

Abstract
Targeting cannabinoid-2 (CB2) receptors with selective agonists may represent a novel therapeutic avenue in various inflammatory diseases, but the mechanisms by which CB2 activation exerts its anti-inflammatory effects and the cellular targets are elusive. Here, we investigated the effects of CB2-receptor activation on TNF-α-induced signal transduction in human coronary artery endothelial cells in vitro and on endotoxin-induced vascular inflammatory response in vivo. TNF-α induced NF-κB and RhoA activation and upregulation of adhesion molecules ICAM-1 and VCAM-1, increased expression of monocyte chemoattractant protein, enhanced transendothelial migration of monocytes, and augmented monocyte-endothelial adhesion. Remarkably, all of the above-mentioned effects of TNF-α were attenuated by CB2 agonists. CB2 agonists also decreased the TNF-α- and/or endotoxin-induced ICAM-1 and VCAM-1 expression in isolated aortas and the adhesion of monocytes to aortic vascular endothelium. CB1 and CB2 receptors were detectable in human coronary artery endothelial cells by Western blotting, RT-PCR, real-time PCR, and immunofluorescence staining. Because the above-mentioned TNF-α-induced phenotypic changes are critical in the initiation and progression of atherosclerosis and restenosis, our findings suggest that targeting CB2 receptors on endothelial cells may offer a novel approach in the treatment of these pathologies.
Keywords: endothelial activation, inflammation, RhoA, adhesion molecules
Atherosclerosis is the leading cause of common cardiovascular disorders such as coronary artery disease, stroke, and various forms of heart failure, abdominal aortic aneurysms, and ischemic gangrene, which are the principal causes of death in Western countries. Recent studies have revealed important cross-talk between inflammation, generation of reactive oxygen, nitrogen species, and lipid metabolism in the pathogenesis of atherosclerosis and vascular remodeling following injury (10, 12, 30, 31). Bacterial endotoxin(s) and proinflammatory cytokines (e.g., TNF-α), which mediate, at least in part, their proatherogenic effects by eliciting NF-κB activation in endothelial cells (17, 21, 24, 39, 48, 50), are considered to play pivotal roles in vascular inflammation associated with atherosclerosis and development of coronary arterial disease (4, 20, 23, 27, 35, 39, 43, 44). The activation of this pathway leads to induction of adhesion molecules and chemokines, e.g., VCAM-1, ICAM-1 (48), which promote monocyte adhesiveness to the endothelium, and the release of a variety of factors that promote smooth muscle migration and proliferation (from the medium into the intima), which then synthesize and deposit extracellular matrix (10, 14, 28). There is considerable evidence suggesting that disruption of the cytokine-induced NF-κB signaling pathway confers significant vasculoprotective effects by attenuating vascular inflammation (24, 36), which delays or prevents atherogenesis in animal models (4, 18, 42) of disease.
To date, two cannabinoid (CB) receptors have been identified by molecular cloning: the CB1 receptor, which is highly expressed in the brain but is also present in peripheral tissues including the heart, vascular tissues, and liver, and the CB2 receptor, previously thought to be expressed predominantly by immune and hematopoietic cells (reviewed in Refs. 22, 29). However, more recent studies have also demonstrated CB2 receptors in brain (45), myocardium (26), cardiomyoblasts (26, 34), and endothelial cells of various origins (3, 9, 25, 52) (reviewed in Refs. 19, 29).
The natural ligands of these receptors are lipid-like substances called endocannabinoids, which include arachidonoyl ethanolamide or anandamide and 2-arachidonoylglycerol (22, 29). Endocannabinoids and their synthetic analogs exert various central nervous system, cardiovascular, and anti-inflammatory effects through CB1 and CB2 receptors (19, 29).
A recent study has demonstrated that orally administered cannabis constituent Δ9-tetrahydrocannabinol, which is a mixed weak CB1/2-receptor agonist with psychoactive property, inhibited atherosclerosis progression in a mouse model of disease, an effect that could be blocked by a selective CB2-receptor antagonist (38). However, the role of CB2 receptor in vascular endothelial cell activation and inflammatory response is still unexplored.
Herein, we evaluated the effects of CB2-receptor activation on TNF-α-induced NF-κB and RhoA activation, upregulation of adhesion molecules ICAM-1 and VCAM-1, expression of monocyte chemoattractant protein in human coronary artery endothelial cells (HCAECs), transendothelial migration (TEM) of monocytes, monocyte-endothelial adhesion in vitro, and endotoxin-induced vascular inflammatory response in vivo. Because these pathological events are pivotal in the initiation and progression of atherosclerosis and restenosis, our findings may have important clinical implications and underscore the role of CB2 receptors as a novel therapeutic target in various vascular diseases associated with inflammation.
MATERIALS AND METHODS
Materials
The selective CB2 receptor agonist JWH-133 was either purchased from Tocris Bioscience (Ellisville, MO) or synthesized as described earlier (13). The selective CB2-receptor agonist HU-308 (11) was from Cayman Europe (Tallinn, Estonia). The CB1-receptor antagonist AM-281 and CB2-receptor antagonist AM-630 were purchased from Tocris Bioscience (Ellisville, MO). The CB1 antagonist SR-141716A and CB2 antagonist SR-144528 were from NIDA Drug Supply (Research Triangle Park, NC). Human recombinant TNF-α was obtained from R&D Systems. Sources of all the other reagents are mentioned in the text where appropriate.
Cell Culture
HCAECs and the growth medium were purchased from Cell Applications (San Diego, CA). The human monocytic cell line THP-1 was obtained from American Type Culture Collection; cells were grown in RPMI 1640 medium supplemented with 2 mM L-glutamine, 10 mM HEPES, 10% FBS, 100 U of penicillin, and 100 μg of streptomycin/ml (Invitrogen).
CB1 and CB2 Expression in HCAECs
Immunofluorescence staining
CB1 expression and CB2 expression in the human endothelial cells were determined by immunofluorescence staining technique. In brief, HCAECs were grown to confluence in chamber slides (Nalgene-Nunc, Lab-Tek). Growth medium was aspirated, and cells were washed three times with PBS and then fixed with 4.0% paraformaldehye for 20 min at 4°C. After cells were washed with PBS, cells were permeabilized with 0.2% Triton X-100 in PBS for 15 min at room temperature. Subsequently, CB1 expression and CB2 expression in the human endothelial cells were determined by immunofluorescence staining technique using anti-CB1 (rabbit polyclonal; Cayman Chemical) or anti-CB2 (rabbit polyclonal; Cayman Chemical), respectively, used at 1:100 dilution for 6 h at 4°C. After the cells were rinsed with PBS, the cells were probed with goat anti-rabbit FITC (1:250; Pierce) for 1 h at room temperature. The nucleus was counterstained with 4′,6-diamidino-2-phenylindole (Molecular Probes). Images were obtained with a fluorescent microscope (Olympus IX 81) at ×20 objective with ×1.5 optical zoom. To rule out the nonspecific staining, we used the corresponding blocking peptides for CB1 (catalog no. 10006591, Cayman Chemical) or CB2 (catalog no. 301550, Cayman Chemical). In brief, the blocking peptides were mixed with corresponding antibodies in a 1:1 ratio and incubated for 1 h at room temperature. This preabsorbed antibody was then used for the staining process. This procedure essentially blocks the antibody-antigen (protein) formation during the immunofluorescence staining and aids in determining the specificity of the staining.
Conventional RT-PCR and quantitative real-time RT-PCR
Total RNA was isolated from the cells using Trizol LS reagent (Invitrogen) according to manufacturer’s instruction. The RNA was treated with RNase-free DNase (Ambion) to remove traces of genomic DNA contamination. Total RNA was then reverse transcribed to cDNA using SuperScript II (Invitrogen), and the target genes were amplified with the standard PCR kit (Bio-Rad). The PCR conditions were as follows: after initial denaturation at 95°C for 2 min, 35 cycles were performed at 95°C for 30 s and at 60°C for 30s. Primers used were as follows: for CB1, 5′-TTCCCTCTTGTGAAGGCACTG-3′ (forward) and 5′-TCTTGACCGTGCTCTTGATGC-3′ (reverse); for CB2, 5′-TTTGCTTTCTGCTCCATGCTG-3′ (forward) and 5′-TTCTTTT-GCCTCTGACCCAAG-3′ (reverse); for β-actin, 5′-ATTGCCGA-CAGGATGCAGAAG-3′ (forward) and 5′-TAGAAGCATTTGCG-GTGGACG-3′ (reverse).
The amplified products were separated on 1.5% agarose gels, stained with ethidium bromide, and documented using the Typhoon system (GE Healthcare).
In a separate set of experiments, real-time PCR was performed in identical conditions, except amplification and quantification of the target gene expression were performed with the iTaq Sybr green mix (Bio-Rad) and Bio-Rad chromo4/opticon system (data not shown). Relative quantification was performed with the relative comparative threshold (CT) method.
Western immunoblot assay
HCAECs were grown to confluence in 100-mm culture dishes coated with 0.2% gelatin. Cells were suspended in RIPA lysis buffer (Pierce) supplemented with protease inhibitors (Roche). Cell lysates were then prepared by sonication 15k for 20 s) on ice. The lysates then were clarified to remove the cellular debris by centrifuging at 10,000 rpm for 15 min at 4°C. Protein content in the lysates was determined with the Lowry assay (Bio-Rad). Protein (30 μg) was resolved in 12% SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare). Blocking was performed for 2 h at room temperature with 5% nonfat skimmed milk powder prepared in PBS containing 0.1% Tween 20 (PBST; Sigma). After they were washed with PBST, membranes were probed with either rabbit polyclonal CB1 (Cayman Chemical; 1:1,000 dilution or an antibody raised against the last 15 residues of rat CB1) or CB2 antibody (Cayman Chemical; 1:1,000 dilution) overnight at 4°C. After a subsequent washing with PBST, the secondary antibody goat anti-rabbit horseradish peroxidase (Pierce) and incubated at room temperature for 1 h. The membranes were then developed using chemiluminescence detection kit (SuperSignal-west Pico substrate, Pierce). To confirm uniform loading, membranes were stripped and reprobed with β-actin (Chemicon). Proteins from mouse brain extract and human monocytic cell line (THP-1) lysate were used as appropriate positive controls for CB1 and CB2 receptors, respectively.
Cell Surface ICAM-1 and VCAM-1 Expression Assay
Cell surface expression of ICAM-1 and VCAM-1 was measured by in situ ELISA as described (2). In brief, HCAECs were grown in 96-well plates coated with 0.2% gelatin. After treatments, in situ ELISA was performed with anti-human ICAM-1 or VCAM-1 monoclonal antibodies (1:1,500 dilution; R&D Systems) and by measuring the absorbance at colorimetrically at 450 nm using the horseradish peroxidase-3,3′,5,5′-tetramethylbenzidine developing system (Sigma). Each treatment was performed in triplicate, and the experiments were repeated three times.
Monocyte-Endothelial Cell Adhesion Assay
Monocyte adhesion to endothelial cells was performed as described with modifications (47). In brief, HCAECs were grown to confluence in 24-well plates and treated with TNF-α ± CB2 agonists/antagonists. THP-1 monocytes were then labeled with 1.5 μM calcein-AM (Molecular Probes-Invitrogen) for 1 h at 37°C in RPMI 1640 containing 1% FBS. HCAECs were washed twice with HCAEC basal medium and incubated with 400 μl of basal medium. Then 105 cells/100 μl of labeled THP-1 cells were overlaid on HCAECs and incubated for 1 h at 37°C. After incubation, the monolayer was carefully washed with PBS to remove the unbound monocytes. The adherent monocytes to the endothelial cells were documented with an Olympus IX 81 fluorescent microscope using ×10 objective with ×1.5 optical zoom. Individual treatments were preformed in duplicate, and the entire set of experiments was repeated at least three times.
Monocyte Chemoattractant Protein-1 Expression
Levels of monocyte chemoattractant protein-1 (MCP-1) expression in the HCAECs were determined using an ELISA kit (R&D Systems).
Monocyte Transendothelial Migration Assay
HCAECs were allowed to reach confluence on 0.2% gelatin-coated 3.0-μm polyethylene terepthalate track-etched cell culture inserts (BD Biosciences). TEM assays were then performed essentially as described with modifications (5). In brief, THP-1 cells were labeled with 2 μM Cell Tracker green 5-chloromethylfluorescein diacetate (Molecular Probes). Labeled THP-1 cells (3 × 105/200 μl) were added to the upper compartment of the cell culture insert, and the lower compartment of the insert contained 0.5 ml serum-free RPMI 1640. The monocytes were then allowed to transmigrate for 4 h at 37°C in 5% CO2 incubator. After incubation, fluorescence at the lower chamber was measured with the Victor-Wallac Multilabel counter (Perkin Elmer) at excitation of 492 nm and emission of 571 nm. Each treatment was performed in duplicate, and experiment sets were repeated two times. The monocyte TEM was expressed as percent cell migrated.
RhoA Activation Assay
Rho activation in HCAECs was performed with commercially available kits (Pierce). In brief, RhoA activation assay was performed by pull-down assay, with Rhotekin-Rho binding domain fused with glutathione S-transferase. The active or GTP-RhoA pulled down from the cell lysates was analyzed by Western blot assays using anti-Rho antibody.
NF-κB Activation
NF-κB activation by TNF-α and the effect of CB2 agonists/antagonists were analyzed by Western imunoblot and immunofluorescence assays, respectively.
In Vivo Vascular Inflammation Model
Protocols involving the use of experimental animals were approved by the author’s institutional animal care and use committee and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male C57Bl/6J mice (6–8 wk) were administered 3 mg/kg LPS (E. coli serotype O197:B8; Sigma) in a single intraperitoneal dose. In some experiments, 1 h before LPS administration, experimental animals were injected with HU-308 or JWH-133 (10 mg/kg ip) ± AM-630 (3 mg/kg ip). After 4 h, animals were euthanized, and aortas were dissected and snap frozen for the determination of ICAM-1 and VCAM-1 expressions.
Monocyte Adhesion to Aortic Vascular Endothelium
Monocyte-enriched peripheral blood mononuclear cells were isolated from rats, and binding of BCECF-labeled (5 μmol/l final concentration; Molecular Probes) monocytes to the vascular endothelium was determined as previously described (6). In brief, rat aortic segments were treated with HU-308 or JWH-133 (3 μM) ± AM-630 (1 μM) ± TNF-α (50 ng/ml, for 4 h). The vessels were cut open (en face preparation) and incubated with BCECF-loaded monocytes. After a 1-h incubation at 37°C, unbound monocytes were washed out. Bound monocytes were quantified by counting the cells under a fluorescent microscope. Representative images were captured using a fluorescent microscope (Olympus IX 81; original magnification: ×10; endothelial cell’s nuclei were counterstained with Hoechst 33258 for orientation).
In another set of experiments, rats were injected with a single dose of LPS (3 mg/kg ip) with or without the pretreatment with HU-308 or JWH-133 (10 mg/kg) ± AM-630 (3 mg/kg) for 1 h; 6 h later aortas were isolated, and monocyte adhesion to vascular endothelium was performed. Monocyte adhesion to the vascular endothelium was then determined as described in the previous paragraph.
Statistical Analysis
All values are means ± SE. Statistical significance of the data was assessed by one-way ANOVA with Tukey’s post hoc test (GraphPad-Prism4). P < 0.05 was considered significant.
RESULTS
CB1 and CB2 Receptors Are Expressed in HCAECs
As shown in Figs. 1 and 2, CB1 and CB2 receptors are expressed in endothelial cells, at basal conditions as demonstrated by immunofluorescence assays (Fig. 1A), conventional RT-PCR (Fig. 1, B and C), and Western blot (Fig. 1, D and E). From quantitative analysis of Western blot, it was revealed that mice brain extracts had ~4.5-fold increased CB1-receptor expression compared with HCAECs (Fig. 1D). CB2 expression was ~5.5-fold higher in THP-1 monocytes than in HCAECs (Fig. 1E). Protein from mouse brain extracts and THP-1 cell lysates were used as appropriate positive controls for CB1 and CB2 receptors, respectively. To rule out the nonspecific staining for CB1 or CB2 expression, we preabsorbed either CB1 or CB2 with the corresponding blocking peptides supplied with the primary antibodies. Next, we used the preabsorbed anti-body for the detection of CB1 and CB2 in HCAECs simultaneously with the neat antibodies. Our results indicated that preabsorbed antibodies failed to stain the CB1 or CB2 receptors in HCAECs, suggesting that the antibody specifically recognizes CB1 or CB2 receptors in HCAECs (Fig. 1A). Similar results were obtained with Western blot assays (data not shown).
Fig. 1.
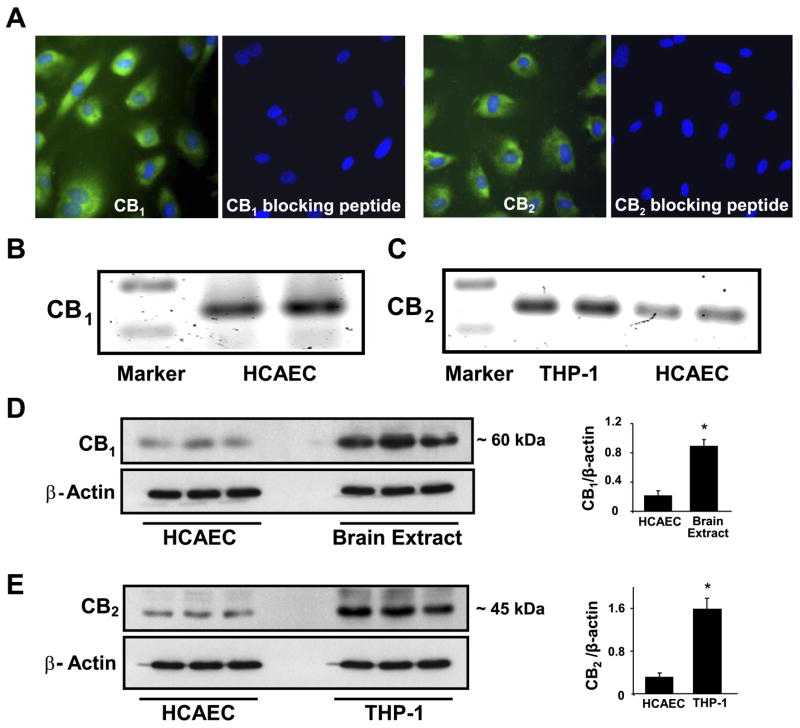
Cannabinoid-1 (CB1) and cannabinoid-2 (CB2) receptors are expressed in human coronary artery endothelial cells (HCAECs). A: expression of CB1 and CB2 receptors in HCAECs demonstrated by immunofluorescence staining. CB1 expression and CB2 expression in the endothelial cells were detected with rabbit polyclonal anti-CB1 and anti-CB2 antibodies (Cayman Chemical) and detected by secondary antibody goat anti-rabbit FITC (Pierce); the nucleus was counterstained with 4′,6-diamidino-2-phenylindole (Molecular Probes, Invitrogen). B and C: RT-PCR analysis of CB1 (B) and CB2 (C) receptor expression in endothelial cells. D and E, left: Western immunoblot demonstrating expression of CB1 (D) and CB2 (E) in endothelial cells. Six samples from 3–5 passages from HCAECs or THP-1 and lysates from six brain tissues from wild-type C57Bl/6J mice were used to evaluate CB1 and CB2 expression in human endothelial cells. D and E, right: the quantification of CB1 and CB2 expression in HCAECs; n = 6 samples. *P < 0.05 vs. brain extract or THP-1 cells.
Fig. 2.

CB2 agonists decrease TNF-α-induced ICAM-1 and VCAM-1 expression in HCAECs. A and B: ICAM-1 (A) and VCAM-1 (B) expression. Cells were treated with either TNF-α or CB2 agonists for 6 h or with CB2 agonists with the indicated concentrations followed by treatment with TNF-α for 6 h. Cell surface ELISA was then performed by measuring the absorbance at 450 nm as described in MATERIALS AND METHODS to determine cell surface ICAM-1 or VCAM-1 expression; n = 9 samples. *P < 0.05 vs. control; #P < 0.05 vs. TNF-α. C and D: cells were pretreated with CB1 or CB2 antagonists followed by treatment with TNF-α ± CB2 agonists as indicated for 6 h or pretreated with CB1 or CB2 antagonists (1 μM each) followed by treatment with either TNF-α alone or in combination with CB2 agonists. Cell surface ELISA was then performed; n = 9 samples. OD, optical density. *P < 0.05 vs. control; #P < 0.05 vs. TNF-α; †P < 0.05 vs. TNF-α + HU-308 or JWH-133.
CB2 Agonists Inhibit TNF-α-Induced ICAM-1 and VCAM-1 Expression In Vitro
TNF-α (50 ng/ml) treatment of HCAECs for 6 h, resulted in marked upregulation of ICAM-1 and VCAM-1 expressions, which were dose dependently diminished by HU-308 or JWH-133 (Fig. 2, A and B). These effects of CB2 agonists were attenuated by CB2 (SR-144528 and AM-630) but not by CB1 (SR-141716 and AM-251) antagonists (Fig. 2, C and D). CB1 or CB2 antagonists by themselves had no effect on TNF-α-induced adhesion molecule expression (Fig. 2, C and D).
CB2 Agonists Attenuate TNF-α-Induced Monocyte Adhesion and MCP-1 Expression In Vitro and Ex Vivo
Pretreatment of HCAECs with HU-308 or JWH-133 (3 μM) from 1 h before and during the complete TNF-α exposure inhibited TNF-α-induced monocyte adhesion (Fig. 3, A and B) and MCP-1 expression (Fig. 3C), and these effects were attenuated by CB2 antagonists (Fig. 3). HU-308 also inhibited TNF-α-induced monocyte adhesion in aortas ex vivo (Fig. 4A).
Fig. 3.

CB2 agonists decrease TNF-α-induced monocyte chemoattractant protein-1 (MCP-1) expression in HCAECs and monocyte adhesion to HCAECs. SR2, SR-144528; T, TNF-α. A: representative fields of monocytes adhered to HCAECs with representative treatments as indicated. B: quantification of data for monocyte adherence to endothelial cells when incubated with TNF-α ± HU-308 or JWH-133 (4 μM) for 6 h or pretreated with CB2 antagonists (1 μM) for 1 h followed by treatment with TNF-α ± HU-308 or JWH-133 for 6 h; n = 9 samples. *P < 0.05 vs. control; #P < 0.05 vs. TNF-α; †P < 0.05 vs. TNF-α + HU-308 or JWH-133. C: MCP-1 expression in HCAECs for treatments indicated; n = 6 samples. *P < 0.05 vs. control; #P < 0.05 vs. TNF-α; †P < 0.05 vs. TNF-α + HU-308 or JWH-133.
Fig. 4.

CB2 agonists mitigate TNF-α- and/or endotoxin (LPS)-induced ICAM-1 and VCAM-1 expression and monocyte adhesion to aortic endothelium of rats ex vivo or in vivo. A: CB2 agonist HU-308 mitigates adhesion of monocytes to aortic vascular endothelium of isolated aortas pretreated with TNF-α; n = 4 samples. *P < 0.05 vs. vehicle [control (Co)]; #P < 0.05 vs. TNF-α; †P < 0.05 vs. LPS + HU-308 or JWH-133. B: mice were treated with LPS ± HU-308 or JWH-133 ± AM-630 as indicated; n = 6 samples. Aortas were then isolated, and ICAM-1 or VCAM-1 expressions were determined by ELISA (R&D Systems).*P < 0.05 vs. vehicle-treated (Co) mice; #P < 0.05 vs. LPS; †P < 0.05 vs. LPS + HU-308 or JWH-133. Protein estimation in the tissue lysates was determined by Lowry assay (Bio-Rad). Values are expressed as ng/mg protein. C: CB2 agonist HU-308 mitigates endotoxin-induced monocyte adhesion to aortic vascular endothelium of isolated aortas of rats treated with endotoxin; n = 4 samples. *P < 0.05 vs. vehicle-treated (Co) mice; #P < 0.05 vs. LPS-treated rats; †P < 0.05 vs. LPS + HU-308 or JWH-133.
CB2 Agonists Attenuate Endotoxin (LPS)-Induced ICAM-1 and VCAM-1 Expression In Vivo and Adhesion of Monocytes to the Aortic Segments
Endotoxin administration to mice has been shown to induce massive vascular inflammation and characterized by increase in adhesion molecules and NF-κB activation (40, 51). Because in vitro CB2-receptor stimulation blunted cytokine-induced adhesion molecule expression and NF-κB activation, we tested whether CB2-receptor stimulation could ameliorate LPS-induced vascular inflammation in vivo. We found that LPS administration markedly induced ICAM-1 and VCAM-1 (Fig. 4B) expression in the aortas (~5.6- and 4.8-fold increase compared with mice treated with vehicle alone). HU-308 or JWH-133 pretreatment markedly reduced the expression of both ICAM-1 and VCAM-1 (Fig. 4B), which could be attenuated by the CB2 antagonist AM-630.
CB2 agonist HU-308 also significantly inhibited monocyte adhesion to the aortic segments prepared from rats treated with LPS (Fig. 4C). This effect was attenuated by CB2 antagonist AM-630 (Fig. 4C), suggesting that CB2-receptor activation could mitigate the endotoxin-induced vascular inflammation.
CB2 Agonists Inhibit TNF-α-Induced Monocyte TEM and RhoA Activation in HCAECs
TNF-α treatment of HCAECs markedly increased monocyte TEM ~4–3 fold vs. control. This was attenuated by HU-308 or JWH-133 (3 μM; Fig. 5A). Furthermore, TNF-α treatment induced RhoA activation (~4.0-fold vs. control), and this activation was attenuated by treatment with HU-308 or JWH-133 (Fig. 5B). These effects of CB2 agonists were attenuated by CB2 antagonist.
Fig. 5.
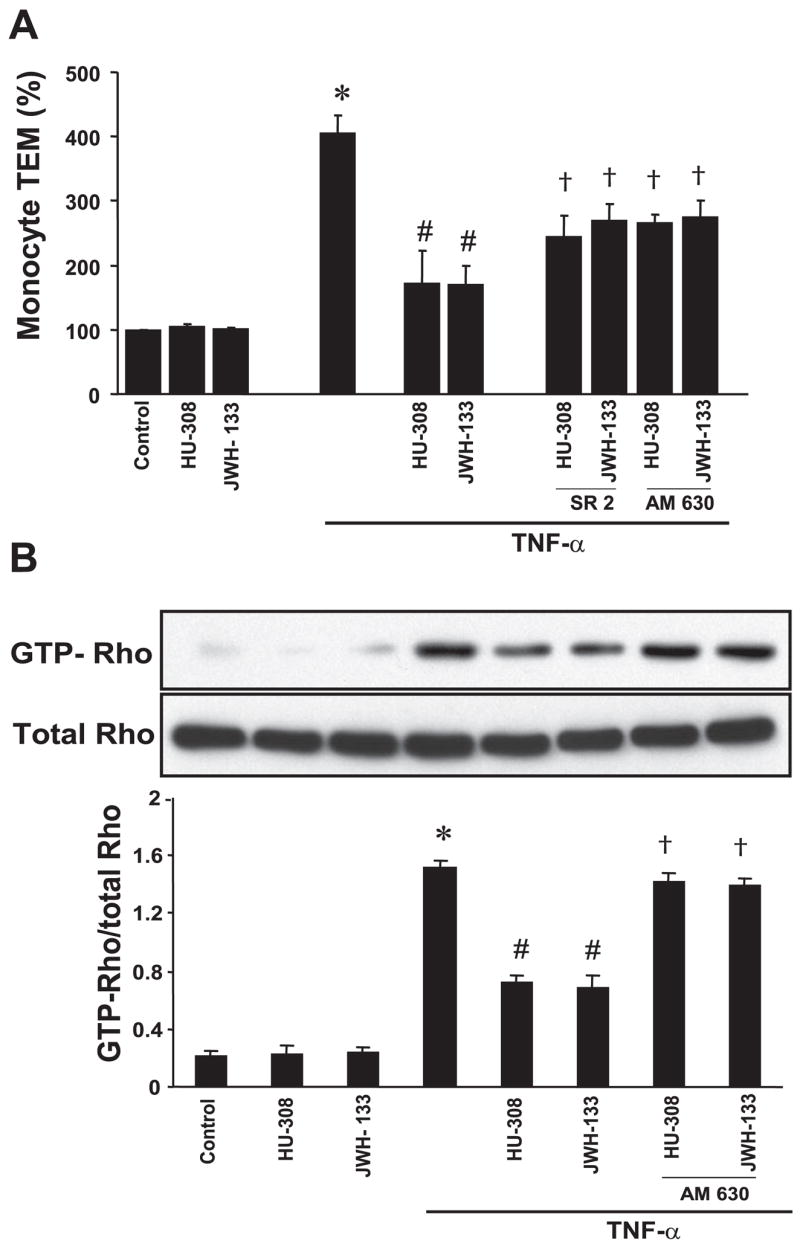
CB2 agonists decrease TNF-α-induced monocyte transendothelial migration (TEM) and RhoA activation in HCAECs. A: HCAECs were treated as indicated, and then monocyte TEM assays were performed as described in MATERIALS AND METHODS; n = 4 samples. *P < 0.05 vs. controls; #P < 0.05 vs. TNF-α; †P < 0.05 vs. TNF-α + HU-308 or JWH-133. B: HCAECs were grown in 100-mm culture dishes and treated in identical manner as described above, 500 μg cell lysates were incubated with Rhotekin-Rho binding domain-glutathione S-transferase fusion protein, and pull-down assays were performed (Pierce), with active RhoA detected with the use of anti-RhoA antibody; n = 3 samples. *P < 0.05 vs. controls; #P < 0.05 vs. TNF-α; †P < 0.05 vs. TNF-α + HU-308 or JWH-133.
CB2 Agonists Decrease TNF-α-Induced NF-κB Activation in HCAECs
TNF-α treatment leads to marked NF-κB activation, characterized by increased translocation to the nucleus as demonstrated by Western blot (Fig. 6A). We observed ~2.5-fold increase in nuclear translocation of p65 (NF-κB) (Fig. 6A); likewise, a marked increase in immunofluorescence was also observed above the nuclei of cells treated with TNF-α (Fig. 6B). These effects were significantly decreased by the pretreatment with HU-308 or JWH-133, and the latter could be attenuated by CB2-antagonist AM-630.
Fig. 6.
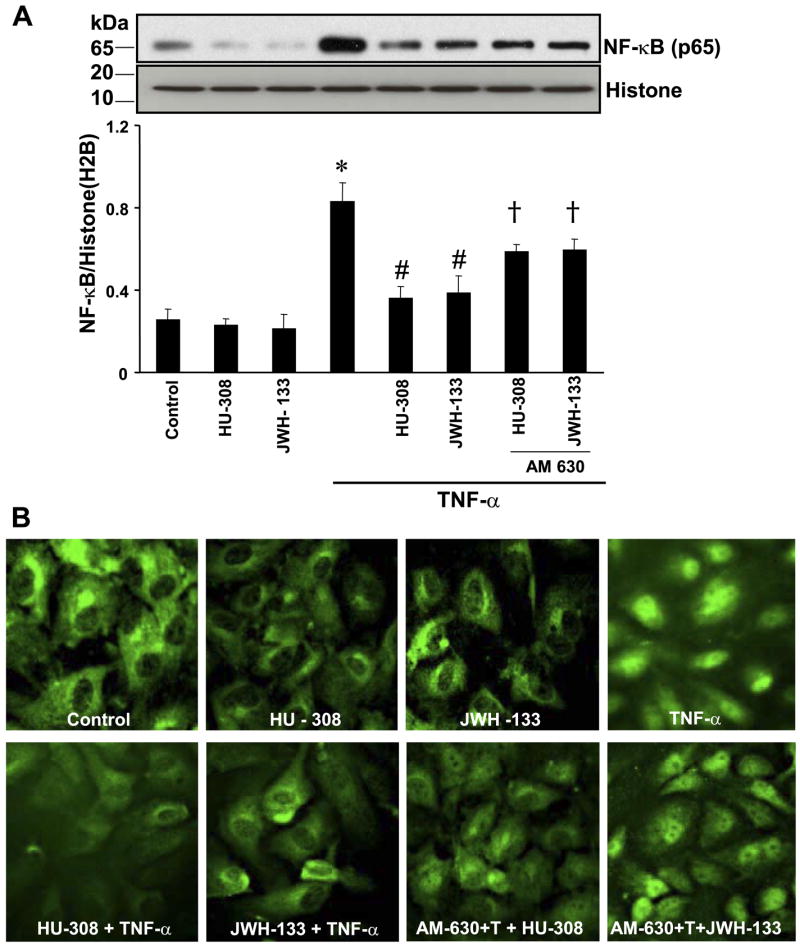
CB2 agonists decrease TNF-α-induced NF-κB activation in HCAECs. A: cells were treated with TNF-α ± HU-308 or JWH-133 (3 μM) for 6 h or pretreated with CB2 antagonists (1 μM) followed by treatment with TNF-α ± HU-308 or JWH-133 for 6 h. Nuclear extracts were then prepared, and NF-κB expression was determined by Western immunoblot assay; n = 3 samples. *P < 0.05 vs. control; #P < 0.05 vs. TNF-α; †P < 0.05 vs. TNF-α + HU-308 or JWH-133. B: activation of NF-κB by TNF-α in HCAECs, and CB2 agonists inhibit NF-κB activation. Note the intense nuclear staining of NF-κB (p65) in cells exposed to TNF-α and the faint staining indicating that CB2 agonists diminished TNF-α activation of NF-κB. Shown are representative images from 3 separate experiments.
DISCUSSION
The novel and definitive findings emerging from our study is that HCAECs express both CB1 and CB2 receptors under basal/physiological conditions. Importantly, our study also demonstrates that CB2-receptor activation attenuates TNF-α-triggered NF-κB and RhoA activation, upregulation of adhesion molecules ICAM-1 and VCAM-1, expression of monocyte chemoattractant protein, TEM of monocytic THP-1 cells, and monocyte-endothelial adhesion in HCAECs. Furthermore, CB2-receptor activation attenuates TNF-α-induced adhesion of monocytes to aortic endothelium ex vivo, the endotoxin-induced ICAM-1 and VCAM-1 expression and adhesion of monocytes to aortic endothelium of isolated aortas of endotoxin-treated rats.
Presently, it is thought that CB1 receptors are primarily expressed in the brain and some peripheral tissues, including the heart, vascular tissues, adipocytes, and liver (22, 29). On the other hand, the CB2 receptor was previously considered to be expressed predominantly by immune and hematopoietic cells (22, 29). However, more recent studies have also found CB2 receptors in brain (45), myocardium (26), cardiomyoblasts (26, 34), and endothelial cells of various origins (3, 9, 25, 52). Here, we report expression of CB1 and CB2 receptors in HCAECs under basal/physiological conditions. Interestingly, the expression of CB2 receptors could be enhanced by the proinflammatory cytokine TNF-α in endothelial cells (data not shown). A similar phenomenon was previously proposed during the microglia activation (46).
The hallmark for the development of atherosclerosis is the adhesion of monocytes to the endothelium, followed by TEM of monocytes (10). It has been well-established that TNF-α and/or endotoxin(s) induces NF-κB-dependent upregulation of the expression of ICAM-1, VCAM-1, and MCP-1 in endothelial cells, contributing to the increased adhesion of monocytes to the endothelium migration and TEM (43). The TNF-α-induced NF-κB activation has been shown to involve RhoA activation (21), and RhoA activation was also implicated in monocyte TEM (33). Likewise, MCP-1 has been shown to be involved in monocyte TEM (41) and endothelial barrier disruption through RhoA activation (37). Consistent with the above-mentioned studies, we found that TNF-α increased NF-κB and RhoA activation, upregulation of adhesion molecules ICAM-1 and VCAM-1, expression of MCP-1, monocyte-endothelial adhesion in HCAECs, and TEM of monocytes. We demonstrate that selective CB2-receptor agonists JWH-133 and HU-308 markedly decrease TNF-α-increased NF-κB activation, upregulation of adhesion molecules ICAM-1 and VCAM-1, expression of MCP-1, monocyte-endothelial adhesion in HCAECs, and TEM of monocytic THP-1 cells in a CB2-dependent manner, since these effects of CB2 agonists could be attenuated by CB2 antagonists. Consistent with our observation, it has been recently demonstrated that JWH-015, another less potent CB2 agonist inhibited the TEM of T lymphocytes stimulated by chemokines such as CXCL12 and CXCR4 (8).
Adhesion molecules also mediate the initial attachment of neutrophils to the activated endothelium, which is a crucial early event in ischemia-reperfusion injury (15, 16). On reperfusion, inflammatory cytokines (e.g., TNF-α) act as continuous stimuli for neutrophil infiltration and upregulate the production of chemokines, which may also contribute to the upregulation of cell adhesion molecules and neutrophil activation (15, 16). Numerous recent studies have demonstrated that CB2-receptor activation may also protect against myocardial (7), cerebral (49), and hepatic ischemia-reperfusion (1, 32) injuries by decreasing the endothelial cell activation, the expression of adhesion molecules ICAM-1 and VCAM-1, TNF-α, and chemokine (MIP-1α and MIP-2) levels, neutrophil infiltration, lipid peroxidation, and apoptosis.
Collectively, our results suggest that selective CB2-receptor agonists may offer a novel approach in the treatment of a variety of inflammatory diseases, including atherosclerosis. Thus attenuation of TNF signaling, coupled with the absence of psychoactive effects associated with CB2-receptor stimulation, makes this a particularly encouraging therapeutic approach.
Acknowledgments
GRANTS
This study was supported by the Intramural Research Program of National Institute on Alcohol Abuse and Alcoholism (to P. Pacher), National Institute on Drug Abuse (NIDA) Grant DA-11322 (to K. Mackie), NIDA Grant DA-03590 (to J. W. Huffman), and American Heart Association Grants 0430108N and 0435140N (to A. Csiszar and Z. Ungvari).
References
- 1.Batkai S, Osei-Hyiaman D, Pan H, El-Assal O, Rajesh M, Mukhopadhyay P, Hong F, Harvey-White J, Jafri A, Hasko G, Huffman JW, Gao B, Kunos G, Pacher P. Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. FASEB J. 2007;21:1788–1800. doi: 10.1096/fj.06-7451com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhunia AK, Arai T, Bulkley G, Chatterjee S. Lactosylceramide mediates tumor necrosis factor-alpha-induced intercellular adhesion molecule-1 (ICAM-1) expression and the adhesion of neutrophil in human umbilical vein endothelial cells. J Biol Chem. 1998;273:34349–34357. doi: 10.1074/jbc.273.51.34349. [DOI] [PubMed] [Google Scholar]
- 3.Blazquez C, Casanova ML, Planas A, Del Pulgar TG, Villanueva C, Fernandez-Acenero MJ, Aragones J, Huffman JW, Jorcano JL, Guzman M. Inhibition of tumor angiogenesis by cannabinoids. FASEB J. 2003;17:529–531. doi: 10.1096/fj.02-0795fje. [DOI] [PubMed] [Google Scholar]
- 4.Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-α reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:2137–2142. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee S, Wei H. Roles of glycosphingolipids in cell signaling: adhesion, migration, and proliferation. Methods Enzymol. 2003;363:300–312. doi: 10.1016/S0076-6879(03)01059-0. [DOI] [PubMed] [Google Scholar]
- 6.Csiszar A, Ahmad M, Smith KE, Labinskyy N, Gao Q, Kaley G, Edwards JG, Wolin MS, Ungvari Z. Bone morphogenetic protein-2 induces proinflammatory endothelial phenotype. Am J Pathol. 2006;168:629–638. doi: 10.2353/ajpath.2006.050284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Filippo C, Rossi F, Rossi S, D’Amico M. Cannabinoid CB2 receptor activation reduces mouse myocardial ischemia-reperfusion injury: involvement of cytokine/chemokines and PMN. J Leukoc Biol. 2004;75:453–459. doi: 10.1189/jlb.0703303. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh S, Preet A, Groopman JE, Ganju RK. Cannabinoid receptor CB2 modulates the CXCL12/CXCR4-mediated chemotaxis of T lymphocytes. Mol Immunol. 2006;43:2169–2179. doi: 10.1016/j.molimm.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Golech SA, McCarron RM, Chen Y, Bembry J, Lenz F, Mechoulam R, Shohami E, Spatz M. Human brain endothelium: coexpression and function of vanilloid and endocannabinoid receptors. Brain Res. 2004;132:87–92. doi: 10.1016/j.molbrainres.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 10.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 11.Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RG, Ross RA, Mechoulam R, Fride E. HU-308: a specific agonist for CB2, a peripheral cannabinoid receptor. Proc Natl Acad Sci USA. 1999;96:14228–14233. doi: 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 13.Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR. 3-(1′,1′-Dimethylbutyl)-1-deoxy-δ8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem. 1999;7:2905–2914. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- 14.Ip JH, Fuster V, Badimon L, Badimon J, Taubman MB, Chesebro JH. Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol. 1990;15:1667–1687. doi: 10.1016/0735-1097(90)92845-s. [DOI] [PubMed] [Google Scholar]
- 15.Jaeschke H. Cellular adhesion molecules: regulation and functional significance in the pathogenesis of liver diseases. Am J Physiol Gastrointest Liver Physiol. 1997;273:G602–G611. doi: 10.1152/ajpgi.1997.273.3.G602. [DOI] [PubMed] [Google Scholar]
- 16.Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol. 1997;61:647–653. doi: 10.1002/jlb.61.6.647. [DOI] [PubMed] [Google Scholar]
- 17.Javaid K, Rahman A, Anwar KN, Frey RS, Minshall RD, Malik AB. Tumor necrosis factor-α induces early-onset endothelial adhesivity by protein kinase Cζ-dependent activation of intercellular adhesion molecule-1. Circ Res. 2003;92:1089–1097. doi: 10.1161/01.RES.0000072971.88704.CB. [DOI] [PubMed] [Google Scholar]
- 18.Jawien J, Gajda M, Mateuszuk L, Olszanecki R, Jakubowski A, Szlachcic A, Korabiowska M, Korbut R. Inhibition of nuclear factor-κB attenuates artherosclerosis in apoE/LDLR-double knockout mice. J Physiol Pharmacol. 2005;56:483–489. [PubMed] [Google Scholar]
- 19.Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev. 2005;5:400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA, Tabas I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6: model of NF-κB- and MAP kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280:21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 21.Lockyer JM, Colladay JS, Alperin-Lea WL, Hammond T, Buda AJ. Inhibition of nuclear factor-κB-mediated adhesion molecule expression in human endothelial cells. Circ Res. 1998;82:314–320. doi: 10.1161/01.res.82.3.314. [DOI] [PubMed] [Google Scholar]
- 22.Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 23.Maier W, Altwegg LA, Corti R, Gay S, Hersberger M, Maly FE, Sutsch G, Roffi M, Neidhart M, Eberli FR, Tanner FC, Gobbi S, von Eckardstein A, Luscher TF. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation. 2005;111:1355–1361. doi: 10.1161/01.CIR.0000158479.58589.0A. [DOI] [PubMed] [Google Scholar]
- 24.Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92:1866–1874. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mestre L, Correa F, Docagne F, Clemente D, Guaza C. The synthetic cannabinoid WIN 55,212-2 increases COX-2 expression and PGE2 release in murine brain-derived endothelial cells following Theiler’s virus infection. Biochem Pharmacol. 2006;72:869–880. doi: 10.1016/j.bcp.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 26.Mukhopadhyay P, Batkai S, Rajesh M, Czifra N, Harvey-White J, Hasko G, Zsengeller Z, Gerard NP, Liaudet L, Kunos G, Pacher P. Pharmacological inhibition of cannabinoid receptor-1 protects against doxorubicin-induced cardiotoxicity. J Am Coll Cardiol. 2007;50:528–536. doi: 10.1016/j.jacc.2007.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, Fujii H, Saito K, Sekikawa K, Seishima M. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005;180:11–17. doi: 10.1016/j.atherosclerosis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 28.Osterud B, Bjorklid E. Role of monocytes in atherogenesis. Physiol Rev. 2003;83:1069–1112. doi: 10.1152/physrev.00005.2003. [DOI] [PubMed] [Google Scholar]
- 29.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel RP, Moellering D, Murphy-Ullrich J, Jo H, Beckman JS, Darley-Usmar VM. Cell signaling by reactive nitrogen and oxygen species in atherosclerosis. Free Radic Biol Med. 2000;28:1780–1794. doi: 10.1016/s0891-5849(00)00235-5. [DOI] [PubMed] [Google Scholar]
- 32.Rajesh M, Pan H, Mukhopadhyay P, Batkai S, Osei-Hyiaman D, Hasko G, Liaudet L, Gao B, Pacher P. Cannabinoid-2 receptor agonist HU-308 protects against hepatic ischemia/reperfusion injury by attenuating oxidative stress, inflammatory response and apoptosis. J Leukoc Biol. doi: 10.1189/jlb.0307180. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seasholtz TM, Brown JH. RHO SIGNALING in vascular diseases. Mol Interv. 2004;4:348–357. doi: 10.1124/mi.4.6.8. [DOI] [PubMed] [Google Scholar]
- 34.Shmist YA, Goncharov I, Eichler M, Shneyvays V, Isaac A, Vogel Z, Shainberg A. δ-9-Tetrahydrocannabinol protects cardiac cells from hypoxia via CB2 receptor activation and nitric oxide production. Mol Cell Biochem. 2006;283:75–83. doi: 10.1007/s11010-006-2346-y. [DOI] [PubMed] [Google Scholar]
- 35.Skoog T, Dichtl W, Boquist S, Skoglund-Andersson C, Karpe F, Tang R, Bond MG, de Faire U, Nilsson J, Eriksson P, Hamsten A. Plasma tumour necrosis factor-α and early carotid atherosclerosis in healthy middle-aged men. Eur Heart J. 2002;23:376–383. doi: 10.1053/euhj.2001.2805. [DOI] [PubMed] [Google Scholar]
- 36.Spiecker M, Darius H, Liao JK. A functional role of IκB-epsilon in endothelial cell activation. J Immunol. 2000;164:3316–3322. doi: 10.4049/jimmunol.164.6.3316. [DOI] [PubMed] [Google Scholar]
- 37.Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of MCP-1 in endothelial cell tight junction “opening”: signaling via Rho and Rho kinase. J Cell Sci. 2003;116:4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- 38.Steffens S, Veillard NR, Arnaud C, Pelli G, Burger F, Staub C, Karsak M, Zimmer A, Frossard JL, Mach F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature. 2005;434:782–786. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- 39.Stoll LL, Denning GM, Weintraub NL. Endotoxin, TLR4 signaling and vascular inflammation: potential therapeutic targets in cardiovascular disease. Current Pharm Design. 2006;12:4229–4245. doi: 10.2174/138161206778743501. [DOI] [PubMed] [Google Scholar]
- 40.Stoll LL, Denning GM, Weintraub NL. Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:2227–2236. doi: 10.1161/01.ATV.0000147534.69062.dc. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi M, Masuyama J, Ikeda U, Kasahara T, Kitagawa S, Takahashi Y, Shimada K, Kano S. Induction of monocyte chemoattractant protein-1 synthesis in human monocytes during transendothelial migration in vitro. Circ Res. 1995;76:750–757. doi: 10.1161/01.res.76.5.750. [DOI] [PubMed] [Google Scholar]
- 42.Takeda R, Suzuki E, Satonaka H, Oba S, Nishimatsu H, Omata M, Fujita T, Nagai R, Hirata Y. Blockade of endogenous cytokines mitigates neointimal formation in obese Zucker rats. Circulation. 2005;111:1398–1406. doi: 10.1161/01.CIR.0000158482.83179.DB. [DOI] [PubMed] [Google Scholar]
- 43.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 44.Tzoulaki I, Murray GD, Lee AJ, Rumley A, Lowe GD, Fowkes FG. C-reactive protein, interleukin-6, and soluble adhesion molecules as predictors of progressive peripheral atherosclerosis in the general population: Edinburgh Artery Study. Circulation. 2005;112:976–983. doi: 10.1161/CIRCULATIONAHA.104.513085. [DOI] [PubMed] [Google Scholar]
- 45.Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 46.Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. 2003;23:1398–1405. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber C, Erl W, Pietsch A, Weber PC. Aspirin inhibits nuclear factor-κB mobilization and monocyte adhesion in stimulated human endothelial cells. Circulation. 1995;91:1914–1917. doi: 10.1161/01.cir.91.7.1914. [DOI] [PubMed] [Google Scholar]
- 48.Wung BS, Ni CW, Wang DL. ICAM-1 induction by TNF-α and IL-6 is mediated by distinct pathways via Rac in endothelial cells. J Biomed Sci. 2005;12:91–101. doi: 10.1007/s11373-004-8170-z. [DOI] [PubMed] [Google Scholar]
- 49.Zhang M, Martin BR, Adler MW, Razdan RK, Jallo JI, Tuma RF. Cannabinoid CB(2) receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J Cereb Blood Flow Metab. 2007;27:1387–1396. doi: 10.1038/sj.jcbfm.9600447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang WJ, Frei B. α-Lipoic acid inhibits TNF-α-induced NF-κB activation and adhesion molecule expression in human aortic endothelial cells. FASEB J. 2001;15:2423–2432. doi: 10.1096/fj.01-0260com. [DOI] [PubMed] [Google Scholar]
- 51.Zhang WJ, Wei H, Hagen T, Frei B. α-Lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci USA. 2007;104:4077–4082. doi: 10.1073/pnas.0700305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zoratti C, Kipmen-Korgun D, Osibow K, Malli R, Graier WF. Anan-damide initiates Ca2+ signaling via CB2 receptor linked to phospholipase C in calf pulmonary endothelial cells. Br J Pharmacol. 2003;140:1351–1362. doi: 10.1038/sj.bjp.0705529. [DOI] [PMC free article] [PubMed] [Google Scholar]