

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) protein is a cAMP-regulated epithelial Cl− channel that, when defective, causes cystic fibrosis. Screening of a collection of 100,000 diverse small molecules revealed four novel chemical classes of CFTR inhibitors with Ki < 10 μM, one of which (glycine hydrazides) had many active structural analogues. Analysis of a series of synthesized glycine hydrazide analogues revealed maximal inhibitory potency for N-(2-naphthalenyl) and 3,5-dibromo-2,4-dihydroxyphenyl substituents. The compound N-(2-naphthalenyl)-[(3,5-dibromo-2,4-dihydroxyphenyl)methylene]glycine hydrazide (GlyH-101) reversibly inhibited CFTR Cl− conductance in <1 min. Whole-cell current measurements revealed voltage-dependent CFTR block by GlyH-101 with strong inward rectification, producing an increase in apparent inhibitory constant Ki from 1.4 μM at +60 mV to 5.6 μM at −60 mV. Apparent potency was reduced by lowering extracellular Cl− concentration. Patch-clamp experiments indicated fast channel closures within bursts of channel openings, reducing mean channel open time from 264 to 13 ms (−60 mV holding potential, 5 μM GlyH-101). GlyH-101 inhibitory potency was independent of pH from 6.5–8.0, where it exists predominantly as a monovalent anion with solubility ∼1 mM in water. Topical GlyH-101 (10 μM) in mice rapidly and reversibly inhibited forskolin-induced hyperpolarization in nasal potential differences. In a closed-loop model of cholera, intraluminal GlyH-101 (2.5 μg) reduced by ∼80% cholera toxin–induced intestinal fluid secretion. Compared with the thiazolidinone CFTR inhibitor CFTRinh-172, GlyH-101 has substantially greater water solubility and rapidity of action, and a novel inhibition mechanism involving occlusion near the external pore entrance. Glycine hydrazides may be useful as probes of CFTR pore structure, in creating animal models of CF, and as antidiarrheals in enterotoxic-mediated secretory diarrheas.
Keywords: cystic fibrosis, diarrhea, high-throughput screening, patch-clamp, drug discovery
INTRODUCTION
The cystic fibrosis transmembrane conductance regulator (CFTR) protein is a cAMP-activated Cl− channel expressed in secretory and absorptive epithelia in the airways, pancreas, intestine, testis, and other tissues (Pilewski and Frizzell, 1999). Mutations in CFTR that reduce or inhibit its function cause the genetic disease cystic fibrosis (CF), which is characterized by chronic lung infection and pancreatic insufficiency, with progressive deterioration in lung function and death. There is considerable interest in the development and characterization of CFTR inhibitors as probes of the CFTR Cl− transport mechanism, and as reagents to reduce CFTR function in vivo for creation of animal models of CF and as clinical antidiarrheals in CFTR-dependent secretory diarrheas caused by Vibrio cholera and Escherichia coli (Thiagarajah and Verkman, 2003). CFTR inhibitors also have potential applications in polycystic kidney disease, as male contraceptives, and in reducing airway glandular secretions in infectious and allergic bronchitis/rhinitis (Gong et al., 2001; Nakanishi et al., 2001; Thiagarajah et al., 2004b).
Several CFTR inhibitors have been introduced, though most having weak potency and lacking CFTR specificity. The oral hypoglycemic agent glibenclamide inhibits CFTR Cl− conductance from the intracellular side by an open channel blocking mechanism (Sheppard and Robinson, 1997; Zhou et al., 2002) at high micromolar concentrations where it affects other Cl− and cation channels (Sturgess et al., 1988; Rabe et al., 1995; Schultz et al., 1999). Other nonselective anion transport inhibitors, including diphenylamine-2-carboxylate (DPC), 5-nitro-2(3-phenylpropyl-amino)benzoate (NPPB), and flufenamic acid, also inhibit CFTR at high concentrations by occluding the pore at an intracellular site (Dawson et al., 1999; McCarty, 2000).
Our laboratory developed a high-throughput screening assay for discovery of CFTR activators and inhibitors (Galietta et al., 2001). CFTR halide transport function is quantified from the time course of fluorescence in response to an iodide gradient in cells coexpressing a green fluorescent protein–based halide sensor (Jayaraman et al., 2000; Galietta et al., 2001) and wild-type CFTR or a CF-causing CFTR mutant. The assay was used to identify small-molecule activators of wild type and ΔF508-CFTR with activating potencies down to 100 nM (Ma et al., 2002b; Yang et al., 2003). A thiazolidinone class of CFTR inhibitors was identified by screening of a collection of 50,000 small, drug-like molecules (Ma et al., 2002a). The lead compound CFTRinh-172 inhibited CFTR Cl− conductance in a voltage-independent manner, probably by binding to the NBD1 domain at the cytoplasmic surface of CFTR (Ma et al., 2002a; Taddei et al., 2004). In intact cells, CFTR Cl− channel function was 50% inhibited at CFTRinh-172 concentrations of 0.3–3 μM depending on cell type and membrane potential. CFTRinh-172 inhibited intestinal fluid secretion in response to cholera toxin and heat-stable (STa) E. coli toxin in rodents (Thiagarajah et al., 2004a), and resulted in the secretion of viscous, CF-like fluid from submucosal glands in pig and human trachea (Thiagarajah et al., 2004b).
Although thiazolidinones are potentially useful as antidiarrheals and for the creation of CF animal models, they have limited water solubility (∼20 μM) and inhibit CFTR by binding to its cytoplasmic-facing surface, requiring cell penetration with consequent systemic absorption when administered orally. The purpose of this work was to identify CFTR inhibitors with high water solubility that occlude the CFTR pore by binding to a site at its external surface. A low stringency, high-throughput screen of 100,000 small molecules was performed to identify novel chemical scaffolds with CFTR inhibitory activity. We identified several new classes of CFTR inhibitors, one of which was highly water soluble, blocked CFTR by occlusion of the CFTR pore near its external surface, and inhibited CFTR function in vivo in rodent models.
MATERIALS AND METHODS
High-throughput Screening for Identification of CFTR Inhibitors
Screening was performed using an integrated system (Beckman Coulter) consisting of a 3-m robotic arm, CO2 incubator, plate washer, liquid handling work station, barcode reader, delidding station, plate sealer, and two fluorescence plate readers (Optima; BMG Lab Technologies), each equipped with two syringe pumps and HQ500/20X (500 ± 10 nm) excitation and HQ535/30M (535 ± 15 nm) emission filters (Chroma Technology Corp.). 100,000 small molecules (most 250–550 D) were selected for screening from commercial sources (ChemBridge and ChemDiv) using algorithms designed to maximize chemical diversity and drug-like properties. Compounds were stored frozen as 2.5 mM stock solutions in DMSO. Fisher rat thyroid (FRT) cells stably expressing wild-type human CFTR and YFP-H148Q were cultured on 96-well black-wall plates as described previously (Ma et al., 2002b). For screening, cells in 96-well plates were washed three times, and then CFTR halide conductance was activated by incubation for 15 min with an activating cocktail containing 10 μM forskolin, 20 μM apigenin, and 100 μM IBMX. Test compounds (25 μM final) were added 5 min before assay of iodide influx in which cells were exposed to a 100 mM inwardly directed iodide gradient. YFP fluorescence was recorded for 2 s before and 12 s after creation of the iodide gradient. Initial rates of iodide influx were computed from the time course of decreasing fluorescence after the iodide gradient (Yang et al., 2003).
Apical Cl− Current and Short-circuit Current Measurements
FRT, T84, and human airway epithelial cells were cultured on Snapwell filters with 1 cm2 surface area (Corning-Costar) to resistances >1,000 Ω·cm2 as described previously (Ma et al., 2002b). Filters were mounted in an Easymount Chamber System (Physiologic Instruments). For apical Cl− current measurements on FRT cells, the basolateral hemichamber was filled with buffer containing (in mM) 130 NaCl, 2.7 KCl, 1.5 KH2PO4, 1 CaCl2, 0.5 MgCl2, 10 Na-HEPES, 10 glucose (pH 7.3). The basolateral membrane was permeabilized with amphotericin B (250 μg/ml) for 30 min before measurements. In the apical solution, 65 mM NaCl was replaced by sodium gluconate, and CaCl2 was increased to 2 mM. For short-circuit current measurements in (nonpermeabilized) T84 and human airway cells, both hemichambers contained Kreb's solution (in mM): 120 NaCl, 25 NaHCO3, 3.3 KH2PO4, 0.8 K2HPO4, 1.2 MgCl2, 1.2 CaCl2, and 10 glucose (pH 7.3). Solutions were bubbled with 95% O2/5% CO2 and maintained at 37°C. For studies in mouse intestine, ileal segments were isolated, washed with ice-cold Kreb's buffer, opened longitudinally through the mesenteric border, and mounted in a micro-Ussing chamber (0.7 cm2 aperture area; World Precision Instruments). Hemichambers were filled with Kreb's solutions containing 10 μM indomethacin. Apical Cl−/short-circuit current was recorded using a DVC-1000 voltage-clamp (World Precision Instruments) with Ag/AgCl electrodes and 1 M KCl agar bridges.
Patch-clamp Analysis
Patch-clamp experiments were performed at room temperature on FRT cells stably expressing wild-type CFTR. Cell-attached and whole-cell configurations were used (Hamill et al., 1981). The cell membrane was clamped at specified voltages using an EPC-7 patch-clamp amplifier (List Medical). Data were filtered at 500 Hz and digitized at 2000 Hz. For whole-cell experiments, the pipette solution contained (in mM): 120 CsCl, 10 TEA-Cl, 0.5 EGTA, 1 MgCl2, 40 mannitol, 10 Cs-HEPES, and 3 mM MgATP (pH 7.3). For cell-attached experiments, EGTA was replaced with 1 mM CaCl2. The bath solution for whole-cell experiments contained (in mM): 150 NaCl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 mannitol, 10 Na-TES (pH 7.4). In some experiments bath solution NaCl was reduced to 20 mM (mannitol added to maintain osmolality). In cell-attached experiments, the bath solution contained (in mM): 130 KCl, 2 NaCl, 2 CaCl2, 2 MgCl2, 10 glucose, 20 mannitol, and 10 K-Hepes (pH 7.3). Inhibitors were applied by extracellular perfusion. CFTR Cl− channel activity in cell-attached patches was analyzed as described previously (Taddei et al., 2004). The number of CFTR channels present in each patch was estimated as the maximum number of simultaneous channel openings detected in the presence of 5 μM forskolin in a continuous recording of at least three minutes.
Nasal Potential Difference Measurements in Mice
After anesthesia with intraperitoneal ketamine (90–120 mg/kg) and xylazine (5–10 mg/kg), the airway was protected by orotracheal intubation with a 21-gauge angiocatheter as previously described (Salinas et al., 2004). A PE-10 cannula pulled to a tip diameter of 0.3 mm was inserted into one nostril 5 mm distal to the anterior nares and connected though a 1 M KCl agar bridge to a Ag/AgCl electrode and high-impedance digital voltmeter (IsoMillivolt Meter; World Precision Instruments). The nasal cannula was perfused at 50 μL/min using dual microperfusion pumps serially with PBS, low chloride PBS (chloride lowered to 4.7 mM by substitution with gluconate), low chloride PBS containing forskolin (10 μM) without and then with GlyH-101 (10 μM), and then PBS. In some studies GlyH-101 (10 μM) or DIDS (100 μM) was present in all solutions. The reference electrode was a PBS-filled 21-gauge needle inserted in the subcutaneous tissue in the abdomen and connected to a second Ag/AgCl electrode by a 1 M KCl agar bridge.
Intestinal Fluid Secretion Measurements
Mice (CD1 strain, 25–35 g) were deprived of food for 24 h and anaesthetized with intraperitoneal ketamine (40 mg/kg) and xylazine (8 mg/kg). Body temperature was maintained at 36–38°C using a heating pad. Following a small abdominal incision three closed ileal loops (length 20–30 mm) proximal to the cecum were isolated by sutures. Loops were injected with 100 μl of PBS or PBS containing cholera toxin (1 μg) without or with GlyH-101 (2.5 μg). The abdominal incision was closed with suture and mice were allowed to recover from anesthesia. At 4 h the mice were anaesthetized, intestinal loops were removed, and loop length and weight were measured to quantify net fluid secretion. Mice were killed by an overdose of ketamine and xylazine. All protocols were approved by the UCSF Committee on Animal Research.
Synthesis Procedures
Procedures were developed for synthesis of glycine hydrazide analogues (see Table I and Fig. 3 B). All synthesized compounds had >98% purity (TLC/HPLC) and were confirmed by mass and 1H nmr spectrometry (typical data for GlyH-101 given below).
TABLE I.
Structure–Activity Relationships of Glycine Hydrazides (See Fig. 4 for Structures)
| Group 1 | Glycine hydrazides (GlyH, R2 = CH2)Oxamic acid hydrazides (OxaH, R2 = CO) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Active compounds | |||||||||
| Compound | R1 | R2 | R3 | R4 | Ki (μM) | % inhibition at 50 μM | |||
| GlyH-101 | 2-naphthalenyl | CH2 | 3,5-di-Br-2,4-di-OH-Ph | H | 4.3 | 95 | |||
| GlyH-102 | 2-naphthalenyl | CH2 | 3,5-di-Br-4-OH-Ph | H | 4.7 | 98 | |||
| GlyH-103 | 2-naphthalenyl | CH2 | 3,5-di-Br-2-OH-4-OMe-Ph | H | 20 | 56 | |||
| GlyH-104 | 1-naphthalenyl | CH2 | 3,5-di-Br-2,4-di-OH-Ph | H | 12 | 86 | |||
| GlyH-105 | 1-naphthalenyl | CH2 | 3,5-di-Br-4-OH-Ph | H | 15 | 87 | |||
| GlyH-106 | 2-naphthalenyl | CHCH3 | 3,5-di-Br-2,4-di-OH-Ph | H | 6 | 91 | |||
| GlyH-107 | 2-naphthalenyl | CHCH3 | 3,5-di-Br-4-OH-Ph | H | 10 | 80 | |||
| GlyH-108 | 2-naphthalenyl | CH2 | 3,5-di-Br-2,4-di-OH-Ph | CH3 | 10 | 81 | |||
| GlyH-109 | 2-naphthalenyl | CH2 | 3,5-di-Br-4-OH-Ph | CH3 | 4.7 | 100 | |||
| OxaH-110 | 2-naphthalenyl | CO | 3,5-di-Br-2,4-di-OH-Ph | H | 2.7 | 86 | |||
| OxaH-111 | 2-naphthalenyl | CO | 3,5-di-Br-4-OH-Ph | H | 5 | 80 | |||
| OxaH-112 | 2-naphthalenyl | CO | 3,5-di-Br-2,4-di-OH Ph | CH3 | 3 | 95 | |||
| OxaH-113 | 2-naphthalenyl | CO | 3,5-di-Br-4-OH-Ph | CH3 | 3 | 90 | |||
| GlyH-114 | 4-Cl-Ph | CH2 | 3,5-di-Br-4-OH-Ph | H | 5 | 95 | |||
| GlyH-115 | 4-Cl-Ph | CH2 | 3,5-di-Br-2,4-di-OH Ph | H | 6.7 | 91 | |||
| GlyH-116 | 4-Me-Ph | CH2 | 3,5-di-Br-2,4-di-OH Ph | H | 10 | 79 | |||
| Weakly active compounds (Ki ∼ 25–50 μM) | |||||||||
| Compound | R1 | R2 | R3 | R4 | |||||
| GlyH-117 | 2-Me-Ph | CH2 | 3,5-di-Br-2,4-di-OH Ph | H | |||||
| GlyH-118 | 1-naphthalenyl | CH2 | 3-Br-4-OH-Ph | H | |||||
| GlyH-119 | 2-naphthalenyl | CH2 | 2,4-di-OH-Ph | H | |||||
| GlyH-120 | 2-naphthalenyl | CH2 | 4-Br-Ph | H | |||||
| GlyH-121 | 2-naphthalenyl | CH2 | 4-carboxy-Ph | H | |||||
| GlyH-122 | 4-Cl-Ph | CH2 | 3,5-di-Br-2-OH-4-OMe-Ph | H | |||||
| GlyH-123 | 4-Cl-Ph | CH2 | 2,4-di-OH-Ph | H | |||||
| GlyH-124 | 2-anthracenyl | CH2 | 3,5-di-Br-2,4-di-OH Ph | H | |||||
| GlyH-125 | 2-anthracenyl | CH2 | 3,5-di-Br-4-OH-Ph | H | |||||
| GlyH-126 | 6-quinolinyl | CH2 | 3,5-di-Br-2,4-di-OH Ph | H | |||||
| GlyH-127 | 6-quinolinyl | CH2 | 3,5-di-Br-4-OH-Ph | H | |||||
| Inactive compounds (Ki > 50 μM) | |||||||||
| R1 = Ph, Monosubstituted-Ph: Alkyl, halo, alkoxy Disubstituted-Ph: Dihalo, hydroxy+alkoxy Trisubstituted-Ph: Trihalo, dihalo+alkyl |
R2 = CH2 | R3 = Ph, Monosubstituted-Ph: alkyl, halo, alkoxy, aryloxy, aryl, nitro, hydroxy, dialkylamino Disubstituted-Ph: dihalo, dihydroxy, dialkyl, halo+alkyl, hydroxy+alkoxy, dialkoxy Trisubstituted-Ph: alkyl/alkoxy+halo+hydroxy |
|||||||
| Group 2 | |||||||||
| Compound | R1 | R2 | R3 | Ki (μM) | % inhibition at 50 μM | ||||
| GlyH-201 | 2-naphthalenyl | CH2 | 3,5-di-Br-2,4-di-OH Ph | 20 | 65 | ||||
| GlyH-202 | 2-naphthalenyl | CH2 | 3,5-di-Br-4-OH-Ph | 22 | 57 | ||||
| OxaH-203 | 2-naphthalenyl | CO | 3,5-di-Br-2,4-di-OH Ph | >50 | |||||
| OxaH-204 | 2-naphthalenyl | CO | 3,5-di-Br-4-OH-Ph | >50 | |||||
| Group 3 | |||||||||
| Compound | R1 | R2 | R3 | Ki (μM) | % inhibition at 50 μM | ||||
| GlyH-301 | 2-naphthalenyl | CH2 | 3,5-di-Br-2,4-di-OH Ph | ∼50 | 50 | ||||
| GlyH-302 | 2-naphthalenyl | CH2 | 3,5-di-Br-4-OH-Ph | ∼50 | 55 | ||||
| OxaH-303 | 2-naphthalenyl | CO | 3,5-di-Br-2,4-di-OH Ph | 10 | 70 | ||||
| OxaH-304 | 2-naphthalenyl | CO | 3,5-di-Br-4-OH-Ph | 12 | 78 | ||||
| Group 4 | Acetic acid hydrazides (AceH) | ||||||||
| Active compounds | |||||||||
| Compound | R1 | R2 | R3 | R4 | Ki (μM) | % inhibition at 50 μM | |||
| AceH-401 | 2-naphthoxy | 3,5-di-Br-2,4-di-OH Ph | 21 | 84 | |||||
| AceH-402 | 2-naphthoxy | 3,5-di-Br-4-OH-Ph | 17 | 86 | |||||
| AceH-403 | 4-Me-Ph | 3,5-di-Br-2,4-di-OH Ph | 10 | 54 | |||||
| AceH-404 | 4-Me-Ph | 3,5-di-Br-4-OH-Ph | 15 | 63 | |||||
Ki is compound concentration giving 50% inhibition of apical membrane Cl− current in CFTR-expressing FRT cells. See Fig. 4 for generic structure of compound groups 1–4.
Figure 3.

Glycine hydrazide (GlyH) CFTR inhibitors. (A) Classes of GlyH-101 analogues prepared for analysis of structure–activity relationships showing sites of modification (brackets). Substitutions to benzaldehyde phenyl rings not shown. (B) Reaction schemes for the synthesis of GlyH-101, 126, 201, and 301. Reagents and conditions (see materials and methods for details): (a) ICH2COOEt, NaOAc, 95°C; (b) N2H4.H2O, EtOH/reflux; (c) 3,5-di-Br-2,4-di-OH-Ph-CHO, EtOH/reflux; (d) 3,5-di-Br-2,4-di-OH-Ph-COCl, pyridine, 22°C; (e) N2H4.H2O, Pd/C (10%), DMF/reflux; (f) glyoxalic acid, 10°C; (g) Na2BH3CN/CH3CN, 48 h; dry HCl, EtOH. (C) Structure (left) and apical Cl− current analysis (right, done as in Fig. 1 C) of OxaH-110 in FRT cells.
N-2-naphthalenyl-[(3,5-dibromo-2,4-dihydroxyphenyl) methylene] Glycine Hydrazide (GlyH-101) and Related Glycine Hydrazides (GlyH-102–109, 114–127) and Acetic Acid Hydrazides (AceH-401–404)
A mixture of 2-naphthylamine (compound I, see Fig. 3 B) (1.43 g, 10 mmol), ethyl iodoacetate (2.14 g, 10 mmol), and sodium acetate (1.64 g, 20 mmol, dissolved in 2 ml of water) was stirred at 90°C for 3 h. The solid material obtained upon cooling was filtered and recrystallized from hexane to yield 1.5 g ethyl N-(2-naphthalenyl)glycinate (II, yield, 65%, mp 83–84°C) (Ramamurthy and Bhatt, 1989). A solution of above product (2.29 g, 10 mmol) in ethanol (10 ml) was refluxed with hydrazine hydrate (0.6 g, 12 mmol) for 10 h. Solvent and excess reagent were distilled under vacuum. The product was recrystallized from ethanol to yield 1.8 g of N-(2-naphthalenyl)glycine hydrazide (compound III) (82%, mp 147–148°C). A mixture of compound III (2.15 g, 10 mmol) and 3,5-dibromo-2,4-dihydroxybenzaldehyde (3 g, 10 mmol) in ethanol (5 ml) was refluxed for 3 h. The hydrazone that crystallized upon cooling was filtered, washed with ethanol, and recrystallized from ethanol to give 3.8 g (78%) of GlyH-101. Melting point (mp) >300°C, ms (ES−): M/Z 492 (M-H)−1; 1H nmr (DMSO-d6): δ 4.1(s, 2H, CH2), 6.5–7.5 (m, 9H, aromatic, NH), 8.5 (s, 1H, CH=N), 10.4 (s, 1H, NH-CO), 11.9 (s, 1H, OH), 12.7(s, 1H, OH). Compounds GlyH-102–109, GlyH-114–127, and AceH-401–404 were synthesized similarly by condensing appropriate hydrazides with substituted benzaldehydes or acetophenones.
N-(6-quinolinyl)-[(3,5-dibromo-2,4-dihydroxyphenyl) methylene] Glycine Hydrazide (GlyH-126) and Related Quinolinyl-Glycine Hydrazides
To a stirred solution of 6-aminoquinoline (compound IV) (0.72 g, 5 mmol) in acetonitrile (20 ml) was added 33% aqueous glyoxylic acid (1.85 g, 20 mmol) solution. A solution of NaBH3CN (0.64 g, 10.2 mmol) in acetonitrile (20 ml) was then added at 3°C over 20 min and the reaction mixture was warmed to room temperature and stirred for 48 h. Acetonitrile was evaporated under vacuum, water (20 ml) was added to the residue, the solution was alkalinized to pH 9.5, and unreacted amine was extracted with ether. Concentrated HCl (25 ml) was added to the aqueous solution and the mixture was stirred at 25°C for 1 h. Solvent was evaporated under vacuum. The resultant residue of N-(6-quinolinyl)glycine was dissolved in dry ethanol (50 ml) saturated with dry HCl, stirred overnight and then refluxed for 3 h. Ethanol was evaporated, the ester hydrochloride was suspended in dry ether, and ammonia gas was bubbled. The ammonium chloride was filtered and ether was removed by evaporation to give ethyl N-(6-quinolinyl)glycinate (0.5 g, 87%, mp 122–123°C, Ramamurthy and Bhatt, 1989). N-(6-quinolinyl)glycine hydrazide (compound VI), synthesized by hydrazinolysis of the above ester, was reacted with 3,5-dibromo-2,4-dihydroxybenzaldehyde to give GlyH-126. Similar procedures were used for synthesis of GlyH-127.
Oxamic Hydrazides (OxaH-110–113)
The oxamic hydrazides were synthesized by heating a mixture of 2-naphthaleneamine with diethyl oxalate in toluene. The resultant N-substituted oxamic acid ethyl ester was treated with hydrazine hydrate followed by condensation with substituted benzaldehydes or acetophenones to yield compounds OxaH-110–113.
3,5-Dibromo-4-hydroxy-[2-(2-naphthalenamine)aceto] Benzoic Acid Hydrazide (GlyH-202) and Related GlyH-201 and Oxa-203–204
N-(2-naphthalenyl)glycine hydrazide (compound III, 2.15 g, 10 mmol) was reacted with 3,5-dibromo-4-hydroxybenzoyl chloride (3.14 g, 10 mmol) (Gilbert et al., 1982) in pyridine (10 ml) for 5 h. Pyridine was removed and the residue was diluted with water. The product was recrystallized from ethanol to yield a gray powder 3.8 g (77%), mp > 300°C. Compounds GlyH-201 and Oxa-203–204 were synthesized by similar procedure.
N-2-naphthalenyl-[(3,5-dibromo-2,4-dihydroxyphenyl)methyl] Glycine Hydrazide (GlyH-301) and Related Glycine Hydrazides (GlyH-302, OxaH-303–304)
A mixture of GlyH-101 (1.5 g, 3 mmol), hydrazine hydrate (0.15 ml, 3 mmol), and Pd/C catalyst (0.1 g, 10% Pd) in 5 ml of dimethylformamide was refluxed for 6–8 h (Verma et al., 1984). The reaction mixture was filtered, diluted with cold water, and extracted with diethyl ether. GlyH-301 was crystallized from ether to yield 0.9 g (60%), mp 258–260°C. Compounds GlyH-302 and OxaH-303–304 were prepared similarly.
RESULTS
Discovery of Novel Classes of CFTR Inhibitors
A collection of 100,000 small, drug-like compounds with high chemical diversity was screened to identify new CFTR inhibitors. As diagrammed in Fig. 1 A, compounds were screened at 25 μM in a cell-based assay of iodide influx after CFTR activation by an agonist mixture containing a cAMP agonist (forskolin, 10 μM), phosphodiesterase inhibitor (IBMX, 100 μM), and flavone-type direct CFTR activator (apigenin, 20 μM). Initial rates of iodide influx were computed from the kinetics of fluorescence decrease after chloride replacement by iodide. Four compounds (Fig. 1 B) reducing iodide influx by >50% were identified, none of which are related structurally to known CFTR activators or inhibitors. 12 compounds reduced iodide influx by 25–50%, most of which were related structurally to the compounds in Fig. 1 B or to the thiazolidinones.
Figure 1.

Identification of novel classes of CFTR inhibitors by high-throughput screening. (A) Screening procedure. FRT cells coexpressing a halide-sensing yellow fluorescent protein (YFP-H148Q) and human wild-type CFTR were incubated with activating cocktail for 15 min before addition of test compounds at 25 μM. Iodide influx was induced by rapid addition of an iodide-containing solution. (B) Structures of novel classes of CFTR inhibitors identified by screening of a collection of 100,000 drug-like small molecules. (C) Dose inhibition of the glycine hydrazide GlyH-101 determined by fluorescence assay (left) and apical Cl− current analysis (right). Apical Cl− current was induced by 100 μM CPT-cAMP. CPT-cAMP and GlyH-101 were added to both apical and basolateral solutions.
To select inhibitor(s) for further evaluation, dose–response measurements were done for the compounds in Fig. 1 B, and CFTR inhibition was confirmed electrophysiologically by measuring apical Cl− current. Ki was ∼7, 5, 5, and 5 μM for compounds a–d, respectively. Fig. 1 C shows representative fluorescence (left) and apical Cl− current (right) data for compound d. We next screened 100–250 commercially available analogues of each compound class to determine whether active structural analogues exist, an important prerequisite for follow-up compound optimization by synthesis of targeted analogues. Whereas few or no active analogues of compounds a, b and c were found, initial screening of 285 analogues of compound d (substituted glycine hydrazides, GlyH) revealed 34 analogues that inhibited CFTR-mediated iodide influx by >25% at 25 μM.
Prior to extensive structure–activity analysis of synthesized GlyH analogues and characterization of inhibition mechanism, we determined the time course of GlyH-101 action and reversibility, and whether inhibition was effective for different CFTR-activating mechanisms. Fig. 2 A shows prompt inhibition of iodide influx in the fluorescence and apical Cl− current (inset) assays upon GlyH-101 addition. Interestingly ∼50% of the inhibition occurred within the ∼1-s addition/mixing time, with further inhibition over ∼1 min. Fig. 2 B indicates complete reversal of inhibition after GlyH-101 washout with >75% reversal over 5 min. Fig. 2 C shows effective CFTR inhibition by GlyH-101 after activation by different types of agonists, including potent direct activators of CFTR that do not elevate cytosolic cAMP or inhibit phosphatase activity (CFTRact-02, 08, and 10; Ma et al., 2002b).
Figure 2.
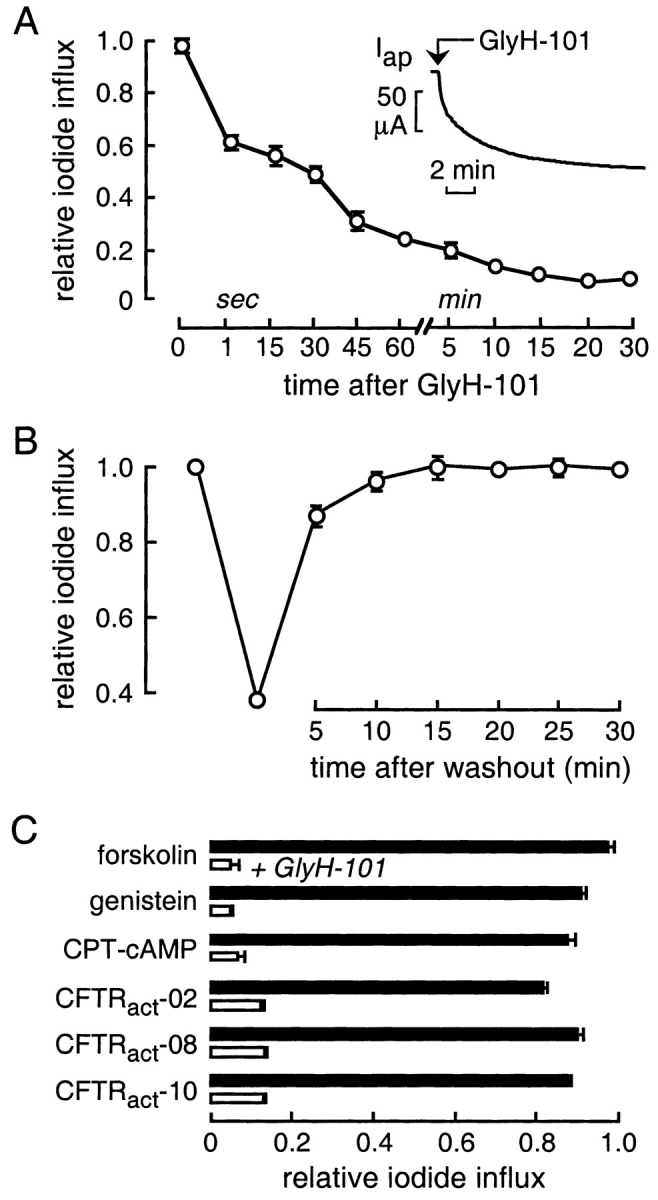
CFTR inhibition by the glycine hydrazide GlyH-101. Studies were done in FRT cells coexpressing human wild-type CFTR and a halide-sensing yellow fluorescent protein (YFP). (A) Kinetics of CFTR inhibition. After CFTR stimulation by a mixture of activators (10 μM forskolin, 100 μM IBMX, 20 μM apigenin), iodide influx (mean ± SEM, n = 3) was measured by the fluorescence assay at indicated times after addition of GlyH-101 (10 μM). Inset shows reduction in apical Cl− current (activated by 100 μM CPT-cAMP) after rapid addition of GlyH-101 (5 μM). (B) Kinetics of reversal of inhibition after GlyH-101 washout. FRT cells were incubated with the mixture of activators containing 5 μM GlyH-101 for 5 min. GlyH-101 was removed and cells were washed three times with PBS. Iodide influx was measured at specified times after GlyH-101 washout in the presence of the activator mixture. (C) Inhibition of iodide influx (mean ± SEM, filled bars) by GlyH-101 (50 μM, open bars) after CFTR stimulation by indicated agonists (all 50 μM, except for forskolin 10 μM, CPT-cAMP 500 μM).
GlyH-101 specificity was tested in an assay measuring the activity of MDR-1, a drug transporter of the ATP binding cassette family with structural homology to CFTR. Cells overexpressing MDR-1 (9HTEo-/Dx) do not incorporate rhodamine 123 as a consequence of MDR-1–mediated extrusion. GlyH-101 at 20 μM did not increase rhodamine 123 uptake, whereas the MDR-1 inhibitor verapamil at 100 μM increased rhodamine 123 incorporation by greater than fivefold (not depicted). As measured by short-circuit current in T84 cells after simulation by 5 μM thapsigargin (in the presence of 10 μM CFTRinh-172 to block CFTR), GlyH-101 at 5 μM did not inhibit calcium-induced Cl− channel activity, whereas inhibition of >50% was seen at 50 μM GlyH-101 (not depicted).
Chemistry and Structure–Activity Relationships of Glycine Hydrazides
The GlyH-101 structure was modified systematically to establish structure–activity relationships and to identify analogues with improved CFTR inhibitory activity. Fig. 3 A shows the various classes of structural analogues that were synthesized and tested for CFTR inhibition. Structural modifications were done on both ends of the glycine hydrazide backbone (Fig. 3 A, left, top, and middle). Replacing the glycine methylene group by a carbonyl group and replacing nitrogen by oxygen generated oxamic acid hydrazides (OxaH, right, top) and acetic acid hydrazides (AceH, right, middle), respectively. The hydrazone group modification produced two important series of compounds (middle, bottom and right, bottom). Also shown are compounds containing an additional methyl group at the hydrazone bond (top, middle), and containing a 6-quinolinyl group replacing the naphthalenyl group (left, bottom).
Fig. 3 B shows the reaction schemes developed for synthesis of the different classes of glycine hydrazide analogues (see materials and methods). Synthesis of GlyH-101 involves reaction of 2-naphthalenamine with ethyl iodoacetate followed by reactions with hydrazine hydrate and 2,4-dihydroxy-3,5-dibromobenzaldehyde. A similar procedure was used for most of the remaining glycine hydrazide derivatives (listed in Table I). The heteroaromatic analogues containing a 6-quinolinium group required different synthetic route in which 6-aminoquinoline was condensed with glyoxalic acid, and reduced using sodium cyanoborohydride (yielding N-[6-quinolinyl]glycine; Ramamurthy and Bhatt, 1989), which was further esterified and reacted with hydrazine hydrate and benzaldehyde. The oxamic acid hydrazides were synthesized starting from aromatic amines and diethyl oxalate.
Modifications were made initially on the N-aryl (R1) and benzaldehyde (R3) positions (see Table I and Fig. 4 for definition of Ri and CFTR inhibition). Good CFTR inhibition was found when R3 contained 3,5-dibromo and at least one hydroxyl substituent at the 4-position (GlyH-102, 105, 114); addition of a second hydroxyl group increased inhibition (GlyH-101, 104, 115–116). Inhibition was greatly reduced when R3 contained 4-bromophenyl or 4-carboxyphenyl substituents (GlyH-120–121). In addition, the 4-hydroxyl group in GlyH-101 was important for inhibition since its 4-methoxy analogue, GlyH-103, had little activity. Similar structure–activity results were found for GlyH-115 and GlyH-122.
Figure 4.

General structures of the synthesized compounds. See Table I for list of substituents and CFTR inhibition activities.
R1 group modifications were performed, maintaining R3 as 2,4-dihydroxy-3,5-dibromophenyl and 3,5-dibromo-4-hydroxyphenyl. Analogues with R1 as 2-naphthalenyl were much better inhibitors than R1 as 4-chlorophenyl or 4-methylphenyl. Replacement of the 2-naphthalenyl of GlyH-101 by 1-naphthalenyl (GlyH-104) decreased inhibition activity substantially, supporting the requirement of the 2-naphthalenyl substituent. GlyH-124–125, containing a 2-anthacenyl group, were weakly active. Replacement of 2-naphthalenyl group in GlyH-101 and GlyH-102 by more polar heteroaromatic rings, such as 6-quinolinyl, gave compound with little activity (Gly-126–127), as did the 2-naphthoxy analogues AceH-401 and AceH-402.
R2 was next modified (replacing methylene), keeping 2-naphthalenyl as R1 and dibromo-dihydroxyphenyl as R3. Introduction of a carbonyl group in GlyH-101 and GlyH-102 at R2, giving OxaH-110 and OxaH-111, gave two to threefold greater inhibitory potency. Replacement of CH2 by CHCH3 (GlyH-106–107) had minimal effect on CFTR inhibition. In another structural variation, addition of a methyl group at R4 to GlyH-102, yielding GlyH-109, gave improved CFTR inhibition. Modification of the N=C group in GlyH-101 and GlyH-102 to NH-CH2 in GlyH-301 and GlyH-302, or to NH-CO in GlyH-201 and GlyH-202, reduced CFTR inhibitory potency.
Fig. 3 C shows apical Cl− current analysis of CFTR inhibition in FRT cells for the most active analogue OxaH-110. Dose–response data for some of the most potent CFTR inhibitors gave Ki values (in μM, ±SEM, n = 3) of 2.7 ± 0.3 (OxaH-110), 4.3 ± 0.9 (GlyH-101), 4.7 ± 0.9 (GlyH-102), 4.7 ± 0.3 (GlyH-109), and 6.7 ± 0.9 (GlyH-115).
Patch-clamp Analysis of CFTR Inhibition Mechanism
The mechanism of CFTR block by GlyH-101 was studied using the whole-cell configuration of the patch-clamp technique. After maximal activation of CFTR in stably transfected FRT cells by 5 μM forskolin, current–voltage relationships were measured at GlyH-101 concentrations from 0 to 50 μM. Representative original current recordings are shown in Fig. 5 A. In the absence of inhibitor (left), membrane current increased linearly with voltage and did not show relaxation phenomena, as expected for pure CFTR Cl− currents. Extracellular perfusion with 10 μM GlyH-101 produced a reduction in current that was strongly dependent on membrane potential (Fig. 5 A, right). At more positive membrane potentials, outward positive currents (Cl− movement into the cell) were reduced compared with inward currents. Fig. 5 B shows current–voltage relationships for GlyH-101 concentrations of 0 (control), 10, and 30 μM, and after washout of 10 μM GlyH-101 (recovery). Data for the thiazolidinone CFTRinh-172 (5 μM) is shown for comparison. The current–voltage relationship was linear in the absence of inhibitor, after GlyH-101 washout, and after inhibition by CFTRinh-172, whereas GlyH-101 inhibition at submaximal concentrations produced inward rectification. Fig. 5 C summarizes percentage CFTR current block as a function of GlyH-101 concentration at different membrane voltages. GlyH-101 inhibitory potency was reduced at more negative voltages, with apparent Ki of 1.4, 3.8, 5.0, and 5.6 μM for voltages of +60, +20, −20, and −60 mV, respectively (Hill coefficients, nH = 0.5, 0.7, 1.3, 1.8). In additional studies to investigate whether the GlyH-101 binds to a site in the CFTR pore, Cl− flux in the CFTR pore was altered by reducing extracellular Cl− concentration to 20 mM. The potency of GlyH-101 inhibition of CFTR Cl− current was significant reduced at the low extracellular Cl− (Fig. 5 D).
Figure 5.

Patch-clamp analysis of GlyH-101 inhibition mechanism. (A) Superimposed whole-cell membrane currents evoked by voltages from −100 to +100 mV (20-mV steps) in CFTR-expressing FRT cells after maximal CFTR stimulation by 5 μM forskolin. Holding potential was −100 mV and interpulse duration was 4 s. Data shown before (left) and after (right) 10 μM GlyH-101. (B) Current–voltage relationships in the absence of inhibitors (control, open circles), after addition of 10 μM (filled squares) and 30 μM (open squares) GlyH-101, after washout of 10 μM GlyH-101 (recovery, shaded circles), and after addition of 5 μM CFTRinh-172 (filled circles). (C) Dose–response for inhibition of CFTR Cl− current by GlyH-101 at indicated membrane potentials. Each point is the mean ± SEM of four to five experiments. Data were fitted to the Hill equation. Fitted Ki: 1.4 ± 0.4, 3.8 ± 0.2, 5.0 ± 0.3, and 5.6 ± 0.4 μM for voltages of +60, +20, −20, and −60 mV, respectively. Ki at −20 and −60 mV significantly greater than at +20 (P < 0.05) and +60 mV (P < 0.02). (D) Effect of reducing extracellular Cl− concentration on apparent GlyH-101 potency. GlyH-101 dose–response in high (150 mM) vs. low (20 mM) extracellular Cl− (mean ± SEM, four sets of experiments). Membrane potential was +20 mV.
Cell-attached patch-clamp experiments were performed to investigate the mechanism of GlyH-101 block of CFTR Cl− current at the single-channel level. Fig. 6 A (representative traces and amplitude histograms) shows a reduction in apparent CFTR channel activity without a change in single channel current at GlyH-101 concentrations up to 5 μM. Mean channel open time was remarkably reduced, with the appearance of brief closures during the open bursts whose frequency increased with GlyH-101 concentration (Fig. 6 B).
Figure 6.

Single channel analysis of CFTR inhibition by GlyH-101. (A) Representative traces (left) and corresponding amplitude histograms (right) obtained from a cell-attached patch. Pipette potential (Vp) was −60 mV. CFTR was stimulated with forskolin (5 μM) in the absence and presence of GlyH-101 at indicated concentrations. Dashed lines show zero current level (channels closed) with downward deflections indicating channel openings (Cl− ions moving from pipette into the cell). Apparent open channel probability decreased from 0.48 in the absence of inhibitor to 0.14 at 5 μM GlyH-101. (B) Mean channel open times (mean ± SEM, five sets of experiments) as a function of GlyH-101 concentration from cell-attached patch-clamp experiments (*, P < 0.05; **, P < 0.01 vs. control).
Physical Properties of Glycine Hydrazides
Interpretation of the voltage-dependent inhibition mechanism requires knowledge of the GlyH-101 ionic species that interacts with CFTR. Apical Cl− current studies indicated that the Ki for GlyH-101 inhibition of CFTR Cl− current was independent of pH in the range 6–8 (not depicted), where the compound is highly water soluble (0.8–1.3 mM in water, 22°C). The possible titrable groups on GlyH-101 in the pH range 3–10 include the secondary glycinyl amine and the resorcinolic hydroxyls. Spectrophotometric titration of GlyH-101 indicated at least two protonation–deprotonations at pH between 4 and 9 (Fig. 7 A, top). To assign pKa values, GlyH-101 analogues that lacked one or more titrable groups were synthesized. Removal of the secondary amine (AceH-403) had little effect on the titration, with only a minor left shift of the ascending portion of the curve, suggesting a pKa of ∼5.5 for titration of the first phenolic hydroxyl. Removal of one ortho hydroxyl (GlyH-102) eliminated the descending portion of the curve, confirming the pKa of ∼5.5 for the first para hydroxyl and ∼8.5 for the second ortho hydroxyl. Removal of the aromatic ring containing the resorcinolic hydroxyls (ethyl N-[2-naphthalenyl] glycinate; Fig. 7 A, bottom) indicated a pKa ∼4.7 for the residual secondary amine. From these data, the deduced equilibria among the ionic forms of GlyH-101 is shown in Fig. 7 B. GlyH-101 exists primarily as a singly charged anion at pH between 6 and 8.
Figure 7.

Determination of pH-dependent ionic equilibria of GlyH-101 by spectrophotometric titration of GlyH-101 analogues. (A) Chemical structures (left) and corresponding pH-dependent absorbance changes (right) of compounds (10 μM) in NaCl (100 mM) containing MES, HEPES, boric acid, and citric acid (each 10 mM) titrated to indicated pH using HCl/NaOH. Absorbance changes measured at wavelengths of 346, 348, 346, and 236 nm (top to bottom). (B) Deduced ionic equilibria of GlyH-101 showing pKa values.
CFTR Inhibition in Mice In Vivo
Inhibition of CFTR-dependent airway epithelial Cl− current in vivo was demonstrated by nasal PD measurements in mice. Nasal PDs were measured continuously in response to serial solution exchanges in which amiloride was added (to block ENaC Na+ channels) followed by Cl− replacement by gluconate (to induce Cl−-dependent hyperpolarization), forskolin addition (to activate CFTR), and GlyH-101 addition (to inhibit CFTR). The representative PD recording in Fig. 8 A (left) shows hyperpolarizations (more negative PDs) following low Cl− and forskolin solutions, representing CFTR-independent and dependent Cl− currents, respectively. Topical application of GlyH-101 in the perfusate rapidly reversed the forskolin-induced hyperpolarization. Averaged results from a series of measurements are summarized in Fig. 8 A (right). Paired analysis of PD changes (ΔPD; Fig. 8 B) indicated ∼4 mV hyperpolarization after forskolin with depolarization of similar magnitude after GlyH-101; for comparison, data are shown for CFTRinh-172 from a previous study (Salinas et al., 2004). In a separate series of experiments, nasal PDs were measured as in A except that all solutions contained DIDS or GlyH-101. Fig. 8 C shows partial inhibition by DIDS of the (CFTR-independent) hyperpolarization produced by low Cl− (left), and substantial inhibition by GlyH-101 of the forskolin-induced hyperpolarization (right). Together these results indicate rapid inhibition of upper airway CFTR Cl− conductance by topical GlyH-101.
Figure 8.

GlyH-101 inhibits forskolin-induced hyperpolarization in nasal potential difference in mice. (A, left) Nasal PD recording showing responses to amiloride (100 μM) and low Cl− (4.7 mM) solutions. Where indicated, the low Cl− solutions contained forskolin (10 μM) without or with GlyH-101 (10 μM). (A, right) Averaged PD values (mean ± SEM, n = 5). (B) Paired analysis of experiments as in A showing PD changes (ΔPD) after forskolin (10 μM), CFTRinh-172 (20 μM), and GlyH-101 (10 μM). (C) A series of experiments was done in which all solutions contained DIDS (100 μM) or GlyH-101 (10 μM). ΔPD (mean ± SEM) for low Cl− and forskolin-induced hyperpolarizations. *, P < 0.005 for reduced ΔPD compared with control. CFTRinh-172 data taken from Salinas et al. (2004).
The efficacy of GlyH-101 in inhibiting cAMP/cholera toxin–induced intestinal fluid secretion was also evaluated. Short-circuit current was measured in different cell types and in intact mouse ileum under nonpermeabilized conditions and in the absence of a Cl− gradient. In each case, CFTR was activated by CPT-cAMP after ENaC inhibition by amiloride. Fig. 9 shows similar Ki ∼ 2–5 μM for inhibition of cAMP-stimulated short-circuit current by GlyH-101 in T84 cells (A), primary human bronchial cell cultures (B), and intact mouse ileum (C). Inhibition was ∼100% at higher GlyH-101 concentrations. Cholera toxin–induced intestinal fluid secretion was measured in an in vivo closed-loop model in which loops for each mouse were injected with saline (control), cholera toxin (1 μg), or cholera toxin (1 μg) + GlyH-101 (2.5 μg). GlyH-101 was added to the lumen (rather than systemically) based on initial studies showing poor intestinal absorption and little effect of systemically administered compound. Referenced to the saline control, the cholera toxin–induced increase in fluid secretion over 4 h, quantified from loop weight-to-length ratio, was ∼80% reduced by GlyH-101 (Fig. 9 D). To rule out the possibility that GlyH-101 inhibited intestinal fluid secretion by blocking the binding of cholera toxin to its cell receptor, T84 cell fluorescence was measured after 1 h incubation with FITC-labeled cholera toxin B subunit (50 μg/ml) at 37°C. GlyH-101 at 50 μM did not inhibit cholera toxin binding/uptake (not depicted).
Figure 9.

GlyH-101 inhibits cholera toxin/cAMP-dependent intestinal fluid secretion. (A–C) Inhibition of short-circuit current by GlyH-101 after CFTR stimulation in T84 cells (A), human airway cells (B), and mouse ileum (C). Following constant baseline current, amiloride (10 μM, apical solution) and CPT-cAMP (0.1 mM, both solutions) were added, followed by indicated concentrations of GlyH-101 (both solutions). Indomethacin (10 μM) was present in all solutions in ileum studies. Experiments representative of three to five measurements. (D) Closed intestinal loop model of cholera toxin–induced fluid secretion. Intestinal luminal fluid, shown as loop weight/length (g/cm, SEM, six mice), measured at 4 h after injection of saline (control), cholera toxin (1 μg), or cholera toxin given together with GlyH-101 (2.5 μg). *, P < 0.01.
DISCUSSION
The glycine hydrazides were discovered using an assay designed to identify rapidly acting inhibitors of CFTR that interact with the CFTR pore or a critical part of the CFTR molecule involved in anion conductance. The kinetic and electrophysiological data suggest that glycine hydrazides block CFTR Cl− conductance by occluding the CFTR anion pore at or near the external membrane surface. Unlike all other CFTR inhibitors, including the thiazolidinone CFTRinh-172, CFTR block by the glycine hydrazide GlyH-101 produced inwardly rectifying CFTR Cl− currents. Compared with CFTRinh-172, GlyH-101 is >50-fold more water soluble and is rapidly acting/reversible when added to or removed from the extracellular solution, consistent with its action at the external-facing surface of CFTR. Structure–activity analysis of a series of targeted glycine hydrazide analogues defined the structural determinants for CFTR inhibition and provided analogues with greater CFTR inhibitory potency, the best being OxaH-110 with Ki ∼2 μM. Although the most potent thiazolidinone CFTRinh-172 has Ki of 0.2–0.3 μM in permeabilized cell preparations, its Ki is 2–5 μM in many intact epithelial cells because of the interior negative membrane potential that reduces its concentration in cytoplasm (Thiagarajah et al., 2004a). Thus, the glycine hydrazides are as or more potent than the thiazolidinones, and like the thiazolidinones, they block CFTR in nasal and intestinal epithelia in vivo.
Patch-clamp studies indicated that CFTR inhibition by GlyH-101 is sensitive to membrane potential. At sub-maximal concentrations of GlyH-101, there was marked inward rectification in the CFTR current–voltage relationship, indicating that Cl− flux from the extracellular to the intracellular side of the membrane is more strongly blocked than that in the opposite direction. The apparent Ki increased approximately fourfold as applied potential was varied from +60 to −60 mV. Since GlyH-101 is negatively charged at pH 6–8, the simplest interpretation of these data is that GlyH-101 inhibition involves direct interaction with the channel pore at the extracellular side of the membrane. Accordingly, negative membrane potentials reduce the inhibitory efficacy of the negatively charged GlyH-101 by electrostatic repulsion, which drives the compound away from the pore. In contrast, the open channel blocker glibenclamide, which is thought to act from the intracellular side of the CFTR pore (Sheppard and Robinson, 1997), produces outward rectification of CFTR current–voltage relationship (Zhou et al., 2002). The reduced GlyH-101 potency at low extracellular Cl− concentration provided further evidence that GlyH-101 binds to a site at the external CFTR pore.
Analysis of GlyH-101 dose–response data also revealed an increase in apparent Hill coefficient at more negative membrane potentials, suggesting the possibility of more than one inhibitor binding site within the pore and/or cooperative interaction between inhibitor molecules, as reported previously for other ion channels (Pottosin et al., 1999; Brock et al., 2001). In support of the hypothesis that GlyH-101 is an open channel blocker, cell-attached patch-clamp experiments revealed fast closures within bursts of channel openings. The frequency of fast closures increased with GlyH-101 concentration, producing a reduction in mean channel open time as found for glibenclamide (Sheppard and Robinson, 1997). The appearance of closure events on the millisecond time scale classifies GlyH-101 as an “intermediate”-type channel blocker, similar to glibenclamide; in contrast, “fast” blockers reduce apparent single channel conductance, and “slow” blockers cause closures of many seconds duration (Hille, 1992). In whole-cell patch-clamp and apical Cl− current experiments, CFTR Cl− conductance was nearly completely inhibited at high concentrations (≥30 μM) of GlyH-101. Together these results suggest that the GlyH-101 inhibition mechanism involves direct CFTR pore occlusion at a site at or near the extracellular-facing pore surface.
Synthesis and characterization of a series of glycine hydrazide analogues indicated the important structural determinants for CFTR inhibition. Synthesis work was directed to alter polarity, planarity, and hydrophilic/hydrophobic properties of the GlyH-101, systematically modifying different portions of the molecule as diagrammed in Fig. 3 A. Slight changes in planarity by replacing the glycinyl methylene group (R2) with carbonyl group improved inhibition activity, whereas reduction of Schiff base group (N=C) reduced activity. An extensive set of modifications of the benzaldehyde moiety (R3) indicated the requirement of two bromines separated by one para-hydroxyl group. Replacement of the 2-naphthalenyl group by (hetero)aromatic groups reduced inhibition activity. Many GlyH-101 analogues having substitutions that reduced overall polarity were inactive, suggesting that the presence of a hydrophobic group at one end at R1 and an anion group at R3 are key requirements for CFTR inhibition activity. Molecular docking computations should be informative when structural information about the CFTR pore becomes available.
In summary, the glycine hydrazides represent a new class of potent CFTR inhibitors with a novel external pore-occluding mechanism producing inward rectification and a reduction in mean channel open time. The large series of structural analogues with varying activities should permit the synthesis of engineered analogues with specified ADME (administration, distribution, metabolism, excretion) and other properties, such as membrane impermeability. The antidiarrheal efficacy of GlyH-101 when added in the intestinal lumen rather than systemically is particularly interesting, and suggests the possibility of developing a nonabsorbable drug for reducing intestinal fluid loss in secretory diarrheas produced by Vibrio cholera and Escherichia coli.
Acknowledgments
This work was supported by grants HL73854, EB00415, EY13574, DK35124, and DK43840 from the National Institutes of Health, and Drug Discovery and Research Development Program grants from the Cystic Fibrosis Foundation (A.S. Verkman) and the Telethon-Italy (L.J. Galietta).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: AceH, acetic acid hydrazide; CFTR, cystic fibrosis transmembrane conductance regulator; FRT, Fisher rat thyroid; GlyH, glycine hydrazide; OxaH, oxamic hydrazide.
References
- Brock, M.W., C. Mathes, and W.F. Gilly. 2001. Selective open-channel block of Shaker (Kv1) potassium channels by s-nitrosodithiothreitol (SNDTT). J. Gen. Physiol. 118:113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, D.C., S.S. Smith, and M.K. Mansoura. 1999. CFTR: mechanism of anion conduction. Physiol. Rev. 79:S47–S75. [DOI] [PubMed] [Google Scholar]
- Galietta, L.J.V., S. Jayaraman, and A.S. Verkman. 2001. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am. J. Physiol. Cell Physiol. 281:C1734–C1742. [DOI] [PubMed] [Google Scholar]
- Gilbert, R., P. Maurits, I. Henri, D. Edmond, P. Maurice, D. Marcel, B. Jacques, R. Jean, and T. Chantal. 1982. Research in the indolizines series. IV. Effect of substitution in the neighborhood of ether function in the butoprozine series. Eur. J. Med. Chem. 17:581–588. [Google Scholar]
- Gong, X.D., J.C. Li, K.H. Cheung, G.P. Leung, S.B. Chew, and P.Y. Wong. 2001. Expression of the cystic fibrosis transmembrane conductance regulator in rat spermatids: implication for the site of action of antispermatogenic agents. Mol. Hum. Reprod. 7:705–713. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1992. Ionic channels in excitable membranes. 2nd ed. Sinauer Associates, Inc. Sunderland, MA. 607 pp.
- Jayaraman, S., P. Haggie, R. Wachter, S.J. Remington, and A.S. Verkman. 2000. Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J. Biol. Chem. 275:6047–6050. [DOI] [PubMed] [Google Scholar]
- Ma, T., J.R. Thiagarajah, H. Yang, N.D. Sonawane, C. Folli, L.J. Galietta, and A.S. Verkman. 2002. a. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera-toxin induced intestinal fluid secretion. J. Clin. Invest. 110:1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, T., L. Vetrivel, H. Yang, N. Pedemonte, O. Zegarra-Moran, L.J.V. Galietta, and A.S. Verkman. 2002. b. High-affinity activators of CFTR chloride conductance identified by high-throughput screening. J. Biol. Chem. 277:37235–37241. [DOI] [PubMed] [Google Scholar]
- McCarty, N.A. 2000. Permeation through the CFTR chloride channel. J. Exp. Biol. 203:1947–1962. [DOI] [PubMed] [Google Scholar]
- Nakanishi, K., W.E. Sweeney, K. Macrae, C.U. Cotton, and E.D. Avner. 2001. Role of CFTR in autosomal recessive polycystic kidney disease. J. Am. Soc. Nephrol. 12:719–725. [DOI] [PubMed] [Google Scholar]
- Pilewski, J.M., and R.A. Frizzell. 1999. Role of CFTR in airway disease. Physiol. Rev. 79:S215–S255. [DOI] [PubMed] [Google Scholar]
- Pottosin, I.I., O.R. Dobrovinskaya, and J. Muñiz. 1999. Cooperative block of the plant endomembrane ion channel by ruthenium red. Biophys. J. 77:1973–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe, A., J. Disser, and E. Fromter. 1995. Cl− channel inhibition by glibenclamide is not specific for the CFTR-type Cl− channel. Pflugers Arch. 429:659–662. [DOI] [PubMed] [Google Scholar]
- Ramamurthy, B., and M.V. Bhatt. 1989. Synthesis of antitubercular activity of N-(2-naphthyl)glycin hydrazide analogues. J. Med. Chem. 32:2421–2426. [DOI] [PubMed] [Google Scholar]
- Salinas, D.B., N. Pedemonte, C. Muanprasat, W.F. Finkbeiner, D.W. Nielson, and A.S. Verkman. 2004. CFTR involvement in nasal potential differences in mice and pigs studied using a thiazolidinone CFTR inhibitor. Am. J. Physiol. In press. [DOI] [PubMed] [Google Scholar]
- Schultz, B.D., A.K. Singh, D.C. Devor, and R.J. Bridges. 1999. Pharmacology of CFTR chloride channel activity. Physiol. Rev. 79:S109–S144. [DOI] [PubMed] [Google Scholar]
- Sheppard, D.N., and K.A. Robinson. 1997. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J. Physiol. 503:333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgess, N.C., R.Z. Kozlowski, C.A. Carrington, C.N. Hales, and M.L. Ashford. 1988. Effects of sulphonylureas and diazoxide on insulin secretion and nucleotide-sensitive channels in an insulin-secreting cell line. Br. J. Pharmacol. 95:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei, A., C. Folli, O. Zegarra-Moran, P. Fanen, A.S. Verkman, and L.J. Galietta. 2004. Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett. 558:52–56. [DOI] [PubMed] [Google Scholar]
- Thiagarajah, J.R., and A.S. Verkman. 2003. CFTR pharmacology and its role in intestinal fluid secretion. Curr. Opin. Pharmacol. 3:594–599. [DOI] [PubMed] [Google Scholar]
- Thiagarajah, J., T. Broadbent, E. Hsieh, and A.S. Verkman. 2004. a. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology. 126:511–519. [DOI] [PubMed] [Google Scholar]
- Thiagarajah, J.R., Y. Song, P. Haggie, and A.S. Verkman. 2004. b. A small-molecule CFTR inhibitor produces cystic fibrosis-like submucosal gland fluid secretions in normal airways. FASEB J. 18:875–877. [DOI] [PubMed] [Google Scholar]
- Verma, M., V.R. Gujrati, M. Sharma, T.N. Bhalla, A.K. Saxena, J.N. Sinha, K.P. Bhargava, and K. Shanker. 1984. Syntheses and anti-inflammatory activities of substituted arylamino-(N'-benxylidene) acetohydrazides and derivatives. Arch. Pharm. (Weinheim). 317:890–894. [DOI] [PubMed] [Google Scholar]
- Yang, H., A.A. Shelat, R.K. Guy, V.S. Gopinath, T. Ma, K. Du, G.L. Lukacs, A. Taddei, C. Folli, N. Pedemonte, et al. 2003. Nanomolar affinity small-molecule correctors of defective ΔF508-CFTR chloride channel gating. J. Biol. Chem. 278:35079–35085. [DOI] [PubMed] [Google Scholar]
- Zhou, Z., S. Hu, and T.C. Hwang. 2002. Probing an open CFTR pore with organic anion blockers. J. Gen. Physiol. 120:647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]