100 mV was 88 ± 2% (n = 10) of that at −100 mV. Based on these data, we hypothesized that voltage might alter the gating behavior of human CFTR. Using linear three-state kinetic schemes, we demonstrated that voltage has marked effects on channel gating. Membrane depolarization decreased both the duration of bursts and the interburst interval, but increased the duration of gaps within bursts. However, because the voltage dependencies of the different rate constants were in opposite directions, voltage was without large effect on the open probability (Po) of human CFTR. In contrast, the Po of murine CFTR was decreased markedly at positive voltages, suggesting that the rectification of murine CFTR is stronger than that of human CFTR. We conclude that inward rectification of CFTR is caused by a reduction in i and changes in gating kinetics. We suggest that inward rectification is an intrinsic property of the CFTR Cl− channel and not the result of pore block.
Keywords: ATP-binding cassette transporter, cystic fibrosis, chloride ion channel, channel gating, voltage dependence
INTRODUCTION
The cystic fibrosis transmembrane conductance regulator (CFTR; Riordan et al., 1989) is a phosphorylation-regulated Cl− channel that plays a central role in transepithelial fluid and electrolyte transport (Gadsby and Nairn, 1999; Sheppard and Welsh, 1999). The characteristic behavior of CFTR is determined by the function of the different domains from which CFTR is assembled. These include the two membrane-spanning domains (MSDs) that are each composed of six transmembrane segments, the two nucleotide-binding domains (NBDs) that each contain motifs which interact with ATP, and the unique R (regulatory) domain that contains multiple consensus phosphorylation sites and many charged amino acids. The MSDs assemble to form the Cl−-selective pore while the NBDs and R domain control CFTR channel gating (Gadsby and Nairn, 1999; Sheppard and Welsh, 1999).
In cell-attached membrane patch recordings, the single-channel current-voltage (I-V) relationship of CFTR outwardly rectifies (Berger et al., 1991; Tabcharani et al., 1991). Berger et al. (1991) attributed this outward rectification to Goldman-type rectification caused by the Cl− concentration gradient. However, subsequent studies have offered alternative explanations. First, Overholt et al. (1993) proposed that rectification is a function of the concentration and permeability of anions within the cell. Second, Fischer and Machen (1994) attributed rectification to high frequency gating of the CFTR Cl− channel at negative voltages. Third, studies by a number of investigators have demonstrated that large anions cause a voltage-dependent block of the CFTR Cl− channel when they are present in the intracellular solution (McDonough et al., 1994; Linsdell and Hanrahan, 1996; Sheppard and Robinson, 1997; Zhou et al., 2002). These anions bind within a deep wide vestibule at the intracellular end of the CFTR pore to prevent Cl− permeation (Linsdell and Hanrahan, 1996; Sheppard and Robinson, 1997; Hwang and Sheppard, 1999; Zhou et al., 2002). This block of the CFTR Cl− channel by large intracellular anions is reminiscent of the effect of intracellular cations on inward-rectifier K+ channels (Kir channels; Hille, 2001). Inward rectification of these K+ channels is caused by voltage-dependent block by Mg2+ and polyamines found in the cytoplasm of the cell (Vandenberg, 1987; Lopatin et al., 1994). Thus, as first proposed by Tabcharani et al. (1991), the outward rectification of CFTR Cl− channels observed in cell-attached membrane patches is likely caused by voltage-dependent block by large anions found in the cytoplasm of the cell (Linsdell and Hanrahan, 1998a, 1999).
When inside-out membrane patches excised from cells expressing wild-type human CFTR are bathed in symmetrical Cl−-rich solutions, the single-channel I-V relationship of CFTR is linear (e.g., Berger et al., 1991). However, we previously observed inward rectification of wild-type CFTR Cl− currents in excised membrane patches from C127 cells expressing wild-type human CFTR (Lansdell et al., 2000; Cai and Sheppard, 2002). At negative voltages, the I-V relationship was linear, whereas at positive voltages, the I-V relationship exhibited inward rectification that was most marked at voltages above +50 mV (Lansdell et al., 2000; Cai and Sheppard, 2002). Using excised membrane patches from baby hamster kidney (BHK) cells expressing wild-type human CFTR, Linsdell and colleagues observed similar results in some studies (Linsdell and Hanrahan, 1999; Linsdell and Gong, 2002), but not in others (e.g., Linsdell et al., 1998; Linsdell and Hanrahan, 1998b). Moreover, Zhao et al. (1996) reported inward rectification of CFTR Cl− channels reconstituted into planar lipid bilayers. In this study, we investigate the mechanism of inward rectification of the CFTR Cl− channel. We employ voltage-ramp and -step protocols to study macroscopic and single-channel currents in excised inside-out membrane patches from cells expressing wild-type human and murine CFTR and kinetic modeling to analyze channel gating.
MATERIALS AND METHODS
Cells and Cell Culture
For this study, we used mouse mammary epithelial (C127) cells stably expressing wild-type human CFTR and Chinese hamster ovary (CHO) cells stably expressing wild-type murine CFTR. C127 and CHO cells were gifts of Dr. C.R. O'Riordan (Genzyme, Framingham, MA) and Dr. S.J. Delaney and Professor B.J. Wainwright (University of Queensland, Brisbane, Australia), respectively. Cells were cultured as described previously (Sheppard and Robinson, 1997; Lansdell et al., 1998a). For experiments using excised inside-out membrane patches, cells were seeded onto glass coverslips and used within 48 h.
Electrophysiology
CFTR Cl− channels were recorded in excised inside-out membrane patches using an Axopatch 200A patch-clamp amplifier (Axon Instruments, Inc.) and pCLAMP data acquisition and analysis software (version 6.04; Axon Instruments, Inc.) as described previously (Hamill et al., 1981; Sheppard and Robinson, 1997). The established sign convention was used throughout; currents produced by positive charge moving from intra- to extracellular solutions (anions moving in the opposite direction) are shown as positive currents.
Both the pipette (extracellular) and bath (intracellular) solutions contained (mM): 140 NMDG, 3 MgCl2, 1 CsEGTA, and 10 TES, adjusted to pH 7.3 with HCl, ([Cl−], 147 mM; free [Ca2+], <10−8 M). Patch pipettes had resistances of 10–30 MΩ when filled with this solution. The intracellular solution was maintained at 37°C using a temperature-controlled microscope stage (Brook Industries).
After excision of inside-out membrane patches, we added the catalytic subunit of PKA (75 nM) and ATP (1 mM) to the intracellular solution within 5 min of patch excision to activate CFTR Cl− channels. To prevent the rundown of CFTR Cl− channels in excised membrane patches, we added PKA to all intracellular solutions. Unless otherwise indicated, membrane patches were voltage-clamped at −50 mV.
To investigate the voltage dependence of CFTR, we used either membrane patches containing large numbers of active channels or membrane patches containing five or less active channels. The number of channels in a membrane patch was determined from the maximum number of simultaneous channel openings observed during the course of an experiment, as previously described (Lansdell et al., 1998a). Using multichannel patches, we generated macroscopic I-V relationships by averaging currents generated by 15–30 ramps of voltage each of 2-s duration; holding voltage was −50 mV. Basal currents with no active CFTR Cl− channels recorded in the absence of PKA (75 nM) and ATP (1 mM) were subtracted from those recorded in the presence of PKA and ATP to generate the I-V relationship of CFTR Cl− currents. To generate ensemble currents, we applied the same voltage protocol used with multichannel patches to membrane patches containing five or less active channels 50–100 times. We subtracted basal currents recorded in the absence of PKA (75 nM) and ATP (1 mM) from currents recorded in the presence of PKA and ATP before averaging subtracted currents to generate the ensemble current. For single-channel studies of the voltage dependence of CFTR, voltage was stepped from −100 to +100 mV in 20-mV increments of 30–60 s duration. Alternatively, voltage was clamped at a single voltage for 2–4-min periods to acquire sufficient data for analyses of gating kinetics.
CFTR Cl− currents were initially recorded on digital audiotape using a digital tape recorder (model DTR-1204, Biologic Scientific Instruments; Intracel Ltd.) at a bandwidth of 10 kHz. On playback, records were filtered with an eight-pole Bessel filter (model 902LPF2, Frequency DevicesTM; SCENSYS Ltd.) at a corner frequency of 500 Hz and acquired using a Digidata 1200 interface (Axon Instruments, Inc.) and pCLAMP software at sampling rates of either 1.0 kHz (voltage-ramp protocols) or 5 kHz (single-channel studies). To determine whether a sampling rate of 1 kHz was suitable for the construction of I-V relationships, we compared the effects of acquiring voltage-ramp protocols at sampling rates of 1 and 5 kHz. Fig. 1 B demonstrates that identical I-V relationships were obtained using the two different sampling rates.
Figure 1.

I-V relationship of CFTR Cl− currents. (A) Current traces from an excised inside-out membrane patch from a C127 cell expressing wild-type human CFTR. The recordings were made in the absence (middle) and presence (bottom) of PKA (75 nM) and ATP (1 mM) in the intracellular solution. The basal recording is the current in response to a single ramp of voltage with no active CFTR Cl− channels, whereas the recording in the presence of PKA + ATP is the average current of 30 ramps of voltage. Holding voltage was −50 mV and the membrane patch was bathed in symmetrical 147 mM NMDGCl solutions. The voltage ramp protocol used is shown and currents were acquired at a sampling rate of 1 kHz as described in the materials and methods. (B) I-V relationship constructed by subtracting the basal trace from the PKA + ATP trace shown in A. The inset shows the I-V relationship of the same data acquired at a sampling rate of 5 kHz (abscissa: −100 to +100 mV; ordinate: −30 to +30 pA). (C) I-V relationship of CFTR Cl− currents. Data are means ± SEM (n = 10) at each voltage calculated by expressing individual current values measured from −100 to +100 mV in 10-mV increments as a percentage of the current value at −100 mV. Error bars are smaller than symbol size. The continuous line is the fit of a second order regression to the data. The dotted line shows the predicted ohmic I-V relationship. Other details as in A.
To measure single-channel current amplitude (i), Gaussian distributions were fit to current amplitude histograms. Chord conductance was calculated by dividing unitary current by the difference between the applied voltage and the reversal potential. For open probability (Po) and kinetic analyses, lists of open open and closed times were created using a half-amplitude crossing criterion for event detection. Transitions <1 ms in duration were excluded from the analyses. Single-channel open and closed time histograms were created using logarithmic x-axes with 10 bins decade−1. Histograms were fit with one or more component exponential functions using the maximum likelihood method. To determine which component function fitted best, the log-likelihood ratio test was used and considered statistically significant at a value of 2.0 or greater (Winter et al., 1994). Burst analysis was performed as described by Carson et al. (1995a), using a burst delimiter (tc; the time that separates interburst closures from intraburst closures) determined by analyses of closed time histograms (Fig. 5, A and B). Closures longer than tc were considered to define interburst closures, whereas closures shorter than this time were considered gaps within bursts. The mean interburst interval (TIBI) was calculated using the equation:
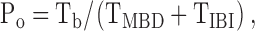 |
(1) |
where Tb = (mean burst duration) × (open probability within a burst). Mean burst duration (TMBD) and open probability within a burst (Po(burst)) were determined directly from experimental data using pCLAMP software. TMBD was determined from histograms of burst duration data plotted with logarithmic x-axes with 10 bins decade−1, Po(burst) was calculated using burst duration and closed times within bursts data, and Po was calculated as described previously (Lansdell et al., 2000). Only membrane patches that contained a single active channel were used for single-channel kinetic analyses.
Figure 5.

Analysis of the dwell time histograms of a single CFTR Cl− channel. (A and B) Representative open and closed time histograms for a single CFTR Cl− channel recorded at −75 and +75 mV, respectively, using the conditions described in Fig. 3. For open time histograms, the continuous line is the fit of a one-component exponential function. For closed time histograms, the continuous line is the fit of a two-component exponential function. The dotted lines show the individual components of the exponential functions. Logarithmic x-axes with 10 bins decade−1 were used for both open and closed time histograms. (C) Burst duration (top), interburst interval (middle), and Po (bottom) of CFTR at the indicated voltages. Columns and error bars indicate means ± SEM (n = 10) at each voltage. For the Po data, filled circles connected by lines represent individual experiments and the open circles are means ± SEM (n = 10). The asterisks indicate values that are significantly different from the −75 mV data (P < 0.05). Burst duration and interburst interval were calculated as described in materials and methods. Other details as in Fig. 1.
Modeling of Single-channel Kinetics
To perform maximum likelihood analysis and develop kinetic models of CFTR channel gating, we used the QuB software suite (www.qub.buffalo.edu; Qin et al., 1997) to analyze data from membrane patches that contained only a single active channel as described previously (Cai and Sheppard, 2002). In brief, digitized current records generated by pCLAMP software were imported with no further filtering and baseline corrected (program PRE). Using a recursive Viterbi algorithm (program SKM), idealized currents were produced. Finally, rate constants for kinetic models were calculated from the idealized current dwell time sequence using a maximum likelihood approach (program MIL). For consistency with analyses using pCLAMP software, transitions <1 ms were excluded.
To investigate the voltage dependence of rate constants, we first calculated rate constants at equivalent positive and negative voltages from individual membrane patches. If rate constants are voltage dependent, the relationship between rate constant and voltage is described by the single exponential function:
 |
(2) |
where k is the unknown rate constant, k0 is the rate constant at unitary ligand concentration and zero voltage, P is the ligand concentration, and V is voltage. The term k1 is defined as:
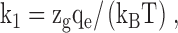 |
(3) |
where zg is the equivalent gating charge (defined as the product of the unit charge moved (z) times the fraction of the electric field (δ)), qe is the elementary charge, kB is the Boltzmann's constant, and T is the absolute temperature. The parameters k1 and k0 are intrinsic properties of the channel that do not vary with experimental conditions (Qin et al., 2000). To determine k0 and k1, we simultaneously fitted combined sets of data obtained at equivalent positive and negative voltages (e.g., ±75 mV) from the same membrane patch. Because the ligand concentration (P) is constant under the experimental conditions employed ([ATP]i = 1 mM), the value of P is assumed to be 1. Finally, using values of k0 and k1, we simulated the effects of voltage on rate constants over a wide range of voltages (program SIM; Qin et al., 1997, 2000).
Reagents
The catalytic subunit of PKA was purchased from Promega UK. ADP (disodium salt), ATP (disodium salt), pyrophosphate (tetrasodium salt), and TES were obtained from Sigma-Aldrich. All other chemicals were of reagent grade.
Statistics
Results are expressed as means ± SEM of n observations. To compare sets of data, we used either a one-way analysis of variance (ANOVA) or Student's paired t test. Differences were considered statistically significant when P < 0.05. All tests were performed using SigmaStatTM (version 2.03; Jandel Scientific GmbH).
Online Supplemental Material
To investigate the possibility that inward rectification of CFTR Cl− currents is caused by a component of the recording solutions, we examined the effect on CFTR of a number of factors in the bath and pipette solutions. These included the nature of the biological buffer (Fig. S1) and the monovalent cation used in our recording solutions (Figs. S2 and S3). Supplemental figures and text is available at http://www.jgp.org/cgi/content/full/jgp.200308921/DC1.
RESULTS
Voltage Dependence of the CFTR Cl− Channel
To learn how voltage regulates the activity of CFTR, we studied CFTR Cl− currents in excised inside-out membrane patches from C127 cells expressing wild-type human CFTR. We bathed membrane patches in symmetrical 147 mM Cl− solutions and recorded membrane currents over the voltage range ±100 mV using a voltage-ramp protocol. Fig. 1 A demonstrates that under basal conditions, the voltage ramp protocol evoked only tiny membrane currents. However, after the activation of CFTR Cl− currents by cAMP-dependent phosphorylation, the voltage-ramp protocol elicited large membrane currents (Fig. 1 A). To identify the membrane currents generated by CFTR, we subtracted the membrane currents recorded under basal conditions from those recorded following the addition of PKA (75 nM) and ATP (1 mM) to the intracellular solution. Fig. 1 B shows the I-V relationship of these CFTR Cl− currents. At negative voltages, the I-V relationship is linear, whereas at positive voltages, the I-V relationship exhibits weak inward rectification (Fig. 1 B). This inward rectification of CFTR Cl− currents is more clearly demonstrated in Fig. 1 C, where data are expressed as a percentage of the current value at −100 mV. At voltages above +50 mV, current values diverge noticeably from the I-V relationship of an ion channel that obeys Ohm's law (Fig. 1 C). These data indicate that CFTR Cl− currents exhibit weak inward rectification when bathed in symmetrical Cl−-rich solutions.
To further investigate the inward rectification of CFTR, we studied single Cl− channels. We reasoned that if the I-V relationship of individual CFTR Cl− channels exhibits inward rectification, summation of single-channel currents should reproduce the rectification of macroscopic CFTR Cl− currents. To test this hypothesis, we applied the same voltage protocol used to study macroscopic CFTR Cl− currents to membrane patches containing five or less active channels. Fig. 2 A shows the activity of a single CFTR Cl− channel evoked by five ramps of voltage from −100 to +100 mV. Visual inspection of these single-channel records suggests that current amplitude at negative voltages is larger than that at positive voltages. Consistent with this idea, the ensemble current generated by averaging the single-channel currents elicited by 70 voltage ramps inwardly rectifies (Fig. 2 B). Importantly, the extent of the inward rectification observed for the ensemble current agrees closely with that of macroscopic CFTR Cl− currents (Figs. 1 C and 2 C). For example, at +100 mV the magnitude of the macroscopic CFTR Cl− current was 74 ± 2% (n = 10) of that at −100 mV while that of the ensemble current was 79 ± 3% (n = 5) of that at −100 mV (P > 0.1). These data indicate that voltage-dependent changes in the single-channel activity of CFTR account for the inward rectification of macroscopic CFTR Cl− currents.
Figure 2.

Summation of single-channel currents reproduces the rectification of macroscopic CFTR Cl− currents. (A) Representative recordings of a single CFTR Cl− channel in an excised inside-out membrane patch elicited by a depolarizing voltage ramp from −100 to +100 mV. ATP (1 mM) and PKA (75 nM) were continuously present in the intracellular solution. The dotted line indicates the zero current level. For the purpose of illustration, single-channel records were digitally refiltered at 100 Hz. (B) Ensemble current obtained by averaging 70 ramps of voltage from the same experiment as that shown in A. The continuous line is the fit of a second-order regression to the data. (C) I-V relationship expressed as a percentage of the current value at −100 mV. Data are means ± SEM (n = 5) at each voltage. Other details as in A and Fig. 1.
Effect of Voltage on the Single-channel Activity of CFTR
In principal, the inward rectification of CFTR Cl− currents might be caused by a voltage-dependent change in first, the number of active channels; second, current flow through open channels; third, channel gating and hence, Po. To discriminate between these different possibilities, we investigated the effect of voltage on the single-channel activity of CFTR. Fig. 3 compares the activity of a single CFTR Cl− channel phosphorylated by PKA and ATP at −75 and +75 mV. These single-channel records suggest that inward rectification was not caused by a decrease in the number of active channels present at positive voltages (n = 6; Fig. 3). On the contrary, the single-channel data suggest that inward rectification is caused by both a reduction in i and changes in gating behavior at positive voltages (Fig. 3).
Figure 3.

Effect of voltage on the single-channel activity of CFTR. Representative recordings of a single CFTR Cl− channel at −75 mV (top) and +75 mV (bottom). ATP (1 mM) and PKA (75 nM) were continuously present in the intracellular solution. Dotted lines indicate the closed channel state. Downward and upward deflections correspond to channel openings at −75 mV and +75 mV, respectively. Each trace is 10-s long. For the purpose of illustration, single-channel records were filtered at 500 Hz and digitized at 1 kHz. Other details as in Fig. 1 A.
The decrease in i at positive voltages is not readily apparent by visual inspection of single-channel records (Fig. 3). However, it is evident from single-channel current amplitude histograms and the I-V relationship of CFTR (Fig. 4, A and B) . Fig. 4 A shows histograms of the distribution of current values at ±50 and ±75 mV. When compared with i values at negative voltages, the reduction in i at +50 mV was very slight and that at +75 mV was small (Fig. 4 A). Nevertheless, like that of macroscopic CFTR Cl− currents, the single-channel I-V relationship of CFTR weakly inwardly rectified at positive voltages (Fig. 4 B). Consistent with these data, the chord conductance of CFTR decreased from 11.4 ± 0.3 pS at −100 mV to 10.0 ± 0.3 pS at +100 mV (n = 10; P < 0.01; Fig. 4 C).
Figure 4.

Single-channel conductance of CFTR. (A) Single-channel current amplitude histograms of a single CFTR Cl− channel at the indicated voltages recorded using the conditions described in Fig. 3. At negative voltages (top), the closed-channel amplitude is shown on the right, whereas at positive voltages (bottom), the closed channel amplitude is shown on the left. Linear x-axes with 20 bins decade−1 were used for the histograms and the continuous lines represent the fit of Gaussian distributions to the data. The vertical dashed lines indicate the position of the open and closed channel levels at negative voltages. (B) Single-channel I-V relationship of CFTR. (C) Relationship between chord conductance and voltage for the data shown in B. Chord conductance was calculated as described in materials and methods. Other details as in Fig. 1.
Comparison of the macroscopic and single-channel I-V relationships of CFTR (Figs. 1 C and 4 B) suggests that the decrease in i at positive voltages is insufficient to account for the rectification of macroscopic CFTR Cl− currents. For example, at +100 mV single-channel current amplitude (i) = 88 ± 2% of that at −100 mV (n = 10), whereas at +100 mV macroscopic current (I) = 74 ± 2% of that at −100 mV (n = 10; P < 0.001). In agreement with these data, Fig. 3 demonstrates that the pattern of channel gating at +75 mV differs strikingly from that observed at −75 mV. At negative voltages the gating behavior of CFTR was characterized by bursts of channel activity, interrupted by brief flickery closures, separated by longer closures between bursts (Fig. 3, top). In contrast, at positive voltages both the duration of bursts as well as the long closed periods separating bursts were decreased in length (Fig. 3, bottom).
To quantify the changes in gating behavior at positive voltages, we used membrane patches that contained only a single active channel. We analyzed histograms of open and closed times at ±75 mV to determine how voltage alters the distribution of open and closed times. At both −75 and +75 mV, open and closed time histograms of CFTR were best fitted with one- and two-component functions, respectively (Fig. 5, A and B , and Table I) . However, at positive voltages the distribution of open and closed times was altered in several ways. First, the open time constant (τO1) was decreased by 52% (Fig. 5, A and B, and Table I). Second, the fast closed time constant (τC1) that describes the flickery closures which interrupt channel openings was increased by 79%, while its share of the closed time distribution expanded slightly from 61 to 66% (Fig. 5, A and B, and Table I). Third, the slow closed time constant (τC2) that describes the long closures which separate channel openings was decreased by 27%, while its share of the closed time distribution contracted slightly from 39% to 34% (Fig. 5, A and B, and Table I). As a consequence of these changes in channel gating, the number of events per minute at +75 mV was double that at −75 mV (Table I).
TABLE I.
Effect of Voltage on the Open and Closed Time Constants of Wild-type CFTR Cl− Channels
| Voltage (mV) | −75 | +75 |
|---|---|---|
| τO1 (ms) | 72.13 ± 4.45 | 34.97 ± 3.50 |
| τC1 (ms) | 1.78 ± 0.03 | 3.19 ± 0.14 |
| τC2 (ms) | 119.05 ± 12.15 | 86.73 ± 10.32 |
| Area under curve τC1 | 0.61 ± 0.02 | 0.66 ± 0.02 |
| Area under curve τC2 | 0.39 ± 0.02 | 0.34 ± 0.02 |
| Events per minute | 965 ± 49 | 1,929 ± 262 |
| Total time (s) | 939 | 614 |
Open and closed time constants were measured at the indicated voltages by fitting one- and two-component exponential functions to open and closed time histograms as described in materials and methods. Area under curve indicates the proportion of the total closed time distribution that corresponds to either τC1 or τC2. Events per minute represent the number of transitions between the open and closed states within one minute. The total time analyzed at each voltage is shown, and in each patch ∼2,000 events were analyzed at each voltage. Values are means ± SEM of n = 10 at each voltage. Measurements were made using the conditions described in Fig. 3.
To further investigate the changes in gating behavior at positive voltages, we performed an analysis of bursts using a burst delimiter (tc) determined as the nadir between the two populations of channel closures (Fig. 5, A and B, and Table I; Carson et al., 1995a; Lansdell et al., 1998b). At −75 mV, tc = 11.26 ± 0.32 ms (n = 10), whereas at +75 mV, tc = 18.97 ± 0.85 ms (n = 10). The large difference between the fast and slow time constants at both negative and positive voltages, suggests that errors caused by misclassification of bursts should be rare. Fig. 5 C demonstrates that both burst duration and interburst interval were significantly decreased at +75 mV (P < 0.001). As a result, the Po of CFTR at +75 mV was only slightly decreased compared with the value at −75 mV, although the difference was statistically significant (P = 0.002; Fig. 5 C). These data suggest that voltage alters the gating behavior of the CFTR Cl− channel without noticeably changing Po.
Thus, our data suggest that the single-channel behavior of CFTR accounts for the rectification of CFTR Cl− currents. CFTR Cl− current is determined by the product of the number of CFTR Cl− channels in the membrane patch (N), the single-channel current amplitude (i), and the probability (Po) that a single channel is open: ICFTR = N × i × Po. If we set each of these variables to 100% for the −100 mV data, we can compare the CFTR Cl− current generated at −100 and +100 mV. Table II presents values of each variable, the predicted CFTR Cl− current determined by calculating N × i × Po and the observed value of CFTR Cl− current. Differences between the observed and predicted values are likely accounted for by using data at ±75 mV and not ±100 mV to calculate Po and the subtraction of basal currents.1 Nevertheless, the predicted and observed values agree closely.
TABLE II.
Comparison of Predicted CFTR Cl− Current, N × i × Po, and Measured CFTR Cl− Current at −100 and +100 mV
| V | N | i | Po | N × i × Po | ICFTR |
|---|---|---|---|---|---|
| mV | % | % | % | % | % |
| −100 | 100 | 100 | 100 | 100 | 100 |
| +100 | 100 | 88 | 93 | 77 | 74 |
N, the number of CFTR Cl− channels in the membrane patch; i, single-channel current amplitude; Po, open-probability. To estimate N, we used data from Fig. 3, and to estimate Po, we used data from Fig. 5C, where voltage was either −75 or +75 mV. The predicted CFTR Cl− current has been corrected for the outward rectification of basal currents (see footnote 1).
Voltage Changes the Gating Kinetics of CFTR
To understand better how voltage changes the pattern of channel gating, we used maximum likelihood analysis and kinetic modeling. Using this approach, Winter et al. (1994) demonstrated that the gating kinetics of single phosphorylated wild-type CFTR Cl− channels are described equally well by the linear three-state schemes shown in Figs. 6 A and 7 A. In these schemes, C1 represents the long duration closed state separating channel openings, C2 the brief flickery closures that interrupt channel openings, and O the open state. Transitions between the three states are described by the rate constants β1, β2, α1 and α2. Using the C1↔C2↔O kinetic scheme, intracellular ATP regulates CFTR at the transition between C1 and C2: as the ATP concentration is raised, β1 increases (Winter et al., 1994). In contrast, none of the other transition rates were altered significantly by ATP (Winter et al., 1994). Similar analyses of our own data support this model of ATP-dependent regulation of CFTR channel gating (Cai and Sheppard, 2002). They also suggest that using the C1↔O↔C2 kinetic scheme, intracellular ATP regulates CFTR at the transition between C1 and O with β1 increasing at elevated ATP concentrations while the other rate constants remain unchanged (unpublished data). As the gating behavior of CFTR is equally well described by the kinetic schemes C1↔C2↔O and C1↔O↔C2, we used both schemes to determine how voltage regulates CFTR channel gating.
Figure 6.

Effect of voltage on the rate constants of the C1↔C2↔O kinetic scheme. (A) The C1↔C2↔O kinetic scheme that describes CFTR channel gating (Winter et al., 1994). States C1, C2, and O represent two closed states and one open state, respectively, while β1, β2, α1, and α2 represent the rate constants describing transitions between these states. States enclosed within the dashed box represent the bursting state. (B) Rate constants at the indicated voltages determined by the maximum likelihood fit to the model shown in A. Data are means ± SEM (n = 6) at each voltage. The asterisks indicate values that are significantly different from the −75 mV data (P < 0.05). (C) The relationship between the rate constants β1 (filled circles), β2 (filled diamonds), α1 (open circles) and α2 (open diamonds) and voltage for the C1↔C2↔O model. Values are means ± SEM (n = 5–6) at each voltage. The continuous lines represent fits of the single exponential function k=k0Pexpk1V (Eq. 2) as described in the materials and methods. Other details as in Fig. 1 and Table III.
Fig. 6 B shows the rate constants for the C1↔C2↔O kinetic scheme (Fig. 6 A and Winter et al., 1994) at −75 and +75 mV calculated using the QuB software suite (www.qub.buffalo.edu; Qin et al., 1997). When compared with the −75 mV data, α1 was decreased by 53% at +75 mV, α2 was increased by 88%, β2 was decreased by 24%, but β1 was unchanged. The duration of bursts is influenced first by α1, the rate constant that determines the velocity with which the channel leaves the bursting state, and second by α2 and β2, the rate constants that control opening and closing transitions within the bursting state (see Eq. 4). The decrease in α1 delays the exit from the bursting state and hence, increases the duration of bursts at +75 mV. In contrast, the decrease in β2 and particularly the increase in α2 both act to decrease the duration of bursts at +75 mV. Thus, the data suggest that depolarized voltages produce reciprocal changes in α1 and the rate constants within the bursting state (i.e., α2 and β2) that tend to offset each other.
Fig. 7 B shows the rate constants for the C1↔O↔C2 kinetic scheme (Fig. 7 A and Winter et al., 1994) at −75 and +75 mV. When compared with the −75 mV data, α1 was increased by 27% at +75 mV, β1 was increased by 29%, β2 was increased by 118% and α2 was decreased by 34%. As with the C1↔C2↔O kinetic scheme, the duration of bursts in the C1↔O↔C2 model is influenced by α1, α2, and β2 (see Eq. (5)). However, unlike the C1↔C2↔O kinetic scheme, the changes in α2 and β2 have little effect on burst duration at +75 mV while the increase in α1 accelerates the rate of exit from the bursting state and hence, decreases the duration of bursts at +75 mV. Similarly, the increase in β1, the rate constant that determines the velocity with which the channel enters the bursting state, causes a reduction in the interburst interval at +75 mV.
Figure 7.

Effect of voltage on the rate constants of the C1↔O↔C2 kinetic scheme. (A) The C1↔O↔C2 kinetic scheme that describes CFTR channel gating (Winter et al., 1994). (B) Rate constants at the indicated voltages determined by the maximum likelihood fit to the model shown in A. (C) The relationship between the rate constants β1 (filled circles), β2 (filled diamonds), and α1 (open circles) and voltage for the C1↔O↔C2 scheme. The inset shows the relationship between the rate constant α2 and voltage for the same scheme. Other details as in Fig. 6 and Table IV.
To investigate whether the changes in rate constants counteract each other, we used the rate constants for the C1↔C2↔O and C1↔O↔C2 kinetic schemes to calculate values of (i) burst duration, (ii) mean duration of gaps within bursts, (iii) interburst interval, and (iv) Po and compared these values with those derived using pClamp software. First, using the kinetic scheme C1↔C2↔O, mean burst duration is given by the equation (Sakmann and Trube, 1984):
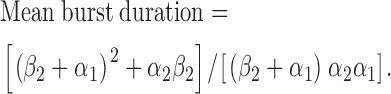 |
(4) |
Using Eq. 4, mean burst duration decreased from 158 ± 17 ms (n = 6) at −75 mV to 130 ± 18 ms (n = 6) at +75 mV, a decrease of 18% (P < 0.05). Using the kinetic scheme C1↔O↔C2, mean burst duration is given by the equation (Sakmann and Trube, 1984):
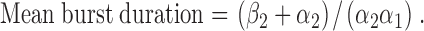 |
(5) |
Using Eq. 5, mean burst duration decreased from 145 ± 18 ms (n = 6) at −75 mV to 120 ± 16 ms (n = 6) at +75 mV, a reduction of 17% (P < 0.05). These data are in close agreement with the mean burst duration values determined using rate constant data for the C1↔C2↔O kinetic scheme and pClamp software (19% decrease; Fig. 5 C). They indicate that burst duration decreases at positive voltages.
Second, using the kinetic scheme C1↔C2↔O, mean duration of gaps within bursts is given by the equation (Sakmann and Trube, 1984):
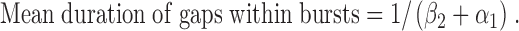 |
(6) |
Using Eq. 6, mean duration of gaps within bursts increased from 1.92 ± 0.07 ms (n = 6) at −75 mV to 3.02 ± 0.17 ms (n = 6) at +75 mV, an increase of 57% (P < 0.05). Using the kinetic scheme C1↔O↔C2, mean duration of gaps within bursts is given by the equation (Sakmann and Trube, 1984):
 |
(7) |
Using Eq. 7, mean duration of gaps within bursts increased from 1.93 ± 0.07 ms (n = 6) at −75 mV to 2.93 ± 0.15 ms (n = 6) at +75 mV, an increase of 52% (P < 0.05). These data are in reasonable agreement with the values of mean duration of gaps within bursts calculated using rate constant data for the C1↔C2↔O kinetic scheme and excellent agreement with values determined using pClamp software (−75 mV, 2.14 ± 0.07 ms (n = 10); +75 mV, 3.25 ± 0.25 ms (n = 10); 52% increase; P < 0.05). They indicate that bursts become interrupted by longer brief closures at positive voltages.
Third, using the kinetic scheme C1↔C2↔O, an approximate value of interburst interval can be determined using the equation (Sakmann and Trube, 1984):
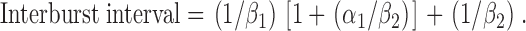 |
(8) |
Using Eq. 8, interburst interval decreased from 113 ± 18 ms (n = 6) at −75 mV to 84 ± 11 ms (n = 6) at +75 mV, a decrease of 26% (P < 0.05). Using the kinetic scheme C1↔O↔C2, interburst interval is given by the equation (Sakmann and Trube, 1984):
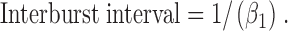 |
(9) |
Using Eq. 9, interburst interval decreased from 106 ± 18 ms (n = 6) at −75 mV to 80 ± 11 ms (n = 6) at +75 mV, a reduction of 25% (P < 0.05). These data are in very good agreement with the values of interburst interval calculated using rate constant data for the C1↔C2↔O kinetic scheme, but only reasonable agreement with those values determined using pClamp software (16% decrease; Fig. 5 C). They indicate that interburst interval decreases at positive voltages.
Fourth, using the kinetic scheme C1↔C2↔O, Po is given by the equation (Sakmann and Trube, 1984):
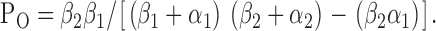 |
(10) |
Using Eq. 10, Po decreased slightly from 0.57 ± 0.04 (n = 6) at −75 mV to 0.56 ± 0.04 (n = 6) at +75 mV, a decrease of 2% (P > 0.05). Using the kinetic scheme C1↔O↔C2, Po is given by the equation (Sakmann and Trube, 1984):
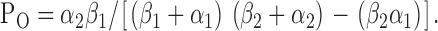 |
(11) |
Using Eq. 11, Po decreased marginally from 0.57 ± 0.04 (n = 6) at −75 mV to 0.56 ± 0.04 (n = 6) at +75 mV, a reduction of 2% (P > 0.05). These data are in very good agreement with the values of Po calculated using rate constant data for the C1↔C2↔O kinetic scheme and good agreement with values determined using pClamp software (5% decrease; Fig. 5 C). However, the decrease in Po calculated using rate constant data was not statistically significant, unlike that calculated using pClamp software. We attribute this subtle effect of voltage on Po to the changes in mean burst duration and interburst interval counterbalancing each other.
To understand better the voltage dependence of CFTR channel gating, we calculated rate constants for both kinetic schemes at −50, −20, +20, and +50 mV and fitted the data using the single exponential function k=k0Pexpk1V (Eq. 2; Figs. 6 C and 7 C). To determine whether rate constants changed with voltage, we performed one-way ANOVAs. Considering first the C1↔C2↔O kinetic scheme, this test indicated that β1 and β2 did not change significantly with voltage (Fig. 6 C; P > 0.05).2 In contrast, a significant difference was found between voltage and α2 and α1 (Fig. 6 C; P < 0.05).2 Considering next the C1↔O↔C2 kinetic scheme, one-way ANOVAs indicated that α1 and β2 did not change significantly with voltage (Fig. 7 C; P > 0.05). In contrast, a significant difference was found between voltage and β1 and α2 (Fig. 7 C; P < 0.05). Thus, in both kinetic schemes voltage has profound effects on the gating behavior of the CFTR Cl− channel. However, because in both kinetic schemes the effects of voltage on the rate constants tend to oppose one another, CFTR Cl− currents exhibit only weak voltage-dependence.
Finally, using the data in Figs. 6 C and 7 C, we derived values of equivalent gating charge for each of the rate constants in the two kinetic schemes (Tables III and IV) . For the C1↔C2↔O kinetic scheme, the total equivalent gating charge for CFTR is 0.32e (Table III) while for the C1↔O↔C2 kinetic scheme it is 0.28e (Table IV). These values of gating charge are threefold lower than that of ClC-0, the voltage-dependent Cl− channel of Torpedo electric organ (Hanke and Miller, 1983; Pusch et al., 1995; Chen and Miller, 1996) and about fortyfold lower than those of voltage-gated Na+, K+ and Ca2+ channels (Hille, 2001).
TABLE III.
Summary of the Fits to the Rate Constants for the C1↔C2↔O Kinetic Scheme
| Rate | k0 | k1 | zg |
|---|---|---|---|
| s−1 | V−1 | e− charges | |
| β1 | 18.22 ± 2.18 | 1.06 ± 0.47 | 0.03 |
| α1 | 141.04 ± 8.02 | −5.09 ± 0.61 | −0.14 |
| β2 | 281.58 ± 20.86 | −1.93 ± 0.27 | −0.05 |
| α2 | 24.44 ± 3.65 | 3.64 ± 0.69 | 0.10 |
Values of k0, k1, and zg (equivalent gating charge) derived from exponential fits to the rate constants for the C1↔C2↔O kinetic scheme using dwell-time data acquired using the conditions described in Fig. 3. Rate constants were fitted with the single exponential function k=k0Pexpk1V (Eq. 2) as described in the materials and methods. For k0 and k1, data are means ± SEM (n = 6). The absolute value of the total equivalent gating charge derived from addition of the individual charges for each rate constant is 0.32e.
TABLE IV.
Summary of the Fits to the Rate Constants for the C1↔O↔C2 Kinetic Scheme
| Rate | k0 | k1 | zg (e− charges) |
|---|---|---|---|
| s−1 | V−1 | ||
| β1 | 11.48 ± 1.17 | 2.11 ± 0.39 | 0.06 |
| α1 | 8.17 ± 0.76 | 1.98 ± 0.26 | 0.05 |
| β2 | 17.05 ± 4.30 | 3.99 ± 0.76 | 0.11 |
| α2 | 404.43 ± 18.86 | −2.11 ± 0.83 | −0.06 |
Values of k0, k1, and zg (equivalent gating charge) derived from exponential fits to the rate constants for the C1↔O↔C2 kinetic scheme using dwell-time data acquired using the conditions described in Fig. 3. The absolute value of the total equivalent gating charge derived from addition of the individual charges for each rate constant is 0.28e. Other details as in Table III.
Effect of Voltage on the Regulation of Channel Gating by the NBDs
Following cAMP-dependent phosphorylation, the gating behavior of CFTR is controlled by cycles of ATP binding and hydrolysis at the NBDs (Gadsby and Nairn, 1999; Sheppard and Welsh, 1999). Previous work indicates that ADP inhibits the activity of CFTR by antagonizing ATP-dependent channel opening to prolong greatly the duration of long closures separating channel openings (Winter et al., 1994). Conversely, the inorganic phosphate analogue, pyrophosphate (PPi) stimulates the activity of CFTR by increasing the frequency and prolonging dramatically the duration of channel openings (Carson et al., 1995b). We were especially interested in the effects of PPi on the voltage dependence of CFTR channel gating, because PPi abolished the asymmetric permeability of CFTR to large organic anions (Linsdell and Hanrahan, 1998b). We speculated that PPi might convert CFTR from an inwardly rectifying channel to a linear channel. To test this hypothesis, we examined the effect of PPi on the inward rectification of CFTR Cl− currents using voltage ramp protocols. As a control, we tested the effect of ADP, which is without effect on the asymmetric permeability of CFTR to large organic anions (Linsdell and Hanrahan, 1998b).
Fig. 8 A shows I-V relationships of CFTR Cl− currents recorded in the absence and presence of PPi (5 mM). PPi (5 mM) increased CFTR Cl− currents by similar amounts at both negative and positive voltages (Fig. 8 B), indicating that PPi-stimulation of CFTR is voltage-independent. Similarly, Fig. 9, A and B , demonstrates that CFTR inhibition by ADP (1 mM) was voltage independent. Consistent with these data, neither PPi (5 mM) nor ADP (1 mM) altered the inward rectification of CFTR Cl− currents (Figs. 8 C and 9 C).
Figure 8.

Pyrophosphate stimulation of CFTR Cl− currents is voltage independent. (A) I-V relationships of CFTR Cl− currents recorded in the absence and presence of pyrophosphate (PPi; 5 mM) in the intracellular solution. (B) Effect of voltage on the fraction of CFTR Cl− current stimulated by PPi (5 mM). Values are means ± SEM (n = 4) at each voltage. The continuous line is the fit of a first order regression to the data. (C) I-V relationships of CFTR Cl− currents recorded in the absence (open circles) and presence (filled circles) of PPi (5 mM) expressed as a percentage of the current value at −100 mV. Data are means ± SEM (n = 4) at each voltage. Other details as in Fig. 1.
Figure 9.

ADP inhibition of CFTR Cl− currents is voltage independent. (A) I-V relationships of CFTR Cl− currents recorded in the absence and presence of ADP (1 mM) in the intracellular solution. (B) Effect of voltage on the fraction of CFTR Cl− current inhibited by ADP (1 mM). (C) I-V relationships of CFTR Cl− currents recorded in the absence (open circles) and presence (filled circles) of ADP (1 mM) expressed as a percentage of the current value at −100 mV. Other details as in Fig. 8.
Voltage Dependence of the Murine CFTR Cl− Channel
In previous work, we demonstrated that wild-type murine CFTR Cl− channels have a reduced single-channel conductance, a dramatically decreased Po and an altered pattern of channel gating compared with those of wild-type human CFTR (Lansdell et al., 1998a,b). To explore further the voltage dependence of CFTR, we investigated the effect of voltage on the activity of wild-type murine CFTR Cl− channels in excised inside-out membrane patches. Fig. 10 A demonstrates that at −80 mV the pattern of gating of murine CFTR is characterized by transitions between a closed state and two open states: sustained openings to a subconductance state and brief openings to a full open state (Lansdell et al., 1998b). At +80 mV, the gating behavior of CFTR was also characterized by prolonged openings to a subconductance state and brief openings to a full open state (Fig. 10 A). However, the frequency and duration of openings to the full open state appeared to be reduced compared with those at −80 mV (Fig. 10 A).
Figure 10.

Effect of voltage on the murine CFTR Cl− channel. (A) Recordings of a single murine CFTR Cl− channel at −80 mV (top) and +80 mV (bottom) made using the conditions described in Fig. 3. (B) Single-channel I-V relationships of human (open circles) and murine (filled circles) CFTR. Data are means ± SEM (n = 8–10) at each voltage. Other details as in Fig. 1. (C) Relationship between chord conductance and voltage for the data shown in B. (D) Effect of voltage on the Po of human (left ordinate) and murine (right ordinate) CFTR. Note the change in scale. Columns and error bars indicate means ± SEM (human, n = 6; murine, n = 10). For human CFTR, voltage was −75 and +75 mV, whereas for murine voltage was −80 and +80 mV. The asterisks indicate values that are significantly different from the data at negative voltages (P < 0.05). Other details as in Fig. 4.
To quantify the voltage dependence of the murine CFTR Cl− channel, we measured i and Po of the full open state. (We did not investigate the voltage dependence of the subconductance state because of its tiny single-channel current amplitude.) Fig. 10 B compares the single-channel I-V relationship of murine CFTR with that of human CFTR. For murine CFTR, at +100 mV i = 73 ± 4% (n = 7) of that at −100 mV while for human CFTR at +100 mV i = 88 ± 2% (n = 10) of that at −100 mV (P < 0.01). Consistent with these data, the chord conductance of murine CFTR decreased by 25% over the voltage range −100 to +100 mV, whereas that of human CFTR decreased by only 12% (Fig. 10 C). Moreover, at each voltage tested, the chord conductance of murine CFTR was reduced compared with that of human CFTR (Fig. 10 C).
Fig. 10 D compares the Po of human and murine CFTR at negative and positive voltages. These data demonstrate two important differences between human and murine CFTR: first, the Po of the full open state of murine CFTR is much reduced compared with that of human CFTR (Fig. 10 D). Second, the Po of murine CFTR is greatly decreased at positive voltages, whereas that of human CFTR is little changed (Fig. 10 D). For murine CFTR, the Po value at +80 mV decreased by 49 ± 10% (n = 10) when compared with that at −80 mV (P < 0.01). In contrast, for human CFTR, the Po value at +75 mV was decreased by only 5 ± 1% (n = 6) when compared with that at −75 mV (P < 0.05). Thus, these single-channel data suggest that inward rectification of murine CFTR is stronger than that of the human CFTR Cl− channel.
DISCUSSION
The goal of this study was to investigate the inward rectification of macroscopic CFTR Cl− currents in excised inside-out membrane patches. Using three independent methods, we demonstrated that CFTR exhibits inward rectification when bathed in symmetrical Cl−-rich solutions. The data indicate that this inward rectification is caused both by a voltage-dependent decrease in single-channel chord conductance and by changes in the kinetics of channel gating at large positive voltages. They also suggest that inward rectification of the murine CFTR Cl− channel is stronger than that of human CFTR.
In principal, inward rectification of the CFTR Cl− channel might either result from channel block by a soluble factor or be an intrinsic property of the CFTR channel protein. Studies of other ion channels provide strong support for both hypotheses. For example, the inward rectification of Kir channels, NMDA receptors, and cyclic nucleotide–gated channels is caused by voltage-dependent block of outward current flow by extrinsic cations. In the case of Kir channels the blocking cations are intracellular Mg2+ and polyamines (Vandenberg, 1987; Lopatin et al., 1994), whereas for NMDA receptors and cyclic nucleotide–gated channels the blocking cations are extracellular Ca2+ and Mg2+ (Nowak et al., 1984; Zimmerman and Baylor, 1986). In contrast, inward rectification of KAT1, an inward-rectifier K+ channel from the plant Arabidopsis thaliana, results from an intrinsic gating mechanism (Zei and Aldrich, 1998). Like voltage-gated K+ channels (Hille, 2001), KAT1 possesses a voltage-sensor, the S4 segment that uses the energy of voltage to control channel gating (Zei and Aldrich, 1998).
Several lines of evidence suggest that channel block is not likely to account for inward rectification of the CFTR Cl− channel in excised inside-out membrane patches. First, changes in the composition of the intra- and extracellular solutions did not abolish inward rectification (for data and discussion, see the online supplemental material, available at http://www.jgp.org/cgi/content/full/jgp.200308921/DC1). Second, inward rectification has been observed using cells expressing either native or recombinant wild-type human CFTR (e.g., Quinton and Reddy, 2000; Lansdell et al., 2000; Linsdell and Gong, 2002; this study), indicating that inward rectification is independent of the expression system used to investigate CFTR. Third, inward rectification of CFTR Cl− channels reconstituted into planar lipid bilayers was unaffected by changes in the composition of the lipid bilayer (Zhao et al., 1996), suggesting that the surface charge of the membrane lipid does not affect inward rectification. Fourth, the effects of voltage on the function of the murine CFTR Cl− channel (present study) indicate that inward rectification is a common characteristic of human and murine CFTR. Fifth, mutation of several residues within the MSDs alters the shape of the I-V relationship of CFTR. Inward rectification of CFTR Cl− currents is accentuated by mutation of R334, T338, T339, and S341 (sixth transmembrane segment (M6); Gong and Linsdell, 2003; Linsdell et al., 1998; Gong et al., 2002), S1118 (M11; Zhang et al., 2000), and T1134 and N1138 (M12; Gupta et al., 2001). In contrast, mutation of V317 (M5; Zhang et al., 2002) causes outward rectification of CFTR Cl− currents. Based on these data, we speculate that inward rectification is an intrinsic property of the CFTR Cl− channel. Because ADP and PPi, two agents that interact with the NBDs, are without effect on inward rectification and because site-directed mutations in the MSDs enhance inward rectification, we propose that the CFTR pore determines this property of the CFTR Cl− channel.
For the CFTR pore, itself, to be responsible for inward rectification, the architecture of the pore should facilitate the flow of Cl− from the intra- to the extracellular side of the membrane, but hinder Cl− flow in the opposite direction. To transport ions at maximal translocation rates, ion channels have evolved charged lined vestibules that concentrate and funnel ions toward the selectivity filter (Green and Andersen, 1991; Dutzler et al., 2002, 2003). Based on these considerations, inward rectification of current flow through the CFTR pore might be achieved by asymmetries in the geometry of the intra- and extracellular vestibules, differences in the distribution of Cl−-binding sites between the two vestibules or a combination of these factors.
Evidence for asymmetries in the topology of the intra- and extracellular vestibules is provided by studies of anion permeation and channel block. Anion permeation studies indicate that the narrowest part of the CFTR pore is ∼0.53–0.60 nm in diameter (Cheung and Akabas, 1996; Linsdell et al., 1997), widening under certain circumstances to a diameter of ∼1.3 nm (Linsdell and Hanrahan, 1998b). This constriction, which likely represents the selectivity filter, occurs in the region of F337 and T338 in M6 because mutation of these residues altered dramatically the anion permeability sequence of CFTR, whereas mutation of other residues in M6 had less marked effects on anion permeation (Linsdell et al., 1998, 2000; McCarty and Zhang, 2001; Gong et al., 2002; Gupta and Linsdell, 2003). On the intra- and extracellular sides of this constriction, the pore enlarges. The voltage dependence of channel block by large organic anions suggests that the CFTR pore contains a wide intracellular vestibule that funnels blocking anions deep into the pore where they bind, occlude the pore, and block Cl− permeation (Linsdell and Hanrahan, 1996; Sheppard and Robinson, 1997; Hwang and Sheppard, 1999; Zhou et al., 2002). Less is known about the topology of the extracellular end of the CFTR pore. Based on the ability of methanethiosulphonate reagents in the extracellular solution to react with M6 residues toward the cytosolic side of the membrane (Cheung and Akabas, 1996), there might also be a wide extracellular vestibule. However, the inability of open-channel blockers to reach their binding sites when added to the extracellular solution and the short length of extracellular loops 3 and 6 led McCarty (2000) to propose that CFTR has a small extracellular vestibule. Consistent with this idea, Linsdell and Hanrahan (1998a)(b) demonstrated that CFTR exhibits an asymmetric permeability to large organic anions (e.g., galacturonate, glutathione, and lactobionate). During the normal gating cycle, flow of large organic anions through the CFTR pore is only permitted in the intra- to extracellular direction (Linsdell and Hanrahan, 1998a,b).
The sixth transmembrane segment plays a crucial role in determining the pore properties of CFTR (Sheppard and Welsh, 1999; McCarty, 2000; Gong et al., 2002). Within M6, the residues R334 (Smith et al., 2001; Gong and Linsdell, 2003), K335 (Anderson et al., 1991), F337 (Linsdell et al., 2000), T338 (Linsdell et al., 1998), S341 (McDonough et al., 1994), I344 (Gong et al., 2002), and possibly R352 (Guinamard and Akabas, 1999; Gong et al., 2002) contribute to Cl−-binding sites. Assuming that the narrowest part of the CFTR pore occurs in the region of F337 and T338 (see above and Linsdell et al., 1998, 2000; McCarty and Zhang, 2001; Gong et al., 2002; Gupta and Linsdell, 2003), the data suggest that three Cl−-binding sites (S341, I344, and R352) might be located in a spacious intracellular vestibule, whereas two Cl−-binding sites (R334 and K335) might be located in a more confined extracellular vestibule. This simple model suggests that current flow through the CFTR pore might inwardly rectify. However, an important caveat of this model is that present knowledge of the mechanism of anion permeation by the CFTR Cl− channel is incomplete and it is quite likely that residues in other transmembrane segments contribute to Cl−-binding sites. Nevertheless, asymmetries in the size of the intra- and extracellular vestibules and/or the distribution of Cl−-binding sites within the CFTR pore are plausible explanations for the inward rectification of CFTR Cl− currents.
Besides the architecture of the CFTR pore, voltage-dependent gating might make some contribution to the observed inward rectification of current flow through the CFTR Cl− channel. Work by other investigators and ourselves demonstrate that CFTR channel gating exhibits voltage dependence despite the lack of a marked effect of voltage on Po. First, using cell-attached membrane patches from NIH 3T3 fibroblasts expressing wild-type human CFTR, Fischer and Machen (1994) demonstrated that slow (ATP dependent) gating is voltage independent, whereas fast (flickery intraburst) gating is voltage dependent, becoming the dominant gating mode at strong negative voltages. Second, using excised membrane patches from Xenopus oocytes expressing wild-type human CFTR, Zhang et al. (2002) demonstrated that burst duration increases at negative voltages, whereas interburst interval is unaffected by voltage. Third, to investigate the flickery closures that interrupt bursts of channel openings, Zhou et al. (2001) analyzed the gating kinetics of the CFTR variant K1250A whose prolonged openings facilitate the discrimination of fast and slow gating events. Zhou et al. (2001) found that the rate of transition from the short-lived closed state to the open state exhibits voltage dependence and is sensitive to permeant anions in the extracellular solution, suggesting that brief flickery closures represent the voltage-dependent occupancy of an anion-binding site within the CFTR pore by unknown intracellular anions.
To elucidate the effects of voltage on CFTR channel gating, we adopted a different strategy to other investigators. We employed kinetic modeling to analyze the gating behavior of single wild-type human CFTR Cl− channels in excised membrane patches bathed in symmetrical Cl−-rich solutions at 37°C and lightly filtered our data. Using this approach, we determined the effect of voltage on both fast and slow gating events. Our data indicate that voltage has significant effects on CFTR channel gating. Membrane depolarization decreased both the duration of bursts and the interburst interval, but increased the duration of gaps within bursts. However, because the voltage dependencies of the different rate constants were in opposite directions, Po was largely unaffected by voltage. The voltage independence of Po and the lack of effect of ADP and PPi on the inward rectification of CFTR Cl− currents argue that changes in CFTR channel gating do not contribute to the rectification of macroscopic CFTR Cl− currents. However, other data suggest the contrary. First, the predicted CFTR Cl− current (N × i × Po) at +100 mV, which is accounted for by reductions in both i and Po, agrees closely with the measured CFTR Cl− current at this voltage. Second, the Po of murine CFTR is decreased markedly at positive voltages. Third, comparison of the data of Smith et al. (2001) and Gong and Linsdell (2003) suggests that the strong inward rectification of the CFTR variant R334C is not accounted for by voltage-dependent changes in i. Based on these data, we argue that voltage-dependent changes in CFTR channel gating contribute to the inward rectification of macroscopic CFTR Cl− currents.
Our data indicate that the total equivalent gating charge for CFTR is only ∼0.30e. The small size of the gating charge of CFTR is consistent with the lack of marked voltage dependence of CFTR channel gating. The data suggest that residues with charged and uncharged polar side chains move only a very small distance within the transmembrane electric field. These residues might be located either along the full-length of the CFTR pore or restricted to a specific region of the permeation pathway. Alternatively, linear three-state kinetic schemes might be too simple to describe the voltage dependence of CFTR channel gating. Because the total equivalent gating charge of CFTR is comparable to the amount of charge movement intrinsic to the voltage-dependent Cl− channel ClC-0 (Chen and Miller, 1996), we favor the former idea. The amino acid sequence of ClC-0 lacks a motif equivalent to the S4 segment, the voltage sensor of voltage-gated cation channels (Hille, 2001). Instead, permeant anions in the extracellular solution act as the source of the gating charge (Pusch et al., 1995; Chen and Miller, 1996). The binding of Cl− ions to a Cl−-binding site located at the extracellular end of the ClC-0 pore causes a conformational change that precedes channel opening and inward Cl− flow (Chen and Miller, 1996). Because bound Cl− ions traverse the transmembrane electric field, gating is both voltage and Cl− concentration dependent (Pusch et al., 1995; Chen and Miller, 1996). Excitingly, the structural basis of this gating mechanism has been elucidated following the determination of high-resolution crystal structures of bacterial CLC proteins (Dutzler et al., 2002, 2003). In these bacterial CLC channels, the Cl−-binding site closest to the extracellular end of the selectivity filter is gated by the carboxyl group of glutamate (E)148 that protrudes into the CLC channel pore: when the carboxyl group of E148 is bound to the Cl−-binding site the pore is closed, but when a Cl− ion binds the pore opens to allow Cl− permeation (Dutzler et al., 2002, 2003).
The regulation of CLC channel gating by localized motions of an amino acid side chain (Dutzler et al., 2003) raises the question as to whether a similar mechanism might operate in the CFTR Cl− channel. However, anion permeation through the CFTR pore is tightly linked to the function of the NBDs that power channel gating. For example, Kogan et al. (2003) recently demonstrated that glutathione permeation through the CFTR pore is controlled by ATP binding rather than ATP binding and hydrolysis as in the case of Cl− permeation. These data suggest that global conformation changes in the structure of the CFTR protein control Cl− permeation, not local changes in the orientation of amino acid side-chains. If this idea were correct, it would suggest that CFTR channel gating shares similarities with activation gating in K+ channels that involves the reorientation of transmembrane α-helices (Jiang et al., 2003). Future studies should test this possibility.
Supplemental Material
Acknowledgments
We thank Professor M.J. Welsh, Drs. M.E. Krouse and M.C. Winter, and our departmental colleagues, especially Dr. R.W. Meech, for valuable discussions, and Dr. T.-C. Hwang for a pre-print. We thank Dr. C.R. O'Riordan (Genzyme, Framingham, MA) for the gift of C127 cells expressing wild-type human CFTR, Dr. S.J. Delaney and Professor B.J. Wainwright (University of Queensland, Brisbane, Australia) for the gift of CHO cells expressing wild-type murine CFTR, and Dr. M.D. Amaral and F. Mendes (Universidade de Lisboa, Lisboa, Portugal) for the gift of BHK cells expressing wild-type human CFTR.
This work was supported by the Cystic Fibrosis Trust and the National Kidney Research Fund.
David C. Gadsby served as editor.
The online version of this paper contains supplemental material.
Abbreviations used in this paper: CFTR, cystic fibrosis transmembrane conductance regulator; M, transmembrane segment; MSD, membrane-spanning domain; NBD, nucleotide-binding domain; PPi, pyrophosphate.
Footnotes
Although very small, the membrane currents evoked by the voltage ramp protocol under basal conditions outwardly rectified. For example, at −100 mV basal current was −0.88 ± 0.20 pA and at +100 mV basal current was 2.00 ± 0.37 pA (n = 10). When basal currents were subtracted from those recorded in the presence of ATP (1 mM) + PKA (75 nM), the outward rectification of basal current accounted for 6 ± 2% (n = 10) of the inward rectification of CFTR Cl− current at +100 mV.
In contrast to analyses of rate constant values at ±75 mV using Student's paired t test, some analyses of rate constant data over the range −75 to +75 mV using one-way ANOVAs failed to achieve statistical significance. We attribute this difference to the fact that paired data were used for analyses using Student's paired t test, but pooled data were used for one-way ANOVAs.
References
- Anderson, M.P., R.J. Gregory, S. Thompson, D.W. Souza, S. Paul, R.C. Mulligan, A.E. Smith, and M.J. Welsh. 1991. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 253:202–205. [DOI] [PubMed] [Google Scholar]
- Berger, H.A., M.P. Anderson, R.J. Gregory, S. Thompson, P.W. Howard, R.A. Maurer, R.C. Mulligan, A.E. Smith, and M.J. Welsh. 1991. Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J. Clin. Invest. 88:1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Z., and D.N. Sheppard. 2002. Phloxine B interacts with the cystic fibrosis transmembrane conductance regulator at multiple sites to modulate channel activity. J. Biol. Chem. 277:19546–19553. [DOI] [PubMed] [Google Scholar]
- Carson, M.R., S.M. Travis, and M.J. Welsh. 1995. a. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J. Biol. Chem. 270:1711–1717. [DOI] [PubMed] [Google Scholar]
- Carson, M.R., M.C. Winter, S.M. Travis, and M.J. Welsh. 1995. b. Pyrophosphate stimulates wild-type and mutant cystic fibrosis transmembrane conductance regulator Cl− channels. J. Biol. Chem. 270:20466–20472. [DOI] [PubMed] [Google Scholar]
- Chen, T.-Y., and C. Miller. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, M., and M.H. Akabas. 1996. Identification of cystic fibrosis transmembrane conductance regulator channel-lining residues in and flanking the M6 membrane-spanning segment. Biophys. J. 70:2688–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler, R., E.B. Campbell, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E.B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Fischer, H., and T.E. Machen. 1994. CFTR displays voltage dependence and two gating modes during stimulation. J. Gen. Physiol. 104:541–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby, D.C., and A.C. Nairn. 1999. Control of cystic fibrosis transmembrane conductance regulator channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 79:S77–S107. [DOI] [PubMed] [Google Scholar]
- Gong, X., S.M. Burbridge, E.A. Cowley, and P. Linsdell. 2002. Molecular determinants of Au(CN)2 − binding and permeability within the cystic fibrosis transmembrane conductance regulator Cl− channel pore. J. Physiol. 540:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, X., and P. Linsdell. 2003. Molecular determinants and role of an anion binding site in the external mouth of the CFTR chloride channel pore. J. Physiol. 549:387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, W.N., and O.S. Andersen. 1991. Surface charges and ion channel function. Annu. Rev. Physiol. 53:341–359. [DOI] [PubMed] [Google Scholar]
- Guinamard, R., and M.H. Akabas. 1999. Arg352 is a major determinant of charge selectivity in the cystic fibrosis transmembrane conductance regulator chloride channel. Biochemistry. 38:5528–5537. [DOI] [PubMed] [Google Scholar]
- Gupta, J., A. Evagelidis, J.W. Hanrahan, and P. Linsdell. 2001. Asymmetric structure of the cystic fibrosis transmembrane conductance regulator chloride channel pore suggested by mutagenesis of the twelfth transmembrane region. Biochemistry. 40:6620–6627. [DOI] [PubMed] [Google Scholar]
- Gupta, J., and P. Linsdell. 2003. Extent of the selectivity filter conferred by the sixth transmembrane region in the CFTR chloride channel pore. Mol. Membr. Biol. 20:45–52. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Hanke, W., and C. Miller. 1983. Single chloride channels from Torpedo electroplax: activation by protons. J. Gen. Physiol. 82:25–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. 3rd ed. Sinauer Associates, Inc., Sunderland, MA. 814 pp.
- Hwang, T.-C., and D.N. Sheppard. 1999. Molecular pharmacology of the CFTR Cl− channel. Trends Pharmacol. Sci. 20:448–453. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B.T. Chait, and R. MacKinnon. 2003. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Kogan, I., M. Ramjeesingh, C. Li, J.F. Kidd, Y. Wang, E.M. Leslie, S.P.C. Cole, and C.E. Bear. 2003. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. 22:1981–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell, K.A., S.J. Delaney, D.P. Lunn, S.A. Thomson, D.N. Sheppard, and B.J. Wainwright. 1998. a. Comparison of the gating behaviour of human and murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in mammalian cells. J. Physiol. 508:379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell, K.A., J.F. Kidd, S.J. Delaney, B.J. Wainwright, and D.N. Sheppard. 1998. b. Regulation of murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in Chinese hamster ovary cells. J. Physiol. 512:751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell, K.A., Z. Cai, J.F. Kidd, and D.N. Sheppard. 2000. Two mechanisms of genistein inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in murine cell line. J. Physiol. 524:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell, P., J.A. Tabcharani, J.M. Rommens, Y.-X. Hou, X.-B. Chang, L.-C. Tsui, J.R. Riordan, and J.W. Hanrahan. 1997. Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. J. Gen. Physiol. 110:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell, P., S.-X. Zheng, and J.W. Hanrahan. 1998. Non-pore lining amino acid side chains influence anion selectivity of the human CFTR Cl− channel expressed in mammalian cell lines. J. Physiol. 512:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell, P., A. Evagelidis, and J.W. Hanrahan. 2000. Molecular determinants of anion selectivity in the cystic fibrosis transmembrane conductance regulator chloride channel pore. Biophys. J. 78:2973–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell, P., and X. Gong. 2002. Multiple inhibitory effects of Au(CN)2 − ions on cystic fibrosis transmembrane conductance regulator Cl− channel currents. J. Physiol. 540:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell, P., and J.W. Hanrahan. 1996. Disulphonic stilbene block of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a mammalian cell line and its regulation by a critical pore residue. J. Physiol. 496:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell, P., and J.W. Hanrahan. 1998. a. Glutathione permeability of CFTR. Am. J. Physiol. Cell Physiol. 275:C323–C326. [DOI] [PubMed] [Google Scholar]
- Linsdell, P., and J.W. Hanrahan. 1998. b. Adenosine triphosphate-dependent asymmetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. J. Gen. Physiol. 111:601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell, P., and J.W. Hanrahan. 1999. Substrates of multidrug resistance-associated proteins block the cystic fibrosis transmembrane conductance regulator chloride channel. Br. J. Pharmacol. 126:1471–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatin, A.N., E.N. Makhina, and C.G. Nichols. 1994. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 372:366–369. [DOI] [PubMed] [Google Scholar]
- McCarty, N.A. 2000. Permeation through the CFTR chloride channel. J. Exp. Biol. 203:1947–1962. [DOI] [PubMed] [Google Scholar]
- McCarty, N.A., and Z.-R. Zhang. 2001. Identification of a region of strong discrimination in the pore of CFTR. Am. J. Physiol. Lung Cell Mol. Physiol. 281:L852–L867. [DOI] [PubMed] [Google Scholar]
- McDonough, S., N. Davidson, H.A. Lester, and N.A. McCarty. 1994. Novel pore-lining residues in CFTR that govern permeation and open-channel block. Neuron. 13:623–634. [DOI] [PubMed] [Google Scholar]
- Nowak, L., P. Bregestovski, P. Ascher, A. Herbet, and A. Prochiantz. 1984. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 307:462–465. [DOI] [PubMed] [Google Scholar]
- Overholt, J.L., M.E. Hobert, and R.D. Harvey. 1993. On the mechanism of rectification of the isoproterenol-activated chloride current in guinea-pig ventricular myocytes. J. Gen. Physiol. 102:871–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch, M., U. Ludewig, A. Rehfeldt, and T.J. Jentsch. 1995. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature. 373:527–531. [DOI] [PubMed] [Google Scholar]
- Qin, F., A. Auerbach, and F. Sachs. 1997. Maximum likelihood estimation of aggregated Markov processes. Proc. R. Soc. Lond. B. Biol. Sci. 264:375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, F., A. Auerbach, and F. Sachs. 2000. A direct optimization approach to hidden Markov modeling for single channel kinetics. Biophys. J. 79:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton, P.M., and M.M. Reddy. 2000. CFTR, a rectifying, non-rectifying anion channel? J. Korean Med. Sci. 15:S17–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan, J.R., J.M. Rommens, B.-S. Kerem, N. Alon, R. Rozmahel, Z. Grzelczak, J. Zielenski, S. Lok, N. Plavsic, J.-L. Chou, et al. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245:1066–1073. [DOI] [PubMed] [Google Scholar]
- Sakmann, B., and G. Trube. 1984. Voltage-dependent inactivation of inward-rectifying single-channel currents in the guinea-pig heart cell membrane. J. Physiol. 347:659–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard, D.N., and K.A. Robinson. 1997. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J. Physiol. 503:333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard, D.N., and M.J. Welsh. 1999. Structure and function of the cystic fibrosis transmembrane conductance regulator chloride channel. Physiol. Rev. 79:S23–S45. [DOI] [PubMed] [Google Scholar]
- Smith, S.S., X. Liu, Z.-R. Zhang, F. Sun, T.E. Kriewall, N.A. McCarty, and D.C. Dawson. 2001. CFTR: covalent and noncovalent modification suggests a role for fixed charges in anion conduction. J. Gen. Physiol. 118:407–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabcharani, J.A., X.-B. Chang, J.R. Riordan, and J.W. Hanrahan. 1991. Phosphorylation-regulated Cl− channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 352:628–631. [DOI] [PubMed] [Google Scholar]
- Vandenberg, C.A. 1987. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc. Natl. Acad. Sci. USA. 84:2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, M.C., D.N. Sheppard, M.R. Carson, and M.J. Welsh. 1994. Effect of ATP concentration on CFTR Cl− channels: a kinetic analysis of channel regulation. Biophys. J. 66:1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zei, P.C., and R.W. Aldrich. 1998. Voltage-dependent gating of single wild-type and S4 mutant KAT1 inward rectifier potassium channels. J. Gen. Physiol. 112:679–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.-R., S.I. McDonough, and N.A. McCarty. 2000. Interaction between permeation and gating in a putative pore domain mutant in the cystic fibrosis transmembrane conductance regulator. Biophys. J. 79:298–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.-R., S. Zeltwanger, S.S. Smith, D.C. Dawson, and N.A. McCarty. 2002. Voltage-sensitive gating induced by a mutation in the fifth transmembrane domain of CFTR. Am. J. Physiol. Lung Cell Mol. Physiol. 282:L135–L145. [DOI] [PubMed] [Google Scholar]
- Zhao, J., B. Zerhusen, J. Xie, M.L. Drumm, P.B. Davis, and J. Ma. 1996. Rectification of cystic fibrosis transmembrane conductance regulator chloride channel mediated by extracellular divalent cations. Biophys. J. 71:2458–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z., S. Hu, and T.-C. Hwang. 2001. Voltage-dependent flickery block of an open cystic fibrosis transmembrane conductance regulator (CFTR) channel pore. J. Physiol. 532:435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z., S. Hu, and T.-C. Hwang. 2002. Probing an open CFTR pore with organic anion blockers. J. Gen. Physiol. 120:647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, A.L., and D.A. Baylor. 1986. Cyclic GMP-sensitive conductance of retinal rods consists of aqueous pores. Nature. 321:70–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.