

Abstract
Osmotic swelling of cardiac myocytes and other types of cells activates an outwardly rectifying, tamoxifen-sensitive Cl− current, ICl,swell, but it is unclear whether Cl− currents also are activated by direct mechanical stretch. We tested whether specific stretch of β1-integrin activates a Cl− current in rabbit left ventricular myocytes. Paramagnetic beads (4.5-μm diameter) coated with mAb to β1-integrin were applied to the surface of myocytes and pulled upward with an electromagnet while recording whole-cell current. In solutions designed to isolate anion currents, β1-integrin stretch elicited an outwardly rectifying Cl− current with biophysical and pharmacological properties similar to those of ICl,swell. Stretch-activated Cl− current activated slowly (t1/2 = 3.5 ± 0.1 min), partially inactivated at positive voltages, reversed near ECl, and was blocked by 10 μM tamoxifen. When stretch was terminated, 64 ± 8% of the stretch-induced current reversed within 10 min. Mechanotransduction involved protein tyrosine kinase. Genistein (100 μM), a protein tyrosine kinase inhibitor previously shown to suppress ICl,swell in myocytes, inhibited stretch-activated Cl− current by 62 ± 6% during continued stretch. Because focal adhesion kinase and Src are known to be activated by cell swelling, mechanical stretch, and clustering of integrins, we tested whether these tyrosine kinases mediated the response to β1-integrin stretch. PP2 (10 μM), a selective blocker of focal adhesion kinase and Src, fully inhibited the stretch-activated Cl− current as well as part of the background Cl− current, whereas its inactive analogue PP3 (10 μM) had no significant effect. In addition to activating Cl− current, stretch of β1-integrin also appeared to activate a nonselective cation current and to suppress IK1. Integrins are the primary mechanical link between the extracellular matrix and cytoskeleton. The present results suggest that integrin stretch may contribute to mechano-electric feedback in heart, modulate electrical activity, and influence the propensity for arrhythmogenesis.
Keywords: stretch-activated channels, swelling-activated channels, mechano-electric feedback, protein tyrosine kinase, arrhythmia
INTRODUCTION
A volume-sensitive Cl− current, ICl,swell, is broadly distributed throughout the heart (Sorota, 1999; Hume et al., 2000; Baumgarten and Clemo, 2003) and other tissues (Nilius et al., 1994). ICl,swell in cardiac cells is activated by osmotic swelling (Sorota, 1992; Tseng, 1992) or hydrostatic pressure-induced cell inflation (Hagiwara et al., 1992). The current is time independent over most of the physiological voltage range, partially inactivates at positive potentials, and exhibits outward rectification with physiologic or symmetric Cl− gradients. Activation of ICl,swell in both cardiac and noncardiac cells occurs over several minutes and is regulated by multiple signaling molecules, including PKC (Duan et al., 1995; Clemo and Baumgarten, 1998), protein tyrosine kinase (PTK) (Sorota, 1995), phosphatidylinositol-3-kinase (PI-3K) (Shi et al., 2002), small GTP binding proteins such as Rho (Tilly et al., 1996; Nilius et al., 1999), and protein phosphatases (Clemo et al., 1999a; Duan et al., 1999).
Questions have been raised as to whether ICl,swell is truly a mechanosensitive current (Hu and Sachs, 1997; Cazorla et al., 1999). Cell swelling is a nonspecific stimulus. Besides its mechanical effects, it reduces ionic strength, dilutes cytoplasmic ions and macromolecules, and relieves macromolecular crowding, thereby activating signaling molecules. These perturbations, rather than mechanical stretch, may be responsible, at least in part, for the activation of ICl,swell upon cell swelling. In support of this idea, directly stretching cardiac myocytes generally activates cation currents but not ICl,swell (Hu and Sachs, 1997; Kamkin et al., 2003).
Ideally, studies of cardiac stretch-activated channels would make use of the structures that normally transmit forces to myocytes within the intact heart. Mechanical forces are transmitted by integrins (Wang et al., 1993), a family of heterodimeric (αβ), noncovalently associated, transmembrane glycoproteins that act as extracellular matrix (ECM) receptors and physically link the ECM and cytoskeleton (Ross, 2002). The β1D-splice variant is the principal β isoform expressed in adult heart (Zhidkova et al., 1995). In myocytes, integrins are a component of costameres, macromolecular complexes in register with Z-lines that are specialized for force transmission (Danowski et al., 1992; Borg et al., 2000). The cytoskeletal proteins paxillin, talin, muscle integrin binding protein, and α-actinin bind directly to the cytoplasmic tail of β1-integrin, and additional cytoskeletal proteins, including vinculin, melusin, and desmin, are colocalized within the costamere (Ross and Borg, 2001).
Integrins also participate in cell signaling (Borg et al., 2000; Ross and Borg, 2001; Parsons, 2003). A variety of signaling molecules, adaptor proteins, and other cell surface receptors are colocalized with integrins and cytoskeletal proteins within costameres. Focal adhesion kinase (FAK) and members of the Src kinase family are the principal upstream PTKs activated by clustering of integrin receptors. FAK binds to the cytoplasmic domain of β1-integrin, resulting in autophosphorylation of FAK at Y397. Src binds via its SH2 domain to Y397 of FAK. Once activated, FAK and Src recruit a number of other proteins, including PKC, PI-3K, protein phosphatases, small GTPases, and GTPase-activating proteins, and orchestrate a network of downstream signaling cascades (Parsons, 2003). It is striking that the signaling cascades initiated by integrins also are activated by both mechanical stretch and cell swelling (Sadoshima et al., 1996; Sadoshima and Izumo, 1997). Furthermore, some of these signaling molecules are known to regulate ICl,swell.
The aims of the present study were to determine whether Cl− currents are activated by direct and specific mechanical stretch of integrins in cardiac myocytes and to explore the mechanism of mechanotransduction. Paramagnetic beads coated with mAb to β1-integrin were employed to apply force. Stretch of integrins activated an outwardly rectifying, tamoxifen-sensitive Cl− current resembling ICl,swell. The mechanotransduction pathway includes the PTK family members, FAK and/or Src. Preliminary reports appeared previously (Browe and Baumgarten, 2003a,b).
MATERIALS AND METHODS
Ventricular Myocyte Isolation
Left ventricular myocytes were freshly isolated from adult New Zealand white rabbits (∼3 kg) of either gender by enzymatic dissociation. Hearts were excised and retrogradely perfused via the aorta with oxygenated, modified Tyrode solutions at 37°C. Successively, Ca-containing and Ca-free Tyrode solutions were run for 5 min each, and then, enzyme solution was run for 30 min. At selected intervals, portions of the left ventricle were dissected, placed in test tubes, and gently agitated. After filtering through 210 μm monofilament nylon mesh (Small Parts), myocytes were washed and stored in a modified KB solution. The yield of rod-shaped, quiescent myocytes typically was >70%. All electrophysiological recordings were performed within 10 h of cell isolation. Single, rod-shaped myocytes that were quiescent displayed clear striations, and were free of membrane blebbing or other morphological irregularities were chosen for study.
Tyrode solution for cell isolation contained (mM): 130 NaCl, 5 KCl, 1.8 CaCl2, 0.4 KH2PO4, 3 MgCl2, 5 HEPES, 15 taurine, 5 creatine, 10 glucose, pH 7.25. For Ca-free Tyrode solution, CaCl2 was replaced with 0.1 mM Na2EGTA. For enzyme solution, 1.5– 1.75 mg/ml bovine serum albumin (Sigma-Aldrich), 0.5 mg/ml collagenase (type II; Worthington), and 0.05 mg/ml pronase (type XIV; Sigma-Aldrich) were added to Ca-free Tyrode. KB solution contained (mM): 120 K-glutamate, 10 KCl, 10 KH2PO4, 0.5 K2EGTA, 10 taurine, 1.8 MgSO4, 10 HEPES, 20 glucose, 10 mannitol, pH 7.2.
Experimental Solutions and Drugs
Ventricular myocytes were dispersed over a poly-l-lysine-coated glass-bottomed chamber (∼0.3 ml) mounted on an inverted microscope (Diaphot; Nikon) and were visualized with Hoffman modulation optics (40×; NA = 0.55) and a high-resolution TV camera (CCD72; Dage-MTI). Bath solution was superfused at 2–3 ml/min at room temperature (22–23°C). Recordings were made in physiological bath solution containing (mM): 140 NaCl, 5 KCl, 2.5 MgCl2, 1.8 CaCl2, 10 HEPES, 5 glucose, pH 7.4 with a pipette containing (mM): 104 K-aspartate, 10 KCl, 10 NaCl, 2.5 MgATP, 8 K2EGTA, 0.1 CaCl2, 10 HEPES, pH 7.1 (liquid junction potential, −8.3 mV). To isolate anion currents, bath Na+ and K+ were replaced with N-methyl-d-glucamine, and Ca2+ was replaced with Mg2+; in the pipette, Na+ and K+ were replaced with Cs+ (liquid junction potential, −13.2 mV). For one set of experiments, a high Cl− pipette solution was employed in which all aspartate was replaced with an equimolar Cl− (liquid junction potential, −5.1 mV). Pipette-free Ca2+ was ∼35 nM (WinMAXC ver 2.4; www.stanford.edu/~cpatton/maxc.html).
Tamoxifen (20 mM; Sigma-Aldrich), genistein (100 mM), PP2 (10 mM), and PP3 (10 mM; Calbiochem) were prepared as stock solutions in DMSO and kept frozen (−4°C) in small aliquots until use.
Paramagnetic Bead Method
Mechanical force was applied specifically to β1-integrins using mAb-coated paramagnetic beads and an electromagnet, a method derived from those of Wang et al. (1993) and Glogauer et al. (1995)(Glogauer and Ferrier, 1998). Mouse anti–human IgG1 mAb for the β1 subunit of integrin (MAB2250; Chemicon), which cross-reacts with rabbit β1-integrin, was attached to the surface of uniform, 4.5 ± 0.2 μm diameter (±SD), superparamagnetic, polystyrene beads containing iron oxides (Dynabeads M-450 Pan Mouse IgG, 110.22; Dynal Biotech). These beads are supplied covalently bound to a mAb (human IgG4) for mouse IgG Fc that facilitates attachment of primary mAb to the beads. After resuspending the Dynabeads (4.0 × 108 beads/ml), 200 μl of the suspension was transferred to a sterile 0.5-ml plastic vial, washed three times, and resuspended in PBS. Then, 8 μl of MAB2250 (1 mg/ml) was added to the vial, and the vial was mounted on the drum of a rotary tumbler (Lortone) and slowly rotated for 30 min at 4°C. After washing three times to remove unbound MAB2250, the MAB2250-coated beads (∼0.1 pg MAB2250/bead) were resuspended in PBS to a final volume of 200 μl and stored at 4°C.
For control experiments, mouse anti–human IgG1 mAb for transferrin receptors (CBL 137; Chemicon) was attached to the beads. The procedures for attaching antitransferrin receptor mAb to the beads, mAb dilutions, and storage conditions were identical to those used for the β1-integrin mAb.
For each experiment, mAb-coated beads were resuspended, and a 20 μl aliquot was diluted to 2 ml with bath solution to give ∼4,000 beads/μl. Generally, 75–200 μl of this final dilution of coated beads was added to myocytes in the chamber with the flow turned off and allowed to randomly settle on cells from above. After ∼5 min, unbound beads were washed away by restoring superfusion. Typically 3–5 coated beads were firmly attached to myocytes selected for study, each presumably to multiple β1 integrins.
The electromagnet consisted of a water-cooled coil connected to a constant current circuit (Fig. 1) . The coil was made from 475 turns of 18 AWG copper magnet wire wrapped around a 1.1-mm thick conical copper form and was sealed with thermally conductive epoxy.
Figure 1.

Paramagnetic bead method for stretching β1-integrins. (A) Schematic diagram of the apparatus. A water-cooled electromagnet coil was placed directly on top of the bath. Energizing the coil with a constant current source, I, generated a magnetic flux density gradient of 2,400 G/m. The resulting force vector for each bead pointed upward toward the plane of the coil and perpendicular to the long axis of the myocyte. (B) Ventricular myocyte with 4.5-μm diameter beads coated with anti-β1 integrin mAb attached to the upper surface. Out-of-focus beads are near chamber floor.
The electromagnet was placed directly on top of the bath, and patch pipettes passed through an ellipsoidal opening (23 × 12 mm) at its base. Measurements with a Gauss meter (5080; F.W. Bell) indicated that the magnetic flux density (B) was 35 Gauss (G) and was uniform in the x-y plane occupied by the myocytes, 4 mm below the face of the electromagnet. In contrast, B varied along the z-axis. Based on a plot of B versus distance, the magnetic flux density gradient, ∂B z/∂z, was 2,400 G/m at the level of the myocytes. This magnetic field induced a magnetic dipole moment within the Dynabeads that was parallel to the z-axis of the coil. As a result, the force vector exerted on the β1-integrin–bound beads was oriented upward toward the coil and perpendicular to the long axis of the myocyte. The force per bead (F) was estimated as 1.2 pN based on the equation:
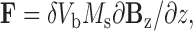 |
where δ is the fraction of bead volume, V b, occupied by iron oxide, M s is the saturated magnetization of iron oxide, and ∂B z/∂z is as defined above. δ was taken as 20% (according to manufacturer's data), V b was calculated from the nominal bead diameter, M s was 4.8 ×105 A/m (Glogauer and Ferrier, 1998), and ∂B z/∂z was measured.
Electrophysiological Recordings
Pipettes were pulled using thin-walled borosilicate glass capillary tubing and then fire polished. The final pipette tip diameter was 3–4 μm, and the pipette resistance in bath solution was 2–3 MΩ. A 150 mM KCl agar bridge was used as the ground.
Membrane currents were recorded with an EPC-7 amplifier (List-Medical) using the whole-cell configuration of the patch clamp technique. Seal resistances of 5–30 GΩ were typically achieved, and the membrane patch was ruptured by application of negative pressure or by a brief, 500-mV zapping pulse. Myocytes were dialyzed for 10 min before recordings commenced. Voltage-clamp protocols and data acquisition were governed by a Digidata 1200B A/D board and pClamp 8.0 (Axon Instruments, Inc.). Successive 500-ms voltage steps were taken from a holding potential of −60 mV to test potentials ranging from −100 to +40 mV in +10-mV increments. Voltages were corrected for the measured liquid junction potential before forming a seal. Membrane currents were low-pass filtered at 2 kHz (8-pole Bessel 902; Frequency Devices) and digitized at 10 kHz. For presentation, selected records were filtered at 50 Hz. Under conditions designed to isolate anion currents, the integrin stretch–induced current exhibited strong voltage-dependent inactivation, and I-V curves were constructed using the average current at 20–35 ms. In experiments with physiologic bath Na+, steady-state I-V curves were plotted using the current at the end of the 500-ms pulse.
Statistics
Data are reported as mean ± SEM; n denotes the number of cells. Mean currents are expressed as current density (pA/pF) to account for differences in myocyte surface membrane area. For multiple comparisons, a one-way repeated measures ANOVA or a two-way ANOVA was performed, as appropriate, and the Student-Newman-Keuls test was used to compare groups. For simple comparisons of two groups, a one-tailed paired Student's t test was administered. Statistical analyses were done using SigmaStat 2.03 (SPSS), and P < 0.05 was considered significant.
RESULTS
β1-Integrin Stretch Activates a Cl− Current
The paramagnetic bead method was used to test whether specific stretch of β1-integrins activates a Cl− current (Cl− SAC) in rabbit left ventricular myocytes. Fig. 2 illustrates the effect of β1-integrin stretch on membrane current under conditions designed to isolate Cl− currents. With anti–β1-integrin IgG1 mAb-coated paramagnetic beads attached to the myocyte but no stretch stimulus (control), a small background current that partially inactivated at potentials positive to +10 mV was observed (Fig. 2 A). The I-V relationship for the control current exhibited mild outward rectification and a reversal potential of about −50 mV (Fig. 2 D), and its magnitude was not correlated with the number of beads on the myocyte. An outwardly rectifying background Cl− current has been reported previously under similar experimental conditions and was attributed to ICl,swell (Sorota, 1992; Duan et al., 1995, 1997).
Figure 2.

Stretch of β1-integrins activated a Cl− current (Cl− SAC) in bath and pipette solutions designed to isolate anion currents. Membrane potential was stepped from −60 mV to between −100 and +40 mV for 500 ms. Families of currents before stretch (A, Control), after 5 min of integrin stretch (B, Stretch), and the stretch-induced difference current (C, Difference) obtained by digital subtraction. Horizontal bar denotes 0 current. I-V relationships for membrane current (D) before (•) and after (○) integrin stretch, and (E) for the stretch-induced difference current (▪). The background current in the absence of stretch was outwardly rectifying, reversed near ECl (−52 mV), and underwent partial inactivation at positive potentials. Stretch markedly augmented outward currents, and the resulting Cl− SAC exhibited much stronger outward rectification, reversed near ECl, and partially inactivated at positive potentials. Cl− SAC elicited by β1-integrin stretch was 1.13 ± 0.10 pA/pF at + 40 mV (n = 34).
Static stretch of bead-attached β1-integrins achieved by applying a magnetic flux density gradient of 2,400 G/m progressively increased the Cl− current. Cl− current after 5 min of static stretch partially inactivated at potentials positive to +10 mV (Fig. 2 B), as previously shown for the control current, and the I-V relationship exhibited strong outward rectification (Fig. 2 D). The reversal potential was unchanged at −50 mV, however. To more clearly define the current activated by integrin stretch, difference currents were calculated by subtracting the control current from the corresponding current recorded after stretch (Fig. 2 C). The stretch-induced difference currents partially inactivated at positive potentials, and their I-V relationship (Fig. 2 E) displayed strong outward rectification and reversed at −50 mV, close to the calculated ECl of −52 mV. The difference currents emphasize that stretch caused a much more prominent increase in outward than inward current.
A stretch-induced, outwardly rectifying Cl− current was observed in 75% of myocytes tested (34 out of 45). In these cells, stretch increased outward current by 1.13 ± 0.10 pA/pF at +40 mV (n = 34, P < 0.0005), from 2.89 ± 0.77 to 4.01 ± 0.85 pA/pF. In contrast, inward current increased only −0.23 ± 0.06 pA/pF at −100 mV (n = 34, P < 0.0005), from −1.98 ± 0.88 to −2.21 ± 0.93 pA/pF. The reversal potential was −47 ± 2 mV (n = 4). A small subset of these myocytes displayed a larger increase of inward current (−1.0 ± 0.3 pA/pF, n = 4) that began after a delay of 4–6 min after the onset of β1-integrin stretch.
When pipette solution was switched to high Cl−, the reversal potential shifted to −19 ± 1 mV (n = 4), 28 mV more positive than with the 20.2 mM Cl− pipette solution, and the stretch-induced currents still exhibited strong outward rectification. The shift in reversal potential gives a PAsp/PCl ratio of 0.2, similar to the value of 0.1 reported previously for ICl,swell (Vandenberg et al., 1994).
Cl− SAC activation required both anti-β1 integrin mAb and the application of stretch. Current was unaffected by application of a magnetic field alone without beads (n = 4, ns) and by untreated anti-IgG beads that adhered to myocytes in a nonspecific manner (n = 4, ns). Furthermore, Cl− SAC also was not elicited by specific stretch of cardiac transferrin receptors (Parkes et al., 2000; Upton et al., 2003) using antitransferrin receptor IgG1 mAb–coated paramagnetic beads attached to the myocyte (n = 5, ns).
Activation of ICl,swell by osmotic swelling or pressure-induced inflation occurs over 3–5 min (Hagiwara et al., 1992; Tseng, 1992). Activation of Cl− SAC also occurred slowly, and the time course at +40 mV is illustrated in Fig. 3 . The time course of activation was sigmoidal with a half-time of 3.5 ± 0.1 min (n = 5), and steady-state was reached in ∼10 min.
Figure 3.

Time course of Cl− SAC activation by β1-integrin stretch. Stretch-induced currents at +40 mV were recorded at 1-min intervals and normalized by the current at 10 min to obtain fractional activation. Activation of Cl− SAC followed a sigmoidal time course with a half time of 3.5 ± 0.1 min (n = 5).
With the ruptured patch technique, ICl,swell elicited by hypoosmotic swelling or positive pipette pressure only partially turns off after return to control conditions (Hagiwara et al., 1992; Sorota, 1992; Tseng, 1992). Similarly, activation of Cl− SAC was only partially reversed after termination of integrin stretch. Fig. 4 shows the background (Fig. 4 A) and outwardly rectifying Cl− current induced by 6 min of stretch (Fig. 4 B) and their I-V relationships (Fig. 4 D). When the magnetic field was turned off, current slowly decreased in amplitude, but recovery was incomplete at 6 min (Fig. 4, C and D) and remained incomplete even after 10 min. Comparable behavior was observed in each myocyte in which reversibility was tested. On average, stretch significantly increased Cl− current at +40 mV from 1.56 ± 0.22 to 4.48 ± 1.01 pA/pF (P < 0.05), and the current significantly decreased to 2.61 ± 0.35 pA/pF after termination of stretch (P < 0.05), a recovery of 64 ± 8%. Although recovery appeared to be incomplete, the control and postrecovery currents were not significantly different. These data suggest that activation of Cl− SAC is due to a reversible process rather than to mechanical damage of the membrane or to a nonspecific change in leak conductance. Recovery of inward Cl− current at −100 mV tended to behave similarly to recovery of outward current. Inward current increased from −0.36 ± 0.19 to −0.58 ± 0.28 pA/pF after stretch and recovered to −0.46 ± 0.24 pA/pF after termination of stretch. The currents at −100 mV were not significantly different from each other, however.
Figure 4.

Cl− SAC activation partially reversed upon termination of integrin stretch. Families of currents before (A, Control) and after (B, Stretch) activation of Cl− SAC by 6 min of integrin stretch and after a recovery period of 10 min with the magnet turned off (C, Recovery). (D) I-V relationships for control (•), stretch (○), and recovery (⋄). At +40 mV, Cl− SAC decreased by 64 ± 8% (n = 4) during the recovery period.
Both the background Cl− current and the Cl− current recorded after stretch underwent partial inactivation at positive potentials in ∼60% (20 of 34) of myocytes. Fig. 5 compares the extent of inactivation of the currents before and after stretch and their voltage dependencies. Inactivation significantly increased at positive potentials. At +40 mV, control current inactivated by 52 ± 3% (n = 20) and after stretch by 59 ± 3% (n = 20). In contrast, at 0 mV inactivation was only 4 ± 1% (n = 20) and 4 ± 3% (n = 20), respectively. A two-way ANOVA confirmed a significant voltage dependence (P < 0.001), but stretch did not significantly alter inactivation. In the remaining myocytes (14 of 34), neither the background current nor the Cl− SAC exhibited prominent inactivation at positive potentials. The similarity of the extent of inactivation and whether or not it occurs suggests that the channels responsible for the background Cl− current also are responsible for the Cl− SAC.
Figure 5.

Stretch did not alter the voltage dependence of steady-state inactivation. The percentage of current undergoing inactivation was determined at 0, +20, and +40 mV both before and after 5–8 min of integrin stretch (n = 20). Steady-state inactivation was significantly dependent on voltage (P < 0.001) but was not significantly affected by stretch (P = 0.137). Because the overall effect of stretch was not significant (2-way ANOVA), comparison of stretch and control at individual test potentials was precluded. The Cl− SAC did not undergo inactivation at negative potentials before or after stretch.
Tamoxifen Blocks Cl− SAC
Under conditions that isolate Cl− currents, tamoxifen is regarded as a selective and potent inhibitor of ICl,swell and has been used to distinguish this current from the other major Cl− currents (Hume et al., 2000; Baumgarten and Clemo, 2003). Tamoxifen also blocks several cation currents and gap junctions, however (for references see, Baumgarten and Clemo, 2003). Fig. 6 shows that 10 μM tamoxifen blocks both Cl− SAC and the background Cl− current present before stretch. In the experiment illustrated, a relatively large, outwardly rectifying, background Cl− current was recorded before application of stretch (Fig. 6, A and D), and 6 min of integrin stretch elicited additional outward current (Fig. 6, B and D). Fig. 6, C and D, illustrates the effect of tamoxifen (10 μM, 6 min) in the presence of continued stretch. Tamoxifen inhibited both the Cl− SAC and a large fraction of the background Cl− current.
Figure 6.

Tamoxifen, an inhibitor of ICl,swell, blocks both Cl− SAC and background Cl− current. Families of currents before (A, Control) and after (B, Stretch) activation of Cl− SAC by 6 min of integrin stretch and after application of 10 μM tamoxifen for 6 min with continued stretch (C, +Tamoxifen). (D) I-V relationships for A-C. Each reversed near ECl. Tamoxifen blocked all of the Cl− SAC and a large fraction of the background Cl− current. At +40 mV, tamoxifen blocked 116 ± 5% (n = 5) of the stretch-induced current.
Similar results were obtained in each myocyte tested using both physiologic and high Cl− pipette solutions. With physiologic Cl− pipette solution, stretch increased the Cl− current at +40 mV from 1.79 ± 0.44 to 2.91 ± 0.59 pA/pF, and tamoxifen (10 μM, 6–8 min) significantly decreased the current to 1.63 ± 0.48 pA/pF. Thus, tamoxifen blocked 116 ± 5% of the Cl− SAC at +40 mV (n = 5, P < 0.01). At −100 mV, the effects of both stretch and tamoxifen on Cl− current were small and not statistically significant. With high Cl− pipette solution, tamoxifen blocked 150 ± 19% of the Cl− SAC at +40 mV (n = 3, P < 0.005).
Protein Tyrosine Kinases Regulate Cl− SAC
Although the data indicate that mechanical stretch elicits a Cl− current, the time course of activation is slow enough to allow for participation of signaling molecules in the transduction process. Moreover, cell swelling (Sadoshima et al., 1996), mechanical stretch (Sadoshima and Izumo, 1997), and integrin clustering (Clark and Brugge, 1995; Giancotti and Ruoslahti, 1999) activate protein tyrosine kinases (PTKs), and PTKs have been implicated in the activation of ICl,swell in heart (Sorota, 1995) and other tissues (Voets et al., 1998; Shi et al., 2002). Therefore, we tested whether mechanical activation of Cl− SAC involved signaling by PTK.
Fig. 7 shows the effect of genistein, a broad-spectrum PTK inhibitor. A small, outwardly rectifying background current that partially inactivated at positive potentials was present before stretch (Fig. 7, A and D), and 6 min of integrin stretch significantly increased the Cl− current and the degree of rectification (Fig. 7, B and D). Superfusion of genistein (100 μM, 8 min) partially inhibited the Cl− current (Fig. 7, C and D). At +40 mV, genistein inhibited 62 ± 6% of the stretch-induced current (n = 4, P < 0.01); stretch increased the Cl− current from 2.25 ± 1.00 to 3.21 ± 1.14 pA/pF, and genistein reduced the current to 2.62 ± 1.05 pA/pF. The effect of genistein at −100 mV was similar, but the currents were smaller and the changes were not statistically significant. These data suggest that PTK is involved in the activation of Cl− current by integrin stretch. On the other hand, genistein appeared to enhance the Cl− current in one additional myocyte studied.
Figure 7.

The PTK inhibitor genistein partially blocks Cl− SAC. Current records before (A, Control) and after (B, Stretch) activation of Cl− SAC by 6 min of integrin stretch and after application of 100 μM genistein for 8 min with continued stretch (C, +Genistein). (D) I-V relationships for A–C. Each reversed near −50 mV. At +40 mV, genistein blocked 62 ± 6% (n = 4) of the stretch-induced current.
FAK and Src are the two principal upstream PTKs activated by both mechanical stretch and integrin clustering (Sadoshima and Izumo, 1997; Parsons, 2003). PP2, a selective inhibitor of FAK and Src (Hanke et al., 1996), was used to test whether these PTKs regulate Cl− SAC. As previously shown, integrin stretch activated an outwardly rectifying Cl− current (Fig. 8, A, B, and D) . Then, while maintaining stretch, the myocyte was superfused with 10 μM PP2 for 10 min. PP2 inhibited both the Cl− SAC and a large fraction of the background Cl− current (Fig. 8, C and D).
Figure 8.

PP2, a selective inhibitor of Src and FAK, blocks both Cl− SAC and background Cl− current. Current records before (A, Control) and after (B, Stretch) activation of Cl− SAC by 5 min of integrin stretch and after application of 10 μM PP2 for 10 min with continued stretch (C, +PP2). (D) I-V relationships for A–C. Each reversed near −50 mV. At +40 mV, PP2 blocked 134 ± 13% (n = 6) of the stretch-induced current.
At +40 mV, PP2 (10 μM, 8–10 min) blocked 134 ± 13% of the Cl− SAC (n = 6, P < 0.001); stretch increased Cl− current from 1.61 ± 0.28 to 2.49 ± 0.28 pA/pF, and PP2 reduced the current to 1.28 ± 0.27 pA/pF. The current after PP2 was significantly less than the control current (P < 0.05). Similar behavior was observed at −100 mV, but the currents and effect of PP2 were smaller, and the differences were not statistically significant.
To verify that the profound inhibitory action of PP2 was not due to a nonspecific effect, PP3, an inactive analogue of PP2, also was tested. Although integrin stretch activated Cl− current (Fig. 9, A, B, and D) , exposure to 10 μM PP3 for 10 min while maintaining stretch did not significantly affect the current (Fig. 9, C and D). Similar results were obtained in all four myocytes examined, and block at +40 mV was 1 ± 11% (n = 4); stretch increased Cl− current from 1.31 ± 0.21 to 2.36 ± 0.14 pA/pF, and after PP3, Cl− current was 2.34 ± 0.19 pA/pF. Together, these results suggest that FAK and/or Src, members of the PTK family, play a key role in the activation of Cl− SAC by integrin stretch.
Figure 9.

PP3, an inactive analogue of PP2, did not affect Cl− SAC or background Cl− current. Current records before (A, Control) and after (B, Stretch) activation of Cl− SAC by 5 min of integrin stretch and after application of 10 μM PP3 for 10 min with continued stretch (C, +PP3). (D) I-V relationships for A–C. Each reversed near −50 mV. At +40 mV, PP3 did not significantly alter the stretch-induced current, −1 ± 11% (n = 4).
β1-Integrin Stretch Modulates Cation Currents
The effect of integrin stretch also was examined in physiological Tyrode solution in order to determine whether cation SAC is activated by integrin stretch. Steady-state I-V relationships are shown in Fig. 10 A. In physiological Tyrode solution, K+ conductance dominates, and the steady-state I-V relationship reversed at −77 mV. After 5 min of integrin stretch, the steady-state I-V relationship was more linear, reversed at −38 mV, and appeared to be rotated counter-clockwise, except at very negative potentials. Whereas outward current positive to −20 mV was enhanced, inward current at −100 mV was inhibited. The stretch-induced difference current (Fig. 10 B) crossed the voltage axis twice, at −91 and −23 mV, indicating that stretch of integrins modulated more than one membrane current. On average, integrin stretch in physiological bathing media shifted the reversal potential positive by 42.7 ± 4.7 mV (n = 3, P < 0.01), from −73.0 ± 2.1 to −30.3 ± 6.2 mV. Outward current at +40 mV increased by 5.30 ± 1.56 pA/pF (n = 3, P < 0.05), from 4.02 ± 2.87 to 9.33 ± 2.45 pA/pF. This enhancement of steady-state current was four to five times greater than the enhancement of peak current in solutions designed to isolate anion currents. At intermediate voltages, −40 to −80 mV, stretch shifted the current in an inward direction. At −60 mV, for example, the stretch-induced current was −3.37 ± 0.65 pA/pF (n = 3, P < 0.05). In contrast, inward current at −100 mV was inhibited by 3.86 ± 1.12 pA/pF (n = 3, P < 0.05), decreasing from −18.51 ± 0.51 to −14.65 ± 1.24 pA/pF. This behavior is distinct from the small increase in inward anion current at −100 mV noted previously and suggests IK1 is inhibited by integrin stretch. A similar stretch-induced inhibition of IK1 was reported recently by Isenberg et al. (2003) in guinea pig ventricular myocytes stretched with a glass stylus. In a fourth myocyte studied under physiological conditions, reversal potential did not shift, and changes in the I-V curve were similar to those seen in solutions designed to isolate anion currents. Based on these data, one can infer that integrin stretch in physiological bathing media modulates cation currents in ∼75% of myocytes.
Figure 10.

β1-integrin stretch activates a nonselective cation current and suppresses IK1. Recordings were made using physiological bath and pipette solutions using the same voltage-clamp protocol as in previous figures. (A) I-V relationships for steady-state current before (Control, •) and after (Stretch, ○) 5 min of integrin stretch, and (B) for the stretch-induced difference current (Difference, ▪). Integrin stretch increased current over most of the voltage range, attenuated inward rectification, and shifted reversal potential positive. The difference current (B) reversed at −91 and −23 mV, suggesting that stretch modulated multiple currents. This is consistent with suppression of inwardly rectifying IK1 (appears inverted; difference current = stretch − control) and augmentation of a linear poorly selective current. (C) To characterize the stretch-activated cation component and minimize contributions from IK1, steady-state I-V relationships were obtained between −60 and +40 mV in physiological solutions (n = 3; Total, ○) and in solutions designed to isolate anion currents (n = 3; Anion, •). Stretch-activated total current was significantly greater than Cl− SAC both at +40 mV (5.30 ± 1.56 vs. 1.19 ± 0.17 pA/pF; P < 0.05) and −60 mV (−3.37 ± 0.65 vs. −0.02 ± 0.01 pA/pF; P < 0.02). (D) The stretch-activated cation current (Cation, □) was estimated as the difference between the total and anion currents in C assuming independence. The I-V relationship for the cation SAC was linear and reversed at −10 mV.
Fig. 10 C compares the steady-state I-V relationship after stretch in physiological bath solution to that under conditions that isolate Cl− currents. Assuming independence, the contribution of cations to the stretch-activated current was obtained by subtracting the curves. Voltages between −60 and +40 mV were considered to minimize contributions from IK1 in physiological Tyrode solution. The difference I-V relationship (Fig. 10 D) was nearly linear and reversed at −10 mV, suggesting integrin stretch activated a nonselective cation current. A nonselective stretch–activated cation current with a linear I-V relationship previously was recorded in cardiac myocytes in response to longitudinal stretch using two concentric pipettes or two patch electrodes, or by means of a glass stylus (Zeng et al., 2000; Zhang et al., 2000; Isenberg et al., 2003).
DISCUSSION
Contrary to previous claims (Hu and Sachs, 1997; Cazorla et al., 1999), cardiac Cl− channels can be activated by a mechanical stimulus that does not alter cell volume or dilute the cytoplasmic contents. In solutions designed to isolate anion currents, stretch of β1-integrins with mAb-coated paramagnetic beads elicited a Cl− current in ∼75% of rabbit left ventricular myocytes. The current was outwardly rectifying, partially inactivated at positive voltages, and was blocked by tamoxifen; its reversal potential followed ECl. When stretch was terminated, the Cl− current largely returned to its control value. In contrast, application of a magnetic field either with beads that had not been coated with primary mAb or with beads coated with antitransferrin receptor mAb of the same class and subtype (mouse IgG1) as the anti-β1 integrin mAb failed to stimulate the current, and the magnetic field alone failed to stimulate the current. These data suggest that Cl− channels were activated by a specific stretch of β1 integrins rather than by some other nonspecific process or as a general response to myocyte stretch. Because mechanical stretch rather than volume changes serve as the stimulus, the current should be considered a Cl− SAC. The conclusion that certain cardiac Cl− channels are stretch activated is supported by observations that amphiphiles that cause membrane curvature without cell swelling (crenators) activate Cl− current (Tseng, 1992). The same amphiphiles are thought to activate the mechanosensitive 2-P K+ channels, TRAAK and TREK, via membrane curvature (Patel et al., 2001). In addition, the open probability of cardiac Cl− channels is increased by applying negative pressure to inside-out and positive pressure to outside-out patches from human atrial cells (Sato and Koumi, 1998).
One important difference between this and previous attempts to provoke Cl− currents with mechanical perturbations is the means of applying force to myocytes. The paramagnetic bead method takes advantage of the specificity of mAb to apply force in a defined, reproducible, and quantifiable manner. Because integrins are an innate mechanical link between the ECM and cytoskeleton and function as mechanosensors in multiple cell types (Salter et al., 1997; Mobasheri et al., 2002; Shyy and Chien, 2002), it is appealing to think that stretch of β1 integrins represents a physiologically relevant means of mechanical stimulation. On the other hand, ECM normally interact simultaneously with both the α and β subunits of the integrin heterodimer (Clark and Brugge, 1995). Therefore, there may be differences in the information delivered and the responses evoked by stretching ECM in vivo or just β1 integrins. Furthermore, cardiac myocytes express other β-integrin isoforms, including β3 and β5 (Ross and Borg, 2001), and force also is transmitted from the ECM to the cytoskeleton by the dystrophin–dystroglycan complex (Petrof et al., 1993). These molecules and the attached cytoskeleton also are potential mechanotransducers for SAC. By coupling various mAb and ECM components to beads and applying either a static or time-dependent magnetic field, the paramagnetic bead method should prove useful in unraveling how external mechanical forces ultimately regulate cellular function.
Identity of the Cl− SAC
The main Cl− channels identified in cardiac myocytes are the PKA-dependent, cardiac variant of the cystic fibrosis transmembrane conductance regulator (ICFTR,cardiac or ICl,PKA), the calcium-dependent transient outward Cl− current (ICl,Ca or Ito2), and the volume-sensitive Cl− current (ICl,swell or ICl,vol) (Sorota, 1999; Hume et al., 2000). All three of these currents are outwardly rectifying with a physiological Cl− gradient, as was the Cl− SAC. Nevertheless, based on biophysical and pharmacological characteristics, it seems unlikely that the Cl− SAC recorded here is due to either ICFTR,cardiac or ICl,Ca. Cl− SAC partially inactivated at positive potentials, whereas ICFTR,cardiac is time independent at all voltages (Shuba et al., 1996; Hume et al., 2000). ICl,Ca is triggered by elevation of cytoplasmic Ca2+ and displays both inactivation at positive potentials and a bell-shaped I-V relationship when Ca2+ handling is normal (Zygmunt and Gibbons, 1991). When cytoplasmic Ca2+ is clamped at an elevated level, however, ICl,Ca is time independent (Zygmunt, 1994). In contrast, Cl− SAC inactivated and exhibited outward rectification in Ca2+-free bathing media with 8 mM EGTA in the pipette solution, conditions that reduced cytoplasmic-free Ca2+ to ∼35 nM and minimized Ca2+ transients. Furthermore, 10 μM tamoxifen blocked Cl− SAC, but ICFTR,cardiac (Vandenberg et al., 1994) and ICl,Ca (Valverde et al., 1993) are insensitive to tamoxifen. It also is unlikely that Cl− SAC is due to ClC-2, which yields an inwardly rectifying Cl− current in heart (Duan et al., 2000), or P-glycoprotein, which is insensitive to tamoxifen (Ehring et al., 1994).
Although activation of a novel Cl− channel by integrin stretch rigorously cannot be ruled out, the characteristics of Cl− SAC resemble those of ICl,swell. Like ICl,swell (Sorota, 1999; Hume et al., 2000; Baumgarten and Clemo, 2003), the Cl− SAC activated slowly, was outwardly rectifying, partially inactivated at positive potentials, was blocked by tamoxifen, was poorly permeant to aspartate, and was not observed in every myocyte. On the other hand, several quantitative differences should be noted. The current density for Cl− SAC, 1.13 ± 0.10 pA/pF at +40 mV, was less than that typically reported for ICl,swell in mammalian ventricle, 2 to 10 pA/pF (Vandenberg et al., 1996). This could be due to the fact that Cl− SAC was activated by stretching only localized patches of integrin receptors, whereas ICl,swell arises from swelling of the entire cell. Second, in ∼60% of the myocytes Cl− SAC inactivated to a greater degree than is typical for cardiac ICl,swell (Tseng, 1992; Duan et al., 1995; cf., Shuba et al., 1996). The reason why Cl− SAC exhibited prominent inactivation in some cells but not in others is unclear. Nevertheless, both the extent of inactivation at positive potentials, if any, and its voltage dependence were identical for the background Cl− current and Cl− SAC in each cell studied. The similarity of inactivation is consistent with the idea that the Cl− SAC and the background Cl− current, which has been attributed to ICl,swell (Sorota, 1992; Duan et al., 1995, 1997), arise from the same channel. The possibility that Cl− SAC is partially activated by binding of the anti–β1-integrin mAb-coated beads without stretch and contributes to the background current cannot be excluded, however. Finally, the outward rectification of Cl− SAC was greater than for ICl,swell in rabbit ventricular myocytes (Clemo and Baumgarten, 1997) with both physiological and high pipette Cl− concentrations. Perhaps the quantitative differences arise in part from the dilution of an intracellular regulator or blocking ion upon cell swelling but not after integrin stretch.
Stretch and cell volume may regulate the same Cl− channel in other cell types. Plating human neutrophils on surfaces coated with mAb for β2 integrin or the α subunits of LFA-1, CR3, and gp150/95 integrins results in Cl− efflux (Menegazzi et al., 1999) that was markedly inhibited by MK-447 and analogue A, which block ICl,swell in neutrophils (Simchowitz et al., 1993). Moreover, stretch and swelling activate the same anion channel in cultured opossum kidney cells (Ubl et al., 1988).
Mechanism of Mechanotransduction
The slow time course of activation of Cl− SAC and the fact that localized stretch induced a significant whole-cell current indicates the involvement of a signaling cascade in the transduction process. In support of this idea, genistein, a broad-spectrum isoflavone PTK inhibitor, significantly but incompletely blocked Cl− SAC in the presence of continued β1-integrin stretch. Incomplete block may arise from nonspecific effects of this agent. Genistein activates ICFTR,cardiac in a PTK-independent manner at the concentration used (Chiang et al., 1997), and it is likely that the observed incomplete block reflects the balance between block of Cl− SAC and stimulation of ICFTR,cardiac.
Stronger evidence for the involvement of PTK was obtained with PP2, a specific blocker of FAK and Src (Hanke et al., 1996) that competitively antagonizes the binding of ATP to the activated kinase domain (Zhu et al., 1999). PP2, but not its inactive analogue PP3, completely blocked Cl− SAC as well as a significant fraction of the background Cl− current. To our knowledge, this is the first evidence for the involvement of FAK and/or Src in the regulation of cardiac Cl− currents.
The regulation of Cl− SAC by PTK family members is consistent with the idea ICl,swell may be responsible for the Cl− SAC. Genistein blocks ICl,swell in canine atrial myocytes (Sorota, 1995). Moreover, the Src family of PTK regulates ICl,swell in noncardiac cells. Src activator peptide, EPQ(pY)EEIPI, significantly enhances ICl,swell in cultured rabbit nonpigmented ciliary epithelial cells (Shi et al., 2002). In addition, p56lck, a Src kinase, rescues defective activation of ICl,swell in p56lck knockout human T lymphocytes (Lepple-Wienhues et al., 1998) and activates an outwardly rectifying Cl− current independent of CFTR (Lepple-Wienhues et al., 2000). Finally, as previously noted, FAK and Src are upstream PTKs that orchestrate integrin-mediated signaling (Parsons, 2003), and integrins and PTK are activated by both mechanical stretch and swelling of myocytes (Sadoshima et al., 1996; Sadoshima and Izumo, 1997).
Pathophysiological Activation of Cl− SAC
Cardiac disease, including dilated cardiomyopathies, valvular disease, and myocardial infarction, results in chamber dilatation and wall motion abnormalities that ultimately stretch cardiac myocytes. We postulate that global or regional stretch of myocytes in situ by forces transmitted from ECM to integrins will lead to activation of Cl− SAC, at least in part, via integrin signaling involving Src and/or FAK. Because Cl− SAC is outwardly rectifying and reverses between the plateau and resting potential, its activation will tend to generate a small diastolic depolarization, reduce action potential duration (APD), and limit the overall prolongation of APD resulting from the contributions of other ionic currents that are typically altered or remodeled by disease processes. In the ventricle, the effect on APD may be protective by suppressing arrhythmias due to early afterdepolarizations. On the other hand, abbreviation of APD upon stretch would favor reentrant tachycardias.
Activation of FAK and Src in cardiac disease is well established. Acute pressure overload due to aortic constriction results in phosphorylation of FAK and Src (Franchini et al., 2000; Laser et al., 2000; Babbitt et al., 2002), and association of FAK with other components of costameres including Src, Grb2, PI-3K, and actin (Franchini et al., 2000). Similarly, acute volume overload produced by balloon inflation causes phosphorylation of FAK, association of FAK with Src, Grb2, and ERK (Domingos et al., 2002), and activation of Src (Takeishi et al., 2001). Chronic stretch triggers altered β1-integrin expression (Babbitt et al., 2002), and the up-regulation and remodeling of ECM (Bendall et al., 2002; Cleutjens and Creemers, 2002), the natural integrin ligand. Furthermore, β1-integrins (Ross et al., 1998), FAK (Pham et al., 2000; Taylor et al., 2000), and Src (Takeishi et al., 2001) have a role in compensatory hypertrophy, and β1-integrins may also participate in the progression to decompensated heart failure (Goldsmith et al., 1999).
Activation of cardiac Cl− channels in animal models of cardiovascular disease and in man also has been established. ICl,swell is persistently activated under isosmotic conditions in ventricular myocytes from dilated cardiomyopathy induced by rapid pacing (Clemo et al., 1999b) or aortic regurgitation (Clemo and Baumgarten, 1998; Clemo et al., 1999a), in myocytes isolated 30 d after infarction from the infarct and peri-infarct zones (Clemo et al., 2001), and in human atrial myocytes from patients with atrial enlargement or elevated ventricular end-diastolic pressure (Patel et al., 2003). In addition, an outwardly rectifying Cl− current is up-regulated in a pressure overload hypertrophy model (Bénitah et al., 1997), but the identity of the current was not established. It remains to be seen whether the regulation of Cl− currents activated in cardiac disease is the same as for Cl− SAC.
Effect of Integrin Stretch on Cation Currents
In addition to activating Cl− SAC, stretch of β1-integrins appeared to activate a poorly selective cation current with a linear I-V relationship that reversed near −10 mV, and to suppress the inwardly rectifying potassium current, IK1. The decrease in inward current at −100 mV (Fig. 10) underestimates the suppression of IK1 because inward current is simultaneously augmented by the poorly selective cation current. Other means of cardiac myocyte stretch have been shown to activate nonselective cation currents (Zeng et al., 2000; Zhang et al., 2000; Isenberg et al., 2003) and recently to inhibit IK1 (Isenberg et al., 2003). Although these phenomena were not studied in detail, they are likely to be important because the effects of stretch on cation currents were larger in amplitude than those on Cl− currents. On the other hand, stretch was applied locally. The relative efficiency of the resulting signaling cascades to propagate throughout the cell and activate anion and cation SAC and inhibit IK1 are unknown. Therefore, the magnitude of effects on anion and cation currents of mechanical stimuli that stretch integrins over the entire sarcolemma remains uncertain.
Integrin stretch previously has been shown to modulate cation currents in other cell types. For example, stretching integrins with collagen-coated beads triggers Ca2+ transients in fibroblasts that were regulated by the actin cytoskeleton (Wu et al., 1999) and blocked by Gd3+ (Glogauer et al., 1995), a cation SAC channel blocker. Furthermore, integrin ligand–coated beads modulate Ba2+ current through L-type Ca2+ channels in rat smooth muscle cells (Wu et al., 1998) by a process requiring the activity of FAK and Src (Wu et al., 2001). We cannot exclude the possibility that stretch of integrins activates other currents in heart.
Electrophysiological Consequences of Integrin Stretch
Whereas Cl− SAC alone is expected to shorten APD and cause slight diastolic depolarization, the overall effect of integrin stretch is likely to be more complex. Activation of a poorly selective cation SAC and inhibition of IK1 will result in significant diastolic depolarization, a more pronounced shortening of the plateau, and a slowing of the final portion of repolarization. Full analysis of the combined effects must await detailed measurements of anion and cation SAC with widespread rather than localized stretch of integrins, as might occur under physiologic and pathophysiologic conditions. Nevertheless, the predicted electrophysiologic consequences of integrin stretch are likely to contribute to arrhythmias associated, for example, with dilated cardiomyopathies and atrial enlargement.
In summary, this study shows for the first time that specific mechanical stretch of β1-integrins in cardiac myocytes activates a Cl− current with biophysical and pharmacological characteristics that resemble ICl,swell. Mechanotransduction involves PTK, particularly FAK and/or Src. Furthermore, stretch of β1-integrin in cardiac myocytes also appears to activate a poorly selective cation current and inhibit IK1.
Acknowledgments
We thank Jason Hackenbracht for helpful discussions and comments on the manuscript, Justin Hormel for technical assistance, and Peter Robinson for design and fabrication of the magnetic coil and controller.
This work was supported by National Institutes of Health grant HL-46764.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: APD, action potential duration; ECM, extracellular matrix; FAK, focal adhesion kinase; G, Gauss; PI-3K, phosphatidylinositol-3-kinase; PTK, protein tyrosine kinase; SAC, stretch-activated current.
References
- Babbitt, C.J., S.Y. Shai, A.E. Harpf, C.G. Pham, and R.S. Ross. 2002. Modulation of integrins and integrin signaling molecules in the pressure-loaded murine ventricle. Histochem. Cell Biol. 118:431–439. [DOI] [PubMed] [Google Scholar]
- Baumgarten, C.M., and H.F. Clemo. 2003. Swelling-activated chloride channels in cardiac physiology and pathophysiology. Prog. Biophys. Mol. Biol. 82:25–42. [DOI] [PubMed] [Google Scholar]
- Bendall, J.K., C. Heymes, P. Ratajczak, and J.L. Samuel. 2002. Extracellular matrix and cardiac remodelling. Arch. Mal. Coeur Vaiss. 95:1226–1229. [PubMed] [Google Scholar]
- Bénitah, J.P., A.M. Gomez, C. Delgado, P. Lorente, and W.J. Lederer. 1997. A chloride current component induced by hypertrophy in rat ventricular myocytes. Am. J. Physiol. 272:H2500–H2506. [DOI] [PubMed] [Google Scholar]
- Borg, T.K., E.C. Goldsmith, R. Price, W. Carver, L. Terracio, and A.M. Samarel. 2000. Specialization at the Z line of cardiac myocytes. Cardiovasc. Res. 46:277–285. [DOI] [PubMed] [Google Scholar]
- Browe, D.M., and C.M. Baumgarten. 2003. a. Specific stretch of β1 integrin with mAb-coated paramagnetic beads activates ICl,swell in rabbit ventricular myocytes. Biophys. J. 84:22a. [Google Scholar]
- Browe, D.M., and C.M. Baumgarten. 2003. b. Activation of a Cl− current by specific stretch of β1 integrin is mediated by FAK and Src in rabbit ventricular myocytes. J. Gen. Physiol. 122:31a–32a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla, O., C. Pascarel, F. Brette, and J.Y. Le Guennec. 1999. Modulation of ion channel and membrane receptor activities by mechanical interventions in cardiomyocytes: possible mechanisms for mechanosensitivity. Prog. Biophys. Mol. Biol. 71:29–58. [DOI] [PubMed] [Google Scholar]
- Chiang, C.E., S.A. Chen, M.S. Chang, C.I. Lin, and H.N. Luk. 1997. Genistein directly induces cardiac CFTR chloride current by a tyrosine kinase-independent and protein kinase A-independent pathway in guinea pig ventricular. Biochem. Biophys. Res. Commun. 235:74–78. [DOI] [PubMed] [Google Scholar]
- Clark, E.A., and J.S. Brugge. 1995. Integrins and signal transduction pathways: the road taken. Science. 268:233–239. [DOI] [PubMed] [Google Scholar]
- Clemo, H.F., and C.M. Baumgarten. 1997. Swelling-activated Gd3+-sensitive cation current and cell volume regulation in rabbit ventricular myocytes. J. Gen. Physiol. 110:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemo, H.F., and C.M. Baumgarten. 1998. Protein kinase C (PKC) activation blocks ICl,swell and causes myocyte swelling in a rabbit congestive heart failure (CHF) model. Circulation. 98:I–695.
- Clemo, H.F., J.S. Danetz, and C.M. Baumgarten. 1999a. Inhibition of protein phosphatase by okadaic acid blocks ICl,swell in ventricular myocytes from both normal and congestive failure rabbits. Circulation. 100:I–425.
- Clemo, H.F., B.S. Stambler, and C.M. Baumgarten. 1999. b. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ. Res. 84:157–165. [DOI] [PubMed] [Google Scholar]
- Clemo, H.F., J. Rana, A.M. Vaida, G.N. Tseng, R.S. Higgins, and C.M. Baumgarten. 2001. Chronic activation of ICl,swell in canine infarction model suppresses inducibility of early afterdepolarizations. Circulation. 104:II–624. [Google Scholar]
- Cleutjens, J.P.M., and E.E.J.M. Creemers. 2002. Integration of concepts: cardiac extracellular matrix remodeling after myocardial infarction. J. Card. Fail. 8:S344–S348. [DOI] [PubMed] [Google Scholar]
- Danowski, B.A., K. Imanaka-Yoshida, J.M. Sanger, and J.W. Sanger. 1992. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J. Cell Biol. 118:1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingos, P.P., P.M. Fonseca, W. Nadruz, and K.G. Franchini. 2002. Load-induced focal adhesion kinase activation in the myocardium: role of stretch and contractile activity. Am. J. Physiol. Heart Circ. Physiol. 282:H556–H564. [DOI] [PubMed] [Google Scholar]
- Duan, D., S. Cowley, B. Horowitz, and J.R. Hume. 1999. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. J. Gen. Physiol. 113:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, D., B. Fermini, and S. Nattel. 1995. β-adrenergic control of volume-regulated Cl− currents in rabbit atrial myocytes: characterization of a novel ionic regulatory mechanism. Circ. Res. 77:379–393. [DOI] [PubMed] [Google Scholar]
- Duan, D., J.R. Hume, and S. Nattel. 1997. Evidence that outwardly rectifying Cl− channels underlie volume-regulated Cl− currents in heart. Circ. Res. 80:103–113. [DOI] [PubMed] [Google Scholar]
- Duan, D., L. Ye, F. Britton, B. Horowitz, and J.R. Hume. 2000. A novel anionic inward rectifier in native cardiac myocytes. Circ. Res. 86:E63–E71. [PubMed] [Google Scholar]
- Ehring, G.R., Y.V. Osipchuk, and M.D. Cahalan. 1994. Swelling-activated chloride channels in multidrug-sensitive and -resistant cells. J. Gen. Physiol. 104:1129–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini, K.G., A.S. Torsoni, P.H.A. Soares, and M.J.A. Saad. 2000. Early activation of the multicomponent signaling complex associated with focal adhesion kinase induced by pressure overload in the rat heart. Circ. Res. 87:558–565. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Glogauer, G., and J. Ferrier. 1998. A new method for application of force to cells via ferric oxide beads. Pflugers Arch. 435:320–327. [DOI] [PubMed] [Google Scholar]
- Glogauer, M., J. Ferrier, and C.A.G. McCulloch. 1995. Magnetic fields applied to collagen-coated ferric oxide beads induce stretch-activated Ca2+ flux in fibroblasts. Am. J. Physiol. 269:C1093–C1104. [DOI] [PubMed] [Google Scholar]
- Goldsmith, E.C., R. Price, W. Carver, E.O. Weinberg, B. Ding, and T.K. Borg. 1999. The role of α5β1 integrin in the transition from cardiac hypertrophy to failure. Circulation. 100:I–560.
- Hagiwara, N., H. Masuda, M. Shoda, and H. Irisawa. 1992. Stretch-activated anion currents of rabbit cardiac myocytes. J. Physiol. 456:285–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke, J.H., J.P. Gardner, R.L. Dow, P.S. Changelian, W.H. Brissette, E.J. Weringer, B.A. Pollok, and P.A. Connelly. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. J. Biol. Chem. 271:695–701. [DOI] [PubMed] [Google Scholar]
- Hu, H., and F. Sachs. 1997. Stretch-activated ion channels in the heart. J. Mol. Cell. Cardiol. 29:1511–1523. [DOI] [PubMed] [Google Scholar]
- Hume, J.R., D. Duan, M.L. Collier, J. Yamazaki, and B. Horowitz. 2000. Anion transport in heart. Physiol. Rev. 80:31–81. [DOI] [PubMed] [Google Scholar]
- Isenberg, G., V. Kazanski, D. Kondratev, M.F. Gallitelli, I. Kiseleva, and A. Kamkin. 2003. Differential effects of stretch and compression on membrane currents and [Na+]c in ventricular myocytes. Prog. Biophys. Mol. Biol. 82:43–56. [DOI] [PubMed] [Google Scholar]
- Kamkin, A., I. Kiseleva, and G. Isenberg. 2003. Ion selectivity of stretch-activated cation currents in mouse ventricular myocytes. Pflugers Arch. 446:220–231. [DOI] [PubMed] [Google Scholar]
- Laser, M., C.D. Willey, W. Jiang, G. Cooper, D.R. Menick, M.R. Zile, and D. Kuppuswamy. 2000. Integrin activation and focal complex formation in cardiac hypertrophy. J. Biol. Chem. 275:35624–35630. [DOI] [PubMed] [Google Scholar]
- Lepple-Wienhues, A., I. Szabo, T. Laun, N.K. Kaba, E. Gulbins, and F. Lang. 1998. The tyrosine kinase p56lck mediates activation of swelling-induced chloride channels in lymphocytes. J. Cell Biol. 141:281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepple-Wienhues, A., I. Szabo, U. Wieland, L. Heil, E. Gulbins, and F. Lang. 2000. Tyrosine kinases open lymphocyte chloride channels. Cell. Physiol. Biochem. 10:307–312. [DOI] [PubMed] [Google Scholar]
- Menegazzi, R., S. Busetto, E. Decleva, R. Cramer, P. Dri, and P. Patriarca. 1999. Triggering of chloride ion efflux from human neutrophils as a novel function of leukocyte β2 integrins: relationship with spreading and activation of the respiratory burst. J. Immunol. 162:423–434. [PubMed] [Google Scholar]
- Mobasheri, N., S.D. Carter, P. Martin-Vasallo, and M. Shakibaei. 2002. Integrins and stretch activated ion channels; putative components of functional cell surface mechanoreceptors in articular chondrocytes. Cell Biol. Int. 26:1–18. [DOI] [PubMed] [Google Scholar]
- Nilius, B., J. Sehrer, F. Viana, C. De Greef, L. Raeymaekers, J. Eggermont, and G. Droogmans. 1994. Volume-activated Cl− currents in different mammalian non-excitable cell types. Pflugers Arch. 428:364–371. [DOI] [PubMed] [Google Scholar]
- Nilius, B., T. Voets, J. Prenen, H. Barth, K. Aktories, K. Kaibuchi, G. Droogmans, and J. Eggermont. 1999. Role of rho and rho kinase in the activation of volume-regulated anion channels in bovine endothelial cells. J. Physiol. 516:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes, J.G., Y. Liu, J.B. Sirna, and D.M. Templeton. 2000. Changes in gene expression with iron loading and chelation in cardiac myocytes and non-myocytic fibroblasts. J. Mol. Cell Cardiol. 32:233–246. [DOI] [PubMed] [Google Scholar]
- Parsons, J.T. 2003. Focal adhesion kinase: the first ten years. J. Cell Sci. 116:1409–1416. [DOI] [PubMed] [Google Scholar]
- Patel, A.J., M. Lazdunski, and E. Honore. 2001. Lipid and mechano-gated 2P domain K+ channels. Curr. Opin. Cell Biol. 13:422–428. [DOI] [PubMed] [Google Scholar]
- Patel, D.G., R.S. Higgins, and C.M. Baumgarten. 2003. Swelling-activated Cl current, ICl,swell, is chronically activated in diseased human atrial myocytes. Biophys. J. 84:233a. [Google Scholar]
- Petrof, B.J., J.B. Shrager, H.H. Stedman, A.M. Kelly, and H.L. Sweeney. 1993. Proc Natl. Acad. Sci. USA. 90:3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham, C.G., A.E. Harpf, R.S. Keller, H.T. Vu, S.Y. Shai, J.C. Loftus, and R.S. Ross. 2000. Striated muscle-specific β1D-integrin and FAK are involved in cardiac myocyte hypertrophic response pathway. Am. J. Physiol. Heart Circ. Physiol. 279:H2916–H2926. [DOI] [PubMed] [Google Scholar]
- Ross, R.S. 2002. The extracellular connections: the role of integrins in myocardial remodeling. J. Card. Fail. 8:S326–S331. [DOI] [PubMed] [Google Scholar]
- Ross, R.S., and T.K. Borg. 2001. Integrins and the myocardium. Circ. Res. 88:1112–1119. [DOI] [PubMed] [Google Scholar]
- Ross, R.S., C. Pham, S.Y. Shai, J.I. Goldhaber, C. Fenczik, C.C. Glembotski, M.H. Ginsberg, and J.C. Loftus. 1998. β1 integrins participate in the hypertrophic response of rat ventricular myocytes. Circ. Res. 82:1160–1172. [DOI] [PubMed] [Google Scholar]
- Sadoshima, J., Z. Qiu, J.P. Morgan, and S. Izumo. 1996. Tyrosine kinase activation is an immediate and essential step in hypotonic cell swelling-induced ERK activation and c-fos gene expression in cardiac myocytes. EMBO J. 15:5535–5546. [PMC free article] [PubMed] [Google Scholar]
- Sadoshima, J., and S. Izumo. 1997. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu. Rev. Physiol. 59:551–571. [DOI] [PubMed] [Google Scholar]
- Salter, D.M., J.E. Robb, and M.O. Wright. 1997. Electrophysiological responses of human bone cells to mechanical stimulation: evidence for specific integrin function in mechanotransduction. J. Bone Miner. Res. 12:1133–1141. [DOI] [PubMed] [Google Scholar]
- Sato, R., and S. Koumi. 1998. Characterization of the stretch-activated chloride channel in isolated human atrial myocytes. J. Membr. Biol. 163:67–76. [DOI] [PubMed] [Google Scholar]
- Shi, C., S. Barnes, M. Coca-Prados, and M.E.M. Kelly. 2002. Protein tyrosine kinase and protein phosphatase signaling pathways regulate volume-sensitive chloride currents in a nonpigmented ciliary epithelial cell line. Invest. Ophthalmol. Vis. Sci. 43:1525–1532. [PubMed] [Google Scholar]
- Shuba, L.M., T. Ogura, and T.F. McDonald. 1996. Kinetic evidence distinguishing volume-sensitive chloride current from other types in guinea-pig ventricular myocytes. J. Physiol. 491:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyy, J.Y.J., and S. Chien. 2002. Role of integrins in endothelial mechanosensing of shear stress. Circ. Res. 91:769–775. [DOI] [PubMed] [Google Scholar]
- Simchowitz, L., J.A. Textor, and E.J. Cragoe. 1993. Cell volume regulation in human neutrophils: 2-(aminomethyl) phenols as Cl− channel inhibitors. Am. J. Physiol. 265:C143–C145. [DOI] [PubMed] [Google Scholar]
- Sorota, S. 1992. Swelling-induced chloride-sensitive current in canine atrial cells revealed by whole-cell patch-clamp method. Circ. Res. 70:679–687. [DOI] [PubMed] [Google Scholar]
- Sorota, S. 1995. Tyrosine protein kinase inhibitors prevent activation of cardiac swelling-induced chloride current. Pflugers Arch. 431:178–185. [DOI] [PubMed] [Google Scholar]
- Sorota, S. 1999. Insights into the structure, distribution and function of the cardiac chloride channels. Cardiovasc. Res. 42:361–376. [DOI] [PubMed] [Google Scholar]
- Takeishi, Y., Q. Huang, J. Abe, M. Glassman, W. Che, J.D. Lee, H. Kawakatsu, E.G. Lawrence, B.D. Hoit, B.C. Berk, and R.A. Walsh. 2001. Src and multiple MAP kinase activation in cardiac hypertrophy and congestive heart failure under chronic pressure-overload: comparison with acute mechanical stretch. J. Mol. Cell. Cardiol. 33:1637–1648. [DOI] [PubMed] [Google Scholar]
- Taylor, J.M., J.D. Rovin, and J.T. Parsons. 2000. A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J. Biol. Chem. 275:19250–19257. [DOI] [PubMed] [Google Scholar]
- Tilly, B.C., M.J. Edixhoven, L.G. Tertoolen, N. Morii, Y. Saitoh, S. Narumiya, and H.R. de Jonge. 1996. Activation of the osmo-sensitive chloride conductance involves p21rho and is accompanied by a transient reorganization of the f-actin cytoskeleton. Mol. Biol. Cell. 7:1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng, G.N. 1992. Cell swelling increases membrane conductance of canine cardiac cells: evidence for a volume-sensitive Cl channel. Am. J. Physiol. 262:C1056–C1068. [DOI] [PubMed] [Google Scholar]
- Ubl, J., H. Murer, and H.A. Kolb. 1988. Ion channels activated by osmotic and mechanical stress in membranes of opossum kidney cells. J. Membr. Biol. 104:223–232. [DOI] [PubMed] [Google Scholar]
- Upton, R.L., Y. Chen, S. Mumby, J.M. Gutteridge, P.B. Anning, A.G. Nicholson, T.W. Evans, and G.J. Quinian. 2003. Variable tissue expression of transferrin receptors: relevance to acute respiratory distress syndrome. Eur. Respir. J. 22:335–341. [DOI] [PubMed] [Google Scholar]
- Valverde, M.A., G.M. Mintenig, and F.V. Sepulveda. 1993. Differential effects of tamoxifen and I− on three distinguishable chloride currents activated in T84 intestinal cells. Pflugers Arch. 425:552–554. [DOI] [PubMed] [Google Scholar]
- Vandenberg, J.I., A. Yoshida, K. Kirk, and T. Powell. 1994. Swelling-activated and isoprenaline-activated chloride currents in guinea pig cardiac myocytes have distinct electrophysiology and pharmacology. J. Gen. Physiol. 104:997–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg, J.I., S.A. Rees, A.R. Wright, and T. Powell. 1996. Cell swelling and ion transport pathways in cardiac myocytes. Cardiovasc. Res. 32:85–97. [PubMed] [Google Scholar]
- Voets, T., V. Manolopoulos, J. Eggermont, C. Ellory, G. Droogmans, and B. Nilius. 1998. Regulation of a swelling-activated chloride current in bovine endothelium by protein tyrosine phosphorylation and G proteins. J. Physiol. 506:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N., J.P. Butler, and D.E. Ingber. 1993. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 260:1124–1127. [DOI] [PubMed] [Google Scholar]
- Wu, X., J.E. Mogford, S.H. Platts, G.E. Davis, G.A. Meininger, and M.J. Davis. 1998. Modulation of calcium current in arteriolar smooth muscle by αvβ3 and α5β1 integrin ligands. J. Cell Biol. 143:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X., G.E. Davis, G.A. Meininger, E. Wilson, and M.J. Davis. 2001. Regulation of the L-type calcium channel by α5β1 integrin requires signaling between focal adhesion proteins. J. Biol. Chem. 276:30285–30292. [DOI] [PubMed] [Google Scholar]
- Wu, Z., K. Wong, M. Glogauer, R.P. Ellen, and C.A.G. McCulloch. 1999. Regulation of stretch-activated intracellular calcium transients by actin filaments. Biochem. Biophys. Res. Commun. 261:419–425. [DOI] [PubMed] [Google Scholar]
- Zeng, T., G.C.L. Bett, and F. Sachs. 2000. Stretch-activated whole cell currents in adult rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 278:H548–H557. [DOI] [PubMed] [Google Scholar]
- Zhang, Y.H., J.B. Youm, H.K. Sung, S.H. Lee, S.Y. Ryu, S.H. Lee, W.K. Ho, and Y.E. Earm. 2000. Stretch-activated and background non-selective cation channels in rat atrial myocytes. J. Physiol. 523:607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhidkova, N.I., A.M. Belkin, and R. Mayne. 1995. Novel isoform of beta 1 integrin expressed in skeletal and cardiac muscle. Biochem. Biophys. Res. Commun. 214:279–285. [DOI] [PubMed] [Google Scholar]
- Zhu, X., J.L. Kim, J.R. Newcomb, P.E. Rose, D.R. Stover, L.M. Toledo, H. Zhao, and K.A. Morgenstern. 1999. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure Fold. Des. 7:651–661. [DOI] [PubMed] [Google Scholar]
- Zygmunt, A.C. 1994. Intracellular calcium activates a chloride current in canine ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 267:H1984–H1995. [DOI] [PubMed] [Google Scholar]
- Zygmunt, A.C., and W.R. Gibbons. 1991. Calcium-activated chloride current in rabbit ventricular myocytes. Circ. Res. 68:424–437. [DOI] [PubMed] [Google Scholar]