

Abstract
Background
The exact origin of the cause of the Severe Acute Respiratory Syndrome (SARS) is still an open question. The genomic sequence relationship of SARS-CoV with 30 different single-stranded RNA (ssRNA) viruses of various families was studied using two non-standard approaches. Both approaches began with the vectorial profiling of the tetra-nucleotide usage pattern V for each virus. In approach one, a distance measure of a vector V, based on correlation coefficient was devised to construct a relationship tree by the neighbor-joining algorithm. In approach two, a multivariate factor analysis was performed to derive the embedded tetra-nucleotide usage patterns. These patterns were subsequently used to classify the selected viruses.
Results
Both approaches yielded relationship outcomes that are consistent with the known virus classification. They also indicated that the genome of RNA viruses from the same family conform to a specific pattern of word usage. Based on the correlation of the overall tetra-nucleotide usage patterns, the Transmissible Gastroenteritis Virus (TGV) and the Feline CoronaVirus (FCoV) are closest to SARS-CoV. Surprisingly also, the RNA viruses that do not go through a DNA stage displayed a remarkable discrimination against the CpG and UpA di-nucleotide (z = -77.31, -52.48 respectively) and selection for UpG and CpA (z = 65.79,49.99 respectively). Potential factors influencing these biases are discussed.
Conclusion
The study of genomic word usage is a powerful method to classify RNA viruses. The congruence of the relationship outcomes with the known classification indicates that there exist phylogenetic signals in the tetra-nucleotide usage patterns, that is most prominent in the replicase open reading frames.
Keywords: RNA virus, convergent evolution, horizontal gene transfer, factor analysis, SARS
Background
Severe Acute Respiratory Syndrome (SARS), a newly identified infectious disease, has imperilled the health of human population in more than 30 nations. It has claimed over 812 lives and infected more than 8442 (9.61% death rate) by July 2, 2003 [1] since its outbreak in November 2002 in the province of GuangDong, People's Republic of China. By May 15, 2003, the primary etiological agent for SARS was found to fulfil Koch's postulate through experimental infection of cynomolgus macaques (Macaca fascicularis) [2]. Chronicles for the discovery of SARS CoronaVirus (SARS-CoV) can be found in articles [e.g. [3,4]] and websites [e.g. [5]].
A common question is often asked when investigating viral evolution: what hallmark, in term of genome sequence or RNA word usage, could be used to trace back the emergence of a new pathogen in humans/animals? In particular, CoronaViruses are prone to recombination [6,7] and like all other viruses they mutate at a high frequency [8]. This makes extremely hazardous to try to trace the origin of the virus. Nevertheless, this prompted us to investigate their relationships using the RNA word usage hoping to identify some RNA viruses that display similar word usage pattern. Such RNA viruses might hint about the origin of SARS-CoV. This study will contribute to our understanding of the RNA word usage of SARS-CoV and some other pathogenic RNA viruses. In the present study, we explored the relationships of 31 RNA viruses, which are known to cause diseases to their corresponding hosts with either similar symptoms or infectiousness, including SARS-CoV, based on their global tetra-nucleotide usage pattern.
Preliminary analysis of the sequence data indicated that there are 11–14 open reading frames in the SARS-CoV genome [9-11]. The overall gene order for this novel pathogen supported its placement in the family of Coronaviridae which includes the animal/human CoronaViruses. It should be emphasized that the sequence similarity shown is attributed mainly to the large RNA-dependent RNA polymerase (replication enzyme or RdRp) residing in the first two open reading frames (ORFs). These two ORFs constitute more than 65% (>20 kb) of the total genome size and these regions are more conserved in their nucleotide sequences due to their specialized role for viral RNA replication. Therefore, the possible relationship based on the sequence of the replication enzyme alone was also investigated.
Results and Discussion
Mono-nucleotide bias
Table 1 presents the breakdown of the RNA sequence into mononucleotide frequencies for the 31 viral genomes in our dataset. Except for the Rabbit Hemorrhagic disease Virus (RHV) that shows a fair usage of the four nucleotides in approximately equal number, the other RNA viruses have a biased genome composition. Bovine CoronaVirus (BCoV) and Human CoronaVirus 229E (HCoV) favor the U nucleotide (35.5% and 34.6%) at the expense of the C nucleotide (15.3% and 16.7%). Relatively strong nucleotide biases are visible in the other genomes and we will mention a few of the extremes. The highest base count is 28.4% G in the Yellow Fever Virus (YFV), 38.9% A in the Respiratory Syncytial Virus (RSV), 35.5% U in the Bovine CoronaVirus (BCoV) and 28.5% C count in the Foot-and-Mouth disease Virus (FMV). The lowest base counts are 15.8% G in the Human Respiratory syncytial Virus (HRV), 21.2% A in the Equine arteritis Virus (EV1), 20.9% U in the Igbo Ora Virus (IOV) and 13.6% C in the Bovine ephemeral Fever Virus (BFV). The A nucleotide is the most popular base among RNA viruses (ranging from 21.2% to 38.9%), and C is the most variable nucleotide (ranging from 13.6% to 33.1%).
Table 1.
RNA virus in current study.
| Virus Name | Type | Acession Number | DNA Stage | Segment | Acronym | Size (nt) | G | A | U | C | A+U% | ||
| ssRNA positive-strand viruses | 1 | Avian infectious bronchitis virus | ss-RNA | NC_001451 | No | 1 | ABV | 27608 | 21.7 | 28.9 | 33.2 | 16.2 | 62.1 |
| 2 | Bovine coronavirus | ss-RNA | NC_003045 | No | 1 | BCoV | 31028 | 21.8 | 27.4 | 35.5 | 15.3 | 62.9 | |
| 3 | Equine arteritis virus | ss-RNA | NC_002532 | No | 1 | EV1 | 12704 | 26.0 | 21.2 | 27.1 | 25.6 | 48.3 | |
| 4 | Human coronavirus 229E | ss-RNA | NC_002645 | No | 1 | HCoV | 27317 | 21.6 | 27.2 | 34.6 | 16.7 | 61.7 | |
| 5 | Lactate dehydrogenase-elevating virus | ss-RNA | NC_002534 | No | 1 | LDV | 14225 | 25.9 | 23.1 | 28.2 | 22.6 | 51.3 | |
| 6 | Murine hepatitis virus | ss-RNA | NC_001846 | No | 1 | MHV | 31357 | 23.9 | 26.0 | 32.3 | 17.9 | 58.2 | |
| 7 | Porcine epidemic diarrhea virus | ss-RNA | NC_003436 | No | 1 | PDV | 28033 | 22.8 | 24.7 | 33.2 | 19.2 | 58.0 | |
| 8 | Porcine reproductive and respiratory syndrome virus | ss-RNA | NC_001961 | No | 1 | PRV | 15428 | 26.2 | 21.7 | 25.3 | 26.7 | 47.0 | |
| 9 | SARS coronavirus | ss-RNA | NC_004718 | No | 1 | SAR | 29751 | 20.8 | 28.5 | 30.7 | 20.0 | 59.2 | |
| 10 | Feline coronavirus | ss-RNA | AY204704 | No | 1 | FCoV | 9979 | 22.6 | 27.9 | 29.2 | 20.3 | 57.2 | |
| 11 | Simian hemorrhagic fever virus | ss-RNA | NC_003092 | No | 1 | SFV | 15717 | 22.6 | 22.5 | 27.4 | 27.5 | 49.9 | |
| 12 | Transmissible gastroenteritis virus | ss-RNA | NC_002306 | No | 1 | TGV | 28586 | 20.6 | 29.5 | 32.9 | 17.0 | 62.4 | |
| 13 | Avian encephalomyelitis virus | ss-RNA | NC_003990 | No | 1 | AEV | 7055 | 25.7 | 27.0 | 28.3 | 19.0 | 55.3 | |
| 14 | Bovine viral diarrhea virus genotype 2 | ss-RNA | NC_002032 | No | 1 | BDV | 12255 | 25.2 | 32.7 | 22.3 | 19.8 | 54.9 | |
| 15 | Foot-and-mouth disease virus C | ss-RNA | NC_002554 | No | 1 | FMV | 8115 | 25.6 | 24.8 | 21.2 | 28.5 | 45.9 | |
| 16 | Igbo Ora virus | ss-RNA | NC_001924 | No | 1 | IOV | 11821 | 24.1 | 31.1 | 20.9 | 24.0 | 51.9 | |
| 17 | Poliovirus | ss-RNA | NC_002058 | No | 1 | PV1 | 7440 | 23.0 | 29.7 | 24.0 | 23.3 | 53.7 | |
| 18 | Rabbit hemorrhagic disease virus | ss-RNA | NC_001543 | No | 1 | RHV | 7437 | 25.5 | 25.9 | 23.9 | 24.7 | 49.8 | |
| 19 | Tamana bat virus | ss-RNA | NC_003996 | No | 1 | TBV | 10053 | 21.5 | 33.2 | 28.3 | 16.9 | 61.6 | |
| 20 | Yellow fever virus | ss-RNA | NC_002031 | No | 1 | YFV | 10862 | 0.28 | 0.27 | 0.23 | 0.21 | 0.50 | |
| ssRNA negative-strand viruses | 21 | Avian paramyxovirus 6 | ss-RNA | NC_003043 | No | 1 | APV | 16236 | 0.23 | 0.29 | 0.25 | 0.23 | 0.54 |
| 22 | Bovine ephemeral fever virus | ss-RNA | NC_002526 | No | 1 | BFV | 14900 | 0.20 | 0.38 | 0.28 | 0.14 | 0.66 | |
| 23 | Bovine respiratory syncytial virus | ss-RNA | NC_001989 | No | 1 | BRV | 15140 | 0.17 | 0.38 | 0.29 | 0.17 | 0.66 | |
| 24 | Canine distemper virus | ss-RNA | NC_001921 | No | 1 | CDV | 15690 | 0.22 | 0.31 | 0.26 | 0.21 | 0.57 | |
| 25 | Human respiratory syncytial virus | ss-RNA | NC_001781 | No | 1 | HRV | 15225 | 0.16 | 0.39 | 0.28 | 0.18 | 0.67 | |
| 26 | Hantaan virus | ss-RNA | AF345636 | Yes | 2 | HV1 | 11772 | 0.21 | 0.33 | 0.29 | 0.17 | 0.62 | |
| 27 | Influenza B virus | ss-RNA | NC_002208 | Yes | 8 | IBV | 14452 | 0.22 | 0.36 | 0.24 | 0.18 | 0.60 | |
| 28 | Measles virus | ss-RNA | NC_001498 | No | 1 | MV1 | 15894 | 0.24 | 0.29 | 0.23 | 0.24 | 0.53 | |
| 29 | Respiratory syncytial virus | ss-RNA | NC_001803 | No | 1 | RSV | 15191 | 0.16 | 0.39 | 0.28 | 0.18 | 0.67 | |
| 30 | Reston Ebola virus | ss-RNA | NC_004161 | No | 1 | REV | 18891 | 0.20 | 0.31 | 0.28 | 0.21 | 0.59 | |
| 31 | Tioman virus | ss-RNA | NC_004074 | No | 1 | TV2 | 15522 | 0.21 | 0.30 | 0.26 | 0.22 | 0.57 | |
The information about 31 RNA viruses being investigated in this study. Their accession number, abbreviation, genome size, number of segments and whether they undergo DNA stage are tabulated. The breakdown of the RNA nucleic acids and A+U contents are also shown.
From the standpoint of the overall genomic composition analysis, the G+C content is an interesting property for a genome, in that the overall content often correlates with the organism pathogenicity [12]. Most of the pathogens genomes have a low G+C content, while some such as Mycobacterium tuberculosis has a relatively high G+C content. Therefore, as expected in Table 1, we noted that most of the pathogenic viruses are A+U-rich (>50%), except for Porcine reproductive and Respiratory syndrome Virus (PRV), Equine arteritis virus (EV1), Rabbit hemorrhagic disease virus (RHV), Simian hemorrhagic Fever Virus (SFV) and Foot-and-Mouth disease Virus C (FMV).
Di-nucleotide bias
The frequencies of occurrence for di-nucleotides were compared to the random RNA counterparts having the same base proportion in order to compute the z value that reflected their di-nucleotide bias (Table 2). Among the 31 virus sequences examined, the frequencies of occurrence for di-nucleotide were not randomly distributed, with only a few exceptional di-nucleotides starting with a purine residue present at the expected frequencies (ApC, ApG, GpC, |z| < 3). A remarkable deviation from the expected frequencies occurs for the di-nucleotide pairs CpG and UpA (suppression or under-representation, z < -50) as well as di-nucleotides pairs CpA and UpG (enhancement or over-representation, z > 40). These di-nucleotide biases, together with mono-nucleotide bias [13], have a direct impact on the codon usage of viruses. For example, in the codon usage for the 24 protein coding sequences in human CoronaVirus 229E (Table 3), only 2.85% of codons contain the under-represented subword CpG di-nucleotide whereas 11.26% of the codons contain the over-represented CpA di-nucleotide (the aggregate codon usage containing each di-nucleotide subword without mono- and di-nucleotide bias is close to 6.25%).
Table 2.
Di-nucleotide bias for six RNA viruses.
| BCoV | MHV | SARS | ABV | HCoV | PDV |
Average z value across 31 viruses |
|||||||||||||
| Di-nucleotide | N(w) | E(w) | z | N(w) | E(w) | z | N(w) | E(w) | z | N(w) | E(w) | z | N(w) | E(w) | z | N(w) | E(w) | z | |
| CG | 497 | 1034 | -103.81 | 798 | 1342 | -104.39 | 566 | 1235 | -121-14 | 486 | 976 | -109.69 | 487 | 979 | -95.94 | 684 | 1226 | -102.03 | -77.31 |
| GC | 1344 | 1037 | 62.19 | 1694 | 1341 | 62.33 | 1432 | 1236 | 36.12 | 1147 | 970 | 35.96 | 1164 | 976 | 37.86 | 1416 | 1228 | 35.05 | 5.74 |
| AU | 2845 | 3007 | -26.44 | 2499 | 2614 | -19.25 | 2234 | 2594 | -58.91 | 2200 | 2642 | -76.99 | 2092 | 2556 | -81.97 | 1976 | 2296 | -55.43 | -15.54 |
| UA | 2818 | 3000 | -30.10 | 2404 | 2616 | -35.12 | 2080 | 2594 | -87.64 | 2409 | 2641 | -42.42 | 2033 | 2554 | -84.51 | 1965 | 2299 | -53.83 | -52.48 |
| AG | 1824 | 1848 | -4.25 | 1968 | 1941 | 4.77 | 1749 | 1760 | -2.00 | 1844 | 1728 | 21.13 | 1416 | 1601 | -34.78 | 1537 | 1579 | -7.12 | 3.80 |
| GA | 1629 | 1849 | -39.08 | 1745 | 1941 | -32.74 | 1677 | 1764 | -16.43 | 1505 | 1730 | -39.47 | 1397 | 1598 | -36.09 | 1358 | 1581 | -38.05 | -1.33 |
| AC | 1371 | 1303 | 12.93 | 1384 | 1458 | -13.50 | 1978 | 1695 | 50.18 | 1474 | 1292 | 35.28 | 1558 | 1236 | 58.96 | 1594 | 1332 | 50.25 | 5.42 |
| CA | 1594 | 1297 | 56.03 | 1705 | 1453 | 46.19 | 2203 | 1695 | 87.29 | 1603 | 1290 | 59.90 | 1638 | 1234 | 74.68 | 1783 | 1327 | 83.96 | 49.99 |
| CU | 1801 | 1674 | 22.52 | 1874 | 1806 | 12.28 | 2190 | 1814 | 67.50 | 1661 | 1487 | 31.50 | 1724 | 1568 | 28.13 | 1953 | 1784 | 29.95 | 16.50 |
| UC | 1179 | 1674 | -88.35 | 1296 | 1802 | -94.30 | 1552 | 1815 | -46.36 | 1127 | 1482 | -65.41 | 1130 | 1568 | -79.37 | 1410 | 1781 | -67.80 | -17.49 |
| GU | 2449 | 2394 | 9.10 | 2473 | 2402 | 11.92 | 1868 | 1898 | -5.35 | 2154 | 1982 | 29.46 | 2240 | 2044 | 34.60 | 2262 | 2119 | 23.86 | -7.13 |
| UG | 3101 | 2392 | 120.25 | 3146 | 2408 | 128.13 | 2663 | 1897 | 137.30 | 2476 | 1983 | 87.74 | 2898 | 2040 | 152.24 | 2814 | 2117 | 126.99 | 65.79 |
The di-nucleotide bias in six RNA viruses. The z value quantifies the di-nucleotide bias as defined in equation 1. N (w) and E (w) are actual and expected frequency of occurrence for word w. The last column is the average z value across 31 RNA viruses.
Table 3.
Codon usage for Human CoronaVirus 229E (HCoV).
| Amino Acid | Codon | Usage/% | Amino Acid | Codon | Usage/% |
| Arg | CGU | 1.04 | Ile | AUU | 3.34 |
| CGC | 0.41 | AUC | 0.74 | ||
| CGA | 0.17 | AUA | 1.35 | ||
| CGG | 0.13 | Gly | GGU | 4.12 | |
| AGA | 1.23 | GGC | 1.43 | ||
| AGG | 0.36 | GGA | 0.67 | ||
| Leu | UUA | 1.49 | GGG | 0.22 | |
| UUG | 2.96 | Val | GUU | 6.00 | |
| CUU | 2.48 | GUC | 1.23 | ||
| CUC | 0.46 | GUA | 1.09 | ||
| CUA | 0.65 | GUG | 1.90 | ||
| CUG | 0.63 | Lys | AAA | 3.15 | |
| Ser | UCU | 2.70 | AAG | 2.31 | |
| UCC | 0.66 | Asn | AAU | 4.15 | |
| UCA | 1.37 | AAC | 1.82 | ||
| UCG | 0.20 | Gln | CAA | 2.04 | |
| AGU | 1.86 | CAG | 1.17 | ||
| AGC | 0.71 | his | CAU | 1.14 | |
| Thr | ACU | 3.23 | CAC | 0.46 | |
| ACC | 0.76 | Glu | GAA | 2.81 | |
| ACA | 2.21 | GAG | 1.21 | ||
| ACG | 0.29 | Asp | GAU | 3.09 | |
| Pro | CCU | 1.6S | GAC | 1.96 | |
| CCC | 0.35 | Tyr | UAU | 3.00 | |
| CCA | 1.07 | UAC | 1.46 | ||
| CCG | 0.19 | Cys | UGU | 2.26 | |
| Ala | GCU | 3.58 | UGC | 0.95 | |
| GCC | 0.83 | Phe | UUU | 4.59 | |
| GCA | 1.80 | UUC | 1.10 | ||
| GCG | 0.42 |
The relative usage of synonymous codons in the 24 known CDSs of Human Corona Virus 229E (HCoV).
In double stranded DNA genomes the deficiency in di-nucleotide CpG is often supposed to be due to the fact that they are the targets for methyltransferase activity that leads to cytosine deamination [14,15]. It is however unlikely that the mechanism of deamination that alters the genetic contents at the DNA level would affect the viral RNA content of most RNA viruses without a DNA stage. There might exist specific cytosine RNA methylases that could be responsible for this effect [16]. However it is more consistent to propose that, unlike the mechanism of cytosine deamination in the DNA realm, the dominating process is cytosine deamination in RNA viruses, converting cytosine to uracil (C ♦ U) instead of thymine (T). As a consequence of this mechanism, di-nucleotide CpG changes to either di-nucleotide UpG or CpA in the direct/complementary strands of RNA viruses and causes the over-representation in di-nucleotide UpG and CpA (z > 19). Interestingly, there is experimental evidence in vitro that the rate of cytosine deamination is faster (>100 times) in the single stranded than in double-stranded state [17]. Apart from the under-representation in di-nucleotide CpG and over-representation in di-nucleotide CpA and UpG, the reason for the observed di-nucleotide UpA scarcity in RNA may be explained by its chemical lability [18]. The UpA dinucleotide is chemically the most unstable among the 16 dinucleotides. Furthermore, UpA appears to be a preferential target for ribonucleases [19]. This lability would create a selection pressure against di-nucleotide UpA in RNA viruses.
If we choose a critical value for z (|z| = 3.29) that only allows a chance of 1 in 1000 error for classifying a word as biased (over/under-represented), all di-nucleotides show some kind of bias in their usage pattern across 31 different viruses (Table 4, derived from the complete form of Table 2 provided as the additional file 1). The causes for these biases await further investigation.
Table 4.
Overall statistics for biased di-nucleotides and tetra-nucleotides.
| Percentage of di-nucleotide that can be used to discriminate between vi ruses(|z| > 3.29) | Percentage of tetra-nucleotide that can be used to discriminate between vi ruses(|z| > 3.29) | Virus | Percentage of biased di-nucleotide (|z| > 3.29)/% | Percentage of biased tetra-nucleotide (|z| > 3.29)/% |
| 100% | 96.09% | BCoV | 93.8 | 29.7 |
| MHV | 93.8 | 28.1 | ||
| SARS | 81.3 | 34.4 | ||
| ABV | 81.3 | 27.3 | ||
| HCoV | 93.8 | 31.3 | ||
| PDV | 81.3 | 28.5 | ||
| TGV | 87.5 | 31.6 | ||
| LDV | 93.8 | 19.5 | ||
| PRV | 93.8 | 15.6 | ||
| SFV | 93.8 | 16.0 | ||
| FCoV | 75.0 | 11.7 | ||
| EV1 | 87.5 | 14.5 | ||
| TBV | 75.0 | 21.9 | ||
| AEV | 93.8 | 11.7 | ||
| PV1 | 87.5 | 11.7 | ||
| YFV | 93.8 | 29.3 | ||
| BDV | 87.5 | 17.6 | ||
| RHV | 93.8 | 9.4 | ||
| FMV | 87.5 | 12.1 | ||
| IOV | 75.0 | 9.8 | ||
| HV1 | 62.5 | 12.5 | ||
| RSV | 87.5 | 18.8 | ||
| HRV | 87.5 | 19.1 | ||
| BRV | 93.8 | 19.9 | ||
| TV2 | 81.3 | 15.2 | ||
| REV | 87.5 | 18.4 | ||
| MV1 | 81.3 | 15.2 | ||
| CDV | 75.0 | 16.0 | ||
| APV | 93.8 | 11.7 | ||
| BFV | 81.3 | 15.2 | ||
| IBV | 87.5 | 23.4 |
The percentage of biased di-nucleotides and tetra-nucleotides that shows strong biases (lzl > 3.29) in 31 RNA viruses (right). For di-nucleotides, all 16 (100%) of them show strong biases in part of or all 31 RNA viruses. For tetra-nucleotides, 246 (96%) of the tetra-nucleotides show strong biases in part of or all 31 RNA viruses.
Tetra-nucleotide bias
Inspection of the tetra-nucleotide usage pattern for RNA viruses (additional file 2) reveals considerable differences. The frequencies of occurrence for tetra-nucleotides were compared to artificial chromosomes constructed as random RNA sequences having the same nucleotide succession up to order three to compute the z values that reflect their tetra-nucleotide bias in the corresponding virus (Table 5). If we choose a critical value for z (|z| = 3.29) that only allows a chance of 1 in 1000 error for classifying a word as over/under-represented, 96% of the tetra-nucleotides show a strong bias in their usage pattern across 31 viruses (shown in Table 4, derived from the complete form of Table 5 provided as the additional file 1). This indicated strongly that tetra-nucleotides are being used in a different manner between different viruses, providing us with a tool to study the relationships between viruses based on the tetra-nucleotide bias exhibited in their genomes.
Table 5.
Tetra-nucleotide bias for three RNA viruses. The tetra-nucleotide bias in three viruses. z value quantifies the tetra-nucleotide bias, as defined in equation (1). N (w) and E (w) are actual and expected frequency of occurrence for word w.
| BCoV | MHV | SARS | BCoV | MHV | SARS | ||||||||||||||
| Tetra-nucleotide | N(w) | E(w) | z | N(w) | E(w) | z | N(w) | E(w) | z | Tetra-nucleotide | N(w) | E(w) | z | N(w) | E(w) | z | N(w) | E(w) | z |
| AAAA | 148 | 206.2 | -7.4 | 147 | 145.8 | 0.2 | 222 | 216.5 | 0.7 | UAAA | 264 | 226.2 | 4.6 | 187 | 170.9 | 2.2 | 170 | 183 | -1.7 |
| AAAC | 110 | 103.7 | 1.1 | 98 | 91.2 | 1.3 | 154 | 148.1 | 0.9 | UAAC | 78 | 105.1 | -4.8 | 85 | 122.4 | -6.1 | 123 | 128 | -0.8 |
| AAAG | 184 | 169.7 | 2.0 | 173 | 133.6 | 6.2 | 165 | 158.8 | 0.9 | UAAG | 205 | 171.7 | 4.6 | 193 | 165.3 | 3.9 | 107 | 134.4 | -4.3 |
| AAAU | 217 | 220 | -0.4 | 179 | 164.9 | 2.0 | 213 | 200.6 | 1.6 | UAAU | 322 | 309.9 | 1.3 | 245 | 259.8 | -1.7 | 166 | 193.9 | -3.6 |
| AACA | 133 | 114.1 | 3.2 | 113 | 112.1 | 0.2 | 215 | 175.7 | 5.4 | UACA | 178 | 163.7 | 2.0 | 122 | 123.4 | -0.2 | 230 | 200.3 | 3.8 |
| AACC | 76 | 61.3 | 3.4 | 107 | 75.2 | 6.7 | 102 | 92.5 | 1.8 | UACC | 97 | 82.9 | 2.8 | 106 | 98.5 | 1.4 | 118 | 97.1 | 3.8 |
| AACG | 29 | 40.7 | -3.3 | 35 | 61.8 | -6.2 | 44 | 66.3 | -5.0 | UACG | 50 | 54.4 | -1.1 | 58 | 72.6 | -3.1 | 46 | 63.3 | -3.9 |
| AACU | 91 | 121.5 | -5.0 | 84 | 106.5 | -4.0 | 171 | 168.5 | 0.4 | UACU | 196 | 205.2 | -1.2 | 153 | 168.2 | -2.1 | 195 | 192.2 | 0.4 |
| AAGA | 172 | 157.9 | 2.0 | 176 | 136.4 | 6.2 | 184 | 161.8 | 3.2 | UAGA | 128 | 123.4 | 0.8 | 119 | 124.7 | -0.9 | 102 | 119.6 | -2.9 |
| AAGC | 137 | 103.8 | 5.9 | 140 | 103 | 6.6 | 96 | 112.8 | -2.9 | UAGC | 79 | 98.7 | -3.6 | 82 | 118.7 | -6.1 | 71 | 84.8 | -2.7 |
| AAGG | 133 | 121.3 | 1.9 | 159 | 122.4 | 6.0 | 140 | 117.3 | 3.8 | UAGG | 73 | 78.8 | -1.2 | 67 | 121.3 | -9.0 | 74 | 75.6 | -0.3 |
| AAGU | 191 | 180.6 | 1.4 | 179 | 163.1 | 2.3 | 136 | 139.4 | -0.5 | UAGU | 171 | 213 | -5.2 | 161 | 190.7 | -3.9 | 101 | 126.8 | -4.2 |
| AAUA | 189 | 215.2 | -3.2 | 148 | 182.1 | -4.6 | 113 | 154.1 | -6.0 | UAUA | 251 | 237 | 1.7 | 192 | 189 | 0.4 | 99 | 136.5 | -5.8 |
| AAUC | 100 | 104.5 | -0.8 | 75 | 93.2 | -3.4 | 93 | 121.9 | -4.8 | UAUC | 84 | 112.3 | -4.9 | 86 | 99.9 | -2.5 | 84 | 116.1 | -5.4 |
| AAUG | 246 | 229.3 | 2.0 | 234 | 232.1 | 0.2 | 230 | 201.5 | 3.7 | UAUG | 310 | 271.5 | 4.3 | 278 | 238.1 | 4.7 | 189 | 190.3 | -0.2 |
| AAUU | 265 | 265.5 | -0.1 | 212 | 207.8 | 0.5 | 211 | 212 | -0.1 | UAUU | 314 | 345 | -3.0 | 253 | 248.5 | 0.5 | 190 | 211.8 | -2.7 |
| ACAA | 144 | 137.1 | 1.1 | 115 | 114.1 | 0.2 | 269 | 204.1 | 8.3 | UCAA | 131 | 130 | 0.2 | 136 | 117.7 | 3.1 | 202 | 174.1 | 3.8 |
| ACAC | 84 | 66.4 | 3.9 | 88 | 75.4 | 2.6 | 168 | 142.2 | 3.9 | UCAC | 53 | 60.4 | -1.7 | 57 | 67.1 | -2.2 | 130 | 109.6 | 3.5 |
| ACAG | 118 | 105.7 | 2.2 | 108 | 104.9 | 0.5 | 151 | 145.1 | 0.9 | UCAG | 107 | 122.1 | -2.5 | 105 | 106.7 | -0.3 | 110 | 121.4 | -1.9 |
| ACAU | 128 | 123.5 | 0.7 | 106 | 122.7 | -2.7 | 186 | 172.9 | 1.8 | UCAU | 84 | 124.6 | -6.6 | 88 | 117.8 | -5.0 | 153 | 146.7 | 0.9 |
| ACCA | 105 | 76.9 | 5.8 | 116 | 85 | 6.1 | 161 | 117.7 | 7.3 | UCCA | 68 | 73.4 | -1.1 | 74 | 80.6 | -1.3 | 76 | 95.4 | -3.6 |
| ACCC | 56 | 37.3 | 5.6 | 84 | 57.6 | 6.3 | 54 | 60.7 | -1.6 | UCCC | 31 | 37.4 | -1.9 | 45 | 56 | -2.7 | 31 | 44.6 | -3.7 |
| ACCG | 24 | 35.5 | -3.5 | 52 | 57.4 | -1.3 | 31 | 48.8 | -4.6 | UCCG | 15 | 26 | -3.9 | 37 | 55.1 | -4.4 | 19 | 29.7 | -3.5 |
| ACCU | 83 | 77.7 | 1.1 | 97 | 97.7 | -0.1 | 139 | 111.2 | 4.8 | UCCU | 74 | 101.4 | -4.9 | 103 | 102.4 | 0.1 | 80 | 107.1 | -4.8 |
| ACGA | 32 | 44.5 | -3.4 | 31 | 56.5 | -6.2 | 40 | 64.4 | -5.5 | UCGA | 18 | 41.4 | -6.6 | 42 | 50.4 | -2.2 | 43 | 67.5 | -5.4 |
| ACGC | 29 | 34.1 | -1.6 | 49 | 52.5 | -0.9 | 31 | 54.8 | -5.8 | UCGC | 30 | 34.1 | -1.3 | 45 | 54 | -2.2 | 38 | 56.2 | -4.4 |
| ACGG | 26 | 31.3 | -1.7 | 46 | 48.5 | -0.7 | 26 | 41 | -4.2 | UCGG | 19 | 29.1 | -3.4 | 33 | 49.8 | -4.3 | 16 | 39.9 | -6.9 |
| ACGU | 47 | 60.5 | -3.1 | 53 | 72.1 | -4.1 | 53 | 72.6 | -4.2 | UCGU | 51 | 74.1 | -4.9 | 59 | 74.8 | -3.3 | 73 | 84.4 | -2.3 |
| ACUA | 141 | 127.9 | 2.1 | 119 | 121.3 | -0.4 | 166 | 167.3 | -0.2 | UCUA | 116 | 130.2 | -2.3 | 115 | 124.5 | -1.5 | 130 | 140.9 | -1.7 |
| ACUC | 49 | 68.2 | -4.2 | 61 | 68.9 | -1.7 | 119 | 115 | 0.7 | UCUC | 52 | 67.1 | -3.3 | 69 | 69.6 | -0.1 | 82 | 108.4 | -4.6 |
| ACUG | 144 | 131.5 | 2.0 | 126 | 141.4 | -2.4 | 159 | 163.3 | -0.6 | UCUG | 119 | 135.5 | -2.6 | 117 | 135.5 | -2.9 | 133 | 141.4 | -1.3 |
| ACUU | 142 | 160.9 | -2.7 | 116 | 132.1 | -2.5 | 207 | 184.9 | 3.0 | UCUU | 195 | 191.8 | 0.4 | 153 | 142.8 | 1.6 | 219 | 182.6 | 4.9 |
| AGAA | 147 | 141.6 | 0.8 | 162 | 126.6 | 5.7 | 144 | 158.7 | -2.1 | UGAA | 174 | 195.3 | -2.8 | 154 | 176.8 | -3.1 | 164 | 180.2 | -2.2 |
| AGAC | 67 | 71.9 | -1.1 | 87 | 80.6 | 1.3 | 114 | 117.2 | -0.5 | UGAC | 86 | 101.8 | -2.8 | 118 | 127.6 | -1.5 | 153 | 151.9 | 0.2 |
| AGAG | 107 | 88.9 | 3.5 | 115 | 103.4 | 2.1 | 146 | 112.3 | 5.8 | UGAG | 96 | 127.7 | -5.1 | 144 | 167 | -3.2 | 117 | 136.2 | -3.0 |
| AGAU | 177 | 170.4 | 0.9 | 158 | 145.2 | 1.9 | 128 | 141.1 | -2.0 | UGAU | 314 | 311.7 | 0.2 | 243 | 261 | -2.0 | 215 | 196.5 | 2.4 |
| AGCA | 113 | 105.8 | 1.3 | 102 | 112.1 | -1.7 | 105 | 118.8 | -2.3 | UGCA | 181 | 166.3 | 2.1 | 187 | 161.5 | 3.6 | 166 | 182 | -2.2 |
| AGCC | 77 | 54.1 | 5.7 | 91 | 80.7 | 2.1 | 68 | 55.4 | 3.1 | UGCC | 102 | 81.6 | 4.1 | 144 | 122.4 | 3.6 | 114 | 81.6 | 6.5 |
| AGCG | 48 | 44.9 | 0.8 | 62 | 71.2 | -2.0 | 32 | 46.4 | -3.8 | UGCG | 52 | 65.5 | -3.0 | 86 | 97.6 | -2.1 | 58 | 58.5 | -0.1 |
| AGCU | 126 | 122.6 | 0.6 | 132 | 132 | 0.0 | 140 | 146.9 | -1.0 | UGCU | 270 | 218.4 | 6.4 | 254 | 226.8 | 3.3 | 315 | 224.5 | 11.0 |
| AGGA | 116 | 96.4 | 3.6 | 114 | 117.8 | -0.6 | 138 | 99.5 | 7.0 | UGGA | 171 | 154.4 | 2.4 | 187 | 147.4 | 5.9 | 152 | 126 | 4.2 |
| AGGC | 65 | 61.9 | 0.7 | 114 | 104.6 | 1.7 | 92 | 80.3 | 2.4 | UGGC | 144 | 103.8 | 7.2 | 184 | 144.4 | 6.0 | 141 | 103.4 | 6.7 |
| AGGG | 55 | 59.1 | -1.0 | 88 | 79.5 | 1.7 | 53 | 60.7 | -1.8 | UGGG | 81 | 105 | -4.3 | 90 | 118.9 | -4.8 | 59 | 74.4 | -3.2 |
| AGGU | 137 | 143.8 | -1.0 | 128 | 150 | -3.3 | 129 | 119.5 | 1.6 | UGGU | 307 | 302.3 | 0.5 | 260 | 236.3 | 2.8 | 200 | 173.5 | 3.7 |
| AGUA | 137 | 159.2 | -3.2 | 124 | 155.6 | -4.6 | 115 | 116.9 | -0.3 | UGUA | 228 | 233.8 | -0.7 | 202 | 215.5 | -1.7 | 161 | 165.6 | -0.6 |
| AGUC | 62 | 77.6 | -3.2 | 75 | 93.9 | -3.5 | 76 | 87.3 | -2.2 | UGUC | 116 | 116.5 | -0.1 | 159 | 143.6 | 2.3 | 141 | 129.8 | 1.8 |
| AGUG | 152 | 156.2 | -0.6 | 187 | 173.8 | 1.8 | 127 | 120.7 | 1.0 | UGUG | 266 | 246.9 | 2.2 | 300 | 255.8 | 5.0 | 214 | 170.9 | 6.0 |
| AGUU | 222 | 239.6 | -2.1 | 214 | 206 | 1.0 | 126 | 161.7 | -5.1 | UGUU | 498 | 407.8 | 8.1 | 415 | 346.1 | 6.7 | 274 | 252 | 2.5 |
| AUAA | 228 | 220.7 | 0.9 | 189 | 188.5 | 0.1 | 129 | 152.4 | -3.4 | UUAA | 322 | 269.8 | 5.8 | 258 | 235.5 | 2.7 | 195 | 202.8 | -1.0 |
| AUAC | 124 | 129.2 | -0.8 | 100 | 112.8 | -2.2 | 100 | 132.3 | -5.1 | UUAC | 185 | 173.5 | 1.6 | 158 | 155.6 | 0.3 | 186 | 179.8 | 0.8 |
| AUAG | 120 | 141.9 | -3.3 | 120 | 135 | -2.3 | 65 | 91.7 | -5.1 | UUAG | 141 | 177.5 | -5.0 | 131 | 183 | -7.0 | 112 | 119.5 | -1.2 |
| AUAU | 205 | 237.9 | -3.9 | 151 | 185.9 | -4.7 | 99 | 144.4 | -6.9 | UUAU | 397 | 385.6 | 1.1 | 309 | 269.1 | 4.4 | 191 | 226.5 | -4.3 |
| AUCA | 105 | 122 | -2.8 | 77 | 99.2 | -4.1 | 139 | 136.8 | 0.3 | UUCA | 127 | 155 | -4.1 | 132 | 126.1 | 1.0 | 206 | 180 | 3.5 |
| AUCC | 59 | 62.9 | -0.9 | 63 | 65.5 | -0.6 | 54 | 65.9 | -2.7 | UUCC | 66 | 80.1 | -2.9 | 75 | 90.9 | -3.0 | 71 | 87.9 | -3.3 |
| AUCG | 31 | 46.9 | -4.2 | 42 | 57.2 | -3.7 | 31 | 59.6 | -6.7 | UUCG | 33 | 55.3 | -5.5 | 64 | 69.3 | -1.2 | 56 | 71.8 | -3.4 |
| AUCU | 108 | 129.1 | -3.4 | 87 | 109.2 | -3.9 | 108 | 137.1 | -4.5 | UUCU | 193 | 201.6 | -1.1 | 133 | 151.2 | -2.7 | 226 | 189.5 | 4.8 |
| AUGA | 204 | 212.3 | -1.0 | 203 | 202.8 | 0.0 | 189 | 198.6 | -1.2 | UUGA | 237 | 239.3 | -0.3 | 197 | 213.4 | -2.0 | 189 | 186.8 | 0.3 |
| AUGC | 186 | 151.1 | 5.2 | 194 | 164.2 | 4.2 | 179 | 154.3 | 3.6 | UUGC | 197 | 174.1 | 3.2 | 188 | 184.4 | 0.5 | 185 | 162.5 | 3.2 |
| AUGG | 211 | 180.8 | 4.1 | 197 | 179.4 | 2.4 | 185 | 143.1 | 6.4 | UUGG | 213 | 230.6 | -2.1 | 208 | 185.6 | 3.0 | 153 | 143.2 | 1.5 |
| AUGU | 296 | 273.3 | 2.5 | 275 | 269.9 | 0.6 | 218 | 197.4 | 2.7 | UUGU | 415 | 363.3 | 4.9 | 368 | 298 | 7.4 | 245 | 204.3 | 5.2 |
| AUUA | 239 | 253.9 | -1.7 | 191 | 216 | -3.1 | 190 | 192 | -0.3 | UUUA | 407 | 345.3 | 6.0 | 303 | 257.1 | 5.2 | 204 | 204.1 | 0.0 |
| AUUC | 106 | 126.1 | -3.3 | 100 | 110.2 | -1.8 | 127 | 136.8 | -1.5 | UUUC | 141 | 161.3 | -2.9 | 109 | 146.1 | -5.6 | 187 | 162.4 | 3.5 |
| AUUG | 245 | 253.5 | -1.0 | 206 | 211.6 | -0.7 | 208 | 176 | 4.4 | UUUG | 367 | 357.7 | 0.9 | 318 | 271.3 | 5.2 | 207 | 194.8 | 1.6 |
| AUUU | 361 | 337.8 | 2.3 | 287 | 251.6 | 4.1 | 197 | 205.6 | -1.1 | UUUU | 454 | 495.8 | -3.4 | 296 | 325 | -2.9 | 215 | 245.2 | -3.5 |
| GAAA | 118 | 124 | -1.0 | 104 | 111.4 | -1.3 | 142 | 140.8 | 0.2 | CAAA | 128 | 133.8 | -0.9 | 160 | 108.5 | 9.0 | 221 | 182.3 | 5.2 |
| GAAC | 58 | 64.6 | -1.5 | 63 | 75.1 | -2.5 | 89 | 96.9 | -1.5 | CAAC | 83 | 71 | 2.6 | 93 | 74.9 | 3.8 | 166 | 128.5 | 6.0 |
| GAAG | 136 | 125.8 | 1.7 | 153 | 123.2 | 4.9 | 126 | 117.7 | 1.4 | CAAG | 108 | 111 | -0.5 | 135 | 111.5 | 4.0 | 158 | 132.5 | 4.0 |
| GAAU | 118 | 140.8 | -3.5 | 119 | 142.4 | -3.6 | 90 | 125.7 | -5.8 | CAAU | 144 | 147.5 | -0.5 | 126 | 139.1 | -2.0 | 178 | 170.2 | 1.1 |
| GACA | 82 | 83 | -0.2 | 99 | 91 | 1.5 | 162 | 128.5 | 5.4 | CACA | 82 | 78.3 | 0.8 | 84 | 80.6 | 0.7 | 168 | 150.9 | 2.5 |
| GACC | 37 | 39.2 | -0.6 | 61 | 69.1 | -1.8 | 66 | 68.5 | -0.5 | CACC | 59 | 44 | 4.1 | 76 | 59.3 | 3.9 | 98 | 80.1 | 3.6 |
| GACG | 27 | 33 | -1.9 | 46 | 51 | -1.3 | 33 | 52.9 | -5.0 | CACG | 28 | 31.5 | -1.1 | 40 | 46 | -1.6 | 27 | 50.4 | -6.0 |
| GACU | 82 | 86.6 | -0.9 | 88 | 94.7 | -1.3 | 101 | 115.3 | -2.4 | CACU | 108 | 75.3 | 6.8 | 97 | 93.1 | 0.7 | 184 | 151.4 | 4.8 |
| GAGA | 73 | 77.8 | -1.0 | 104 | 100.3 | 0.7 | 105 | 111.8 | -1.2 | CAGA | 125 | 106.4 | 3.3 | 123 | 98.1 | 4.6 | 141 | 128.8 | 2.0 |
| GAGC | 52 | 60.1 | -1.9 | 66 | 90.9 | -4.8 | 83 | 74.9 | 1.7 | CAGC | 96 | 71.9 | 5.2 | 99 | 82.8 | 3.2 | 95 | 96.6 | -0.3 |
| GAGG | 73 | 68.6 | 1.0 | 112 | 108.6 | 0.6 | 95 | 89.1 | 1.1 | CAGG | 94 | 84.9 | 1.8 | 106 | 93.4 | 2.4 | 102 | 93.4 | 1.6 |
| GAGU | 103 | 100.9 | 0.4 | 128 | 134 | -0.9 | 108 | 98.6 | 1.7 | CAGU | 108 | 128.7 | -3.3 | 132 | 127.4 | 0.7 | 98 | 127.2 | -4.7 |
| GAUA | 149 | 172.1 | -3.2 | 127 | 145.4 | -2.8 | 81 | 111 | -5.2 | CAUA | 88 | 110.8 | -3.9 | 92 | 112.5 | -3.5 | 99 | 118.2 | -3.2 |
| GAUC | 70 | 86.1 | -3.2 | 63 | 73 | -2.1 | 55 | 75.7 | -4.3 | CAUC | 49 | 56.5 | -1.8 | 45 | 67.1 | -4.9 | 100 | 91.9 | 1.5 |
| GAUG | 231 | 209.7 | 2.7 | 237 | 199.5 | 4.8 | 198 | 159 | 5.6 | CAUG | 110 | 117.8 | -1.3 | 119 | 143.9 | -3.8 | 153 | 138.3 | 2.3 |
| GAUU | 205 | 201.7 | 0.4 | 159 | 176.8 | -2.4 | 125 | 128.6 | -0.6 | CAUU | 166 | 149 | 2.5 | 160 | 159 | 0.1 | 196 | 173 | 3.2 |
| GCAA | 104 | 114.7 | -1.8 | 137 | 123.3 | 2.2 | 133 | 131 | 0.3 | CCAA | 84 | 81.4 | 0.5 | 126 | 90.8 | 6.7 | 119 | 107.2 | 2.1 |
| GCAC | 70 | 65.3 | 1.1 | 77 | 74.2 | 0.6 | 99 | 92 | 1.3 | CCAC | 71 | 41.7 | 8.2 | 74 | 55.9 | 4.4 | 81 | 77.7 | 0.7 |
| GCAG | 131 | 102.4 | 5.1 | 157 | 113.3 | 7.5 | 80 | 100.5 | -3.7 | CCAG | 67 | 64.5 | 0.6 | 90 | 80.7 | 1.9 | 95 | 77.5 | 3.6 |
| GCAU | 120 | 109.4 | 1.8 | 128 | 140.9 | -2.0 | 112 | 104.2 | 1.4 | CCAU | 81 | 71.9 | 1.9 | 94 | 97.4 | -0.6 | 97 | 91 | 1.1 |
| GCCA | 84 | 57.8 | 6.2 | 111 | 97.4 | 2.5 | 99 | 76.5 | 4.7 | CCCA | 46 | 41.6 | 1.2 | 83 | 62.7 | 4.7 | 56 | 60.1 | -1.0 |
| GCCC | 34 | 34.5 | -0.2 | 75 | 63.8 | 2.5 | 35 | 36.6 | -0.5 | CCCC | 28 | 22.2 | 2.2 | 43 | 47.5 | -1.2 | 18 | 28.8 | -3.6 |
| GCCG | 29 | 29.7 | -0.2 | 51 | 60.5 | -2.2 | 21 | 31.7 | -3.4 | CCCG | 17 | 20.4 | -1.4 | 45 | 39.4 | 1.6 | 16 | 20.1 | -1.6 |
| GCCU | 84 | 66.8 | 3.8 | 122 | 106.2 | 2.8 | 75 | 71.9 | 0.7 | CCCU | 58 | 43.9 | 3.8 | 76 | 78 | -0.4 | 48 | 60.7 | -3.0 |
| GCGA | 30 | 38.9 | -2.6 | 42 | 57.3 | -3.7 | 36 | 43.5 | -2.1 | CCGA | 25 | 27.9 | -1.0 | 45 | 47.1 | -0.6 | 16 | 36.2 | -6.0 |
| GCGC | 31 | 31.7 | -0.2 | 65 | 57.4 | 1.8 | 38 | 41 | -0.8 | CCGC | 20 | 21.8 | -0.7 | 50 | 47 | 0.8 | 21 | 32.1 | -3.6 |
| GCGG | 21 | 31.7 | -3.4 | 43 | 56.8 | -3.3 | 23 | 29.6 | -2.2 | CCGG | 11 | 20.9 | -3.9 | 36 | 44.9 | -2.4 | 13 | 21.2 | -3.2 |
| GCGU | 63 | 55.9 | 1.7 | 87 | 82.1 | 1.0 | 47 | 52.9 | -1.5 | CCGU | 29 | 38.2 | -2.7 | 54 | 68.1 | -3.1 | 37 | 41.8 | -1.3 |
| GCUA | 165 | 131.3 | 5.4 | 162 | 144.3 | 2.7 | 153 | 140.7 | 1.9 | CCUA | 85 | 77 | 1.7 | 83 | 96.5 | -2.5 | 104 | 88.6 | 3.0 |
| GCUC | 58 | 58.8 | -0.2 | 75 | 80.5 | -1.1 | 89 | 98.1 | -1.7 | CCUC | 38 | 40.1 | -0.6 | 79 | 58.6 | 4.8 | 63 | 65.2 | -0.5 |
| GCUG | 136 | 131.5 | 0.7 | 187 | 173.4 | 1.9 | 196 | 145.3 | 7.6 | CCUG | 89 | 80.4 | 1.7 | 118 | 108.1 | 1.7 | 70 | 89 | -3.7 |
| GCUU | 167 | 147.3 | 3.0 | 158 | 162.5 | -0.6 | 180 | 149.5 | 4.5 | CCUU | 86 | 97.4 | -2.1 | 119 | 113.3 | 1.0 | 105 | 104.6 | 0.1 |
| GGAA | 86 | 82.1 | 0.8 | 83 | 103.4 | -3.7 | 103 | 86 | 3.3 | CGAA | 23 | 42.7 | -5.5 | 40 | 58.5 | -4.4 | 37 | 55.5 | -4.5 |
| GGAC | 51 | 48.4 | 0.7 | 57 | 67.7 | -2.4 | 68 | 72.3 | -0.9 | CGAC | 24 | 22.5 | 0.6 | 32 | 34.3 | -0.7 | 27 | 46.9 | -5.3 |
| GGAG | 81 | 66.6 | 3.2 | 109 | 95.9 | 2.4 | 92 | 70.1 | 4.8 | CGAG | 17 | 29.5 | -4.2 | 42 | 53.5 | -2.9 | 35 | 49.8 | -3.8 |
| GGAU | 122 | 127 | -0.8 | 139 | 124.7 | 2.3 | 80 | 83.7 | -0.7 | CGAU | 41 | 63.1 | -5.0 | 46 | 63.3 | -4.0 | 36 | 56 | -4.9 |
| GGCA | 93 | 70 | 5.0 | 142 | 99.4 | 7.7 | 108 | 83.7 | 4.8 | CGCA | 38 | 40.7 | -0.8 | 67 | 63.3 | 0.8 | 46 | 58.2 | -2.9 |
| GGCC | 34 | 33.7 | 0.1 | 74 | 74.8 | -0.2 | 33 | 39.1 | -1.8 | CGCC | 19 | 17.1 | 0.8 | 50 | 45.6 | 1.2 | 15 | 27.1 | -4.2 |
| GGCG | 28 | 32.2 | -1.3 | 57 | 62.9 | -1.4 | 33 | 40.5 | -2.1 | CGCG | 17 | 14.9 | 1.0 | 32 | 39.2 | -2.1 | 21 | 23.4 | -0.9 |
| GGCU | 95 | 88.7 | 1.2 | 135 | 117.9 | 2.9 | 115 | 94.9 | 3.8 | CGCU | 36 | 44.4 | -2.3 | 61 | 73.5 | -2.7 | 46 | 74.1 | -5.9 |
| GGGA | 38 | 53 | -3.7 | 52 | 65.7 | -3.1 | 36 | 48.5 | -3.3 | CGGA | 15 | 26.4 | -4.0 | 35 | 51 | -4.1 | 18 | 33.8 | -4.9 |
| GGGC | 20 | 37.4 | -5.1 | 64 | 68.8 | -1.1 | 36 | 38.3 | -0.7 | CGGC | 21 | 19 | 0.8 | 45 | 47.2 | -0.6 | 20 | 29.3 | -3.1 |
| GGGG | 26 | 41.9 | -4.5 | 23 | 53.8 | -7.6 | 20 | 31.4 | -3.7 | CGGG | 10 | 19.9 | -4.0 | 27 | 39.1 | -3.5 | 12 | 17.5 | -2.4 |
| GGGU | 88 | 95 | -1.3 | 88 | 100.4 | -2.2 | 52 | 63.8 | -2.7 | CGGU | 31 | 50.2 | -4.9 | 52 | 67.2 | -3.4 | 28 | 55.5 | -6.7 |
| GGUA | 147 | 153.8 | -1.0 | 113 | 130.8 | -2.8 | 106 | 102.8 | 0.6 | CGUA | 55 | 53.6 | 0.3 | 52 | 71.6 | -4.2 | 52 | 60.2 | -1.9 |
| GGUC | 51 | 70.4 | -4.2 | 61 | 76.8 | -3.3 | 40 | 71.3 | -6.7 | CGUC | 16 | 24.9 | -3.2 | 29 | 41.9 | -3.6 | 36 | 41.9 | -1.6 |
| GGUG | 160 | 161.8 | -0.3 | 179 | 171.9 | 1.0 | 135 | 119.9 | 2.5 | CGUG | 60 | 64.9 | -1.1 | 84 | 90.6 | -1.3 | 69 | 71.3 | -0.5 |
| GGUU | 205 | 201.3 | 0.5 | 175 | 181.2 | -0.8 | 127 | 123.2 | 0.6 | CGUU | 59 | 83.2 | -4.8 | 88 | 104.4 | -2.9 | 53 | 81.6 | -5.8 |
| GUAA | 165 | 174.4 | -1.3 | 135 | 160.2 | -3.6 | 101 | 130.5 | -4.7 | CUAA | 154 | 145.5 | 1.3 | 128 | 140 | -1.8 | 141 | 153.8 | -1.9 |
| GUAC | 99 | 109.2 | -1.8 | 86 | 110.2 | -4.2 | 143 | 109 | 5.9 | CUAC | 112 | 88.1 | 4.6 | 95 | 87.5 | 1.5 | 160 | 140.2 | 3.0 |
| GUAG | 112 | 118.4 | -1.1 | 104 | 136.9 | -5.1 | 96 | 88.6 | 1.4 | CUAG | 78 | 86.6 | -1.7 | 74 | 103.6 | -5.3 | 75 | 99.2 | -4.4 |
| GUAU | 191 | 195.4 | -0.6 | 166 | 172 | -0.8 | 94 | 118.6 | -4.1 | CUAU | 163 | 148.3 | 2.2 | 182 | 150.6 | 4.7 | 177 | 162 | 2.1 |
| GUCA | 85 | 95.2 | -1.9 | 105 | 98.5 | 1.2 | 114 | 113.3 | 0.1 | CUCA | 59 | 75 | -3.4 | 73 | 82.5 | -1.9 | 137 | 130.2 | 1.1 |
| GUCC | 30 | 52.2 | -5.6 | 59 | 73.4 | -3.1 | 35 | 52.7 | -4.4 | CUCC | 33 | 41 | -2.3 | 62 | 58.7 | 0.8 | 46 | 62.3 | -3.7 |
| GUCG | 33 | 39.6 | -1.9 | 35 | 59 | -5.7 | 40 | 51.3 | -2.9 | CUCG | 21 | 32 | -3.5 | 39 | 46 | -1.9 | 43 | 59.9 | -4.0 |
| GUCU | 97 | 109.1 | -2.1 | 125 | 120.1 | 0.8 | 104 | 112.9 | -1.5 | CUCU | 84 | 82 | 0.4 | 110 | 94.8 | 2.8 | 126 | 136.9 | -1.7 |
| GUGA | 122 | 162.3 | -5.8 | 152 | 163.3 | -1.6 | 131 | 127 | 0.6 | CUGA | 107 | 124.3 | -2.8 | 108 | 139.4 | -4.8 | 141 | 150.6 | -1.4 |
| GUGC | 113 | 115.2 | -0.4 | 149 | 148.5 | 0.1 | 130 | 110.9 | 3.3 | CUGC | 109 | 91.9 | 3.2 | 141 | 110.9 | 5.2 | 159 | 128.2 | 4.9 |
| GUGG | 158 | 146.3 | 1.8 | 180 | 158.8 | 3.1 | 109 | 101 | 1.4 | CUGG | 121 | 98.1 | 4.2 | 136 | 123.2 | 2.1 | 106 | 104.8 | 0.2 |
| GUGU | 245 | 218.3 | 3.3 | 269 | 223.8 | 5.5 | 174 | 129.3 | 7.2 | CUGU | 151 | 157.5 | -0.9 | 164 | 179.7 | -2.1 | 152 | 159.3 | -1.1 |
| GUUA | 255 | 244.6 | 1.2 | 237 | 225.1 | 1.4 | 126 | 168.7 | -6.0 | CUUA | 143 | 152.3 | -1.4 | 125 | 150.3 | -3.8 | 164 | 163.1 | 0.1 |
| GUUC | 104 | 123 | -3.1 | 119 | 116.6 | 0.4 | 97 | 114.8 | -3.0 | CUUC | 68 | 80.6 | -2.5 | 76 | 81.3 | -1.1 | 148 | 124.3 | 3.9 |
| GUUG | 280 | 254.7 | 2.9 | 283 | 248 | 4.0 | 165 | 169.1 | -0.6 | CUUG | 168 | 147.9 | 3.0 | 154 | 152.9 | 0.2 | 190 | 154.7 | 5.2 |
| GUUU | 344 | 316.4 | 2.8 | 253 | 239.5 | 1.6 | 192 | 171 | 2.9 | CUUU | 211 | 212.4 | -0.2 | 191 | 177.1 | 1.9 | 209 | 183.8 | 3.4 |
Approach one – Sequence Relationship of Viruses based on The Correlation of Tetra-nucleotide Bias
Two relationship trees were derived, one from the entire genome and the other from the replication enzyme (Figure 1). The result based on the replication enzyme sequence was included because these regions in RNA viruses are submitted to a strong selective pressure to ensure successful replication of their own RNA in the host cell. The two distance trees can be clustered distinctly into two major groups of viruses. Interestingly, this clustering validates our approach, since these clusters are consistent with biological properties of the viruses: Group #1 corresponds to all positive strand ssRNA viruses while Group #2 corresponds to negative strand ssRNA viruses. Each group must undergo different evolutionary paths which lead to their distinct pattern in tetra-nucleotide usage. The classification for the two main groups of viruses (positive/negative strand ssRNA viruses) demonstrate a level of congruence with the taxonomy of the viruses [20] and indicated that there exists a relationship signal in tetra-nucleotide usage patterns.
Figure 1.

Two Relationship trees based on the correlation coefficients of tetra-nucleotide usage bias The distance tree for 31 RNA viruses based on tetra-nucleotide usage pattern for the entire genome (right) and the replication enzyme (left). The correlation distances are shown on top of each branch.
Inside both relationship trees, Avian Encephalomyelitis Virus (AEV), Lactate Dehydrogenase-elevating Virus (LDV), Porcine Reproductive and respiratory syndrome Virus (PRV), Equine arteritis Virus (EV1), Rabbit Hemorrhagic disease Virus (RHV), Yellow Fever Virus (YFV), are the outermost group of viruses, exhibiting differences in their tetra-nucleotide usage pattern. From the family of positive strand ssRNA viruses, CoronaViruses form the largest cluster. The SARS-CoV is found to be at the basal position of other CoronaVirus types and remains closest to the Transmissible Gastroenteritis Virus (TGV) and Feline CoronaVirus (FCoV). This placement is consistent with the findings from two seminal papers [9,10] where the SARS-CoV was classified in a separate group from the rest of the known CoronaViruses. In addition, both distance trees suggested that the Bovine CoronaVirus (BCoV) and the Mouse Hepatitis Virus (MHV) should be grouped together whereas the Human CoronaVirus 229E (HCoV) is the closest to the Porcine epidemic Diarrhea Virus (PDV). For the family of negative strand ssRNA viruses, there are two obvious classes that have evolved through different branches of word usage pattern. The first class covers Hantaan Virus (HV1), Reston Ebola Virus (REV), Bovine Ephemeral Fever Virus (BFV), Bovine Respiratory syncytial Virus (BRV), Respiratory Syncytial Virus (RSV) and Human Respiratory syncytial Virus (HRV). The second class covers the remaining negative strand ssRNA viruses.
Approach two – Sequence Relationship of Viruses based on The Factors of the Tetra-nucleotide Usage Pattern [21-23]
The overall tetra-nucleotide usage pattern (additional file 2) was decomposed into several eigen-vectors using a factor analysis algorithm. They are the uncorrelated components of the original usage pattern embedded within the overall tetra-nucleotide usage pattern. Three eigen-vectors, which carry 83.3% of the variance for the viral tetra-nucleotide usage patterns, were retained (Figure 2). From the three dimensional figures (Figure 3, Figure 4, Figure 5 and Figure 6) plotted against these retained eigen-vectors, the negative strand ssRNA viruses stemmed clearly out from the positive strand ssRNA viruses. This is most obvious when the axes of projection were the 1st and 3rd eigen-vectors. This indicated that both types of viruses have a complex component of tetra-nucleotide usage patterns and that these patterns changes with different family of viruses.
Figure 2.

Relationship between the number of eigen-vectors retained and the percentage of the variance they represent in the entire usage patterns for 31 viruses. As each consecutive factor is defined to identify a usage pattern that is not captured by the preceding eigen-vectors, each consecutive factors are therefore independent of each other. In addition, the order for the consecutive eigen-vectors is extracted with diminishing importance.
Figure 3.

3-D plot for the vectorial profiling of each virus onto the three eigen-vectors. The tetra-nucleotide usage patterns V for the replicase open reading frame in each virus have been redisplayed on the 1st, 2nd and 3rd eigen-vector axes ('o' represents positive strand ssRNA virus; x represents negative strand ssRNA virus). The two families of viruses clustered into two different regions of the plot.
Figure 4.

2-D plots for Figure 3 with different viewpoint specifications. The tetra-nucleotide usage patterns for the replicase open reading frame in each virus have been redisplayed on the (1st vs 2nd), (1st vs 3rd) and (2nd vs 3rd) eigen-vector axes ('o' represents positive strand ssRNA virus; 'x' represents negative strand ssRNA virus). For the top figure, the order for 'o' is [15,17,12,16,8,14,9,11,13,4,7,3,10,6,2,5,1]* (left to right), whereas 'x' is [24,27,25,28,22,26,18,23,20,19,21]* (left to right). For the middle figure, the order for 'o' is [15,17,12,16,8,14,9,11,13,4,7,3,10,6,2,5,1]* (left to right), whereas 'x' is [24,27,25,28,22,26,18,23,20,19,21]* (left to right). For the bottom figure, the order for 'o' is [15,17,12,16,8,14,9,11,13,4,7,3,10,6,2,5,1]* (left to right), whereas 'x' is [24,27,25,28,22,26,18,23,20,19,21]* (left to right). *The corresponded virus for each number follows Figure 3.
Figure 5.

3-D plot for the vectorial profiling of each virus onto the three eigen-vectors. The tetra-nucleotide usage patterns table in the additional file 2 (entire genome) for each virus have been redisplayed on the 1st, 2nd and 3rd eigen-vector axes ('o' represents positive strand ssRNA virus; 'x' represents negative strand ssRNA virus). The two families of viruses clustered into three different regions of the plot.
Figure 6.

2-D plots for Figure 5 with different viewpoint specifications. The tetra-nucleotide usage patterns table in the additional file 2 (entire genome) for each virus have been redisplayed on the (1st vs 2nd), (1st vs 3rd) and (2nd vs 3rd) eigen-vector axes ('o' represents positive strand ssRNA virus, 'x' represents negative strand ssRNA virus). For the top figure, the order for 'o' is [3,7,1,4,2,5,6,17,13,20,10,16,9,8,11,15,12,14,18,19]* (left to right), whereas 'x' is [26,22,30,23,24,28,31,27,25,29,21]* (left to right). For the middle figure, the order for 'o' is [3,7,1,4,2,5,6,17,13,20,10,16,9,8,11,15,12,14,18,19]* (left to right), whereas 'x' is [26,22,30,23,24,28,31,27,25,29,21]* (left to right). For the bottom figure, the order for 'o' is [3,7,1,4,2,5,6,17,13,20,10,16,9,8,11,15,12,14,18,19]* (left to right), whereas 'x' is [26,22,30,23,24,28,31,27,25,29,21]* (left to right). *The corresponded virus for each number follows Figure 5.
In the result based on replication enzyme sequence (Figure 3 and Figure 4), we observed a clear splitting between two main families of RNA viruses (positive/negative strand ssRNA virus). All viruses that belong to a specific family were clustered together closely. This pointed to an interesting hypothesis that the replication enzyme sequence between closely related RNA viruses adopt a common word usage pattern that are closely linked. In addition, it is clear that the viruses from different family groups adopt different strategy of word usage.
However in Figure 5 and Figure 6, when we project the tetra-nucleotide usage patterns (entire genome) for each virus on the 1st, 2nd and 3rd eigen-vector axes, the separation between viruses showed a different outcome when V was derived from the entire genome. The two main families of viruses were grouped into three clusters, two being allocated to the positive strand ssRNA viruses. It is particularly interesting that all viruses in the upper left corner corresponded to the viruses originating from the CoronaVirus family. Unexpectedly, the Hantaan Virus (HV1) is the only negative strand ssRNA virus to have a high loading on the eigen-vector that corresponded to the tetra-nucleotide usage pattern for the positive strand ssRNA viruses.
It is important to realize what factor analysis will provide and how this analysis is different from the previous method of relationship tree generation using correlation coefficient. For the previous method that is based on correlation coefficient of word usage patterns, it treats the vectorial profiling V for each virus as a whole entity, However, the factor analysis considered the vectorial profiling V as a superposition of many patterns which can be separated into mutually uncorrelated patterns of word usage. Each eigen-vector represents the embedded component of RNA word usage patterns communalised by a group of viruses presumably under the same selection pressures. By projecting the overall usage patterns on these eigen-vectors, it is possible to determine a group of viruses that adopt a common strategy of word usage.
Conclusion
Using the two approaches to study the tetra-nucleotide usage pattern in RNA viruses, we reached the following conclusions:
1. Based on the correlation of the overall tetra-nucleotide usage patterns, the Transmissible Gastroenteritis Virus (TGV) and the Feline CoronaVirus (FCoV) are closest to SARS-CoV.
2. Based on the three most significant eigen-vectors, the genomes of the viruses from the same family conform to a similar tetra-nucleotide usage pattern, irrespective of their genome size.
3. The study of word usage is a powerful method to classify RNA viruses. The congruence of the relationship trees with the known classification indicates that there exist phylogenetic signals in tetra-nucleotide usage patterns, and this signal is most prominent in the replicase open reading frames.
Methods
Dataset
We focused our study on the genomic sequences (their translated strand) of ssRNA viruses (Table 1), which incorporated 20 species from the family of positive strand ssRNA viruses and 11 species from the family of negative strand ssRNA viruses. We are aware of the fact that these viruses constitute completely different species, most probably unrelated to one another. They are included in a common study in order to try to have means to identify relevant features from purely statistical background properties. The coverage included the viruses that are known to cause diseases to their corresponding hosts. The acronym for each virus is shown in the table and is referred to throughout this study. All sequences corresponding to their translated strand were retrieved from GenBank, and the accession numbers and genomic size (in nucleotides) for individual virus were provided for reference. For the present study, two sets of data were generated from the complete sequence for each virus. Dataset 1 covered the entire genome and dataset 2 covered only their replicase open reading frame. The flowchart for studying the tetra-nucleotide usage pattern in 31 viruses is shown in Figure 7.
Figure 7.

Flowchart for studying the tetra-nucleotide usage pattern. The FA and NJ algorithms stand for factor analysis [21-23] and neighbor joining [29] algorithm.
Computer hardware and software
Sun Fire 6800 Server with 24 CPUs (each running with a clock speed of 900 MHz) was employed throughout this study. The computation of correlation coefficient and factor analysis algorithm were implemented using Matlab Technical Programming language.
Method for counting the frequency of occurrence for RNA words
It is necessary to address the question of how we counted the number of time each tetra-nucleotide (for example 'GAGA' or any other tetra-nucleotide), appeared in a given genome. For this study, we adopted the convention of not counting overlapping words [24]. Take a sequence "UAUGAGAGAUCCGAGA' as example. With second or higher overlapping words not counted, the tetra-nucleotide 'GAGA' is counted as occurring only twice, namely in position 4–7 and 13–16. Positions 6–9 are omitted because they overlap with 'GAGA' at position 4–7.
However, when we counted tetra-nucleotide 'UGAG', position 3–6 would also be registered as position 4–6 already recorded when counting tetra-nucleotide 'GAGA'. In short, all frequency counting of tetra-nucleotide were started anew when we changed from counting the frequency of one tetra-nucleotide to another; this was to preserve the correlation of tetra-nucleotides which have overlapping subword (e.g: 'UAGA' and 'GACA'). A table showing the frequencies of tetra-nucleotides is shown in the additional file 2.
Vectorial profiling (V) of the viral RNA genome word usage pattern
The nucleotide composition has being suggested to be a specific characteristic in different virus phylogeny [25]. Because most viral genomes are short, and because we lack a prior information on the tempo and modes of evolution of RNA viruses, we proceeded as follows. We created a vector, V = [C1,C2, ... Ci, ... Ck], with each element representing the frequency for a specific RNA word of length n. The number of components (k) in V increases exponentially with word size (n) - k = 4n. In order to use V for discrimination between viruses, two criteria must be met. First, V must contain sufficient components (di-nucleotide k = 16; tri-nucleotide k = 64; tetra-nucleotide k = 256); second, the frequencies for tetra-nucleotides must show a prominent bias (over/under-representation) that is unique for a family of viruses.
For the first criteria, there are pros and cons for choosing either longer or shorter words. When the shorter words are used, they inherit the problem of inadequate representation of the viral genome because the long motifs will be neglected. But the shorter words have an advantage of saving computational time. On the other hand, when the longer words are used, they cause a problem of computer tractability due to a larger word set to explore (k = 4n). However, the larger words have an advantage of accounting for the correlation of their sub-words. In contrast the number of their occurrences falls down rapidly, preventing accurate statistical analysis. We chose tetra-nucleotides for our study because they provide 256 vector components (additional file 2) and account for correlation of sub-words up to the order three.
For the second criteria, the bias in RNA word usage was examined. The bias in word usage (of size n) is influenced by the bias of word with sizes less than n [26]. Therefore, in order to evaluate the true bias of word size m, it is required to compare the frequencies of word usage in the original sequence to that of model chromosomes that take into account the biases of word size m - 1, m - 2 ... 1. These model chromosomes were generated by obeying the Markov model of the order (m - 1)th. This can be achieved by shuffling m - 1 viral nucleotides as one whole unit so that the nucleotide successions up to order (m - 1)th were being preserved. Several statistical approaches have been proposed for quantifying word biases [27,28]. In this study, we employed the z statistics (Equation 1) for di-nucleotide and tetra-nucleotide biases [27,28]. The z value is a measure of the bias of a word, with values close to zero meaning no bias, negative values meaning under-representation and positive values meaning over-representation of the word w in the RNA text.
![]()
where w is a word of size m; N(w) is observed count in actual viral RNA; E(w) and Var(w) are expected count and variance for w derived from the 100 artificial chromosomes that preserved the nucleotide succession up to order m - 1.
Approach one – sequence relationship of viruses based on the correlation of tetra-nucleotide bias
A scale-invariant parameter, the correlation coefficient r, was employed to compare between word usage patterns of viruses. The correlation coefficient r measures the degree of linear relationship between two vectors. Here, the two vectors are the tetra-nucleotide word usage pattern V corresponding to each viral genome. The magnitude of r would indicate how much of the change of pattern in the tetra-nucleotide word usage in one virus is explained by the change in another. The magnitude of r is always between -1 and +1 and the relationship between the two variables will approach perfect linearity as the magnitude of correlation coefficient approaches to extreme values (+/-1). However, perfect positive correlation (r = 1) does not mean identity of the paired Vi, but, rather, identity up to positive linearity, that is, identity between the paired standardized values. This is a crucial property of r (scale-invariant) that enables the comparison of viral genome despite their differences in genomic sizes. Positive magnitude of r indicates positive association whereas negative magnitude of r indicates negative association between two usage patterns. For this study, correlation coefficient, r, for let say virus 1 and virus 2, is defined as follow:
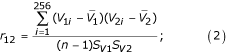
where V1, V2 are vector representing the tetra-nucleotide usage pattern; SV1 and SV2 standard deviation of V1, V2; 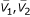 are the mean of V1, V2.
are the mean of V1, V2.
Then, the distance between the tetra-nucleotide usage patterns of two viruses is defined as follows:
Distance Dij = 1 - rij; (3)
where Dij is the distance between the tetra-nucleotide usage patterns of virus i and virus j; rij is the correlation coefficient between the tetra-nucleotide usage patterns of virus i and virus j
Prior to the construction of a relationship tree, the pair-wise distance matrix M of size 31 by 31 was constructed (see additional file 3). Pair-wise distance between two viral genomes is measured by the value of (1 - r). Each row/column corresponds to a specific virus and an entry at the intersection of row X and column Y corresponds to the distance between virus X and virus Y. Such matrix has a diagonal entry of value 0. For the purpose of constructing a relationship tree, only the lower/upper triangular matrix of M is required. After obtaining lower/upper triangular matrix of M, the neighbor-joining method (NJ) algorithm was used to construct the relationship tree (Figure 1). The neighbor-joining method is based on minimum-distance principle. Details of the NJ algorithm are available in [29].
Approach two – sequence relationship of viruses based on the factors of the tetra-nucleotide usage pattern
The factor analysis is a statistical method that reveals simpler patterns within a complex set of tetra-nucleotide usage patterns V (additional file 2). It seeks to discover if the observed usage patterns can be explained in terms of a much smaller number of un-correlated pattern sets called factors (eigen-vectors). Suppose we take a simple case where there are 31 viruses each represented by two components (x,y) in vector V (x,y represent the frequencies of occurrence for two specific tetra-nucleotides). Then, in a scatter-plot we can think of the regression line as the original X-axis, rotated so that it approximates the regression line. This type of rotation maximize the variance of the variables (x,y) on the eigen-vector. The remaining variability around this the first eigen-vector was captured in the subsequent eigen-vectors. In this manner, consecutive eigen-vectors are extracted but with a diminishing importance. What each eigen-vector represents is the embedded RNA word usage patterns communalised by a group of viruses presumably under the same selection pressures.
We implemented the factor analysis algorithm [21-23] in Matlab Technical Programming Language and computed a set of eigen-vectors. Then, the original usage pattern V was re-mapped for each virus onto the new coordinate system based on these derived eigen-vectors. The difference between approach two and approach one is discussed in the results and discussion section.
Authors' contributions
YLY participated in the design and performed the statistical analysis.
AD participated in the design and overall coordination of this study.
XWZ participated in the design of the study.
All authors read and approved the final manuscript.
Supplementary Material
The RNA word biases of different sizes in RNA viruses. These tables show the di-nucleotide, tetra-nucleotide and penta-nucleotide biases for 31 RNA viruses.
Vectorial profiling of tetra-nucleotide usage pattern in seven RNA viruses. The tetra-nucleotide frequencies of occurrence in seven viral genomes. Each column represents a tetra-nucleotide usage pattern Vi for a single virus. We derived correlation coefficient (r) by comparing any two columns simultaneously. This parameter r indicates the likeness of word usage patterns in any two viruses.
The distance matrices. Each entry in matrix M is computed using Equation 3. The correlation coefficient (r) in equation 3 is obtained by comparing any two columns in the tetra-nucleotide usage patterns table in the additional file 2 simultaneously.
Acknowledgments
Acknowledgements
Indispensable support was provided by the doctoral fellowship from The University of Hong Kong (HKU). We wish to thank the Hong Kong Innovation and Technology Fund for supporting work upstream of the present study, that made it possible at a time when the unexpected SARS outbreak reached Hong Kong. Finally, we wish to thank Dr Ralf Altmeyer for his critical interest for this work as he came at the head of the HKU-Pasteur Research Centre.
Contributor Information
Yee Leng Yap, Email: daniely@hkusua.hku.hk.
Xue Wu Zhang, Email: xwzhang@hkucc.hku.hk.
Antoine Danchin, Email: adanchin@pasteur.fr.
References
- Cumulative Number of Reported Probable Cases of SARS http://www.who.int/csr/sarscountry/en/
- Fouchier RA, Kuiken T, Schutten M, Van Amerongen G, Van Doornum GJ, Van Den Hoogen BG, Peiris M, Lim W, Stohr K, Osterhaus AD. Aetiology: Koch's postulates fulfilled for SARS virus. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey J, Maskalyk J. SARS update. CMAJ. 2003;168:1294–5. [PMC free article] [PubMed] [Google Scholar]
- James JS. SARS Web information. AIDS Treat News. 2003. p. 6. [PubMed]
- Situation Updates – SARS http://www.who.int/csr/sars/archive/en/
- Drosten C, Gunther S, Preiser W, Van Der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. Identification of a Novel CoronaVirus in Patients with Severe Acute Respiratory Syndrome. N Engl J Med. 2003;348:1967–76. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Van Vugt JJ, Storgaard T, Oleksiewicz MB, Botner A. High frequency RNA recombination in porcine reproductive and respiratory syndrome virus occurs preferentially between parental sequences with high similarity. J Gen Virol. 2001;82:2615–20. doi: 10.1099/0022-1317-82-11-2615. [DOI] [PubMed] [Google Scholar]
- Lerner DL, Wagaman PC, Phillips TR, Prospero-Garcia O, Henriksen SJ, Fox HS, Bloom FE, Elder JH. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc Natl Acad Sci USA. 92:7480–4. doi: 10.1073/pnas.92.16.7480. 1995 Aug 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, BeRNArd K, Booth TF, Bowness D, Drebot M, FeRNAndo L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. The Genome Sequence of the SARS-Associated CoronaVirus. Science. 2003;300:1399–404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rassmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. Characterization of a Novel CoronaVirus Associated with Severe Acute Respiratory Syndrome. Science. 2003;300:1394–9. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ. A Novel CoronaVirus Associated with Severe Acute Respiratory Syndrome. N Engl J Med. 2003;348:1953–66. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2001;2:376–81. doi: 10.1093/embo-reports/kve097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hemert FJ, Berkhout B. The tendency of lentiviral open reading frames to become A-rich: constraints imposed by viral genome organization and cellular tRNA availability. J Mol Evol. 1995;41:132–40. doi: 10.1007/BF00170664. [DOI] [PubMed] [Google Scholar]
- Hubacek J. Biological function of DNA methylation. Folia Microbiol (Praha) 1992;37:323–9. doi: 10.1007/BF02815658. [DOI] [PubMed] [Google Scholar]
- Karlin S, Doerfler W, Cardon LR. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J Virol. 1994;68:2889–97. doi: 10.1128/jvi.68.5.2889-2897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantt RR, Stromberg KJ, Montes de Oca F. Specific RNA methylase associated with avian myeloblastosis virus. Nature. 1971;234:35–37. doi: 10.1038/234035a0. [DOI] [PubMed] [Google Scholar]
- Frederico LA, Kunkel TA, Shaw BR. A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry. 2001;29:2532–7. doi: 10.1021/bi00462a015. [DOI] [PubMed] [Google Scholar]
- Bibillo A, Figlerowicz M, Ziomek K, Kierzek R. The nonenzymatic hydrolysis of oligoribonucleotides. VII. Structural elements affecting hydrolysis. Nucleosides Nucleotides Nucleic Acids. 2000;19:977–94. doi: 10.1080/15257770008033037. [DOI] [PubMed] [Google Scholar]
- Beutler E, Gelbart T, Han JH, Koziol JA, Beutler B. Evolution of the genome and the genetic code: selection at the dinucleotide level by methylation and polyribonucleotide cleavage. Proc Natl Acad Sci USA. 1989;86:192–6. doi: 10.1073/pnas.86.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regenmortel MHV, van Fauquet CM, Bishop DHL, Carstens EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, McGeoch DJ, Pringle CR, Wickner RB. Virus Taxonomy: Classification and Nomenclature of Viruses Seventh Report of the International Committee on Taxonomy of Viruses. Academic Press, San Diego; 2000. [Google Scholar]
- Bartholomew DJ. Factor Analysis for Categorical Data, Journal of the Royal Statistical Society. Series B (Methodological) 1980. pp. 293–321.
- Kim J, Mueller Charles W. Introduction to factor analysis: What it is and how to do it. Newbury Park, CA: Sage Publications; 1978. [Google Scholar]
- Bartholomew DJ. Factor Analysis for Categorical Data, Journal of the Royal Statistical Society. Series B (Methodological) 1980. pp. 293–321.
- Ewens WJ, Grant GR. Statistical Methods in Bioinformatics. Springer-Verlag New York, Inc., New York; 2001. [Google Scholar]
- Bronson EC, Anderson JN. Nucleotide composition as a driving force in the evolution of retroviruses. J Mol Evol. 1994;38:506–32. doi: 10.1007/BF00178851. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Viari A, Danchin A. Oligo-nucleotide bias in Bacillus subtilis: general trends and taxonomic comparisons. Nucleic Acids Res. 1998;26:2971–80. doi: 10.1093/nar/26.12.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung MY, Marsh GM, Speed TP. Over- and underrepresentation of short DNA words in herpesvirus genomes. J Comput Biol. 1996;3:345–60. doi: 10.1089/cmb.1996.3.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schbath S, Prum B, de Turckheim E. Exceptional motifs in different Markov chain models for a statistical analysis of DNA sequences. J Comput Biol. 1995;2:417–37. doi: 10.1089/cmb.1995.2.417. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing trees. Mol Biol and Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The RNA word biases of different sizes in RNA viruses. These tables show the di-nucleotide, tetra-nucleotide and penta-nucleotide biases for 31 RNA viruses.
Vectorial profiling of tetra-nucleotide usage pattern in seven RNA viruses. The tetra-nucleotide frequencies of occurrence in seven viral genomes. Each column represents a tetra-nucleotide usage pattern Vi for a single virus. We derived correlation coefficient (r) by comparing any two columns simultaneously. This parameter r indicates the likeness of word usage patterns in any two viruses.
The distance matrices. Each entry in matrix M is computed using Equation 3. The correlation coefficient (r) in equation 3 is obtained by comparing any two columns in the tetra-nucleotide usage patterns table in the additional file 2 simultaneously.