

Abstract
The pore properties and the reciprocal interactions between permeant ions and the gating of KCNQ channels are poorly understood. Here we used external barium to investigate the permeation characteristics of homomeric KCNQ1 channels. We assessed the Ba2+ binding kinetics and the concentration and voltage dependence of Ba2+ steady-state block. Our results indicate that extracellular Ba2+ exerts a series of complex effects, including a voltage-dependent pore blockade as well as unique gating alterations. External barium interacts with the permeation pathway of KCNQ1 at two discrete and nonsequential sites. (a) A slow deep Ba2+ site that occludes the channel pore and could be simulated by a model of voltage-dependent block. (b) A fast superficial Ba2+ site that barely contributes to channel block and mostly affects channel gating by shifting rightward the voltage dependence of activation, slowing activation, speeding up deactivation kinetics, and inhibiting channel inactivation. A model of voltage-dependent block cannot predict the complex impact of Ba2+ on channel gating in low external K+ solutions. Ba2+ binding to this superficial site likely modifies the gating transitions states of KCNQ1. Both sites appear to reside in the permeation pathway as high external K+ attenuates Ba2+ inhibition of channel conductance and abolishes its impact on channel gating. Our data suggest that despite the high degree of homology of the pore region among the various K+ channels, KCNQ1 channels display significant structural and functional uniqueness.
Keywords: permeation, ion channels, outer vestibule, inactivation, channel gating
INTRODUCTION
The KCNQ family of voltage-gated K+ channels comprises five members (KCNQ1-5) that are expressed in a wide variety of tissues and play a major role in brain and cardiac excitability (Jentsch, 2000; Robbins, 2001). The channel complex formed by the KCNQ2/Q3 α subunits produces the M-current, a slowly activating, noninactivating K+ current that has profound effects on brain excitability and acts as a brake for repetitive neuronal firing (Wang et al., 1998; Brown and Yu, 2000; Jentsch, 2000; Rogawski, 2000). The KCNQ1 α subunits coassemble with various KCNE proteins, an emerging family of auxiliary β subunits, to produce functionally distinct K+ currents (Abbott and Goldstein, 1998; Melman et al., 2002). The coassembly of KCNQ1 and KCNE1 produces the IKS potassium current that is critical for the late repolarization of the cardiac action potential (Barhanin et al., 1996; Sanguinetti et al., 1996). Consistent with their physiological importance, mutations of human KCNQ genes lead to major cardiovascular and neurological disorders such as the cardiac long QT syndrome or neonatal epilepsy (Lehmann-horn and Jurkat-Rott, 1999; Ashcroft, 2000; Brown and Yu, 2000; Jentsch, 2000; Lerche et al., 2001; Robbins, 2001).
Like all voltage-gated K+ channel α subunits, KCNQ proteins share a common core structure of six transmembrane segments and a P-loop forming the pore region of the channel (Yellen, 2002). They are built from two parts: (a) a voltage-sensing domain, each comprised of S1-S4 that is responsible for controlling gating behavior and for energy transduction; and (b) a pore domain (S5-P-S6) whose structure is likely to be similar to the crystal structures of the bacterial KcsA and MthK K+ channels, with the inner helix S6 lining much of the pore (Doyle et al., 1998; Jiang et al., 2002). Coexpression of KCNQ1 with KCNE1 produces voltage- and time-dependent K+ currents with very slow activation kinetics and with a positive shift in the voltage dependence of activation, in comparison to homomeric KCNQ1 channels (Barhanin et al., 1996; Sanguinetti et al., 1996). There is also an increase in unitary channel conductance, leading to increased macroscopic current amplitude (Pusch, 1998; Sesti and Goldstein, 1998; Yang and Sigworth, 1998). When associated with KCNE3, KCNQ1 channels yield K+ currents that are nearly instantaneous and voltage independent (Schroeder et al., 2000).
Homomeric KCNQ1 channels elicit a delayed-rectifier K+ current that undergoes inactivation (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). Inactivation of homomeric KCNQ1 is invisible from macroscopic inspection and could be revealed by a hook of the tail currents. The onset of KCNQ1 inactivation occurs with an apparent delay and voltage dependence and is markedly prevented by the presence of KCNE1 (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). Though the mechanism of KCNQ1 inactivation is unknown, its biophysical characteristics are different from classical N and C-type inactivation of Shaker-like potassium channels, where K+ ions modulate C-type inactivation at an external pore site and N-type inactivation involves a direct pore occlusion at an internal site (Yellen, 2002).
Barium ions have been widely used to probe the permeation mechanisms of a variety of K+ channels. The similar crystal radius of Ba2+ compared with K+ and its stronger charge allows it to fit tightly into the deep selectivity filter (Jiang and MacKinnon, 2000). When applied from the inside, Ba2+ blocks delayed-rectifiers at a site that is accessible only when the channel is open (Armstrong and Taylor, 1980; Eaton and Brodwick, 1980). In contrast, external Ba2+ block can occur both in the open and closed channel states (Hurst et al., 1995; Harris et al., 1998).
The pore properties and the reciprocal interactions between permeant ions and the gating of the KCNQ channel family are poorly understood. Here we used external barium as a probe to address this issue in homomeric KCNQ1 channels. We determined the Ba2+ binding kinetics and the concentration and voltage dependence of Ba2+ steady-state block. Our results indicate that external barium interacts with the permeation apparatus of KCNQ1 at two main sites: (a) A deep Ba2+ site that produces an occlusion of channel conductance and could be simulated by a model of voltage-dependent block; and (b) a superficial Ba2+ site that has a minor impact on channel block and mainly affects KCNQ1 channel gating by shifting rightward the voltage dependence of activation, slowing activation, speeding up deactivation kinetics, and inhibiting channel inactivation. While high external K+ (50 mM) attenuates the inhibitory effect of Ba2+ on channel conductance, it abolishes its impact on channel gating. The strong impact of Ba2+ on KCNQ1 channel gating could not be described by a simple model of voltage-dependent block.
MATERIALS AND METHODS
Channel Expression into Xenopus Oocytes
Female Xenopus laevis frogs were purchased from Xenopus 1 (Dexter). The procedures followed for surgery and maintenance of frogs were approved by the animal research ethics committee of Tel Aviv University and in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, Washington DC). Frogs were anesthetized with 0.15% tricaine (Sigma-Aldrich). Pieces of the ovary were surgically removed and digested with 1 mg/ml collagenase (type IA, Sigma-Aldrich) in Ca2+-free ND96 for about 1 h, to remove follicular cells. Stage V and VI oocytes were selected for cRNA or DNA injection and maintained at 18°C in ND96 (in mM: 96 NaCl, 2 KCl, 1.8 mM CaCl2, 1 MgCl2, and 5 HEPES titrated to pH = 7.5 with NaOH), supplemented with 1mM pyruvate and 50 μg/ml gentamycin. The human KCNQ1 cDNA (in pGEM vector) was linearized by NotI. Capped complementary RNA was transcribed by the T7 RNA polymerase, using the mMessage mMachine transcription kit (Ambion Corp). The cRNA size and integrity was confirmed by formaldehyde-agarose gel electrophoresis. Homomeric expression of human KCNQ1 was performed by injecting 40 nl per oocyte (5 ng cRNA) using a Nanoject injector (Drummond). Several expression experiments were also performed by microinjecting a recombinant DNA vector (pcDNA3) encoding the human KCNQ1 cDNA directly into Xenopus oocyte nuclei (1 ng into 10 nl). Very similar data were obtained for either cRNA or DNA injections.
Electrophysiology
Standard two-electrode voltage-clamp measurements were performed at room temperature (22–24°C) 2–5 d after cRNA or DNA microinjection. Oocytes were placed into a 100-μl recording chamber and superfused with a modified ND96 solution (containing 0.1 mM CaCl2) under constant perfusion using a fast perfusion system at a rate of 0.48 ml/s (ALA VM8, ALA Scientific Instruments). Whole-cell currents were recorded using a GeneClamp 500 amplifier (Axon Instruments, Inc.). Stimulation of the preparation, and data acquisition were performed using the pCLAMP 6.02 software (Axon Instruments, Inc.) and a 586 personal computer interfaced with a Digidata 1200 interface (Axon Instruments, Inc.). Glass microelectrodes (A-M systems, Inc.) were filled with 3M KCl and had tip resistances of 0.2–1 MΩ. Current signals were digitized at 1 kHz and low pass filtered at 0.2 kHz. The holding potential was −80 mV. Leak subtraction was performed off-line, using steps from −120 to −90 mV, assuming that the channels are closed at −80 mV and below. Errors introduced by the series resistance of the oocytes were not corrected and were minimized by keeping expression of the currents below 10 μA. All BaCl2 solutions were prepared in modified ND96 (containing 0.1mM CaCl2) and were iso-osmotically changed for NaCl. Modified ND96 solutions containing high K+ concentrations (50 mM) were also iso-osmotically changed for NaCl.
Data Analyses
Data analysis was performed using the Clampfit program (pCLAMP 8; Axon Instruments, Inc.), Microsoft Excel 2002 (Microsoft Corporation), SigmaPlot 8.0 (SPSS, Inc.), and Prism (GraphPad software). To analyze the voltage dependence of channel activation, a double exponential fit was applied to the tail currents at −60 or −120 mV and the slow exponential component was extrapolated to the beginning of the repolarizing step. Chord conductance (G) was calculated by using the following equation:
 |
where I corresponds to the extrapolated tail current and Vrev to the reversal potential measured in each experiment and which was −98 ± 2 mV (n = 10). G was estimated at the tail voltage V and then, normalized to a maximal conductance value, Gmax. Activation curves were fitted to a Boltzmann distribution:
 |
where V50 is the voltage at which the current is half-activated and s is the slope factor. For a measure of inactivation kinetics, we used a three-pulse protocol (see Fig. 6 C) where the membrane potential was stepped to a +30-mV conditioning prepulse for increasing durations (200-ms increments) to progressively activate and inactivate the channel; then, a brief (15 ms) hyperpolarizing interpulse to −130 mV was used to allow channel recovery from inactivation before a +30 mV test pulse was applied to reopen and reinactivate KCNQ1 channels. The decaying current of the third test pulse (reinduction of inactivation) could be fitted by a single exponential function. For a quantitative measure of fractional inactivation that takes into account the electrochemical driving force, G was deduced from tail currents at either −60 or −120 mV, where tail relaxations reveal a recovery from inactivation (hook) faster than deactivation. Currents recorded at the voltage V were first converted to conductance G. The time course of the tail conductance was then fitted to a triple exponential function of the form:
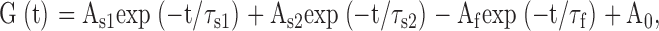 |
where a fast time constant τf reflects the recovery from inactivation at the tail potential and two slow time constants τs1 and τs2 describe the deactivation process and A0 is a steady-state current. At the tail potential, the amplitude Af is directly related to the degree of inactivation and the ratio Af/(As1 + As2) is a good indicator of the probability of the channels being in the inactivated state.
Figure 6.

Effect of Ba2+ on activation gating. (A) Activation kinetics at +30 mV were fitted by a double exponential function, with a fast (left) and slow time constant (right), plotted as a function of external Ba2+ concentrations (n = 10). A sigmoidal dose–response function yielded an EC50 = 0.44 mM for the fast time constant. (B) The fast (left) and slow (right) activation time constant (n = 11) were plotted against voltage steps and were measured in the absence (empty symbols) or the presence of 10mM Ba2+ (solid squares). (C) Representative normalized traces of current recorded at +30 mV before (control) and after application of 10 mM Ba2+. (D) The normalized conductance, deduced from the tail currents was plotted as a function of step voltage for control (closed squares), 0.5 mM Ba2+ (solid triangles), 2 mM Ba2+ (closed circles), and 10 mM Ba2+ (empty triangles). The normalized conductance curves were fitted using a Boltzmann function.
The voltage dependence of Ba2+ block was calculated according to the Woodhull's model (Woodhull, 1973) and the voltage dependence of the dissociation constant is given by:
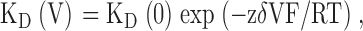 |
where KD(0) is the dissociation constant at zero voltage, z is the ion valency, F is the Faraday constant, δ is the fractional potential drop at the binding site (fractional electrical distance), V is the membrane potential (in volts), R is the gas constant, and T is the absolute temperature. All data were expressed as mean ± SEM. Statistically significant differences between paired groups were assessed by Student's t test.
Model
Analysis in the absence of barium.
The time course of channel activation could be described as a Markov process (Pusch et al., 1998) and was fitted to the following scheme:
in which ks are the rate constants in s−1 and C0 to I are the corresponding state occupancies. Only O1 and O2 are assumed to conduct. Scheme I is sufficient for the description of macroscopic behavior of KCNQ1 channels (Pusch et al., 1998), assuming that the stochastic behavior of a single channel is reflected in the macroscopic behavior of a channel population (Blunck et al., 1998). We generated a set of differential equations based on Scheme I and fitted it to the observed time-course of channel activation in order to obtain the transition rate constants and relative occupancies of each state. For simplicity, we assumed that no openings occur when the cell is voltage-clamped to −80 mV (the holding potential) and consequently we fixed initial values of all states to zero, except C0.
SCHEME I.

According to Scheme I, the open channel probability can be calculated from Eq.1:
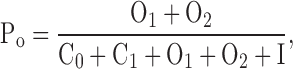 |
(1) |
and the relative state occupancy (φ) is defined as:
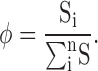 |
(2) |
We fitted the activation time course from the holding potential (−80 mV) to the +30-mV voltage step. We fixed the k4 and k−4 values (which are assumed to be voltage-independent) and also the initial C0 to those obtained from the previous procedure, and then fitted the activation from the holding potential (−80 mV) to the −60-mV voltage step. This analysis was performed for KCNQ1 expressing oocytes that were superfused with low (2 mM) or high (50 mM) extracellular K+ containing ND96 solutions in the absence of barium.
To validate the model, we used the fit data and simulated the tail envelope protocol. For the simulation, we used the averaged values of the rate constants and the relative state occupancies obtained from the fit of the activation time course during the +30-mV voltage step and allowed the simulation to run for predefined time intervals. Thereafter, we used the simulated values and changed the rate constants to those corresponding to the −60-mV voltage step. Based on the simulation of the different prepulse intervals, we generated a curve describing the dependence of the “hook” amplitude on the prepulse time and compared it to experimental data.
Analysis of Ba2+ Block in High External K+ Solutions
For simulating Ba2+ block in high external K+ solutions, we used a simplistic scheme of voltage-dependent block as illustrated in Scheme II .
SCHEME II.

We assumed that the conductance block originates from the existence of one Ba2+-occluding site. Thus, the channel can populate two different kinds of states, assumed to be in equilibrium: a Ba2+-blocked state (B) and a Ba2+-unblocked state. For simplicity, we assumed that all states (C0 to I) bind Ba2+ with equal affinity. Indeed, the affinity of the closed states cannot be estimated from our records. However, the on-kinetics of Ba2+ are clearly dominated by a slow component that allows us to estimate Ba2+ affinity for the open states. To preserve microscopic reversibility, we also assumed that transitions through the blocked states occur with similar rates to their unblocked counterparts. Further analyzing the equilibrium, we defined f, the fractional inhibition as:
 |
(3) |
in which Ihk and Ihk,Ba are the steady-state amplitudes of the recorded currents before and after Ba2+ application (in 50 mM external K+). Consequently, according to the mass action law:
 |
(4) |
in which Su corresponds to unblocked state and Sb to the blocked state, while KD is the dissociation constant of Ba2+. Combining Eqs. 3 and 4 and rearranging gives:
 |
(5) |
thus enabling us to estimate the KD value.
On the other hand, according to the properties of bimolecular reactions:
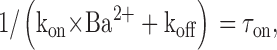 |
(6) |
in which τon is the blocking time constant obtained from experiments. Combining Eqs. 5 and 6 allows us to estimate the rates of block and unblock.
Time-course fitting and simulation were done with Berkeley Madonna software (Version 8.0.2 for Windows; Kagi Shareware).
Online Supplemental Material
A figure illustrating the lack of effects of 10 mM MgCl2 on KCNQ1 currents, with representative current traces, current-voltage relations, normalized conductance curves, quantitative measures of inactivation, and deactivation.
A table summarizing the rate constants of KCNQ1 activation in low and high external K+ (without Ba2+) obtained from the fit to Scheme I and the rate constants of KCNQ1 activation in high external K+ in the presence of Ba2+ obtained from the fit to Scheme II. Online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.200409068/DC1.
RESULTS
Kinetics of External Barium Block
In low external K+ (2 mM), extracellular Ba2+ produced a time-dependent inhibition of homomeric KCNQ1 K+ currents when expressed in Xenopus oocytes. As shown in Fig. 1 A, Ba2+ potently inhibited the K+ currents when applied to opened channels. From a holding potential of -80 mV, the KCNQ1 channels were opened by step depolarization and the oocyte was held constantly at this depolarizing potential until the end of the kinetic experiment. After 4 s of channel opening, Ba2+ was applied for 20 s and thereafter washed out for 16 s at the same depolarizing potential (e.g., at −20 mV in Fig. 1 A). It is clear from the wash-in experiments that Ba2+ on-binding displayed a fast and slow kinetic component (Fig. 1, A–C). For example, 10 mM Ba2+ blocked at −20 mV the KCNQ1 K+ currents with fast and slow time constants τf = 220 ± 43 ms and τs = 1,073 ± 128 ms, respectively (n = 8) (Fig. 1 B). This suggested that Ba2+ binds to two sites. However, the fast kinetic component was too close to the rate of exchange of the bath to determine accurately its time course. This probably explains why the fast time constant did not vary significantly as a function of Ba2+ concentration (Fig. 1 B). The slow time constant increased markedly with depolarized potentials, illustrating the voltage dependence of Ba2+ binding to its slow site (Fig. 2 B). According to the Woodhull model, it was possible to calculate the electrical distance of the slow Ba2+ site and to show that it lies deep in the membrane electric field with δ = 0.32 (Woodhull, 1973). Consistent with a voltage-dependent block at the slow Ba2+ site, the relative amplitude of the slow binding component decreased significantly with depolarized potentials from 0.45 at −30 mV to 0.21 at +20 mV (see also Fig. 3 B). The slow time constant τs (consequently, the slow blocking rate 1/τs) reached saturation at concentrations above 2 mM Ba2+ (Fig. 1 B). This was also accompanied by a saturation of the relative amplitude of the slow blocking component that drops from 0.81 at 0.5 mM Ba2+ to 0.66 at 2 mM Ba2+ and levels off at 0.55 at higher Ba2+ concentrations (unpublished data). This saturation suggests that barium binding does not follow a simple bimolecular reaction with just one Ba2+ binding site.
Figure 1.

Concentration dependence of Ba2+ wash-in and washout kinetics. The kinetic experiments were performed with various extracellular Ba2+ concentrations at −20 mV in low external K+ solutions (2 mM). (A) Representative normalized traces are shown. From a holding potential of −80 mV, the KCNQ1 channels were opened by a −20-mV depolarizing step. After 4 s of channel opening, 0.5 mM or 10 mM Ba2+ was applied for 20 s and thereafter washed out for 16 s at the same depolarizing potential (−20 mV). (B) Ba2+ wash-in kinetics were fitted by a double exponential function with a fast (solid triangles) and slow (solid squares) time constants plotted as a function of Ba2+ concentrations (n = 8). (C) Ba2+ washout kinetics were fitted by a double exponential function with a fast (empty triangles) and slow (empty squares) time constants plotted as a function of Ba2+ concentrations (n = 8).
Figure 2.

Voltage dependence of Ba2+ wash-in and washout kinetics. The kinetic experiments were performed with 10 mM Ba2+ at various depolarizing voltages in low external K+ solutions (2 mM). (A) Representative normalized traces are shown. From a holding potential of −80 mV, the KCNQ1 channels were opened at −30 or +20 mV. After 4 s of channel opening, 10 mM Ba2+ was applied for 20 s and thereafter washed out for 16 s at the same depolarizing potential. (B) Ba2+ wash-in kinetics were fitted by a double exponential function with a fast (solid triangles) and slow (solid squares) time constants plotted as a function of depolarizing voltages (n = 5). (C) Ba2+ washout kinetics were fitted by a double exponential function with a fast (empty triangles) and slow (empty squares) time constants plotted as a function of depolarizing voltages (n = 5).
Figure 3.

Comparison of Ba2+ wash-in kinetics in low and high external K+ solutions. (A) Representative normalized traces of Ba2+ wash-in kinetics performed on the same oocyte in 2 mM [K+]0 and in 50 mM [K+]0. From a holding potential of −80 mV, the KCNQ1 channels were opened by a +30-mV depolarizing step. After 4 s of channel opening, 10 mM Ba2+ was applied for 20 s and thereafter washed out for 16 s at the same depolarizing potential (+30 mV). (B) Relative amplitudes of Ba2+ wash-in fast (empty symbols) and slow (solid symbols) kinetic components in 2 mM [K+]0 (squares) and in 50 mM [K+]0 (triangles) plotted as a function of membrane voltage.
Barium also blocked KCNQ1 channels in the closed state. As already shown for Shaker channels (Hurst et al., 1995; Harris et al., 1998), external Ba2+ entered the pore of KCNQ1 at negative potentials where the channel is closed and remained bound to the deep slow site after washout, as recorded by a subsequent depolarizing step to open the gate and measure the KCNQ1 current block. While holding the membrane potential at −100 mV, 10 mM Ba2+ was extracellularly applied to the oocyte for 2 min (∼70 times the slow time constant of Ba2+ wash-in at −30 mV), after which barium was washed out for 2 min (at −100 mV); then a single depolarizing step to +30 mV was applied to assay the level of current block (Fig. 4, A and B) . Under these conditions, the KCNQ1 current was blocked by 51 ± 7% (n = 6) at the beginning of the depolarizing step (at 200 ms) and by 31 ± 3% (n = 6) at the end of the pulse (at 2,000 ms), thus providing strong evidence that barium could block KCNQ1 channels in the closed state. The slower rise in KCNQ1 current after Ba2+ exposure, as well as the progressive decrease in block during the depolarizing step, reflects the slow Ba2+ unbinding at +30 mV (Fig. 4, A and B). Though this protocol of Ba2+ binding and washout at −100 mV kept Ba2+ bound to its deep slow site, it removed Ba2+ from its fast external site and thus prevented its impact on channel gating as we shall see later (see Fig. 4 and below). Holding constantly the membrane potential at +30 mV, we let Ba2+ dissociate from the open state by washing out the external solution. Results reveal that Ba2+ off-kinetics also comprised a fast and slow component (Figs. 1 and 2). Various concentrations of Ba2+ were applied to open channels at −20 mV until it reached steady-state inhibition after which Ba2+ dissociation was measured. Ba2+ wash-out exhibited significant dependence on blocker concentration with the fast and slow time constants varying, respectively, from τf = 0.63 ± 0.07 s and τs = 3.65 ± 0.33 s at 0.2 mM Ba2+ to τf = 1.85 ± 0.16 s and τs = 8.70 ± 0.47 s at 10 mM (n = 8) (Fig. 1, A and C). The wash-out rate does not only reflect the unbinding rate. Rather, it is the sum of binding and unbinding rate. Therefore, it should be expected to depend on Ba2+ concentration. Ba2+ dissociated faster with increasing depolarization. The fast time constant of Ba2+ unbinding displayed marked dependence on voltage, with τf = 1.66 ± 0.20 s at −20 mV and τf = 0.74 ± 0.06 s at +30 mV (n = 5) (Fig. 2, A and C).
Figure 4.

Effect of barium exposure to closed KCNQ1 channels. (A) Representative traces where from a −100 mV holding potential, KCNQ1 channels were opened for 2 s at +30 mV and repolarized for 0.5 s to a −60-mV tail potential (left trace, before Ba2+); then, 10 mM Ba2+ was bound for 2 min to closed channels at −100 mV and subsequently washed out for 2 min at −100 mV, before channels were opened for 2 s at +30 mV as above (right trace, after 10 mM Ba2+). (B) Superimposed traces showing KCNQ1 current before Ba2+ exposure (black trace), after 10 mM Ba2+ (red trace), and the subtracted trace (green) corresponding to the Ba2+-inhibited current. (C) Fractional inactivation measured from the tail currents at −60 mV, before (empty bars) and after 10-mM Ba2+ application (solid bars) as described in A (n = 8). (D) Fast and slow deactivation time constants measured from the tail currents at −60 mV, before (empty bars) and after 10-mM Ba2+ application (solid bars) as described in A (n = 8).
Steady-state Barium Block
Various concentrations of extracellular Ba2+ were applied and current-voltage relations were constructed at steady-state, allowing normalized activation curves to be deduced from the tail currents. In 2 mM [K+]0, extracellular Ba2+ exerted on homomeric KCNQ1 channels a series of complex effects, including a voltage-dependent pore blockade as well as gating alterations (Fig. 5 A). Like with many K+ channels, Ba2+ block of KCNQ1 currents was voltage-dependent, with weaker inhibition at depolarized potentials (Figs. 5 B and 11 B). For example, 2 mM Ba2+ produced a 90% and 51% block at −50 mV and +30 mV, respectively (Fig. 5 B). The slopes of Ba2+ dose–response curves were shallow at depolarized potentials, suggesting that Ba2+ bound to more than one site. Between −70 and −30 mV, the Ba2+ inhibitory curves could be fitted to one site with apparent KD values of ∼0.30–0.35 mM; however, the Hill coefficients were ranged from 2.8 to 2.1 (at −70 to −40 mV), suggesting the existence of at least two Ba2+ sites of very similar affinity at these negative potentials (Fig. 5 C). At more positive voltages from −20 to +30 mV, the curves became very shallow and needed to be fitted to two binding sites (Fig. 5 D). While the high-affinity site was not sensitive to membrane voltage changes, the low-affinity site was voltage dependent (δ = 0.45) with increasing KD values (from 0.67 to 6.4 mM) at increasing depolarized potentials (from −20 to +30 mV). Though being a rough approximation, application of the Woodhull's model for only one binding site did not fit properly the apparent KD values over all potentials tested (Fig. 5 E). We had to split and fit the Ba2+ KD values into two voltage regions. Between −70 and −20 mV, we got a shallow voltage dependence with a low fractional electrical distance δ1 = 0.06, whereas between −20 and +30 mV there was a steeper sensing of the membrane electric field with a higher electrical distance δ2 = 0.33 (Fig. 5 E). This latter value is in reasonable agreement with the δ value calculated above from the fit of two Ba2+ binding sites and with that of the slow Ba2+ site (0.32), derived from the on-kinetic experiments. As we shall also see below, these results suggest the existence of two Ba2+ sites, a fast superficial binding site that senses weakly the electric field and a slow Ba2+ binding site that lies deep in the membrane electric field.
Figure 5.

Barium produces a concentration- and voltage-dependent channel block. (A) Representative KCNQ1 current traces recorded in the absence (control) or presence of 10 mM Ba2+. From a holding potential of −80 mV, oocytes were stepped for 3 s from −70 to +30 mV in 10-mV increments and then repolarized for 1.5 s at −60-mV tail potential. (B) Current-voltage relations (n = 7) of control (solid squares), 0.5 mM Ba2+ (solid triangles), 2 mM Ba2+ (solid circles), and 10 mM Ba2+ (empty triangles). (C) Ba2+ inhibitory curves expressed as fractional channel unblock versus log10 of Ba2+ concentrations and measured at −70 mV (closed squares), −60 mV (upward closed triangles), −50 mV (downward closed triangles), −40 mV (closed diamonds), and −30 mV (closed circles). The KD values were obtained from the fit of a sigmoidal dose–response function (n = 7). (D) Ba2+ inhibitory curves expressed as in C and measured at −20 mV (open squares), −10 mV (upward open triangles), 0 mV (downward open triangles), +10 mV (open diamonds), +20 mV (open circles), and +30 mV (X labels). The KD values were obtained from the fit of a two site competition function. (E) Semilogarithmic plot of the KD values obtained from one site fit of the Ba2+ inhibitory curves versus membrane voltage (n = 7).
In addition to its voltage-dependent block of the pore, Ba2+ also altered the gating of homomeric KCNQ1 channels, including slowing of the activation kinetics, rightward shift in the voltage dependence of activation, inhibition of channel inactivation and acceleration of deactivation kinetics. Ba2+ produced a +34-mV gating shift in the voltage dependence of activation with V50 = −26.0 ± 0.6 mV (n = 7) and V50 = +8.0 ± 1.3 mV (n = 7) in the absence and presence of 10 mM Ba2+, respectively (Table I , Fig. 6 D). This rightward shift was dose-dependent with an EC50 of 0.85 mM Ba2+ as deduced from the plot of the V50 shift as a function of Ba2+ concentration (Table I). Barium produced a marked slowing of KCNQ1 activation kinetics that is well seen at all potentials (Figs. 5 A and 6). Activation kinetics were best fitted by a double exponential function, with a fast and slow component. Ba2+ increased by ∼3.5-fold the fast time constant, from τf = 88 ± 6 ms to τf = 312 ± 49 ms at +30 mV in the absence and presence of 4 mM Ba2+, respectively (n = 9, P < 0.01; Fig. 6 A). The increase of the fast time constant displayed an EC50 of 0.44 mM Ba2+ at +30 mV (Fig. 6 A) and the slowing of KCNQ1 activation was so pronounced that at 10 mM Ba2+, the fast kinetic component disappeared, leaving only one slow kinetic component (Fig. 6, B and C). The slowing of activation kinetics seems to arise mainly from a gating effect rather than from a slow Ba2+ dissociation at depolarized potentials. First, as already described in the protocol where 10 mM Ba2+ was bound and washed at −100 mV, a channel block was obtained (Fig. 4, A and B). However, only a mild slowing of the activation kinetics was found, which contrasts to the marked slowing found for the same Ba2+ concentration in steady-state experiments (see Figs. 5 A, 6 C, and 4). Second, high external K+ (50 mM) prevented the slowing of KCNQ1 activation produced by 10 mM Ba2+, though Ba2+ washout kinetics were even slower in 50 mM [K+]0 than in 2 mM [K+]0 (Fig. 10 A, Table II and see below). Figs. 7 and 8 depict the effects of external Ba2+ on KCNQ1 deactivation kinetics that could be fitted by a double exponential function. Ba2+ dose-dependently speeded up deactivation, especially at tail potentials comprised from −30 to −70 mV, with an EC50 of 1.4 mM Ba2+ as measured for τfast at −60 mV (Figs. 7 B and 8 A). The acceleration of deactivation was so marked at 10 mM Ba2+ (∼10-fold) that a slow kinetic component could not be anymore measured at −60 mV (Fig. 7, A and B). As shown in Figs. 7 A and 8 A, the speeding effect of Ba2+ on deactivation was weaker at very negative tail potentials (compare tails at −60 and −140 mV). The Ba2+-induced acceleration of tail currents appears to involve a gating effect rather than a rapid block at repolarizing potentials (voltage-dependent block) as speed-up of deactivation kinetics is less prominent at more negative potentials (−110–140 mV), where Ba2+ produces a very potent block (∼70% and 90% inhibition at −60 and −140 mV, respectively). In addition, the impact of Ba2+ on deactivation could be prevented by high external K+ (50 mM) (see Fig. 9 A), a feature similar to the effect of Ba2+ on activation kinetics.
TABLE I.
Voltage Activation Parameters of KCNQ1 Channels in the Presence of Barium
| Slope | V50 | |
|---|---|---|
| mV | mV | |
| Control | 16.6 ± 0.6 | −26.0 ± 0.6 (18) |
| 0.2 mM Ba2+ | 22.1 ± 2.5 | −22.6 ± 1.8 (6) |
| 0.5 mM Ba2+ | 18.5 ± 1.5 | −12.8 ± 1.3 (8) |
| 1 mM Ba2+ | 15.4 ± 1.3 | −5.0 ± 1.4 (8) |
| 2 mM Ba2+ | 16.3 ± 1.2 | −3.2 ± 1.3 (7) |
| 4 mM Ba2+ | 15.2 ± 1.3 | 1.5 ± 1.7 (6) |
| 10 mM Ba2+ | 13.9 ± 0.8 | 8.0 ± 1.3 (7) |
KCNQ1 channels were exposed to the indicated external Ba2+ concentrations until reaching steady-state and then the G-V curves were constructed from the tail currents, normalized, and fitted by a Boltzmann function. The numbers in parentheses represent the number of cells.
Figure 10.

High external K+ relieves the impact of Ba2+ on KCNQ1 activation, inactivation, and deactivation gating. (A) Representative normalized traces of KCNQ1 activation measured at +30 mV before (control) and after application of 10mM Ba2+ in 2 mM [K+]0 (left), or 50 mM [K+]0 (right). (B) Representative normalized traces of KCNQ1 deactivation measured from a +30-mV prepulse at −120-mV tail potential before (control) and after 10 mM Ba2+ in 2 mM [K+]0 (left), or 50 mM [K+]0 (right). (C) Fractional KCNQ1 inactivation measured from the tail currents before (empty bars) and after application of 10 mM Ba2+ (filled bars) in 1 μM, 2 mM, and 50 mM external K+. Tail currents were recorded at −60 mV for 1 μM and 2 mM [K+]0 and at −120 mV for 50 mM [K+]0. The fractional inactivation was corrected for the change in driving force.
TABLE II.
Kinetic Parameters of Barium Binding to KCNQ1 Channels in High External K+ Solutions
| Wash-in | Wash-in | Washout | Washout | |
|---|---|---|---|---|
| Fast τ | Slow τ | Fast τ | Slow τ | |
| ms | ms | ms | ms | |
| 0 mV | 319 ± 37 | 6,472 ± 806 | 511 ± 49 | 11,810 ± 1,879 |
| 10 mV | 368 ± 58 | 7,615 ± 1,269 | 576 ± 88 | 13,774 ± 2,038 |
| 20 mV | 532 ± 149 | 8,173 ± 1,567 | 713 ± 145 | 11,958 ± 1,715 |
| 30 mV | 781 ± 276 | 12,047 ± 1,703 | 1,013 ± 437 | 13,134 ± 1,769 |
On- and off-binding kinetics of extracellular barium were measured in high external K+ solutions (50 mM) by applying 10 mM Ba2+ to KCNQ1 channels previously opened at the indicated potentials. Wash-in and washout kinetics were fitted by a double exponential function and the fast and slow time constants are indicated (mean ± SEM, n = 5).
Figure 7.

Concentration-dependence of Ba2+ action on deactivation and inactivation gating. (A) Representative normalized traces of tail current recorded at −60 mV (left) or at −140 mV (right) after a +30-mV prepulse, before (control) and after application of 10 mM Ba2+. (B) The deactivation kinetics were recorded at −60 mV after a +30-mV prepulse. Tail currents were fitted by a double exponential function, with a fast (left) and slow time constant (right), plotted as a function of external Ba2+ concentrations (n = 7). A sigmoidal dose–response function yielded an EC50 = 1.4 ± 0.2 mM for the fast time constant (n = 7). (C) Dose-dependent inhibition of KCNQ1 inactivation by increasing concentrations of Ba2+ as measured by the hook of the tail currents at −60 mV (n = 7). A sigmoidal dose-response function yielded an EC50 = 0.94 ± 0.07 mM (n = 7).
Figure 8.

Effect of barium on time-dependent inactivation and voltage-dependent deactivation kinetics. (A) Representative tail currents obtained after a 1.5-s prepulse at +30 mV from −140 to −30 mV in 10-mV increments before (left) and after (right) application of 10 mM Ba2+. (B) The deactivation kinetics were determined in the absence (control) or presence of 10 mM Ba2+ at various tail potentials following a 1.5-s prepulse at +30 mV and were fitted by a double exponential function, with a fast (left) and slow (right) time constant (n = 5). (C) Representative current traces (n = 7) where the membrane potential was stepped to +30 mV for increasing durations by increments of 200 ms to progressively activate and inactivate the channel; then, a brief (15 ms) hyperpolarizing interpulse to −130 mV was used to allow channel recovery from inactivation before a +30-mV test pulse was applied to reopen and reinactivate KCNQ1 channels. The decaying current of the third test pulse (reinduction of inactivation) could be fitted by a single exponential function.
Figure 9.

Barium effects in high external K+ solutions. (A) Representative KCNQ1 current traces recorded in high external K+ (50 mM) solution, in the absence (left) or presence (right) of 10 mM Ba2+. From a holding potential of −80 mV, oocytes were stepped for 3 s from −70 to +30 mV in 10-mV increments and then repolarized for 1.5 s at −120-mV tail potential. (B) Current-voltage relations (n = 8) before (control, empty squares) and after application of 10 mM Ba2+ (solid squares) measured at steady-state in high external K+ (50 mM). (C) Current-voltage relations of the same experiments as in B (n = 8), before (control, empty squares) and after application of 10 mM Ba2+ (solid squares) measured at −120 mV tail potential in high external K+ (50 mM). (D) Current-voltage relations (n = 7), before (control, empty squares) and after application of 10 mM Ba2+ (solid squares) measured at −120-mV tail potential in low external K+ (2 mM). (E) The normalized conductance, deduced from the tail currents at −120 mV, was measured before (control, open squares) and after application of 10mM Ba2+ (solid squares) in 1 μM (left), 2 mM (middle), and 50 mM (right) external K+. The normalized conductance curves were fitted using a Boltzmann function.
Homomeric KCNQ1 channels were previously shown to undergo an inactivation process that occurs with an apparent delay and voltage dependence (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). This inactivation is invisible macroscopically but could be revealed by hooked tail currents whose ascending phase reflects recovery from channel inactivation. Kinetic analysis suggested that KCNQ1 could populate two distinct open states and a voltage-independent inactivation state (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). Extracellular Ba2+ dose-dependently depressed KCNQ1 inactivation as revealed by the progressive suppression of the hook in tail currents (Fig. 7, A and C). The fractional inactivation was measured and quantified from the hooked tail currents (see materials and methods and Fig. 7 C). When measured upon a 3-s test potential at +30 mV and from a −60-mV tail potential, 10 mM Ba2+ reduced by ∼61% the fractional inactivation of KCNQ1 from 0.54 ± 0.07 to 0.21 ± 0.03 (n = 7) with an EC50 = 0.94 ± 0.07 mM (Fig. 7 C). Barium virtually suppressed inactivation when measured by a triple pulse protocol (Fig. 8 C). In fact, KCNQ1 inactivation is an intrinsically voltage-independent and fast process with a τinact = 20 ± 3 ms (n = 7) (Fig. 8 C). The decaying current of the third test pulse which reflects the reinduction of inactivation is strongly depressed in the presence of 10 mM Ba2+ (Fig. 8 C).
External Magnesium Does Not Affect KCNQ1 Conductance and Gating
The strong impact of Ba2+ on KCNQ1 gating could in principle result from a surface screening charge effect. Ba2+ may screen diffuse negative charge and thus shift channel gating. Our experiments indicate that this is not the case, for two reasons. First, in case of a surface screening charge effect, one would expect other divalent cations to produce similar effects. External Mg2+ (10 mM) neither blocked KCNQ1 conductance nor affected channel gating (online supplemental material, available at http://www.jgp.org/cgi/content/full/jgp.200409068/DC1). Steady-state experiments indicate that current amplitude was not inhibited by 10 mM Mg2+. When measured at +30 mV, the steady-state current amplitude was 5.0 ± 0.8 μA (n = 6) and 6.1 ± 0.9 μA (n = 6) in the absence and presence of external Mg2+ (10 mM), respectively. In addition, external Mg2+ (10 mM) did not shift KCNQ1 activation curve (V50 = −18.3 ± 0.6 mV and V50 = −22.9 ± 0.6 mV with and without 10 mM Mg2+, respectively, n = 6). External Mg2+ (10 mM) did not affect channel inactivation (fractional inactivation f = 0.58 ± 0.09 and f = 0.55 ± 0.08 with and without 10 mM Mg2+, respectively, n = 6). The activation and deactivation kinetics were not altered by 10 mM Mg2+ (online supplemental material). Similarly, external Ca2+ (10 mM) neither blocked channel conductance nor affected KCNQ1 gating (unpublished data). Second, replacing partially external Na+ by K+ (50 mM) eliminated the impact of 10 mM Ba2+ on KCNQ1 gating, including activation and deactivation kinetics, inactivation and rightward shift of the activation curve (see below). Thus, these results do not stand with a surface screening charge hypothesis.
High External K+ Attenuates Ba2+ Block and Eliminates its Impact on Channel Gating
Previous studies have shown that elevating extracellular K+ can relieve the Ba2+ block of voltage-gated K+ currents, including those of Shaker channels (Armstrong and Taylor, 1980; Wollmuth, 1994; Hurst et al., 1995). To check whether the effects of extracellular Ba2+, including those related to gating, are mediated through its interaction with the permeation pathway and could be competed out by K+, we examined the Ba2+ action at high external K+ (50 mM [K+]0) (Figs. 9 and 10) . The voltage-dependent block of KCNQ1 current produced by external Ba2+ was significantly attenuated in 50 mM [K+]0, as measured in steady-state experiments. For example, at +30 mV 10 mM Ba2+ led to a 54 ± 2% block of KCNQ1 outward currents in 50 mM [K+]0, compared with a 72 ± 3% inhibition in 2 mM [K+]0 (P < 0.01, n = 10; Fig. 9, A and B and Fig. 11, A and B). Similarly, high external K+ attenuated the Ba2+-induced inhibition of inward tail currents when measured at +30-mV test pulse and −120-mV tail potential with a 58 ± 4% block in 50 mM [K+]0, compared with a 89 ± 6% block in 2 mM [K+]0 (P < 0.01, n = 6; Fig. 9, C and D). Like in low external K+, the Ba2+ block in 50 mM [K+]0 was voltage dependent with weaker inhibition at increasing depolarization from −10 mV to +30 mV (Fig. 11, A and B). Extracting the apparent Ba2+ KD values from the fractional current inhibition (see materials and methods and Fig. 11 C) allowed us to derive the electrical distance of the Ba2+ blocking site in high external K+ (50 mM) with δ = 0.28, a value similar to that found above for the Ba2+ slow site (δ = 0.32). In contrast to low external K+, the voltage dependence of the Ba2+ block in 50 mM [K+]0 was different at negative potentials from −40 to −60 mV, with a decreasing inhibition (Fig. 11 A). The increase in apparent KD values at these negative potentials probably reflects the propensity of Ba2+ to dissociate in the inward direction.
Figure 11.

Effect of membrane voltage on the fractional current inhibition produced by barium. (A) Fractional current inhibition produced by 10 mM Ba2+ in high external K+ (50 mM) and plotted as a function of step voltages. (B) Fractional current inhibition produced by 10 mM Ba2+ in low external K+ (2 mM) and plotted as in A. (C) Extraction of the Ba2+ KD values from the fractional current inhibition (see materials and methods) and plotted a function of step voltages.
Increasing external K+ did not only attenuate the voltage-dependent Ba2+ block but also eliminated all Ba2+ effects on channel gating. First, while at low external K+ (1 μM and 2 mM) Ba2+ virtually suppressed KCNQ1 inactivation, Ba2+ left KCNQ1 inactivation intact at 50 mM [K+]0, as revealed by the presence of the hook in tail currents (Fig. 9 A) with a fractional inactivation of ∼0.6 without and with Ba2+ (Fig. 10 C). It is also important to notice that in contrast to C-type inactivation, KCNQ1 inactivation is not affected by high external K+. Second, in 50 mM [K+]0 Ba2+ did not produce a significant gating shift in the voltage dependence of activation that is observed at 1 μM or 2 mM [K+]0 (Fig. 9 E). Ba2+ (10 mM) shifted by only +2.4 mV the KCNQ1 activation curve at 50 mM [K+]0, while it produced a gating shift of +28.2 and +34 mV at 1 μM [K+]0 and 2 mM [K+]0, respectively. Third, high external K+ prevented the slowing of KCNQ1 activation kinetics produced by Ba2+ (Fig. 10 A). This relief did not result from an acceleration of Ba2+ dissociation in high external K+. On the contrary, Ba2+ washout kinetics were even slower in 50 mM [K+]0 compared with 2 mM [K+]0 (Table II). Fourth, elevation of external K+ from 2 to 50 mM prevented the accelerating effect of Ba2+ on deactivation kinetics (Fig. 10 B).
The electrical distance of the Ba2+ blocking site in high external K+ (δ = 0.28) is similar to that of the slow Ba2+ site (δ = 0.32) as deduced from kinetic experiments in low external K+. Thus, we hypothesized that the fast Ba2+ site observed in 2 mM [K+]0 could be involved in the gating effects mediated by Ba2+. Accordingly, we measured the wash-in kinetics of Ba2+ in high external K+ (50 mM) (Fig. 3). Under these conditions, Ba2+ on-binding displayed a fast and slow kinetic component. However, the wash-in time course was dominated by the slow kinetic component (Fig. 3, A and B; Table II). While in low external K+, the relative amplitude of the slow component decreased from 0.45 to 0.24 at potentials between −30 and +30 mV, in high external K+ it predominated at 0.89–0.85 at potentials between 0 and +30 mV (Fig. 3 B). In contrast, the relative amplitude of the fast component had a minor contribution in 50 mM [K+]0, ranging from 0.1 to 0.15 at increasing depolarization (Fig. 3 B). These results suggest that the slow Ba2+ site (δ = 0.32) measured in wash-in kinetics in low external K+ and which predominates in high external K+ mainly accounts for the voltage-dependent block of Ba2+. The fast Ba2+ site (δ = 0.06) that has a minor contribution in high external K+ likely accounts for the gating effects of Ba2+ observed in low external K+ solutions. In support of this hypothesis, the experiment depicted in Fig. 4 indicates that external Ba2+ could bind to a deep occluding site in the closed state (−100 mV) and remained bound to it during washout at −100 mV, thus leading to channel blockade. In contrast, the washout removed Ba2+ from its fast external site, hence preventing its impact on channel gating. Indeed, despite a substantial channel block no effects on inactivation, deactivation, and activation were induced by Ba2+ exposure (Fig. 4, C and D).
Tail Current Analysis and Modeling
Recent studies suggested the presence of multiple open states for homomeric KCNQ1 channels (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). They proposed a sequential gating scheme (Scheme I) assuming two closed states, two voltage-dependent open-states, and a fast, voltage-independent inactivated state (Pusch et al., 1998).
Using a tail current analysis, we tested whether KCNQ1 channels could populate multiple open states in the presence of 10 mM Ba2+ at steady-state in low external K+ (2 mM). We measured the kinetics of current deactivation at −60 mV tail potential for various prepulse durations to +30 mV in the absence and presence of 10 mM Ba2+ (Fig. 12, A and B) . KCNQ1 deactivation kinetics exhibited a fast and slow component and in order to take into account the recovery from inactivation, we fitted the whole tail current time course by a triple exponential function (see materials and methods). The rate of channel deactivation progressively decreased with increasing prepulse durations (Fig. 12, C and D). For example, the fast and slow time constants of KCNQ1 deactivation increased, respectively, from τf = 91 ± 16 ms and τs = 156 ± 22 ms at 0.05 s prepulse (n = 12) to τf = 160 ± 17 ms and τs = 406 ± 41 ms at 2.25-s prepulse (n = 12). It is interesting to note that the tail currents were hooked for prepulse durations >100 ms and the hook amplitude increased with the length of the prepulse (Fig. 12 E). The progressive decrease in deactivation rate as a function of prepulse duration, is indicative of the existence of multiple open states (Tristani-Firouzi and Sanguinetti, 1998). The sigmoidal time course of the tail currents at negative potentials further supports this idea (Fig. 8 A, left). In the presence of 10 mM Ba2+, the rate of channel closure was faster but it did not decrease significantly with increasing prepulse duration (Fig. 12 C). In the presence of Ba2+, the acceleration of deactivation was such that the slow kinetic component disappeared. As shown before, Ba2+ potently depressed inactivation by virtually eliminating the hook of the tail currents (Fig. 12 D). The sigmoidal shape of the tail currents at negative potentials disappeared in the presence of Ba2+ (Fig. 8 A, right). These results suggest that in the presence of Ba2+ and in low external K+, unblocked KCNQ1 channels transit to one open state from which inactivation is not permissible (see discussion).
Figure 12.

Effect of Ba2+ on tail current analysis. (A and B) Representative tail envelopes from the same oocyte showing the kinetics of current deactivation, measured at a −60 mV tail potential after the progressive activation of KCNQ1 channels by various prepulse durations to +30 mV from 50 ms to 2.25 s in 200-ms increments, in the absence (A) and presence of 10 mM Ba2+ (B). (C and D) Fast and slow time constant of channel deactivation before (empty symbols) and after 10-mM Ba2+ application (solid triangles) plotted as a function of prepulse duration (n = 12). (E) Fractional inactivation measured from the same tail current analysis before (empty squares) and after 10-mM Ba2+ application (solid squares) plotted as a function of prepulse duration (n = 12).
For modeling KCNQ1 behavior in low external K+ (2 mM) and in the absence of Ba2+, the time course of channel activation was fitted according to Scheme I, following the previously described gating model (Pusch et al., 1998), where only O1 and O2 are assumed to conduct and for which we assumed for simplicity that no openings could occur when the cell is voltage-clamped to −80 mV. A fitting analysis was performed for KCNQ1 activation in low and high external K+ in the absence of barium (see table of online supplemental material and materials and methods). To validate the model, we used the fit data and simulated a +30-mV activation time course and a tail envelope protocol. The simulated activation time course in low external K+ closely matched the experimental trace (Fig. 13 C). Simulating the tail currents for the different prepulse intervals allowed us to generate a simulated trace that described faithfully the dependence of the “hook” amplitude on the prepulse duration as observed in experimental data (Fig. 13, A and B). Similarly, the simulated time course of KCNQ1 activation in high external K+ and in the absence of Ba2+ closely matched the averaged experimental traces (Fig. 13 D).
Figure 13.

Simulation of KCNQ1 gating. (A) Simulation of the tail envelope protocol as described in Fig. 12. The simulation was done according to Scheme I. All rate constants represent averaged values obtained from the least square fit of currents at +30 mV and −60 mV in the absence of Ba2+ (n = 7). The ordinate represents the calculated open probability (fractional occupancy of the open states). (B) Representative experimental traces of the tail envelope protocol. (C) Simulation (red line) of KCNQ1 activation (+30 mV) and deactivation (−60 mV) according to Scheme I, superimposed on an experimental trace. (D) Scaled simulation (red lines) of KCNQ1 activation in high external K+ (50 mM) according to Scheme II, in the absence and presence of 10mM Ba2+. The simulation was superimposed on experimental average traces obtained from seven oocytes.
Since high external K+ prevented the impact of Ba2+ on channel gating, we used a simplistic model of voltage-dependent block as illustrated in Scheme II in order to simulate Ba2+ block in high external K+ solutions.
We assumed that the conductance reduction originates from the existence of one Ba2+-occluding site. Thus, the channel could populate two different kinds of states, assumed to be in equilibrium: a Ba2+-blocked state (B) and a Ba2+-unblocked state. For simplicity, we assumed that all states (C0 to I) bind Ba2+ with equal affinity (see materials and methods). As the kinetics of Ba2+ in 50 mM [K+]0 are clearly dominated by a slow component, we could estimate Ba2+ affinity for the open states. To preserve microscopic reversibility, we also assumed that transitions through the blocked states occur with similar rates to their unblocked counterparts. Simulating the time course of KCNQ1 activation in the presence of 10 mM Ba2+ and in high external K+ solutions (50 mM) faithfully matched the kinetics of the averaged experimental traces, including the decrease of the current amplitude (Fig. 13 D). In contrast, the simple model of voltage-dependent block failed to simulate the complex Ba2+ actions on KCNQ1 channels in low external K+ conditions (see discussion).
DISCUSSION
The main findings of the present work are that extracellular Ba2+ exerts on homomeric KCNQ1 channels a series of complex effects, including a voltage-dependent pore block as well as unique gating alterations. Our results indicate that external barium interacts with the permeation pathway of KCNQ1 at two distinct sites. (a) A slow deep Ba2+ site that occludes the channel pore and could be simulated by a model of voltage-dependent block. (b) A fast superficial Ba2+ site that barely influences channel block and mainly affects channel gating by shifting rightward the voltage dependence of activation, slowing activation, speeding up deactivation kinetics, and inhibiting channel inactivation. Both sites appear to reside in the permeation pathway as high external potassium attenuates the Ba2+ inhibition of channel conductance and abolishes its impact on channel gating.
External Ba2+ Interacts with the Pore of KCNQ1 at Two Distinct Sites
The existence of at least two distinct Ba2+ binding sites is supported by the biphasic time course of Ba2+ wash-in and washout kinetics. Furthermore, the Ba2+ dose-response curves need to be fitted to two binding sites. Although at negative potentials (−70 to −30 mV) the curves could be fitted assuming one site, the values of the Hill coefficients of ∼2.1–2.8 suggest the existence of at least two Ba2+ sites of very similar affinity (Fig. 5 C). However, with increasing depolarization, the progressive appearance of a lower affinity site reflects the voltage dependence of the slow deep site (Fig. 5 D). The Ba2+ affinities for the two sites probably overlap, especially at negative potentials; however, it is worth noting that the EC50 values obtained for the effects of Ba2+ on channel gating are within the same range (0.44–1.4 mM). A good discrimination between two distinct Ba2+ sites relies on the data obtained in high external K+ solutions. In steady-state experiments, elevating external K+ from 2 to 50 mM substantially preserved the voltage-dependent block of channel conductance though weakening the amplitude of Ba2+ block, but abolished its impact on channel gating. Along the same line, Ba2+ wash-in kinetics performed in high external K+ were dominated by the slow kinetic component while the fast component had a minor contribution. Finally, binding of Ba2+ to the closed channel state adds further support to the existence of two discrete binding sites. Prolonged wash-in and washout of external Ba2+ from closed channels at −100 mV leaves Ba2+ bound to its deep site but removes Ba2+ from its superficial site, leading to a significant channel block and preventing Ba2+ impact on KCNQ1 channel gating. Taken together, our results indicate that external barium interacts with the permeation pathway of KCNQ1 at two sites. (a) A superficial Ba2+ site that mainly affects channel gating and senses weakly the electric field (δ1 = 0.06). It corresponds to the fast kinetic component of Ba2+ wash-in that virtually disappears in high external K+. Knowing the strong rightward shift effect of Ba2+ on KCNQ1 activation curve (+34 mV at 10 mM Ba2+), it is not surprising that the fast kinetic component depresses substantially the current amplitude in wash-in experiments, a feature that in fact reflects the impact of Ba2+ on gating rather than a direct channel block. (b) A deep Ba2+ site that occludes the channel pore and lies profound in the membrane electric field (δ2 = 0.33). It corresponds to the slow kinetic component of Ba2+ wash-in in low external K+ that predominates in high external K+.
It was previously found that external Ba2+ can interact with two distinct and sequential binding sites in Shaker K+ channels (Hurst et al., 1995). In contrast to KCNQ1 channels where the slow deep site is mainly involved in the voltage-dependent Ba2+ block, both fast and slow sites were found to be responsible for the Ba2+ block of Shaker channels with apparent electrical distances of 0.14 and 0.25, respectively. Furthermore, it was suggested that Ba2+ had first to populate a fast external site before being able to populate a slow deep site to produce the voltage-dependent block (Hurst et al., 1995). Consistent with two sites in series, the washout time course of Ba2+ in Shaker channels exhibited only one kinetic component (slow). In KCNQ1 channels, the features of external Ba2+ binding are different and the washout kinetics display a biphasic time course suggesting that the two sites are not sequential and exhibit distinct functional properties. In further support of the latter conclusion, high external K+ preserves the voltage-dependent block at the deep slow site, while virtually abolishing the superficial fast site and the concomitant gating impact of Ba2+. This suggests that one does not need Ba2+ to bind to the external fast site in order to get Ba2+ binding to the slow deep site. The δ value obtained for the deep slow site of Ba2+ block of KCNQ1 is similar to that found for Shaker K+ channels (δ = 0.35) (Harris et al., 1998), but is lower than that obtained for the high affinity Ba2+ block of the inward-rectifier Kir2.1 channels (δ = 0.62) (Alagem et al., 2001) or that found for the two-pore domain K+ channel KCNK0 (δ = 0.60) (Ilan and Goldstein, 2001). Thus, despite the high degree of conservation of the pore region among various classes of K+ channels, subtle differences in the architecture of the permeation apparatus can confer different voltage-sensitivity for Ba2+ block. In other words, the same Ba2+ binding site may sense the voltage drop differently depending on the precise geometry of a given K+ channel pore.
Two external K+ sites located above the deep Ba2+ site were found to affect Ba2+ binding (Neyton and Miller, 1988a,b). One rapidly equilibrating site, of high K+ affinity (lock-in site), whose occupancy by K+ prevents outward Ba2+ dissociation, and one site of lower K+ affinity (enhancement site), whose occupancy by K+ repulses Ba2+ in the inward direction. All three sites were found to be accessible from the external solution to the closed channel state and in Shaker channels the deep Ba2+ site lies between the activation gate and the C-type inactivation gate (Harris et al., 1998). Given the high degree of topological similarity and the strong voltage dependence of Ba2+ block, we assume that the Ba2+ binding site of voltage-gated K+ channels in general, and that of KCNQ channels in particular, is analogous to that of KcsA (Jiang and MacKinnon, 2000). Mutagenesis studies performed on Shaker K+ channels showed that the residue T441, located at the inner boundary of the selectivity filter, may represent the deep high-affinity Ba2+ binding site (Harris et al., 1998). In the inward-rectifier Kir 2.1, an amino acid T141, located just one residue COOH-terminal to the equivalent T441 in Shaker was suggested to stabilize the high affinity Ba2+ binding (Alagem et al., 2001). These results fit well with the crystallographic study performed on KcsA channels (Jiang and MacKinnon, 2000). In KCNQ1, the homologous residue is also a threonine, T311. In Shaker channels, it was suggested that the residue V443 within the selectivity filter facilitates the entry and exit of Ba2+ to and from the deep site, while the amino acid T449 in the external vestibule may represent the external lock-in site (Harris et al., 1998). Preliminary studies performed in V319C KCNQ1 (coexpressed with KCNE1) indicate that the valine mutation V319C located in the external vestibule at the analogous Shaker position T449 potently prevents the voltage-dependent Ba2+ block but does not affect the action of Ba2+ on channel gating (unpublished data). However, knowing the impact of KCNE1 on the biophysical properties of KCNQ1, these data may not be directly applicable to homomeric KCNQ1 V319C channels. Taken together, these results emphasize the structural and functional differences existing between KCNQ1 and other voltage-gated K+ channels in their permeation pathway, despite the high degree of homology of their pore region. They also suggest that in homomeric KCNQ1, the two Ba2+ sites exhibit distinct functional properties.
Barium Affects Channel Gating
Compared with Shaker-like K+ channels and other K+ channels, barium uniquely affects KCNQ1 gating. In addition to its voltage-dependent block of the pore, Ba2+ also alters the gating of homomeric KCNQ1 channels by favoring the closed state. Barium produces a marked rightward shift of the voltage-dependence of activation, a slowing of activation, and an acceleration of deactivation kinetics. In addition, Ba2+ potently depresses inactivation gating of KCNQ1, an effect that opposes current inhibition. This latter impact of Ba2+ on KCNQ1 inactivation appears to be very different from its effects on Shaker channels, where it does not alter C-type inactivation (Harris et al., 1998). In HERG channels, Ba2+ also produced a voltage-dependent block. However, at positive potentials and concomitant with the development of C-type inactivation, Ba2+ block is completely relieved (Weerapura et al., 2000), a feature that is not observed with an inactivation-deficient HERG mutant. In HERG channels, Ba2+ does not prevent inactivation; in fact Ba2+ does not bind to the HERG-inactivated state (Weerapura et al., 2000). This feature contrasts with that obtained for KCNQ1 channels and further suggests that despite the high degree of similarity of the pore region, substantial structural and functional differences exist between the various voltage-gated K+ channel subtypes. Interestingly, all these gating alterations are mediated by the same superficial fast site of Ba2+ binding. The effects of Ba2+ are specific and do not result from a surface-charge–screening effect, as Mg2+ and Ca2+ neither affect the conductance nor alter the gating of KCNQ1. Furthermore, the relief of the impact of Ba2+ on channel gating by high external K+ cannot be accounted for by a charge-screening effect. We believe that the right-shift produced by Ba2+ is not due to a voltage-dependent block: (a) A model of voltage-dependent block does not explain the marked slowing of the activation kinetics that is observed with increasing Ba2+ concentrations (Fig. 6 A, left), as the unblocking rate is not expected to depend on the blocker concentration; (b) in high external K+, there is still a voltage-dependent Ba2+ block (Fig. 11A), precisely under conditions where the right-shift has been lost (Fig. 9 E, right); (c) in low external K+, a simple model of voltage-dependent block was unable to simulate the time course of KCNQ1 activation and deactivation, precisely under conditions where Ba2+ produces the right-shift; (d) the decrease in maximum conductance was much more significant than the shift in V50 produced by all Ba2+ concentrations; and (e) the activation, deactivation, and V50 parameters are not affected to the same extent. For example, 4 mM Ba2+ shifts the fast time constant of the activation kinetics by more than 60 mV (Fig. 6, A and B), while it shifts by only 27 mV the voltage dependence of activation (Table I).
The mechanisms by which Ba2+ affects the gating machinery of KCNQ1 are unknown, however, we suggest that they are coupled to the permeation process as we have shown that the gating effects of Ba2+ are virtually abolished by high external K+. The gating impact of Ba2+ probably involves a fast-equilibrating superficial site of the KCNQ1 external vestibule that remains to be identified. Interestingly, we found that external acidification also alters the gating of homomeric KCNQ1 channels by inhibiting inactivation, slowing activation kinetics, and causing a rightward shift of the voltage dependence of activation (Peretz et al., 2002). It remains to determine whether external protons and Ba2+ ions affect the gating of KCNQ1 by similar mechanisms. In principle, the basic operation of an ion channel relies on the combination of two distinct properties, gating and permeation. However, these processes are not independent. Permeating ions are known to interact with the pore that undergoes conformational changes upon channel opening, inactivation, and closure. Are there regulatory sites within the permeation pathway and remote from the narrow selectivity filter that affect permeation and impact on channel gating? Recent studies have shown that residues of the pore region do interact with the voltage-sensor module of Shaker channels (Li-Smerin et al., 2000). This protein interface may be a crucial determinant for the molecular coupling of the voltage-sensor movement and the opening of the pore. A direct interaction between charges of the voltage sensor and pore residues has already been suggested for the coupling between S4 and the slow inactivation gate of the Shaker channel (Gandhi et al., 2000; Larsson and Elinder, 2000; Loots and Isacoff, 2000; Ortega-Saenz et al., 2000; Elinder et al., 2001). In KCNQ1, it is possible that Ba2+ binds to an external vestibule site that is involved in sensor-gate coupling. Alternatively, the features of the fast superficial Ba2+ site may be also consistent with Ba2+ binding to a site remote from the permeation pathway and residing in a crevice in which K+ but not Mg2+ or Ca2+ can compete with. Along the same line, barium was previously found to directly affect the gating of Shaker channels by speeding up the rate of the off-gating current (Hurst et al., 1996), which is in agreement with the proposal that Ba2+ destabilizes the open state and favors the closed state.
KCNQ1 α subunits produce very different gating phenotypes when coexpressed with the KCNE β subunits (Abbott and Goldstein, 1998; Melman et al., 2002). In the heart, coassembly of KCNQ1 with KCNE1 elicits voltage- and time-dependent K+ currents (IKS) with very slow activation kinetics, a positive shift in the voltage dependence of activation, and an increase in unitary channel conductance, in comparison to homomeric KCNQ1 channels (Barhanin et al., 1996; Sanguinetti et al., 1996). When associated with KCNE3, KCNQ1 channels yield K+ currents that are nearly instantaneous and voltage independent (Schroeder et al., 2000). Regarding the strong impact of KCNE β subunits on the biophysical properties of KCNQ1, it will be important in future experiments to investigate in depth the effects of external Ba2+ on these heteromeric channel complexes. Our preliminary data indicate that compared with homomeric KCNQ1, external Ba2+ has a similar action on KCNQ1/KCNE1 channels and differential effects on KCNQ1/KCNE3 complexes (unpublished data).
Modeling Barium Action
A sequential gating scheme for homomeric KCNQ1 channels was proposed (Scheme I), assuming two closed states, two voltage-dependent open-states, and a fast, voltage-independent inactivated state (Pusch et al., 1998). More recently, inactivation of KCNQ1 was suggested to be related to a fast flicker of the open channel (Seebohm et al., 2003).
Simulating KCNQ1 behavior in low external K+ (2 mM) and in the absence of Ba2+ according to Scheme I generated an activation time course and a tail-current envelope that described faithfully the dependence of the inactivation (hook amplitude) on prepulse duration that is observed in experimental data (Fig. 13, A and B). The progressive decrease in deactivation rate as a function of prepulse duration, further supports the existence of multiple open states (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). Using the same Scheme I, we could simulate the time course of KCNQ1 activation in high external K+ and absence of Ba2+ (Fig. 13 D). This suggests that elevating external K+ does not alter significantly the gating transitions of KCNQ1 channels. In contrast to C-type inactivation, high external K+ does not directly affect the inactivation process of KCNQ1. Paradoxically, high external K+ relieves Ba2+ inhibition of inactivation gating. As high external K+ prevented the impact of Ba2+ on channel gating, we could use a simplistic model of voltage-dependent block, as shown in Scheme II, in order to simulate Ba2+ block in high external K+.
Assuming that the conductance block originates from the existence of one Ba2+-occluding site (i.e., the deep slow Ba2+ site), we could simulate the time course of KCNQ1 activation, including the decrease of current amplitude, in the presence of 10 mM Ba2+ under high external K+ conditions (Fig. 13 D). In contrast, the simple model of voltage-dependent block failed to simulate the complex Ba2+ actions on KCNQ1 channels in low external K+ conditions. On the other hand, inhibition of inactivation gating by Ba2+ in low external K+ cannot be accounted for by a general slowing of the sequential gating Scheme I, since very long depolarizing pulses (+30 mV for 6 min) performed in the presence of 4 mM Ba2+ did not restore KCNQ1 inactivation, as measured by the hooked tail currents (unpublished data). The loss of inactivation does not reflect a lower occupancy of the preceding open state O2 at a given activation potential since when the membrane was stepped to larger depolarizing potentials of up to +80 mV, inactivation did not reappear (unpublished data). The tail current analysis performed in the presence of Ba2+ and in low external K+ conditions reveals that the rate of channel closure was faster but did not decrease with increasing prepulse duration. These results suggest that in the presence of Ba2+ and in low external K+, unblocked KCNQ1 channels transit to an open state from which inactivation is not permissible. Thus, we have to assume that external Ba2+ ions interact with the permeation pathway of KCNQ1 channels at two distinct and nonsequential sites. (a) A deep binding site that occludes the pore and is responsible for the voltage-dependent block of the channel conductance. This Ba2+-bound site leads to nonconducting channels, is well described by Scheme II and is partially competed out by high external K+. (b) A superficial binding site that barely contributes to channel block but produces the gating alterations on KCNQ1 and is totally competed out by high external K+. This latter Ba2+-bound site modifies the gating transitions states of KCNQ1, leads to modified-conducting channels and cannot be accounted for by a model of voltage-dependent block. Assuming a KD = 3 mM for channel block (via occlusion) and a KD = 1 mM for channel modification (via gating), we can predict that in the presence of 10 mM Ba2+ and in low external K+ (2 mM), 21% of KCNQ1 channels are conducting but modified, 7% are blocked, 70% are both modified and blocked and only 2% have no Ba2+ bound (Fig. 14 A). Overall, 77% of KCNQ1 channels are nonconducting (of which 70% are also modified) and 91% are modified (of which 70% are nonconducting). Fig. 14, B–E, illustrates how the relative fractions of nonconducting (blocked) and conducting (modified and nonmodified) channels are predicted to evolve, depending on the KD values for Ba2+ block.
Figure 14.

Schematic calculation of KCNQ1 channel distribution among different populations in the presence of Ba2+. (A) Diagram of the different channel populations in the presence of 10 mM Ba2+, assuming a KD = 3 mM for channel block and a KD = 1 mM for channel gating modification. It is assumed that external Ba2+ interact with the permeation pathway of KCNQ1 channels at two distinct and nonsequential sites. (1) A deep site that accounts for the voltage-dependent block of the pore and leads to blocked nonconducting channels. (2) A superficial site that modifies the gating transitions states of KCNQ1 and leads to modified-conducting channels. (B–E) Illustration of how the relative fractions of nonconducting (blocked), and conducting (modified and nonmodified) channels are predicted to evolve, depending on the KD values for Ba2+ block.
In all, our data suggest that despite the high degree of homology of the pore region of voltage-gated K+ channel subtypes, significant structural and functional differences exist with KCNQ1 channels. Compared with Shaker-like K+ channels, barium uniquely affects KCNQ1 channel gating, especially inactivation.
Supplemental Material
Acknowledgments
We thank Drs. Ilana Lotan, Nathan Dascal, and Jianmin Cui for their helpful comments and a critical reading of the manuscript.
This work was supported by the Israel Science Foundation (grant No: 540/01-1) and by a US-Israel binational Science Foundation grant (No. 2001229).
Olaf S. Andersen served as editor.
The online version of this article contains supplemental material.
References
- Abbott, G.W., and S.A. Goldstein. 1998. A superfamily of small potassium channel subunits: form and function of the MinK-related peptides (MiRPs). Q. Rev. Biophys. 31:357–398. [DOI] [PubMed] [Google Scholar]
- Alagem, N., M. Dvir, and E. Reuveny. 2001. Mechanism of Ba2+ block of a mouse inwardly-rectifying K+ channel: differential contribution by two discrete residues. J. Physiol. 534:381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and S.R. Taylor. 1980. Interaction of barium ions with potassium channels in squid giant axons. Biophys. J. 30:473–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft, F.M. 2000. Ion Channels and Diseases. Academic Press.
- Barhanin, J., F. Lesage, E. Guillemare, M. Fink, M. Lazdunski, and G. Romey. 1996. KvLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 384:78–80. [DOI] [PubMed] [Google Scholar]
- Blunck, R., U. Kirst, T. Riessner, and U. Hansen. 1998. How powerful is the dwell-time analysis of multichannel records? J. Membr. Biol. 165:19–35. [DOI] [PubMed] [Google Scholar]
- Brown, B.S., and S.P. Yu. 2000. Modulation and genetic identification of the M channel. Prog. Biophys. Mol. Biol. 73:135–166. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J. Morais Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Eaton, D.C., and M.S. Brodwick. 1980. Effects of barium on the potassium conductance of squid axon. J. Gen. Physiol. 75:727–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinder, F., R. Mannikko, and H.P. Larsson. 2001. S4 charges move close to residues in the pore domain during activation in a K+ channel. J. Gen. Physiol. 118:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, C.S., E. Loots, and E.Y. Isacoff. 2000. Reconstructing voltage sensor-pore interaction from a fluorescence scan of a voltage-gated K+ channel. Neuron. 27:585–595. [DOI] [PubMed] [Google Scholar]
- Harris, R.E., H.P. Larsson, and E.Y. Isacoff. 1998. A permeant ion binding site located between two gates of the Shaker K+ channel. Biophys. J. 74:1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, R.S., R. Latorre, L. Toro, and E. Stefani. 1995. External barium block of Shaker potassium channels: evidence for two binding sites. J. Gen. Physiol. 106:1069–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, R.S., L. Toro, and E. Stefani. 1996. Molecular determinants of external barium block in Shaker potassium channels. FEBS Lett. 388:59–65. [DOI] [PubMed] [Google Scholar]
- Ilan, N., and S.A.N. Goldstein. 2001. KCNK0: single, cloned potassium leak channels are multi-ion pores. Biophys. J. 80:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch, T.J. 2000. Neuronal KCNQ potassium channels: physiology and role in diseases. Nat. Rev. Neurosci. 1:21–30. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. Mackinnon. 2002. Crystal structure and mechanism of calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., and R. MacKinnon. 2000. The barium site in a potassium channel by X-ray crystallography. J. Gen. Physiol. 115:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, H.P., and F. Elinder. 2000. A conserved glutamate is important for slow inactivation in K+ channels. Neuron. 27:573–583. [DOI] [PubMed] [Google Scholar]
- Lehmann-horn, F., and K. Jurkat-Rott. 1999. Voltage-gated ion channels and hereditary disease. Physiol. Rev. 79:1317–1372. [DOI] [PubMed] [Google Scholar]
- Lerche, H., K. Jurkat-Rott, and F. Lehmann-Horn. 2001. Ion channels and epilepsy. Am. J. Med. Genet. 106:146–159. [DOI] [PubMed] [Google Scholar]
- Li-Smerin, Y., D.H. Hackos, and K.J. Swartz. 2000. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel. Neuron. 25:411–423. [DOI] [PubMed] [Google Scholar]
- Loots, E., and E.Y. Isacoff. 2000. Molecular coupling of S4 to a K+ channel's slow inactivation gate. J. Gen. Physiol. 116:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman, Y.F., A. Krummerman, and T.V. McDonald. 2002. KCNE regulation of KvLQT1 channels: structure-function correlates. Trends Cardiovasc. Med. 12:182–187. [DOI] [PubMed] [Google Scholar]
- Neyton, J., and C. Miller. 1988. a. Discrete barium block as a probe of ion occupancy and pore structure in the high-conductance calcium-activated potassium channel. J. Gen. Physiol. 92:569–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyton, J., and C. Miller. 1988. b. Potassium blocks barium permeation through a calcium-activated potassium channel. J. Gen. Physiol. 92:549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Saenz, P., R. Pardal, A. Castellano, and J. Lopez-Barneo. 2000. Collapse of conductance is prevented by a glutamate residue conserved in voltage-dependent K+ channels. J. Gen. Physiol. 116:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz, A., H. Schottelndreier, L. Ben Aharon-Shamgar, and B. Attali. 2002. Modulation of homomeric and heteromeric KCNQ1 channels by external acidification. J. Physiol. 545:751–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch, M. 1998. Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with mink. Pflugers Arch. 437:172–174. [DOI] [PubMed] [Google Scholar]
- Pusch, M., R. Magrassi, B. Wollnik, and F. Conti. 1998. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, J. 2001. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol. Ther. 90:1–19. [DOI] [PubMed] [Google Scholar]
- Rogawski, M.A. 2000. KCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: implications for therapy. Trends Neurosci. 23:393–398. [DOI] [PubMed] [Google Scholar]
- Sanguinetti, M.C., M.E. Curran, A. Zou, J. Shen, P.S. Spector, D.L. Atkinson, and M.T. Keating. 1996. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 384:80–83. [DOI] [PubMed] [Google Scholar]
- Schroeder, B.C., S. Waldegger, S. Fehr, M. Bleich, R. Warth, R. Greger, and T.J. Jentsch. 2000. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 403:196-199. [DOI] [PubMed] [Google Scholar]
- Seebohm, G., C.M. Sanguinetti, and M. Pusch. 2003. Tight coupling of rubidium conductance and inactivation in human KCNQ1 potassium channels. J. Physiol. 552:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti, F., and S.A.N. Goldstein. 1998. Single-channel characteristics of wild-type Iks channels and channels formed with two mink mutants that cause long QT syndrome. J. Gen. Physiol. 112:651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tristani-Firouzi, M., and M.C. Sanguinetti. 1998. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 510:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H.-S., Z. Pan, W. Shi, B.S. Brown, R.S. Wymore, I.S. Cohen, J.E. Dixon, and D. McKinnon. 1998. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 282:1890–1893. [DOI] [PubMed] [Google Scholar]
- Weerapura, M., S. Nattel, M. Courtemanche, D. Doern, N. Ethier, and T.E. Hebert. 2000. State-dependent barium block of wild-type and inactivation-deficient HERG channels in Xenopus oocytes. J. Physiol. 526:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth, L.P. 1994. Mechanism of Ba2+ block of M-like K channels of rod photoreceptors of Tiger salamanders. J. Gen. Physiol. 103:45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull, A.M. 1973. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., and F.J. Sigworth. 1998. Single-channel properties of Iks potassium channels. J. Gen. Physiol. 112:665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen, G. 2002. The voltage-gated potassium channels and their relatives. Nature. 419:35–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}