

Abstract
Peptide toxins are invaluable tools for studying the structure and physiology of ion channels. Pseudechetoxin (PsTx) is the first known peptide toxin that targets cyclic nucleotide–gated (CNG) ion channels, which play a critical role in sensory transduction in the visual and olfactory systems. PsTx inhibited channel currents at low nM concentrations when applied to the extracellular face of membrane patches expressing olfactory CNGA2 subunits. Surprisingly, 500 nM PsTx did not inhibit currents through channels formed by the CNGA3 subunit from cone photoreceptors. We have exploited this difference to identify the PsTx-binding site on the extracellular face of CNG channels. Studies using chimeric channels revealed that transplantation of the pore domain from CNGA2 was sufficient to confer high affinity PsTx binding upon a CNGA3 background. To further define the binding site, reciprocal mutations were made at 10 nonidentical amino acid residues in this region. We found that two residues in CNGA2, D316 and Y321, were essential for high-affinity inhibition by PsTx. Furthermore, replacement of both residues was required to confer high-affinity PsTx inhibition upon CNGA3. Several other residues, including E325, also form favorable interactions with PsTx. In the CNGA2-E325K mutant, PsTx affinity was reduced by ∼5-fold to 120 nM. An electrostatic interaction with D316 does not appear to be the primary determinant of PsTx affinity, as modification of the D316C mutant with a negatively charged methanethiosulfonate reagent did not restore high affinity inhibition. The residues involved in PsTx binding are found within the pore turret and helix, in similar positions to residues that form the receptor for pore-blocking toxins in voltage-gated potassium channels. Furthermore, biophysical properties of PsTx block, including an unfavorable interaction with permeant ions, also suggest that it acts as a pore blocker. In summary, PsTx seems to occlude the entrance to the pore by forming high-affinity contacts with the pore turret, which may be larger than that found in the KcsA structure.
Keywords: snake venom, neurotoxin, CNG channel, patch-clamp electrophysiology, Xenopus oocyte
INTRODUCTION
Peptide toxins that target ion channels have been obtained from the venoms of many poisonous creatures, including scorpions, snakes, spiders, and cone shells (Olivera et al., 1991, 1995; Garcia et al., 2001; Harvey, 2001; Rash and Hodgson, 2002). Over the past several decades, these peptide toxins have played a crucial role in the study of ion channel structure, function, and physiology. Originally, these toxins were used as specific high-affinity ligands that facilitated the identification and purification of several ion channels. Fluorescent versions of bungarotoxin, charybdotoxin, and apamin have been widely used as tools to study the subcellular distribution and cell biology of nicotinic acetylcholine receptors and calcium-activated potassium channels. Finally, these toxins have been used in structure-function studies to identify structural elements of ion channels and gating mechanisms. In fact, studies with charybdotoxin gave us the first glimpse of the structure of an ion channel pore. By identifying the extracellular residues that comprised the charybdotoxin-binding site on the Shaker potassium channel, MacKinnon, Miller, and Yellen first postulated the existence of a reentrant pore loop (MacKinnon and Miller, 1989; Yellen et al., 1991).
Studies of peptide toxins that target voltage-gated ion channels have resulted in the discovery of toxins with two distinct modes of action. Some ion channel toxins, such as agitoxin, charybdotoxin, and ω-conotoxin-GVIA, are known to function as simple pore blockers that physically occlude the entry of ions into the transmembrane pore (MacKinnon et al., 1990; Ellinor et al., 1994; Garcia et al., 1994; Feng et al., 2001). Other peptide antagonists, such as hanatoxin, grammotoxin, and ω-agatoxin-IVA, are known as gating modifiers because they allosterically inhibit structural rearrangements that are involved in channel activation (Mintz et al., 1992; Lampe et al., 1993; Swartz and MacKinnon, 1995). The mechanism of action of these two types of toxins can be largely predicted by the location of their binding sites. Pore blockers typically bind to the amino acid residues in the pore turret and nearby residues that form the extracellular vestibule at the mouth of the transmembrane pore (MacKinnon and Miller, 1989; MacKinnon et al., 1990; Gross et al., 1994; Gross and MacKinnon, 1996). In contrast, gating-modifier toxins recognize conserved structural features found on the extracellular face of the voltage sensor. In voltage-gated calcium and potassium channels, gating-modifier toxins typically bind to residues located at the extracellular end of the third transmembrane helix (S3) (Swartz and MacKinnon, 1997; Bourinet et al., 1999; Winterfield and Swartz, 2000). Binding of these toxins impairs the structural rearrangements necessary for channel activation, thereby stabilizing the closed or inactivated states of the channel.
CNG ion channels were first discovered in the sensory epithelium of the visual and olfactory systems (for a review see Kaupp and Seifert, 2002). In both photoreceptors and olfactory neurons, CNG channels convert stimulus-induced changes in the intracellular concentration of cyclic nucleotides into changes in membrane potential. In this manner, they control the release of neurotransmitter at the synapse. Currently, six CNG channel subunits are known (Kaupp et al., 1989; Dhallan et al., 1990; Chen et al., 1993; Biel et al., 1994; Bradley et al., 1994; Liman and Buck, 1994; Weyand et al., 1994; Korschen et al., 1995; Gerstner et al., 2000; Bradley et al., 2001). Although they are not activated by changes in membrane voltage, CNG channels are members of the voltage-gated ion channel superfamily on the basis of their subunit structure. Each subunit contains a transmembrane domain with six transmembrane helices (S1–6) and a reentrant pore loop between S5 and S6 (Heginbotham et al., 1992). When expressed individually in oocytes or HEK-293 cells, the CNGA1, 2, and 3 subunits form functional CNG channels. In contrast, CNGA4 and the β-subunits, CNGB1 and CNGB3, do not form functional channels, although they possess a similar structure. However, the β-subunits coassemble with CNGA1–3 to form native channels, and confer distinct properties in terms of ligand affinity, regulation, and ion permeation. The native rod photoreceptor CNG channel has a stoichiometry of 3 A1:1 B1 subunits (Weitz et al., 2002; Zheng et al., 2002; Zhong et al., 2002). The native olfactory CNG channel is composed of three distinct subunit types, including CNGA2, CNGA4, and a splice variant of CNGB1 (Sautter et al., 1998; Bonigk et al., 1999), while cone photoreceptor channels contain CNGA3 and CNGB3 (Gerstner et al., 2000). In the past decade, CNG channels have been discovered in nonsensory tissues through out the body, including the brain, heart, liver, kidneys, and skeletal muscle (Finn et al., 1996). However, the function of CNG channels in these tissues remains obscure, largely due to the lack of specific CNG channel antagonists.
Recently, we purified and cloned pseudechetoxin from the venom of the Australian King Brown snake, Pseudechis australis (Brown et al., 1999; Yamazaki et al., 2002). Pseudechetoxin (PsTx) was the first peptide toxin known to target CNG channels and inhibits CNG channel currents with high affinity when applied to the extracellular face of membrane patches expressing CNGA1 or CNGA2. Surprisingly, in subsequent studies we found that CNG channels formed by the CNGA3 subunit from cone photoreceptors were immune to block by PsTx. In the current study, we have exploited this selectivity to identify residues on the extracellular face of CNG channels that are critical for high-affinity interaction with PsTx. Binding of PsTx is also weakened by an unfavorable interaction with permeant ions, a phenomenon known as “knock-off” (MacKinnon and Miller, 1988). The results indicate that PsTx forms several contacts with the pore turret region of CNG channels and inhibits channel activity by occluding extracellular access to the transmembrane pore.
MATERIALS AND METHODS
Pseudechis australis venom was purchased from Venom Supplies Pty. Ltd. PsTx was purified as described previously (Brown et al., 1999). The plasmid encoding bovine CNGA1 was the gift of Dr. W.N. Zagotta (University of Washington, Seattle, WA). CNGA2 was a gift of Dr. R. Reed (Johns Hopkins University), and CNGB1 was the gift of Dr. Robert Molday (University of British Columbia). Plasmids encoding CNGA3, CNGB1.3, and CNGA4 were the gifts of Drs. M. Biel and A. Gerstner (Ludwig Maximilians University, Munich, Germany). Cyclic nucleotides, including p-chlorophenylthio-cGMP (CPT-cG), were from Sigma-Aldrich. Methanethiosulfonate (MTS) reagents were purchased from Toronto Research Chemicals. All other chemicals were of analytical grade or better.
Construction of Chimeric CNG Channels and Mutants
For the construction of chimeric CNG channels, unique restriction sites were created between each of the extracellular loops using silent mutations. A KpnI site was inserted between transmembrane helices S2 and S3 in both CNGA2 and CNGA3, a Kpn2I site was inserted between S4 and S5, and a BstBI site was inserted after S6 in CNGA3. These two sites were already present in CNGA2. An additional BstBI site was removed from the S3 region of CNGA3. These mutations allowed the creation of several chimeric CNG channels. Splice sites were after S229, I277, and R401 in CNGA2 and after V276, I322, and R446 in CNGA3. Chimeras are named by indicating the source of each of the four channel segments. For instance, the chimera 2-2-3-2 would contain the NH2 terminus and S1–4 of CNGA2, the S5-P-S6 region from CNGA3, and the COOH terminus from CNGA2.
Point mutations were created using the Quick Change kit (Stratagene). After sequence confirmation, the relevant restriction fragment containing the S5-P-S6 region was subcloned into the appropriate CNG channel sequence to prevent second site mutations. Point mutations are named by the number of the parent subunit, followed by the wild-type amino acid, its position, and its replacement. Therefore, the mutant 2-D316N is a point mutation in CNGA2, in which the wild-type D316 is replaced with N, from the corresponding position in CNGA3.
Expression of CNG Channels in Xenopus Oocytes
The coding sequences for CNGA1, A2, and A3 were inserted into the pGEMHE vector, which promotes high expression levels by incorporating flanking sequences from the Xenopus β-globin gene (Liman et al., 1992). For in vitro transcription, the plasmids were linearized with PstI (CNGA1) or NheI (CNGA2 and CNGA3). Complementary RNA was transcribed from the T7 promoter and capped using the mMessage mMachine kit (Ambion). Xenopus oocytes were prepared and injected with cRNA (10–20 ng) by standard methods. After injection, oocytes were maintained at 18°C for 3–7 d in ND 96 media containing 50 μg/ml gentamycin.
Electrophysiology on Oocytes
Recording electrodes were pulled from borosilicate glass (World Precision Instruments) on a P97 Brown-Flaming puller (Sutter) and polished on a microforge (Life Science Resources) to a resistance of 1–2 MΩ. Recordings were made in symmetrical solutions containing 130 mM NaCl, 3 mM HEPES (pH 7.2), and 0.2 mM EDTA. The electrode solution contained 1 mM cyclic GMP to activate CNG channels. Currents were recorded from patches in the outside-out configuration using an Axopatch 200A amplifier (Axon Instruments, Inc.) driven by pClamp 8.0 software via a Digidata 1200 interface (Axon Instruments, Inc.). Typically, 250-ms voltage pulses, ranging from −100 to + 100 mV, were given from a holding potential of 0 mV. Records were typically sampled at 10 kHz and filtered at 2 kHz. Solutions were applied to the extracellular face of the patch via the “sewer pipe” method using an RSC-100 rapid perfusion system (Biologic; Molecular Kinetics). The seal resistance and “leak” currents were estimated after application of a solution containing 10 mM MgCl2, which blocks >98% of the current at –60 mV in all CNG channels studied. Solutions containing toxin were supplemented with 50 μg/ml bovine serum albumin to prevent nonspecific surface adsorption. PsTx concentration was estimated based on the calculated molar extinction coefficient of 21.6 cm−1 for a 10 mg/ml solution (Yamazaki et al., 2002). MTS reagents were tested to ensure their viability based on their ability to shift the cyclic nucleotide sensitivity of CNGA1 by modification of C481 (Brown et al., 1998).
Growth and Transfection of HEK-293 cells
Human embryonic kidney cells (HEK-293) were maintained in DMEM culture medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum and a penicillin/streptomycin mixture at 37°C in 5% CO2. Cells were split 1 d before transfection and grown to 50–70% confluence in a T-25 culture flask. Cells were transfected with plasmids encoding CNG channel subunits by treatment with Effectene (QIAGEN) following manufacturer's instructions. Briefly, the culture media was aspirated and replaced with 1 ml DMEM containing a total of 2 μg of plasmid DNA, 50 μl effectene, and 16 μl effectene enhancer. All transfections contained 0.5 μg of a plasmid encoding EGFP (CLONTECH Laboratories, Inc.), and 1.5 g of plasmid DNA encoding CNG channel subunits. For cotransfection of multiple subunit types, the ratio of DNA encoding the primary subunits (CNGA1, A2, or A3) to CNGA4 and/or B1 was 1:1. After 6 h, the transfection mix was removed by aspiration. The cells were lifted with trypsin/EDTA, and replated at a low density on glass coverslips. To prevent further growth and maintain isolated cells, the media was supplemented with the mitotic blocker FUDR (200 μg/ml 5′ fluoro-2-deoxyuridine and 500 μg/ml uridine). For whole-cell experiments, recording was performed 18–36 h after transfection. Excised patch experiments were performed 24–72 h after transfection to allow expression of larger currents and better adherence to the coverslip substrate.
Electrophysiology on HEK-293 Cells
Coverslips containing transfected cells were rinsed in serum-free media and placed in a recording chamber (Warner Instruments) on the stage of a Nikon inverted microscope. Electrodes had resistances between 2.0–3.5 MΩ when filled with an internal solution containing 145 mM KCl, 5 mM NaCl, 0.5 mM MgCl2 (to maintain seal integrity), and 5 mM HEPES, pH 7.4. In some experiments, the internal solution also contained 1 mM cGMP (Na+ salt; Sigma-Aldrich). Transfected cells were identified under epifluorescent illumination (EGFP filter cube; Chroma Technology Corp.) by green fluorescence due to GFP expression. The initial GΩ seals were obtained in an external solution containing 145 mM NaCl, 5 mM KCl, 5 mM MgCl2, 2 mM CaCl2, and 5 mM Hepes, pH 7.4. For whole-cell recording, the patch was ruptured with a 2-ms pulse of 1.3 V using the Zap feature of the patch-clamp amplifier. After obtaining a whole-cell configuration, the cells were gently lifted from the coverslip and placed in front of a quartz micromanifold (ALA Scientific). The cell capacitance (10–20 pF) and series resistance (5–15 MΩ) were electronically compensated by 60–70%. Cells were perfused using a computer controlled DAD-12 system (ALA Scientific). For the measurement of current through CNG channels, divalent cations were omitted from the external solution described above. CNG channel currents were evoked by perfusion with external solution containing 1 mM CPT-cG, and leak currents were estimated before channel activation. An Axopatch 200A amplifier (Axon Instruments, Inc.) controlled by PClamp 8 software was used to control voltage pulses and acquire current records. Cells were held at 0 mV and given 50–250-ms voltage pulses to potentials ranging from −100 to +100 mV. Records were typically sampled at 10 kHz and filtered at 2 kHz. Similar conditions were used for excised inside-out patch recording except that the compositions of pipet and bath solutions were reversed. The formation of heteromeric channels was confirmed by sensitivity to of l-cis-diltiazem, a hallmark of CNG channels containing CNGB1, and shifts in the dose–response relations for cGMP and cAMP.
Data Analysis
Data were analyzed using the Clampfit program in the pClamp 8.0 suite. Plots were generated using Sigma Plot 2000 (SPSS Science), and figures were composed using Adobe Illustrator (Adobe). Data are reported as mean ± SD. Statistical significance was assessed by the Student's t test. Dose response relations for cyclic nucleotides were fit by the Hill equation. In Fig. 5, PsTx affinity was estimated using a modified form of the Hill equation:
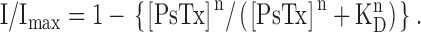 |
Figure 5.

Mutation of several residues in the pore turret of CNGA2 cause modest changes in PsTx affinity. (Top) Current traces obtained at −60 mV are shown for 2-E325K, CNGA2, and 2-S301F. Traces, in order of decreasing magnitude, represent currents measured in control solution, 25 nM Pstx, 50 nM PsTx, 100 nM PsTx, 500 nM PsTx, and 10 mM MgCl2. (Bottom) Dose respose relations for PsTx inhibition of 2-E325K (123 μM), CNGA2 (32 μM), and 2-S301F (16 μM). Dose–response relations were fit by a modified form of the Hill equation with an n value of 1.0.
RESULTS
In previous studies, we identified PsTx as a high-affinity blocker of homomeric CNG channels composed of either CNGA1 or CNGA2. At the outset of this study, we were surprised to discover that PsTx was ineffective at blocking currents through CNG channels composed of CNGA3, the primary subunit of the CNG channel in cone photoreceptors. As shown in Fig. 1 , when 500 nM PsTx was applied to outside-out patches excised from oocytes expressing CNGA1 and CNGA2, it blocked 89% and 98% of the current, respectively, at a test potential of −60 mV. However, when the same concentration of PsTx was applied to patches expressing the CNGA3 subunit, it was almost totally ineffective at blocking the current. In subsequent experiments, we exploited this surprising difference to identify residues that are critical for high-affinity binding of PsTx to the extracellular face of CNG channels.
Figure 1.

Differential inhibition of CNG channels by PsTx. Outside-out patches were excised from Xenopus oocytes expressing homomeric CNG channels composed of CNGA1, 2, or 3 subunits. The intracellular solution contained 1 mM cyclic GMP to activate the channels. Voltage pulses (250 ms) were applied from a holding potential of 0 mV to potentials ranging from −100 to +100 mV in 20-mV increments. Leak currents were estimated by application of 10 mM external MgCl2. Application of 500 nM PsTx blocked 80 ± 6% (n = 5) of current through CNGA1 channels at −60 mV and 94 ± 3% (n = 8) of current through CNGA2 channels. However, currents through CNGA3 channels were virtually unaffected by 500 nM PsTx (2 ± 1% block at −60 mV; n = 3). Bars: 1 nA and 50 ms.
In the first set of experiments, we used chimeric CNG channel constructs to determine if PsTx inhibited channel function by binding to the external vestibule of the channel pore, like a pore-blocking toxin, or if it bound to the periphery of the channel like a gating-modifier toxin. As shown in Fig. 2 , transplantation of the S5-P-S6 region from CNGA2 into a CNGA3 background (chimera 3-3-2-3) was sufficient to restore high-affinity block by PsTx. Conversely, PsTx had little effect on the reciprocal chimera 2-2-3-2. We also tested chimeras in which the NH2-terminal and S1–S4 regions were swapped between CNGA2 and A3 (unpublished data). All of the chimeric channels tested retained the approximate affinity of the channel isoform from which the pore region was derived.
Figure 2.

The pore region of CNG channels is the primary determinant of PsTx affinity. Complementary DNA encoding chimeric CNG channels were created by exchanging a Kpn2I-BstBI restriction fragment between CNGA2 and CNGA3. This fragment encompassed the S5-P-S6 region of the channels as shown (red indicates channel sequences derived from CNGA2, and blue indicates sequences derived from CNGA3). Currents were recorded at −60 mV in the outside-out patch configuration. Trace 1 was recorded in 10 mM MgCl2, trace 2 in control, and trace 3 in 500 nM PsTx. Transplantion of the pore region from CNGA2 into a CNGA3 background caused inhibition by 500 nM PsTx to increase from 2% in wild-type CNGA3 to 90% in the 3-3-2-3 chimera. Conversely, inhibition by 500 nM PsTx decreased from 98% in wild-type CNGA2 to 3% in the 2-2-3-2 chimera.
The differences in PsTx affinity are not likely to be caused by the gross alteration of channel gating properties. The apparent cGMP affinity and cAMP efficacy of both 3-3-2-3 and 2-2-3-2 chimeric channels were similar those of the parent channels, when tested using inside-out patches. For the 2232 chimera (N = 3), the cGMP K1/2 was 10.3 ± 3 μM and n = 2.1 ± 0.3, compared with 3.0 ± 2 μM and 3.2 ± 0.4 for CNGA2. Both 2232 and CNGA2 were activated to a similar extent (0.92 of the maximum current in saturating cGMP) by 10 mM cAMP. A comparison between 3323 and CNGA3 produced similar findings. The 3323 chimera was activated by cGMP with a K1/2 = 20 ± 5 μM and n = 1.8 ± 0.3, similar to the values of K1/2 = 18 ± 4 μM and n = 1.7 for CNGA3 (N = 3). The extent of activation by 10 mM cAMP was similar as well, 0.27 for 3323 versus 0.23 for CNGA3.
Upon examination of the S5-P-S6 region, we noticed 10 positions in the extracellular loop that differed between CNGA2 and CNGA3 (see Fig. 3) . We hypothesized that one or more of these amino acid changes were responsible for the dramatic reduction in PsTx affinity between CNGA2 and CNGA3. To determine which positions were critical for high-affinity PsTx inhibition, amino acids at each of the 10 positions were swapped between the two channel isoforms. As shown in Fig. 4 , substitution at 2 of the 10 positions in CNGA2 caused a dramatic reduction in block by 500 nM PsTx. When D316 or Y321 of CNGA2 was replaced with the corresponding residue in CNGA3 to form the mutants 2-D316N or 2-Y321R, PsTx block was virtually eliminated. Once again, the dramatic decrease in channel block by PsTx cannot be explained by dramatic changes in channel gating. The K1/2 of the 2-D316N channel for cGMP was 3.50 ± 0.3 μM with n = 2.60 ± 0.05 (N = 3); for 2-Y321R, the K1/2 for cGMP was 2.75 μM with n = 2.66 (N = 1). Other mutations in CNGA2 caused little or no change in block by 500 nM PsTx. However, these experiments were not sensitive to small changes in PsTx affinity because this concentration of PsTx is almost 100-fold greater than its Ki for CNGA2. When we measured PsTx dose–response relations for several CNGA2 mutants, we found that other residues in the pore turret made modest contributions to the PsTx-binding site (Fig. 5) . For instance, PsTx affinity was reduced by fourfold in the 2-E325K mutant channel (from 32 to 123 nM using this batch of PsTx). Mutations at other residues, including V306, T308, and T315, had little, if any, effect on PsTx binding. Surprisingly, the mutant channels 2-S301F and 2-T308S displayed a modest increase in PsTx affinity, reducing the Ki for PsTx inhibition to ∼16 nM, compared with 32 nM for wild-type CNGA2 (Fig. 5).
Figure 3.

Sequence comparison of the pore region among CNG channel subunits. Residues that are identical to that found in CNGA2 are shaded. The 10 residues different between CNGA2 and A3 that were mutated in this study are boxed. The black circle represents the site of glycosylation in CNGA1. Structural features (e.g., pore turret and helix, and the selectivity filter [SF]) are predicted based on the structure of the KcsA channel.
Figure 4.

Identification of amino acid residues involved in PsTx binding. Point mutations were made to exchange amino acids differences between CNGA2 and CNGA3. All current was normalized to facilitate comparison. (A) Currents were measured at −60 mV in control (bottom trace), 10 mM MgCl2 (top trace), and 500 nM PsTx (middle trace) for each mutant channel. PsTx blocked all CNGA2 mutants, except 2-D316N and 2-Y321R, with high affinity. Inhibition of 2-D316N and 2-Y321R by 500 nM PsTx was reduced to <5%. No single point mutation was sufficient to confer high affinity PsTx block upon CNGA3. A significant improvement in the block was only observed in the 3-N366Y channel (15 ± 3%). (B) Histogram comparing block of all mutant channels by 500 nM PsTx. Maximum patch currents were >1 nA for all mutant channels, except for 2-D316N and 2-Y321R, which were between 0.5 and 0.8 nA, and 3-R366Y and 3-S374C, which were between 0.1 and 0.3 nA. Error bars indicate standard deviation, and n values are shown.
We then set out to determine the minimum number of mutations required to confer high-affinity block by PsTx upon CNGA3. We found that no single mutation was sufficient to significantly increase block by 500 nM PsTx, although the 3-N361D channel was blocked to a small degree in some patches (Fig. 6) . However, when this mutation was combined with the 3-R366Y mutation to form the double mutant, 3-N361D/R366Y, there was a dramatic increase in PsTx affinity (Fig. 6). The fractional block of 3-R366Y remained almost undetectable at ∼2%, and block of 3-N361D improved only marginally to ∼15%. However, 500 nM PsTx blocked 84% of the current in the double mutant, approaching that seen in the 3-3-2-3 chimera and the wild-type CNGA2 channel. Therefore, it appears that these two residues are both critical for high-affinity inhibition by PsTx. Inclusion of the 3-K370E mutation as well would be predicted to further increase the PsTx affinity, producing a channel with affinity similar to that of wild-type CNGA2.
Figure 6.

Mutations at both N366 and R371 are required to confer high affinity PsTx inhibition upon CNGA3. PsTx inhibition improved slightly in 3-N366D mutant channels (15 ± 3%; n = 9), but remained almost undetectable in 3-R371Y (2 ± 1%), similar to the 2% inhibition of wild-type CNGA3. However, high-affinity inhibition (84 ± 3%; n = 4) was restored in the double mutant, 3-N366D/R371Y, beginning to approach the 94% inhibition seen in CNGA2.
Because PsTx is a highly basic protein (pI = 10.0; Yamazaki et al., 2002) and the 2-D316N channel mutation caused the loss of a negative charge, we set out to determine if this mutation eliminated an important electrostatic interaction. To test this hypothesis, we created a CNGA2 channel with a D316C mutation. PsTx was then applied to the channel after modification of the 2-D316C channel with charged MTS reagents (Fig. 7) . As expected, the D316C mutation significantly reduced the channel's affinity for PsTx; block by 500 nM PsTx was reduced from 98% (on wild-type CNGA2) to ∼33% for the D316C mutant. If electrostatic interactions play a key role in the interaction at D316, we would predict that modification with MTS-ethylsulfonic acid (ES), which donates a negative charge, would restore high affinity binding, whereas modification with MTS-ethyltrimethylamine (ET), which donates a positive charge, would further impair PsTx binding. Instead, we found that modification with either reagent caused a further decrease in PsTx affinity, resulting in block of only 17% and 5%, respectively. This result suggests that the interaction surface between PsTx and the CNGA2 turret is quite tight. Although modification of D316C restores the original negative charge, the extra bulk of the cysteine adduct is poorly tolerated. A similar experimental strategy was used to test the contribution of potential electrostatic interactions at Y321 as well. The PsTx affinity was also reduced in the 2-Y321C channel, such that 500 nM PsTx only blocked 77% of the current. Modification with MTSET had little effect (81% block), but modification with MTSES partially restored PsTx affinity (93% block).
Figure 7.

Modification of 2-D316C and 2-Y321C with charged MTS reagents does not restore high affinity PxTx inhibition. Inhibition of CNG channel current was measured before and after treatment of the patch with the charged MTS reagents, MTSET (positive charge) and MTSES (negative charge).
Next, we determined the efficacy of PsTx on heteromeric CNG channels. For these experiments, the channels were expressed in HEK-293 cells because of the time-dependent decline in current seen when heteromeric channels containing CNGA2 are expressed in Xenopus oocytes (Bradley et al., 1994; Liman and Buck, 1994). Currents were recorded in the whole-cell configuration at −50 mV, and channels were activated by superfusion with 1 mM p-CPT-cGMP, a membrane-permeant analogue of cGMP. Fig. 8 A shows currents recorded before (bottom trace) and after (middle trace) application of 500 nM PsTx. The top trace in every panel shows an initial leak current recorded before channel activation. As expected from the oocyte experiments, 500 nM PsTx effectively blocked currents through homomeric CNGA1 and A2 channels, whereas homomeric CNGA3 channels were insensitive to this concentration of PsTx. This concentration of PsTx also effectively blocked heteromeric CNG channels containing CNGA2+A4, but it was ineffective on channels containing CNGB1, even in combination with CNGA2 or CNGA2+A4. These results are representative of measurements on 3–16 cells for each subunit combination (Fig. 8 B).
Figure 8.

PsTx inhibition of heteromeric CNG channels. (A) Currents were recorded in the whole cell patch-clamp mode from HEK-293 cells transfected with the subunit combinations indicated at the top and side. Currents were recorded at a potential of −60 mV, and the traces indicate the currents recorded in control (top), 500 nM PsTx following channel activation (middle), and 1 mM CPT-cGMP (bottom). Currents have been normalized. (B) Histogram summarizing the effects of CNG channel subunit composition on inhibition by 500 nM PsTx. Error bars indicate standard deviation and N values are noted.
Based on the location of the PsTx-binding site, we hypothesized that PsTx functioned as a pore-blocking toxin, rather than a gating modifier. To further test this hypothesis, we studied several biophysical properties of PsTx block, including voltage dependence, state dependence, and interaction with permeant ions. Inhibition of CNGA2 channels by PsTx was slightly voltage dependent. This voltage dependence of PsTx block was initially noted as the time-dependent increase of current during depolarizing voltage pulses to +80 and + 100 mV (see Fig. 1), which was interpreted to reflect a voltage-dependent relief of block. The voltage dependence was further studied at steady-state by measuring PsTx dose–response relations at different holding potentials. In this way, we determined that PsTx inhibited CNGA2 channels with a significantly higher affinity at negative potentials. The Ki at −60 mV was 24 ± 13 nM (n = 4) compared with 180 ± 52 nM (n = 3) at +60 mV.
To further study the voltage dependence of PsTx block, we measured the kinetics of PsTx wash off at −60 and + 60 mV. In this manner, we determined that the majority of the PsTx affinity voltage dependence is due to a five- to tenfold increase in the off-rate at depolarized voltages. Fig. 9 A shows wash-off of PsTx at holding potentials of −60 and +60 mV. It is apparent that the off-rate is significantly enhanced at +60 mV compared with −60 mV. The traces shown are from consecutive PsTx applications on the same patch. The time course of the PsTx wash-off was fit well by a single-exponential function (gray lines), yielding time constants of 67 s at −60 mV and 7 s at +60 mV. Although there was substantial variability in the measured off-rates among patches, there was always a consistent 5–10-fold difference between the off-rates measured at −60 and +60 mV (n = 5) in solutions containing symmetrical 130 mM NaCl. A possible source of the enhanced off rate at +60 mV is interaction between PsTx and permeant ions within the channel pore, a phenomenon known as “knock-off” (MacKinnon and Miller, 1988). As shown in Fig. 9 B, replacement of 90% of the intracellular Na+ was with the impermeant NMDG+ ion decreased the voltage dependence of the PsTx off-rate. On this patch, the time constant for wash-off was 20 s at −60 mV and 10 s at +60 mV. Although there was considerable patch-to-patch variability in the actual rates, the ratio of the time constants at −60 and +60 mV was always ∼2. Therefore, electrostatic “knock-off” by permeant ions appears to be the primary cause of the voltage dependence of PsTx inhibition of CNG channels.
Figure 9.

Voltage dependence of PsTx dissociation from CNGA2 and interaction with permeant ions. (A) Dissociation of PsTx at holding potentials of −60 and +60 mV in the presence of symmetrical NaCl. Smooth red lines indicate single exponential fits with time constants of 67 s (−60 mV) and 7 s (+60 mV). (B) Dissociation of PsTx at holding potentials of −60 and +60 mV under asymmetrical conditions (13 mM NaCl; 117 NMDG-Cl in the pipette solution). Smooth red lines indicate single exponential fits with time constants of 20 s (−60 mV) and 10 s (+60 mV). Solutions changes are indicated by arrows. C, control solution; P, 500 nM PsTx; M, 10 mM MgCl2.
We also tested the state dependence of CNG channel block by PsTx using outside-out patches containing homomeric CNGA2 channels. The pipette solution contained either 1 mM cGMP, which fully activated the channel current, or 2 μM cGMP, which activated 10–20% of the maximal current when applied to excised inside-out patches. For these experiments, we applied 20 nM PsTx (a concentration near the Ki for PsTx when the channels are fully activated) for increased sensitivity to small changes in PsTx affinity. As shown in Fig. 10 , 20 nM PsTx inhibited ∼50% of the current regardless of the activation state of the CNGA2 channels (percentage inhibition = 48 ± 8% at 1 mM cG (N = 3) and 43 ± 5% at 2 μM cG (N = 4)). In summary, these experiments revealed no state dependence in PsTx inhibition of currents carried by CNGA2, suggesting little or no movement in the pore turret during the channel gating process.
Figure 10.

PsTx inhibition of CNGA2 is not state dependent. Currents were recorded from outside-out patches excised from oocytes expressing CNGA2. Patch electrodes were filled with control solution containing either 1 mM cGMP or 2 μM cGMP as indicated. Traces represent currents measured in 10 mM MgCl2 (top), 20 nM PsTx (middle), and control solution (bottom). In the rightmost panel, the patch current elicited in 2 μM cGMP was scaled to that of the 1 mM to facilitate comparison of PsTx block. Bars: 1 nA and 50 ms.
DISCUSSION
PsTx is a member of the cysteine-rich family of secretory proteins (CRISPs), which are characterized by an abundance of cysteine residues in the COOH-terminal one-third of the primary structure (Yamazaki et al., 2002). Furthermore, PsTx is a highly basic toxin (pI = 10.0) with many of the basic residues clustered near the NH2 and COOH termini. With a molecular weight approaching 25 kD, PsTx is almost an order of magnitude larger than most peptide toxins, such as charybdotoxin and agitoxin, which are rigid molecules ∼30 Å across (Bontems et al., 1991; Krezel et al., 1995). It is difficult to envision how such a large protein can squeeze into the narrow confines of the external vestibule formed by the four pore turrets; however, PsTx may exhibit a domain structure in which a smaller pore-blocking domain is fused to a larger domain of unknown function. At this time little is known about the three-dimensional structure of PsTx, or which residues are involved in its interaction with CNGA2. However, some insights can be gleaned from a comparison with pseudecin, a homologous toxin purified from the venom of P. porphyriacus (Yamazaki et al., 2002). Although pseudecin exhibits 97% identity (203 of 211 residues) with PsTx, its affinity for CNGA2 is reduced by 30-fold, from 15 to 460 nM. The altered residues are clustered in two distinct regions, suggesting that one of these regions may directly interact with CNGA2. The first three differences are located between residues 36 and 57, and the second, tighter cluster is between 167 and 175 in the primary structure. Of these six alterations, four would be expected to result in neutralization of charged residues in PsTx. In the future, we hope to use PsTx as a molecular caliper for probing the complementary structure of the CNG channel extracellular vestibule. Of course, a three-dimensional structure of PsTx, or a homologous protein, is a prerequisite for these studies.
In the current study, we identified six residues in the CNGA2 pore turret region that are involved in PsTx binding. The location these residues suggests that PsTx inhibits CNG channel activity simply by blocking the external entrance to the ion-conducting pore. The location of these residues is similar to that of the residues involved in inhibition of several voltage-dependent potassium channels by agitoxin (Hidalgo and MacKinnon, 1995; Gross and MacKinnon, 1996; Ranganathan et al., 1996). However, there may very well be additional points of contact that were not identified in this study because we only studied residues that were not conserved between CNGA2 and CNGA3. Conserved residues are also likely to be involved in PsTx binding. Several residues on the Shaker potassium channel that lie at the COOH-terminal side of the G-Y-G signature in the pore loop, just before the S6 helix, have been implicated in the binding the pore-blocker agitoxin. In the KcsA structure, these residues lie deeper within the pore and are closer to the ion-conducting pathway. Because these residues are conserved between CNGA2 and CNGA3, we did not test their interaction with PsTx. If these residues were involved in PsTx binding, it would support the idea that a small domain of PsTx may extend deeply into the external vestibule. Further experiments, including more extensive scanning mutagenesis, are needed to examine the interaction between PsTx and CNGA2 in greater detail.
Based on the location of the PsTx-binding site, we propose that PsTx inhibits the flow of current through CNG channels by functioning as a high affinity pore blocker. Subsequent biophysical studies of its mode of action support this conclusion as well. Block of channel current is slightly voltage dependent, largely caused by an unfavorable response to an interaction with permeant ions, rather than an extension of PsTx across a portion of the transmembrane field. Occupancy of the pore by Na+ ions seems to accelerate the dissociation of PsTx at depolarized voltages. When measured in the presence of symmetrical NaCl solutions, the PsTx off-rate is enhanced ∼10-fold by depolarization to +60 mV compared with −60 mV. In contrast, this voltage dependence is greatly reduced when 90% of the intracellular Na+ is replaced with the impermeant NMDG+. This is reminiscent of the “knock-off” phenomenon described for the interaction of charybdotoxin with potassium channels (MacKinnon and Miller, 1988).
PsTx inhibition of channels formed by CNGA2 does not appear to be dependent on the activation state of the channel. That is, PsTx binds equally well to both open and closed channels. This suggests that the regions of the pore turret that form the PsTx receptor do not move significantly during the process of channel activation. In contrast, Liu and Siegelbaum (2000) have reported that the pore helix, a region more proximal to the selectivity filter, rotates as the channel pore opens.
Mutations in CNGA2 that altered PsTx affinity spanned a stretch of 25 amino acids, from S301 to E325. In contrast, residues in the pore turret of Shaker channels that are known to affect binding agitoxin extend from E422 to D431, a span of only 10 residues (Hidalgo and MacKinnon, 1995; Gross and MacKinnon, 1996; Ranganathan et al., 1996). In agreement with this finding, the region forming the pore turret in the KcsA structure is only 10 amino acids long, stretching from E51 to I60 (Doyle et al., 1998). The findings of our current study suggest that the pore turret is more extensive in CNG channels. This hypothesis is supported by the presence of a known glycosylation site at N327 in the CNGA1 channel (Rho et al., 2000), which corresponds to V306 in CNGA2. Therefore, it is highly likely that this residue is found on the extracellular loop between S5 and the reentrant pore loop. If the CNGA1 channel sequence is superimposed directly on the KcsA structure based on an alignment of the pore sequence, this residue falls in the transmembrane region of the S5 helix, a position that would appear untenable for glycosylation. Therefore, we propose that the pore turret of CNG channels is significantly larger than that of KcsA and voltage-gated potassium channels (see Fig. 11) . The proposed size of the CNG channel pore turret is similar to that proposed for human ether-a-go-go channels based on the binding site of the novel scorpion toxin, ErgTx (Pardo-Lopez et al., 2002). The external vestibule of CNG channels may also have a broader opening, facilitating block by PsTx.
Figure 11.

Contrasting models of the CNG channel pore turret. The structure of the left-hand subunit is based on the KcsA structure with a shorter pore turret, and the model that we propose with a longer pore turret is shown on the left. Amino acid residues that were mutated in the current study are indicated as balls on the ribbon backbone. The strength of interaction is shown by a color scale, ranging from blue (undetectable) to yellow (weak) to orange (moderate) to red (strong). The green residue indicates the position homologous to that of a known glycosylation site in CNGA1.
We have reported previously that PsTx is quite ineffective at blocking heteromeric CNG channels containing the rod β-subunit, CNGB1 (Brown et al., 1999). As shown in Fig. 3, the pore turret of CNGB1 has an eight amino acid deletion compared with the sequence of the pore turret in the α-subunits. This deletion removes an extensive portion of the turret that is involved in the interaction with PsTx. The rod channel has recently been proposed to consist of a stoichiometry of three CNGA1 subunits to one CNGB1 (Weitz et al., 2002; Zheng et al., 2002; Zhong et al., 2002). In light of this stoichiometry, we find the dramatic loss of PsTx affinity in the heteromultimeric channel to be quite surprising. If PsTx formed strong contacts with all four subunits in the homomultimer, only one of four interactions would be lost. Likewise, if PsTx interacts strongly with only one of four subunits, only one of four potential orientations would be lost. If one-quarter of the binding energy were lost, the Ki would be predicted to increase from 100 nM in CNGA1 homomultimers to ∼5 μM in the heteromeric channel, whereas the KD of the CNGA1/CNGB1 heteromultimer is >10 μM (Brown et al., 1999). These results suggest that the incorporation of CNGB1 grossly alters the shape of the external vestibule, or that interaction between PsTx and the CNGB1 is quite deleterious. The reduced length of the pore turret region in CNGB1 may pinch off the outer vestibule, sterically occluding the access of PsTx to its primary binding site. In contrast, the PsTx affinity is only slightly reduced in heteromeric channels formed by CNGA2 and CNGA4, a subunit that has a glutamine in the position homologous to D316 and an arginine at the position of Y321. In CNGA2/A4 heteromultimers, block by 500 nM PsTx is only reduced to 85%, suggesting a KD of ∼100 nM, compared with 20 nM for the CNGA2 homomultimer. These results suggest the simple loss of a contact site in CNGA2/CNGA4 heteromultimers, without a gross distortion of the external vestibule.
This study constitutes the first step toward developing a model of the extracellular vestibule of CNG channels. In the short term, our findings will be useful for constructing CNG channels with a differential sensitivity to both cyclic nucleotides and PsTx. These designer channels will be useful as biosensors for monitoring intracellular levels of cAMP and cGMP near the plasma membrane (Rich et al., 2001). In the future, information about the structure of the extracellular vestibule of CNG channels may lead to the design of specific inhibitors of CNG channels, which are sorely needed to further our understanding of their physiological roles throughout the body.
Acknowledgments
We would like to thank Drs. H. Peter Larsson and Jeffrey W. Karpen for helpful discussions and comments on the manuscript.
This work was supported by grants from the National Eye Institute (EY 12,837) and the Oregon Health Science Foundation.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: MTS, methanethiosulfonate; PsTx, pseudechetoxin.
References
- Biel, M., X. Zong, M. Distler, E. Bosse, N. Klugbauer, M. Murakami, V. Flockerzi, and F. Hofmann. 1994. Another member of the cyclic nucleotide-gated channel family, expressed in testis, kidney, and heart. Proc. Natl. Acad. Sci. USA. 91:3505–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonigk, W., J. Bradley, F. Muller, F. Sesti, I. Boekhoff, G.V. Ronnett, U.B. Kaupp, and S. Frings. 1999. The native rat olfactory cyclic nucleotide-gated channel is composed of three distinct subunits. J. Neurosci. 19:5332–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontems, F., C. Roumestand, B. Gilquin, A. Menez, and F. Toma. 1991. Refined structure of charybdotoxin: common motifs in scorpion toxins and insect defensins. Science. 254:1521–1523. [DOI] [PubMed] [Google Scholar]
- Bourinet, E., T.W. Soong, K. Sutton, S. Slaymaker, E. Mathews, A. Monteil, G.W. Zamponi, J. Nargeot, and T.P. Snutch. 1999. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat. Neurosci. 2:407–415. [DOI] [PubMed] [Google Scholar]
- Bradley, J., S. Frings, K.W. Yau, and R. Reed. 2001. Nomenclature for ion channel subunits. Science. 294:2095–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, J., J. Li, N. Davidson, H.A. Lester, and K. Zinn. 1994. Heteromeric olfactory cyclic nucleotide-gated channels: a subunit that confers increased sensitivity to cAMP. Proc. Natl. Acad. Sci. USA. 91:8890–8894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, R.L., S.D. Snow, and T.L. Haley. 1998. Movement of gating machinery during the activation of rod cyclic nucleotide-gated channels. Biophys. J. 75:825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, R.L., T.L. Haley, K.A. West, and J.W. Crabb. 1999. Pseudechetoxin: a peptide blocker of cyclic nucleotide-gated ion channels. Proc. Natl. Acad. Sci. USA. 96:754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T.Y., Y.W. Peng, R.S. Dhallan, B. Ahamed, R.R. Reed, and K.W. Yau. 1993. A new subunit of the cyclic nucleotide-gated cation channel in retinal rods. Nature. 362:764–767. [DOI] [PubMed] [Google Scholar]
- Dhallan, R.S., K.W. Yau, K.A. Schrader, and R.R. Reed. 1990. Primary structure and functional expression of a cyclic nucleotide-activated channel from olfactory neurons. Nature. 347:184–187. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., C.J. Morais, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Ellinor, P.T., J.F. Zhang, W.A. Horne, and R.W. Tsien. 1994. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature. 372:272–275. [DOI] [PubMed] [Google Scholar]
- Feng, Z.P., J. Hamid, G.M. Bosey, T.P. Snutch, and G.W. Zamponi. 2001. Residue Gly1326 of the N-type calcium channel alpha 1B subunit controls reversibility of omega-conotoxin GVIA and MVIIA block. J. Biol. Chem. 276:15728–15735. [DOI] [PubMed] [Google Scholar]
- Finn, J.T., M.E. Grunwald, and K.W. Yau. 1996. Cyclic nucleotide-gated ion channels: an extended family with diverse functions. Annu. Rev. Physiol. 58:395–426. [DOI] [PubMed] [Google Scholar]
- Garcia, M.L., Y. Gao, O.B. McManus, and G.J. Kaczorowski. 2001. Potassium channels: from scorpion venoms to high-resolution structure. Toxicon. 39:739–748. [DOI] [PubMed] [Google Scholar]
- Garcia, M.L., M. Garcia-Calvo, P. Hidalgo, A. Lee, and R. MacKinnon. 1994. Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry. 33:6834–6839. [DOI] [PubMed] [Google Scholar]
- Gerstner, A., X. Zong, F. Hofmann, and M. Biel. 2000. Molecular cloning and functional characterization of a new modulatory cyclic nucleotide-gated channel subunit from mouse retina. J. Neurosci. 20:1324–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, A., T. Abramson, and R. MacKinnon. 1994. Transfer of the scorpion toxin receptor to an insensitive potassium channel. Neuron. 13:961–966. [DOI] [PubMed] [Google Scholar]
- Gross, A., and R. MacKinnon. 1996. Agitoxin footprinting the shaker potassium channel pore. Neuron. 16:399–406. [DOI] [PubMed] [Google Scholar]
- Harvey, A.L. 2001. Twenty years of dendrotoxins. Toxicon. 39:15–26. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., T. Abramson, and R. MacKinnon. 1992. A functional connection between the pores of distantly related ion channels as revealed by mutant K+ channels. Science. 258:1152–1155. [DOI] [PubMed] [Google Scholar]
- Hidalgo, P., and R. MacKinnon. 1995. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 268:307–310. [DOI] [PubMed] [Google Scholar]
- Kaupp, U.B., T. Niidome, T. Tanabe, S. Terada, W. Bonigk, W. Stuhmer, N.J. Cook, K. Kangawa, H. Matsuo, T. Hirose, et al. 1989. Primary structure and functional expression from complementary DNA of the rod photoreceptor cyclic GMP-gated channel. Nature. 342:762–766. [DOI] [PubMed] [Google Scholar]
- Kaupp, U.B., and R. Seifert. 2002. Cyclic nucleotide-gated ion channels. Physiol. Rev. 82:769–824. [DOI] [PubMed] [Google Scholar]
- Korschen, H.G., M. Illing, R. Seifert, F. Sesti, A. Williams, S. Gotzes, C. Colville, F. Muller, A. Dose, M. Godde, et al. 1995. A 240 kDa protein represents the complete beta subunit of the cyclic nucleotide-gated channel from rod photoreceptor. Neuron. 15:627–636. [DOI] [PubMed] [Google Scholar]
- Krezel, A.M., C. Kasibhatla, P. Hidalgo, R. MacKinnon, and G. Wagner. 1995. Solution structure of the potassium channel inhibitor agitoxin 2: caliper for probing channel geometry. Protein Sci. 4:1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe, R.A., P.A. DeFeo, M.D. Davison, J. Young, J.L. Herman, R.C. Spreen, M.B. Horn, T.J. Mangano, and R.A. Keith. 1993. Isolation and pharmacological characterization of omega-grammotoxin SIA, a novel peptide inhibitor of neuronal voltage-sensitive calcium channel responses. Mol. Pharmacol. 44:451–460. [PubMed] [Google Scholar]
- Liman, E.R., J. Tytgat, and P. Hess. 1992. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 9(5):861–871. [DOI] [PubMed] [Google Scholar]
- Liman, E.R., and L.B. Buck. 1994. A second subunit of the olfactory cyclic nucleotide-gated channel confers high sensitivity to cAMP. Neuron. 13:611–621. [DOI] [PubMed] [Google Scholar]
- Liu, J., and S.A. Siegelbaum. 2000. Change of pore helix conformational state upon opening of cyclic nucleotide-gated channels. Neuron. 28:899–909. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., and C. Miller. 1988. Mechanism of charybdotoxin block of the high conductance, Ca2+-activated K+ channel. J. Gen. Physiol. 91:335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon, R., and C. Miller. 1989. Mutant potassium channels with altered binding of charybdotoxin, a pore-blocking peptide inhibitor. Science. 245:1382–1385. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., L. Heginbotham, and T. Abramson. 1990. Mapping the receptor site for charybdotoxin, a pore-blocking potassium channel inhibitor. Neuron. 5:767–771. [DOI] [PubMed] [Google Scholar]
- Mintz, I.M., V.J. Venema, K.M. Swiderek, T.D. Lee, B.P. Bean, and M.E. Adams. 1992. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature. 355:827–829. [DOI] [PubMed] [Google Scholar]
- Olivera, B.M., D.R. Hillyard, M. Marsh, and D. Yoshikami. 1995. Combinatorial peptide libraries in drug design: lessons from venomous cone snails. Trends Biotechnol. 13:422–426. [DOI] [PubMed] [Google Scholar]
- Olivera, B.M., J. Rivier, J.K. Scott, D.R. Hillyard, and L.J. Cruz. 1991. Conotoxins. J. Biol. Chem. 266:22067–22070. [PubMed] [Google Scholar]
- Pardo-Lopez, L., M. Zhang, J. Liu, M. Jiang, G.N. Tseng. 2002. Mapping the bidning site of a human ether-a-go-go-–related gene-specific toxin (ErgTx) to the channel's outer vestibule. J. Biol. Chem. 277:16403–16411. [DOI] [PubMed] [Google Scholar]
- Ranganathan, R., J.H. Lewis, and R. MacKinnon. 1996. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 16:131–139. [DOI] [PubMed] [Google Scholar]
- Rash, L.D., and W.C. Hodgson. 2002. Pharmacology and biochemistry of spider venoms. Toxicon. 40:225–254. [DOI] [PubMed] [Google Scholar]
- Rho, S., H.M. Lee, K. Lee, and C. Park. 2000. Effects of mutation at a conserved N-glycosylation site in the bovine retinal cyclic nucleotide-gated ion channel. FEBS Lett. 478:246–252. [DOI] [PubMed] [Google Scholar]
- Rich, T.C., T.E. Tse, J.G. Rohan, J. Schaack, and J.W. Karpen. 2001. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J. Gen. Physiol. 118:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautter, A., X. Zong, F. Hofmann, and M. Biel. 1998. An isoform of the rod photoreceptor cyclic nucleotide-gated channel beta subunit expressed in olfactory neurons. Proc. Natl. Acad. Sci. USA. 95:4696–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz, K.J., and R. MacKinnon. 1995. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 15:941–949. [DOI] [PubMed] [Google Scholar]
- Swartz, K.J., and R. MacKinnon. 1997. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron. 18:675–682. [DOI] [PubMed] [Google Scholar]
- Weitz, D., N. Ficek, E. Kremmer, P.J. Bauer, and U.B. Kaupp. 2002. Subunit stoichiometry of the CNG channel of rod photoreceptors. Neuron. 36:881–889. [DOI] [PubMed] [Google Scholar]
- Weyand, I., M. Godde, S. Frings, J. Weiner, F. Muller, W. Altenhofen, H. Hatt, and U.B. Kaupp. 1994. Cloning and functional expression of a cyclic-nucleotide-gated channel from mammalian sperm. Nature. 368:859–863. [DOI] [PubMed] [Google Scholar]
- Winterfield, J.R., and K.J. Swartz. 2000. A hot spot for the interaction of gating modifier toxins with voltage-dependent ion channels. J. Gen. Physiol. 116:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki, Y., R.L. Brown, and T. Morita. 2002. Purification and cloning of toxins from elapid venoms that target cyclic nucleotide-gated ion channels. Biochemistry. 41:11331–11337. [DOI] [PubMed] [Google Scholar]
- Yellen, G., M.E. Jurman, T. Abramson, and R. MacKinnon. 1991. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science. 251:939–942. [DOI] [PubMed] [Google Scholar]
- Zheng, J., M.C. Trudeau, and W.N. Zagotta. 2002. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron. 36:891–896. [DOI] [PubMed] [Google Scholar]
- Zhong, H., L.L. Molday, R.S. Molday, and K.W. Yau. 2002. The heteromeric cyclic nucleotide-gated channel adopts a 3A:1B stoichiometry. Nature. 420:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]