

Abstract
κ-Conotoxin-PVIIA (κ-PVIIA) belongs to a family of peptides derived from a hunting marine snail that targets to a wide variety of ion channels and receptors. κ-PVIIA is a small, structurally constrained, 27-residue peptide that inhibits voltage-gated K channels. Three disulfide bonds shape a characteristic four-loop folding. The spatial localization of positively charged residues in κ-PVIIA exhibits strong structural mimicry to that of charybdotoxin, a scorpion toxin that occludes the pore of K channels. We studied the mechanism by which this peptide inhibits Shaker K channels expressed in Xenopus oocytes with the N-type inactivation removed. Chronically applied to whole oocytes or outside-out patches, κ-PVIIA inhibition appears as a voltage-dependent relaxation in response to the depolarizing pulse used to activate the channels. At any applied voltage, the relaxation rate depended linearly on the toxin concentration, indicating a bimolecular stoichiometry. Time constants and voltage dependence of the current relaxation produced by chronic applications agreed with that of rapid applications to open channels. Effective valence of the voltage dependence, zδ, is ∼0.55 and resides primarily in the rate of dissociation from the channel, while the association rate is voltage independent with a magnitude of 107–108 M−1 s−1, consistent with diffusion-limited binding. Compatible with a purely competitive interaction for a site in the external vestibule, tetraethylammonium, a well-known K-pore blocker, reduced κ-PVIIA's association rate only. Removal of internal K+ reduced, but did not eliminate, the effective valence of the toxin dissociation rate to a value <0.3. This trans-pore effect suggests that: (a) as in the α-KTx, a positively charged side chain, possibly a Lys, interacts electrostatically with ions residing inside the Shaker pore, and (b) a part of the toxin occupies an externally accessible K+ binding site, decreasing the degree of pore occupancy by permeant ions. We conclude that, although evolutionarily distant to scorpion toxins, κ-PVIIA shares with them a remarkably similar mechanism of inhibition of K channels.
Keywords: pore blockade, patch clamp, Xenopus oocyte, Conus venom, Shaker K channel
Introduction
Conotoxins are a family of small peptide toxins derived from the venom of marine snails of the Conus, a genus composed of ∼500 predator species (Olivera 1997). These peptides, which bind with high affinity to excitable tissue, are small (8–35 residues) and structurally constrained by intrapeptide disulfide bounds (Myers et al. 1993). The pattern of formation of the disulfide bonds yields a characteristic folding in the peptide family that allows for structural subfamily classification. A set having three disulfide bonds that defines a four-loop framework specifically blocks voltage-gated ion channels (Olivera et al. 1990; Pallaghy et al. 1993). Although members of the four-loop toxins may share little amino-acid similarity among the noncysteine residues, their overall folding can be remarkably similar (Scanlon et al. 1997). Thus, specificity for different voltage-gated channels is believed to be conferred solely by the specific sequence of each peptide. For example, ω-, δ-, and κ-conotoxins are specific for Ca, Na, and K voltage-gated channels, respectively. However, detailed knowledge of their mechanism of inhibition on voltage-gated channels is not yet clear.
κ-Conotoxin-PVIIA (κ-PVIIA)1 is a 27-residue peptide component of the Conus purpurascens venom found to inhibit K channels. This peptide acts rapidly on its target, so it is proposed to have an important role in the quick excitotoxic prey immobilization after the venomous sting (Terlau et al. 1996; Olivera 1997). The structure has recently been resolved by nuclear magnetic resonance methods and revealed as a member of the four-loop family (Scanlon et al. 1997; Savarin et al. 1998). Although κ-PVIIA is ∼10-residues shorter, it shows striking similarities to the space distribution of functionally relevant basic residues of charybdotoxin (CTX), a member of a well-characterized group of K channel pore blockers, the α-KTx, (Miller 1995; Scanlon et al. 1997; Savarin et al. 1998). In CTX, the amino group of the side chain of Lys-27 has been proposed to interact intimately with the K-permeation pathway by occluding the pore. Moreover, its structure shows a lysine paired with an aromatic side chain 6–7 Å away, a conserved dyad in peptide blockers of K channels (Dauplais et al. 1997). Savarin et al. 1998 have proposed that the lysine side chain of this dyad interacts with the K channel pore as Lys-27 does in CTX. In addition to this architectural mimicry to other K channel peptide blockers, there are three lines of evidence suggesting that κ-PVIIA binds to the external vestibule of K channels. First, different pore splice variants bind κ-PVIIA with dissimilar affinity (Kim et al. 1997). Second, κ-PVIIA competes with other putative pore blocker toxins (Savarin et al. 1998). Third, point mutations in the external vestibule of the voltage-gated Shaker K channel modify the extent of inhibition by κ-PVIIA, suggesting that this peptide also inhibits K channels by occluding the permeation pathway (Scanlon et al. 1997; Shon et al. 1998). However, its microsite specificity is different from that of CTX; the same vestibular mutation, Phe425 → Gly, which enhances ∼2,000-fold affinity for α-KTx (Goldstein and Miller 1992), decreases it >20-fold for κ-PVIIA (Scanlon et al. 1997; Shon et al. 1998). On the other hand, recent observations are somehow conflicting with a simple pore blocker mechanism. κ-PVIIA seems to act differently whether or not Shaker K channels carries the N-type of inactivation domain, a portion of the protein that inactivates the open channel conformation from the intracellular side (Hoshi et al. 1990). In the inactivating channel, the toxin-induced inhibition seems to be independent of the voltage applied to activate the channels (Terlau et al. 1996), whereas in the inactivation-removed Shaker, the effect of this toxin is an apparent voltage-dependent modification of the rate of activation (Scanlon et al. 1997). This latter result could be interpreted as if the main toxin effect was to delay the gating mechanism as some spider and scorpion toxins do (see for example Swartz and MacKinnon 1997; Cestèle et al. 1998). In this paper, we attempt to establish the mechanism of action of κ-PVIIA on Shaker K channels.
Using the well-known α-KTx inhibition mechanism on K channels as a paradigm for comparison (Anderson et al. 1988; MacKinnon and Miller 1988; Miller 1995), we made a detailed study of the mechanism by which κ-PVIIA acts on open Shaker K channels. To our knowledge, together with Terlau et al. 1999, these are the first studies in detail of a four-loop conotoxin inhibition mechanism on a voltage-gated ion channel. Here, we present results indicating that (a) κ-PVIIA binding to Shaker K channel is consistent with a 1:1 stoichiometry and diffusion-limited association, (b) a well-known K-pore blocker, tetraethylammonium, reduces the toxin association, but not the dissociation rate, and (c) the peptide toxin interacts, perhaps electrostatically, with K+ from the intracellular side of the channel. We also argue that by binding to the external vestibule, the toxin reduces the occupancy of permeant ions inside the channel pore. Together, these results provide compelling evidence that κ-PVIIA inhibits potassium currents by plugging the pore of K channels in way analogous to scorpion toxins (MacKinnon and Miller 1988).
Methods
Peptide Synthesis
Boc-l amino acids were obtained from Novabiochem or the Peptide Institute, t-Boc-Val-OCH2-PAM-resin (substitution value 0.77 mmol g−1) was obtained from Perkin Elmer. 2-(1H-benzotriazol-1-yl)-1,1,3,3,-tetramethyluronium hexafluorophosphate (HBTU) was obtained from Richelieu Biotechnologies. Other reagents were of peptide synthesis grade from Auspep.
Stepwise synthesis (0.5 mmol scale, 0.649 g resin) was conducted manually using in situ BOC SPPS (Schnolzer et al. 1992), starting from Boc-PAM-Val resin. The average coupling was 99.80, as determined by ninhydrin assay (Sarin et al. 1981). The peptide was cleaved from the resin using HF/p-cresol/p-thiocresol (18:1:1) at −10–0°C for 1 h. Peptide was precipitated with cold ether, collected by filtration on sintered glass, washed with cold ether, and dissolved in 50% AcOH, diluted with water and lyophilized. The crude peptide was purified by preparative chromatography (Vydac C18 column, 2.2 × 25 cm), using a 1% gradient 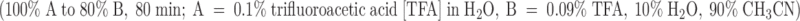 to give reduced peptide in 34% yield.
to give reduced peptide in 34% yield.
The folded product was obtained by dissolving reduced peptide (10 mg) in aqueous 0.33 M NH4OAc/0.5 M GnHCl (154 ml), with pH adjusted to 7.8 using 0.01 M NH4OH. The solution was stirred at 4°C for 5 d, in the presence of reduced and oxidized glutathione (molar ratio of peptide:GSH:GSSG was 1:100:10). Lowering the pH to 2–3 with TFA (5 ml) terminated the oxidation. The reaction mixture was loaded onto a preparative HPLC column (Vydac C18 column, 2.2 × 25 cm) (8 ml min−1) and washed with 0.1% TFA until all oxidation buffer had eluted. A 1% gradient (100% A to 80% B, 80 min) was applied and pure oxidized κ-PVIIA was isolated in 95% yield. Electrospray ionization mass spectra recorded on a PE Sciex API III triple quadrupole mass spectrometer were used to confirm the purity and molecular weights of synthetic peptides.
Heterologous Expression of Shaker K Channels
For electrophysiology, salts of analytical grade were purchased from Baker. Gentamicin, sodium pyruvate, EGTA, HEPES, N-methyl-d-glucamine (NMG), and BSA were from Sigma-Aldrich Química S.A. de C.V.
Females Xenopus laevis (Xenopus One) were anesthetized by immersion in ice. Ovarian lobes were surgically removed and collected in ND96 solution containing (mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, pH 7.6, and 50 μg/ml gentamicin. Type II collagenase, 0.9–1.5 mg/ml, was used for digestion of connective tissue (Worthington Biochemical Corp.). After removing the enzyme, stage IV–VI oocytes were isolated and manually defolliculated in a nominally Ca2+-free ND96 solution.
The inactivation-removed Shaker H4 Δ(6-46) (Hoshi et al. 1990) subcloned in vectors under control of either the cytomegalovirus or the SV40 late promoter were a gift from Dr. Christopher Miller (Brandeis University, Waltham, MA) (Oprian et al. 1989). To expose the nucleus for injection of the nonlinearized plasmids, oocytes were put with the animal pole facing up in a swinging-bucket rotor and spun for 7–12 min at 1,000 g in a Centrific centrifuge (Fisher Scientific). After nuclear injection of 0.05–0.2 ng of cDNA, oocytes were incubated at 18°C in ND96 supplemented with sodium pyruvate (2.5 mM) and bovine serum albumin (0.04%). After 15–72 h incubation, subsequent recordings with two-electrode voltage clamp (TEVC) or outside out patch clamp were made at room temperature (22°–24°C).
Electrophysiology
For whole-cell TEVC recordings, oocytes were positioned in the middle of an ∼500-μl longitudinal recordings chamber (Naranjo and Miller 1996). All measurements were made under continuous perfusion of either a standard recording solution (containing [mM]: 96 NaCl, 2 KCl, 1 MgCl2, 0.3 CaCl2, 10 HEPES-NaOH, pH 7.6, and 25 mg/l BSA) or the same solution plus κ-PVIIA, 30–1,000 nM, added from a stock of 1 mM. Voltage pulse protocols and data acquisition were performed from a personal computer running pClamp 5.5 through a Digidata 1200B acquisition interface (Axon Instruments). Whole oocyte recordings were made with an OC-750C voltage clamp amplifier (Warner Instruments). Recording electrodes, with resistance of 0.3–1 MΩ were made with Ag/AgCl pellet assemblies (Axon Instruments) inside a capillary filled with a solution made of 3 M KCl, 5 mM EGTA, and 10 mM HEPES-KOH, pH 7.0. Voltage pulses of 50–500 ms were applied from a holding potential of −90 mV, and usually ranged from −60 to 50 mV in 10-mV intervals. Because oocytes expressing outward currents >10 μA at 50 mV usually exhibited obvious slower rising times, experiments were performed in oocytes expressing 0.5–8 μA only.
For patch clamp recording, the vitelline membrane was removed after a 10-min incubation in a solution containing (mM): 200 K-aspartate, 10 KCl, 10 EGTA, and 10 HEPES, pH 7.4. After vitelline membrane removal, we followed conventional patch-clamp techniques (Hamill et al. 1981). Outside-out patches were excised from the oocyte membrane and positioned near the outlet of a rapid perfusion system (Liu and Dilger 1991; Naranjo and Brehm 1993). In such a system, a single solenoid movement (225P071; NResearch) performs rapid exchange between two solution streams converging into the tip of the patch pipette. The solenoid simultaneously opens the path of one solution while it closes the path for the other. Solution exchange rate was complete in <5 ms. For most experiments described, patch pipettes (1–4 MΩ) were filled with solutions consisting of (mM) 80 KF, 20 KCl, 1 MgCl2, 10 EGTA, and 10 HEPES-KOH, pH 7.4 (100-K in). For the experiments with reduced internal K+ concentration (15-K in), solution was 90 NMG-F, 10 KF, 1 MgCl2, 10 EGTA, and 10 HEPES-KOH, pH 7.4. External recording solution was 115 NaCl, 1 KCl, 0.2 CaCl2, 1 MgCl2, and 10 HEPES-NaOH, pH7.4 (1-K ex). For all patch clamp experiments shown in this paper, 500 nM κ-PVIIA and 1 mM tetraethylammonium (TEA+) were added to the external patch clamp recording solution. The small inflection of the K current (∼5%) produced by 1 mM TEA+ blockade was used to monitor the position of the patch pipette to obtain the optimal rate of solutions exchange (see Fig. 4, inset). Because of intrinsic variation of the solenoid latency, the TEA+-produced inflection is not often visible in the averaged records. For experiments in nominally zero internal potassium (0-K in), all potassium salts were replaced with NMG-F and the membrane patch was formed and pulled out in the 1-K ex recording solution. Then, the patch was moved to the perfusion system running a solution made of 16 NaCl, 100 KCl, 0.2 CaCl2, 1 MgCl2, and 10 HEPES-KOH, pH 7.6 (100-K ex).
Figure 4.

Comparison between pulse and chronic applications of κ-PVIIA to outside-out patches. A and D show current records from two different outside-out patches elicited by 200-ms long voltage pulses from −60 to +50 mV, with intervals of 10 mV. Bath and pipette K+ concentration were 2 and 15 mM, respectively (see methods). (B) Rapid applications of 1 mM TEA+/500 nM κ-PVIIA to an outside-out patch ∼40 ms after the beginning of the activating voltage pulse. The shaded area (Pulse) indicates the duration of the toxin application. Records shown are the average of four individual records at each voltage. (Inset) A single record taken at 0 mV showing the inflection in the K currents produced by the presence of 1 mM TEA+ in the toxin-containing solution. The TEA+-induced inflection is lost in the average because of the variable latency of the solenoid valve. (E) Chronic application of 1 mM TEA+/500 nM κ-PVIIA to the outside-out patch. The solenoid valve was open while the acquisition lasted. C and F show point-by-point divisions of the leak-subtracted TEA+/toxin records by their respective leak-subtracted controls. Single-exponential fits to each resulting relaxation while the toxin was present were performed (thin lines shown alternately for clarity). The sections including current at holding potential and capacitative transient were eliminated. (F) Exponential functions were extrapolated to the beginning of the voltage pulse and converge to the same value. In A, B, D, and E, the dotted line indicates the zero current level.
For whole oocyte and outside-out currents, off-line leak subtraction was carried out before performing point-by-point division of current records. Curve fitting, statistics, and figure preparation were carried out with Microcal Origin 3.5 (Microcal Inc.).
Elements of Analysis of a Voltage-dependent Blockade with Bimolecular Stoichiometry
Fig. 1 describes the 1:1 toxin binding equilibrium, where Sh, T, and Sh·T correspond to a conducting empty channel, the toxin, and a nonconducting channel–toxin complex, respectively. The rate constants for association and dissociation are k on and k off, respectively. This scheme predicts that in response to a step-like perturbation, the toxin-binding will relax exponentially to a new equilibrium. The relaxation rate, measured as the reciprocal of the relaxation time constant, is a function of the toxin concentration as following:
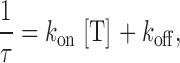 |
1 |
which defines a straight line with slope k on, and k off as intercept. Meanwhile, the fraction of unblocked channels in the new equilibrium is given by:
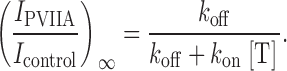 |
2 |
Scheme S1.

Together, and provide a system with two equations and two unknowns, k on and k off. Thus, by fitting the values of τ and
 |
to the macroscopic relaxation in response to each voltage step, a pair of values for k on and k off is obtained. The magnitude of effective valence for the voltage dependence of each rate constant, zδ, was calculated from the expression:
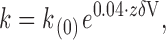 |
3 |
where V is the membrane potential in millivolts, and k (0) is the rate constant at zero applied potential. Because the voltage dependence of macroscopic relaxation depends nonlinearly on the voltage dependence of both k on and k off to describe it, we preferred to use the expression:
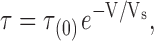 |
4 |
where τ(0) is the time constant at zero applied potential and Vs is the membrane voltage, in millivolts, that produces an e-fold increase in τ.
Results
Whole cell TEVC recordings in oocytes expressing the inactivation removed Shaker (Δ6-46) K channels (Hoshi et al. 1990) were used to examine the effect of κ-PVIIA (Fig. 1). In this set of experiments, the toxin was applied continuously to the recording solution superfusing the oocyte. Fig. 1A and Fig. B, shows K currents elicited by 200-ms depolarizing steps between −60 and +50 mV at 10-mV intervals in the presence and absence of 100 nM κ-PVIIA. The apparent effect of 100 nM κ-PVIIA on the K channels is to make activation kinetics more complex by the introduction of a second, and slower, phase in the rising of the currents elicited by voltage steps (Fig. 1 B). Also, the conductance–voltage relationship measured at the end of the pulse is shifted to the right and is less steep in the presence of the toxin (Fig. 1 C). This voltage-dependent effect is in contradiction with the nearly voltage independent affinity of κ-PVIIA on the inactivating Shaker H4 observed previously (Terlau et al. 1996; see below).
Figure 1.
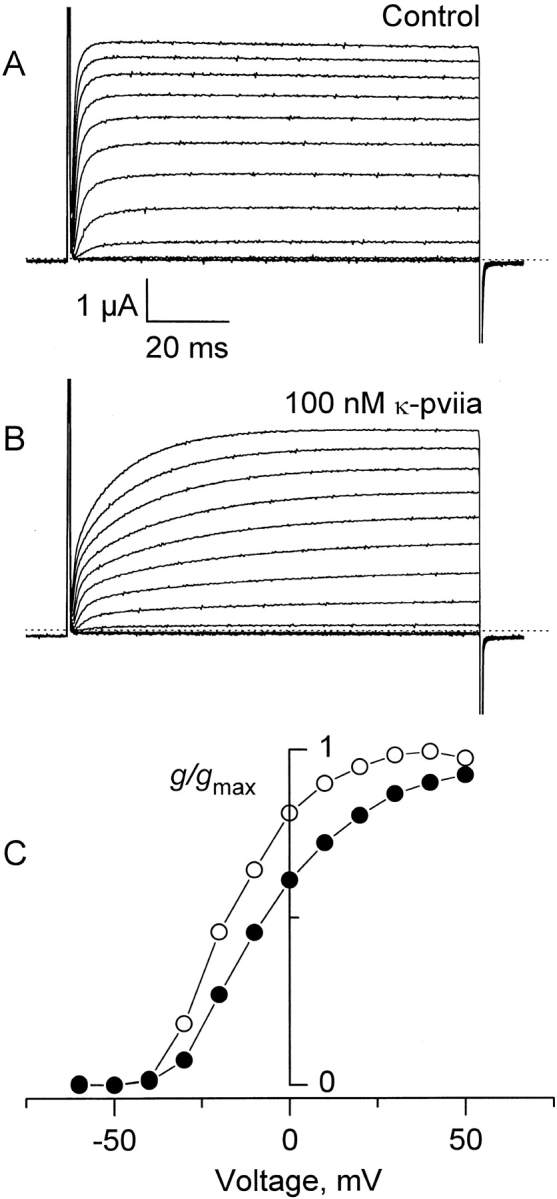
Effect of the κ-PVIIA on Shaker IR. Two-electrode voltage clamp recordings of an oocyte expressing Shaker H4 Δ(6-46) in a control solution (A) and in the presence of 100 nM κ-PVIIA (B). Currents were elicited from a holding voltage of −90 mV by activating pulses of −60 to +50 mV with intervals of 10 mV. Dotted lines indicate the zero current level. (C) Potassium conductance measured from the current at the end of the activating pulse. • and ○ indicate conductance with and without toxin, respectively. Values are taken from the records shown in A and B. After leak subtraction, each value of conductance was calculated assuming a reversal potential of −100 mV. The normalizing value, g max, is the conductance measured at the end of the control record taken at +40 mV.
Results described in Fig. 1 may suggest that the toxin effect on Shaker K channels is to modify the gating mechanism. The toxin would bind preferentially to the closed channel conformation, delaying the rate of activation. Such a mechanism has been proposed for hanatoxin, a component of the spider venom, on other K channels as Kv2.1 (Swartz and MacKinnon 1997). An alternative explanation could be that the toxin binding to its receptor in the channel is voltage dependent. In this latter view, the positive-going voltage pulse used to activate the channels would also destabilize the binding of the toxin. Thus, the slow apparent activation of the currents would represent the time course by which the toxin binding relaxes from one equilibrium at resting potential to one of lower affinity at a more positive voltage. This kind of mechanism would be reminiscent to the “hook” seen in the tail K currents induced by internal tetra-alkylammonium (Armstrong 1971). Data shown in Fig. 2 Fig. 3 Fig. 4 Fig. 5 are from experiments made to distinguish among these two possible mechanisms.
Figure 2.

Voltage dependence of κ-PVIIA inhibition. (A) After leak subtraction, records taken in the presence of 100 nM κ-PVIIA at each pulse voltage (Fig. 1 B) were divided point-by-point by leak-subtracted control record taken at corresponding voltages. Shown are the results of such an operation for the interval 2–100 ms after the beginning of the 0 to +50 activating pulses. For the pulses to −10 and −20 mV, the points intervals 15–100 and 18–100, respectively, are shown. The thin lines correspond to fits of an exponential equation of the from:  , where a, b, and τ correspond to the steady state fractional current, the amplitude extrapolated to the beginning of the pulse, and the time constant, respectively. Note that the I
PVIIA/I
control curves converge to similar values when extrapolated to the beginning of the activating pulse. The mean ± SEM value is: 0.35 ± 0.004 for the −10- to +50-mV pulses. These values correspond to the resting inhibition at holding potential. (B) Plot of the results from the kinetic analysis shown in A. Plotted, as mean ± SEM for four separate experiments, are the time constant and steady state fractional current, (I
PVIIA/I
control)∞, as a function of the activating pulse voltage (a and τ in A). The continuous line over the time constant data (•) was drawn according to with
, where a, b, and τ correspond to the steady state fractional current, the amplitude extrapolated to the beginning of the pulse, and the time constant, respectively. Note that the I
PVIIA/I
control curves converge to similar values when extrapolated to the beginning of the activating pulse. The mean ± SEM value is: 0.35 ± 0.004 for the −10- to +50-mV pulses. These values correspond to the resting inhibition at holding potential. (B) Plot of the results from the kinetic analysis shown in A. Plotted, as mean ± SEM for four separate experiments, are the time constant and steady state fractional current, (I
PVIIA/I
control)∞, as a function of the activating pulse voltage (a and τ in A). The continuous line over the time constant data (•) was drawn according to with  .
.
Figure 3.

Dose dependence of κ-PVIIA inhibition. (A) Four superimposed TEVC records taken with activating pulse to +40 mV in the presence of the indicated nanomolar concentrations of the toxin. Record taken with 300 nM toxin corresponds to a different experiment, but for the sake of comparison was scaled to represent the actual blockade. The dotted line indicates the zero current level. (B) Records divided point-by-point by their respective controls with no toxin added. Thin lines are single exponential fits extrapolated to the beginning of the voltage pulse as described in Fig. 2 A. (Inset) Dose dependence of the inhibition at resting obtained from the values extrapolated to the beginning of the pulse. Measurements made for 30, 100, and 300 nM represent mean ± SEM for three to four oocytes. The value for 1 μM corresponds to a single measurement. The solid line corresponds to a simple inhibition hyperbola with a half-saturation at 65 nM. (C) Rate of relaxation measured as the reciprocal of the time constants obtained from the single exponential fits to data as shown in B. Straight lines are linear regression to the data. For a 1:1 stoichiometry, this rate is represented by . At each voltage, the slope is the second order rate constant, k on, and the intercept at zero toxin corresponds to the dissociation rate, k off. (D) Plot of k on and k off as a function of the applied voltage pulse for each measurement made in C. Solid lines were drawn according to . The values are shown in Table .
Figure 5.

Results from the kinetic analysis to the relaxation resulting in the point-by-point division of the patch clamp experiments shown in Fig. 4. (A) Time constants of the exponential fits to the relaxations are as those shown in Fig. 4C and Fig. F. Data are plotted as mean ± SEM. ○ and • are for pulse and chronic relaxations in the presence of the toxin, respectively. □ are the time constant for recovery resulting from removal of the toxin at the end of the pulse application. Solid lines are best fits to , with:  for pulse applications, and
for pulse applications, and 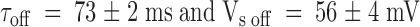 for recovery after toxin removal (
for recovery after toxin removal (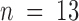 ; pooled data from seven experiments in 100-K
in//1-K
ex, and six in 15-K
in//1-K
ex). For chronic applications: τ = 29 ± 3 ms and Vs = 141 ± 58 mV (
; pooled data from seven experiments in 100-K
in//1-K
ex, and six in 15-K
in//1-K
ex). For chronic applications: τ = 29 ± 3 ms and Vs = 141 ± 58 mV (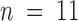 ; pooled data from four experiments with 100-k
in and seven with 15-k
in in the pipette). No significantly different values for the time constant of onset relaxation are detected (○ and •; P > 0.05; paired t test for the data between −20 and +40 mV). (B) Steady state inhibition determined from the asymptotic values of the single exponential fits. The levels of steady state fractional current reached with both protocols are significantly different (P < 0.00001; paired t test for the data between −20 and +40 mV).
; pooled data from four experiments with 100-k
in and seven with 15-k
in in the pipette). No significantly different values for the time constant of onset relaxation are detected (○ and •; P > 0.05; paired t test for the data between −20 and +40 mV). (B) Steady state inhibition determined from the asymptotic values of the single exponential fits. The levels of steady state fractional current reached with both protocols are significantly different (P < 0.00001; paired t test for the data between −20 and +40 mV).
Fig. 2 A shows a point-by-point division of the traces in the presence of the toxin by the control traces at pulse voltages positive to −20 mV shown in Fig. 1. As done before by Scanlon et al. 1997, single exponential fits (thin lines on top of each trace) to the normalized traces were extrapolated to the beginning of the voltage pulse. Regardless of the blocking mechanism, a common origin for the curves indicate the level of the blocking equilibrium at the holding potential. As the voltage pulse is made more positive, the time constant of relaxation gets smaller, and the steady state inhibition is reduced (Fig. 2 B). Good descriptions by single exponential of the traces suggest a voltage-dependent first-order process.
Dose–Response Experiments
A simple bimolecular stoichiometry predicts that the rate of relaxation should increase linearly to the toxin concentration. Fig. 3 A shows records obtained with a 100-ms pulse to +40 mV with 0, 30, 100, and 300 nM κ-PVIIA. In Fig. 3 B, the point-by-point divisions of the traces by their controls at zero toxin are shown. With this operation, the concentration dependence of the blockade at holding potential becomes apparent. From these measurements, a dissociation constant at holding potential of 65 ± 11 nM is obtained (mean ± SEM for four different experiments, Fig. 3 B, inset). This value is in close agreement with the 60 nM measured by Terlau et al. 1996 and with tonic inhibition measured at resting (Terlau et al. 1999). Thus, we can explain the discrepancy between our results and the nearly voltage-independent affinity measured for κ-PVIIA inhibition to the N-inactivating Shaker H4 by Terlau et al. 1996. Because N-type inactivation proceeds much faster than the toxin relaxation (milliseconds versus tens of milliseconds), measurement of fractional inhibition, early during the pulse, at the peak of the current in N-inactivating channels should closely resemble the binding equilibrium at holding potential.
As is apparent from Fig. 3A and Fig. B, the rate of relaxation increases at higher toxin concentrations. Fig. 3 C shows that the rate of relaxation, measured as the reciprocal of the time constant, increases linearly with the concentration of κ-PVIIA at all voltages. Linear regressions to the data show that the intercept changes with the pulse voltage; meanwhile, the slope is almost invariant. This linearity is consistent with a bimolecular stoichiometry for the interaction between κ-PVIIA and the Shaker K channel as shown by Fig. 1 in methods. For such a scheme, states that the slope is k on and the intercept is k off.
Fig. 3 D plots the voltage dependence of both rate constants for the data plotted in C. It is clear that most of the voltage dependence resides in k
off (•). At zero voltage, k
off is 17 ± 1 s−1, with a 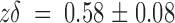 . Meanwhile, the apparent second-order rate constant is nearly voltage independent. The rate constant k
on has a zero-voltage value of 70 ± 9 μM−1 s−1 with
. Meanwhile, the apparent second-order rate constant is nearly voltage independent. The rate constant k
on has a zero-voltage value of 70 ± 9 μM−1 s−1 with 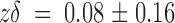 . This value agrees very well with that of 61 μM−1 s−1 for charybdotoxin, a toxin whose binding to potassium channels is proposed to be diffusion limited (Miller 1990; Goldstein et al. 1994). This “on” rate constant seems too fast to be the rate limiting step for a protein conformational change; nevertheless, it is slow enough for a conformational change in the channel promoted by a toxin that is already bound at resting—before the activating voltage pulse is given (see Terlau et al. 1999; Fig. 3 B and inset).
. This value agrees very well with that of 61 μM−1 s−1 for charybdotoxin, a toxin whose binding to potassium channels is proposed to be diffusion limited (Miller 1990; Goldstein et al. 1994). This “on” rate constant seems too fast to be the rate limiting step for a protein conformational change; nevertheless, it is slow enough for a conformational change in the channel promoted by a toxin that is already bound at resting—before the activating voltage pulse is given (see Terlau et al. 1999; Fig. 3 B and inset).
Rapid Application of κ-PVIIA to Outside-Out Patches
Because the use of voltage-step protocols is inherent to the study of Shaker K channels, to differentiate between the effects of the voltage or the open probability of the channels on κ-PVIIA binding, chronic bath application to whole oocytes are inadequate. We compared chronic and rapid applications of the toxin to membrane patches expressing macroscopic currents in the outside-out configuration (Liu and Dilger 1991; Naranjo and Brehm 1993).
Outside-out patches were separated from the oocyte following standard techniques (Hamill et al. 1981). Holding the pipette voltage at −90 mV, the pipette tip was positioned near the opening of the fast perfusion system and 200-ms test voltage pulses from −60 to 50 mV were applied. Fig. 4A and Fig. D, shows two different patches with typical sets of current records obtained with the control recording solution (1-K ex) washing the membrane patch, and with the 15-K in solution in the pipette (see methods). At most test voltages, currents were already fully activated 30 ms after the beginning of the pulse. However, the time courses of the currents are bit more complex than those obtained in whole oocytes recordings. At potentials positives to −20 mV, a time-dependent decrease of the current becomes evident. Such reduction was not apparent in the whole oocyte records. We do not know if this difference from the two-electrode voltage clamp experiments is due to an inactivation process, a local potassium depletion that renders currents smaller, or both. No further attempts to study this phenomenon were made.
To separate gating from toxin kinetics in the chosen interval of pipette voltages, toxin was applied ∼40 ms after the beginning of the test pulse. Visual clues are not usually enough to position the pipette tip where solution exchange is optimally fast. To monitor the optimal positioning of the membrane patch in the rapid application system, together with the toxin, we added 1 mM TEA+ to the test solution. Because this TEA+ concentration produces a fast blockade of a small but measurable fraction of the current, we could monitor the solution exchange rate by monitoring the speed of the inflection produced by the TEA+ effect on K currents on individual records (see Fig. 4 B, inset). Fig. 4 B shows an average of four runs of κ-PVIIA/TEA+. The rapid TEA+ inflection is not visible in averaged records because of inherent variable latency of the solenoid valve (<5 ms). At all voltages, 1 mM TEA+/500 nM κ-PVIIA pulse applications produced a decrease in the current in which the kinetics of the onset and offset of toxin blockade are faster as the voltage is made more positive. As with the whole oocyte recordings, we made point-by-point division of toxin current records by control records at each voltage. As a result of such an operation, we also avoided possible distortions on toxin kinetics resulting from the apparent inactivation, as seen by the complete recovery in the current records taken at more positive voltage pulses (Fig. 4 C). These normalized records show voltage-dependent on and off kinetics in addition to a voltage-dependent steady state inhibition. Single exponential fits applied to each normalized trace are shown as solid lines over some traces. A general result from single exponential fits to the normalized traces is shown in Fig. 5 (open symbols). This figure plots averages of data pooled from a total of 13 outside-out patches with pulse applications of κ-PVIIA. Because we did not find significant difference in the toxin kinetics between experiments obtained with 100-K
in
 or 15-K
in
or 15-K
in
 solutions in the patch pipette (not shown, see Table and discussion), these data also represent the merging together of these two experimental conditions.
solutions in the patch pipette (not shown, see Table and discussion), these data also represent the merging together of these two experimental conditions.
Table 1.
Summary of the Kinetic Parameters of κ-PVIIA Blockade on Shaker IR
| Experimental condition | k on(0) | zδon | k off(0) | zδoff | K d(0) | n | ||
|---|---|---|---|---|---|---|---|---|
| μM−1 s−1 | s−1 | nM | ||||||
| TEVC | ||||||||
| [K+]ext = 2 mM | 70 ± 9 | 0.08 ± 0.16 | 17 ± 1 | 0.58 ± 0.08 | 240 ± 30 | 4 | Fig. 3 | |
| [K+]ext = 2 mM‡ | 52 ± 4 | 0.03 ± 0.04 | 21 ± 2 | 0.61 ± 0.04 | 403 ± 40 | 10 | and | |
| [K+]ext = 100 mM‡ | 36 ± 6 | 0.10 ± 0.05 | 22 ± 2 | 0.66 ± 0.03 | 610 ± 100 | 9 | and | |
| *Patch | ||||||||
| Chronic | 100-K in//1-K ex | 52 ± 3 | 0.03 ± 0.04 | 25 ± 3 | 0.64 ± 0.06 | 480 ± 60 | 4 | and |
| 15-K in//1-K ex | 32 ± 2 | −0.14 ± 0.07 | 10 ± 1 | 0.47 ± 0.08 | 313 ± 37 | 7 | and | |
| Pulse | 100-K in//1-K ex | 44 ± 4 | 0.13 ± 0.4 | 15 ± 1 | 0.52 ± 0.08 | 341 ± 38 | 7 | |
| 15-K in//1-K ex | 48 ± 5 | 0.17 ± 0.09 | 12 ± 1 | 0.56 ± 0.10 | 250 ± 26 | 6 | ||
| 0-K in//100-K ex | ND | ND | 11 ± 1 | 0.28 ± 0.05 | — | 4 | ||
| Pooled patch-clamp data | 43 ± 3 | 0.11 ± 0.05 | 13 ± 2 | 0.53 ± 0.09 | 300 ± 40 | 24 | Fig. 6 | |
Values of the association and dissociation rates, and their voltage dependence for each set of experiments. Values of k on(0) and k off(0) and zδon and zδoff were calculated from (see methods). The zero-voltage dissociation constants were calculated with: K d(0) = k off(0)/k on(0). *Patch clamp experiments were done in presence of 1 mM TEA+, and then the calculated values of k on are underestimated by ∼10% (see Fig. 7). ‡Determinations were done separately in paired experiments.
If the toxin inhibition is produced merely by toxin binding instead of inducing, or preventing, a conformational change, the kinetics of the response to a voltage step in the presence of the toxin should be identical to that induced by pulse applications of toxin at a constant voltage. To the patch whose current record is shown in Fig. 4 D, 1 mM TEA+/500 nM κ-PVIIA was chronically applied, a protocol equivalent to that of the whole-oocyte experiments (Fig. 4 E). As in the whole oocyte experiments, the apparent time course of the current activation is also delayed in these conditions. A point-by-point division of these traces by the controls of Fig. 4 D is shown in F. This family of normalized traces shows that the voltage dependence of the relaxation and steady state inhibition is similar to those with the toxin pulse protocol shown in Fig. 4 C. Thin lines in Fig. 4 F correspond to exponential fits to some of the normalized traces. Average time constant and steady state inhibition for 11 pooled patches are plotted as solid symbols in Fig. 5
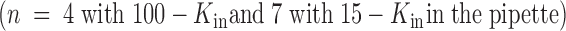 .
.
Fig. 5A and Fig. B, summarizes results of the kinetic analysis of the experiments of κ-PVIIA applications to outside out patches. In Fig. 5 A, time constants for the onset of inhibition and recovery after toxin removal for the pulse protocol experiments are plotted as open symbols; meanwhile, plotted as solid symbols are the time constants of the relaxations in the experiments with chronic application as shown in Fig. 4 F. Fig. 5 B plots the steady state inhibition for pulse (○) and chronic (•) applications. Two lines of evidence strongly suggest that the rate-limiting step for the relaxation of the inhibition is the association of the toxin with the channel and not a conformational change from a closed to open state. (a) Although the levels of steady state fractional current reached with both protocols are significantly different, in average, the systematic difference between both curves accounts for <3% of the inhibition. On the other hand, for both protocols of κ-PVIIA applications, no significantly different values for the time constant of onset relaxation are detected, indicating that both the voltage pulse in the presence of κ-PVIIA and the toxin pulse applied to the open channels promote a nearly identical perturbation on the blockade equilibrium, as expected by a most simple voltage-dependent blocking mechanism. Such a mechanism only requires toxin binding with 1:1 stoichiometry to produce inhibition. (b) Although the toxin binds with moderately higher affinity to the closed state (Terlau et al. 1999), in both protocols, no inflection is observed between values obtained at voltages around 10 mV (Fig. 5 A). At this range of voltages, the open probability of the channels is near its maximum and becomes roughly voltage independent (see Fig. 1 C), thus, reinforcing the idea that the toxin inhibits the open K channels mostly by a voltage-dependent binding. Zero-voltage time constant of the onset of chronic application is 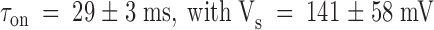 . For pulse application, the zero-voltage onset time constant is
. For pulse application, the zero-voltage onset time constant is 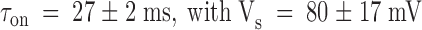 , while for the recovery it is
, while for the recovery it is 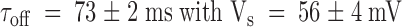 .
.
Rate Constants
In a bimolecular scheme as presented in methods, the time constant of the macroscopic relaxation is governed by . In the absence of toxin, k off can be calculated directly from the reciprocal of the recovery time constant, τoff in the measurements of toxin removal. Also, we have two additional experimental conditions in which the relaxation in the presence of the toxin is seen, chronic and pulse application onset. For a 1:1 stoichiometry, these two types of macroscopic relaxations should reach a steady state level given by .
From the values plotted in Fig. 5, we calculated both k
on and k
off for each pipette potential by using and . Results from such calculations are summarized in Fig. 6. The small open and filled symbols correspond to the calculations of k
on and k
off, respectively. There is a good agreement in the values of k
off measured directly from pulse experiments (Fig. 6, small solid circles) with those calculated from the two-equation system regardless of the experimental protocol. As in the whole oocyte experiments of Fig. 3, most of the voltage dependence appears to be in k
off. An overall average calculated from all three experimental data sets is plotted as the big circles in Fig. 6. The solid line is a single exponential fit to the calculated rate constants. At zero voltage, the second order rate constant, k
on, is 43 ± 3 μM−1 s−1 with 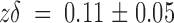 . This value for k
on is only 40% smaller than that measured on the whole-oocyte experiments and is also consistent with a diffusion-limited association (Miller 1990). Similarly, showing good agreement with the whole oocyte experiments, k
off at zero voltage is 13 ± 2 s−1 with
. This value for k
on is only 40% smaller than that measured on the whole-oocyte experiments and is also consistent with a diffusion-limited association (Miller 1990). Similarly, showing good agreement with the whole oocyte experiments, k
off at zero voltage is 13 ± 2 s−1 with 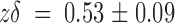 .
.
Figure 6.

Kinetic values of the κ-PVIIA inhibition. Small symbols represent different solutions to the system of and , while the big symbols (○ and •) represent arithmetic averages of the k on and k off values obtained by the different methods (see text). Small open symbols represent k on; ○ and □ result from the pulse application protocol, while the (▵) plot results from the chronic application protocol. Small filled symbols represent k off; • and ▪ from the pulse application protocol, and ▴ from the chronic application protocol. Solid lines are single exponential fits of . The rate constant values that describe the data are shown in Table , under pooled patch clamp data.
The near perfect agreement between the patch clamp and two-electrode voltage clamp experiments reinforce the idea that the κ-PVIIA exerts its inhibition effect on the open K channel by binding to an external site only. Thus, the relaxation of the macroscopic K currents observed in oocytes under chronically applied toxin is analogous to the slow hook seen previously when tetraethylammonium ions dissociate from squid axon potassium channels before rapid deactivation in response to repolarization (Armstrong 1971). In both cases, macroscopic kinetics are determined by a temporal superposition of blocking and gating transitions. Additionally, owing to the noninactivating feature of Shaker-IR, we established that in the relatively slowly perfused whole oocyte preparation we could make high resolution kinetic measurements of a toxin with a resident time in the tens of milliseconds time scale.
Competition with TEA+
Although there is no compelling evidence that κ-PVIIA plugs the pore of K channels, there are several lines of evidence that κ-PVIIA interacts with the Shaker external vestibule. Transference of the H5 segment from a κ-PVIIA–sensitive K channel to an insensitive channel also transfers toxin sensitivity (Shon et al. 1998). Mutations in the H5 segment alters toxin affinity (Scanlon et al. 1997; Shon et al. 1998). Also, externally applied TEA+, a specific pore blocker for K channels, reduces the extent of the blockade, suggesting a competitive interaction with the toxin (Scanlon et al. 1997). As an antecedent to these experiments, TEA+ only reduces the association rate of CTX to the large conductance Ca-activated K channels (MaxiK-channel), without an effect on the dissociation rate (Miller 1988). Thus if κ-PVIIA binds to the Shaker vestibule, similar effects are expected. With two-electrode voltage clamp, κ-PVIIA binding was studied in the presence of TEA+ 0–10 mM, added on top of the recording solution. Fig. 7 shows a summary of such experiments. In the same fashion as we did for the results shown in Fig. 3, we made a point-by-point division of records taken in κ-PVIIA/TEA+ solution from those taken in TEA+ only (not shown). Applying and to the time constants and steady state inhibition obtained from the single exponential fits to the relaxations, apparent k on and k off for the effect of TEA+ were obtained. TEA+ decreases the apparent association rate with little effect on the dissociation rate. This effect seems to be specific for TEA+ because raising Na+ concentration to 10 mM does not significantly affect either k on or k off (Fig. 7, solid lines). Together, these results indicate that the effect of TEA+ on κ-PVIIA binding is not due to the increased ionic strength, but rather to a specific exclusion of the toxin from the external vestibule of Shaker.
Figure 7.

Effect of TEA+ on the kinetic parameters of κ-PVIIA inhibition. Average values of k on (filled symbols) and k off (open symbols) at different TEA+ concentrations. All values shown were normalized by the rates measured at zero applied potential in the absence of TEA+. Symbols represent different concentrations of TEA+: 0 mM (▪ and □), 1 mM (• and ○), 5 mM (♦ and ⋄), and 10 mM (▴ and ▵). The continuous lines correspond to the normalized values of k on and k off measured in 10 mM NaCl added on top of the normal recording solution. Each value represents the average ± SEM from two to five different oocytes.
Internal Potassium Alters Binding to Shaker
To determine whether κ-PVIIA inhibit ionic conduction by occluding the ionic pathway, we analyzed the effect of altering permeant ion concentration in the opposite side of the pore. This strategy had shown that the binding of CTX to the MaxiK-channel is very sensitive to the intracellular K+ (MacKinnon and Miller 1988; Park and Miller 1992b). The rationale for these experiments is that occupancy of the pore by permeant cations coming from the internal side of the channel will repel electrostatically the highly positively charged toxin (+4). In the MaxiK-channel, an approximately fourfold increase in the toxin residence time is expected when the internal K+ concentration is lowered from 110 to ∼10 mM. As with the Ca-activated K channel, half saturation concentration for K conductance in Shaker K channels is near 300 mM, suggesting the presence of a low affinity K-binding site (Eisenmann et al. 1986; Heginbotham and MacKinnon 1993). To our surprise, in outside out patches, preliminary experiments showed a minor effect, either on the association or the dissociation rates, produced by the reduction of the intracellular K+ concentration from 100 to 15 mM (Table ). This result suggests that, if κ-PVIIA occludes the conduction pathway, it is not very sensitive to K occupancy in the pore, or the pore occupancy is not very different in these two distinct conditions. As in the MaxiK-channel, it had already been suggested that in the Shaker K channel there are at least one micromolar affinity potassium binding sites inside the K channel pore (Neyton and Miller 1988a; Baukrowitz and Yellen 1995). Thus, we substituted all the potassium by the nonpermeant cation NMG+ in the patch pipette solution, and we formed and pulled outside-out patches from the oocyte membrane in the 1-K ex recording solution. We attempted to reduce contamination of the patch pipette with K+, and thus minimized occupancy of the pore from internal potassium ions. After obtaining a stable outside-out patch, the pipette was positioned in the rapid perfusion system applying an external solution containing 100 mM K+. Under these conditions, 0-K in//100-K ex and high levels of channel expression, voltage pulses activated inward currents that frequently produced positive feedback responses characteristic of inadequate clamp. This latter effect was avoided by doing recordings with small currents (<200 pA). Fig. 8 A shows 400-ms records with pulse application of a solution that, in addition to the 100 mM KCl, contained 500 nM κ-PVIIA and 1 mM TEA+. Because, at any voltage, this type of record displayed <10% of inactivation (not shown), the rate of dissociation was measured by fitting single exponential functions directly to the time course of the inward currents recovery. Average values (mean ± SEM) for four such experiments are plotted in Fig. 8 B. For the sake of comparison, the best fit to the average dissociation rate made with in whole oocyte TEVC at 100 mM external K+ is shown by dotted lines (Fig. 8, and see Table ). In agreement with the results that Goldstein and Miller 1993 previously obtained for CTX in the nominal absence of internal K+, both the amplitude of the zero-voltage dissociation rate and its effective valence were reduced by half. Such a trans-pore effect of potassium ions is by itself indicative that the mechanism of inhibition κ-PVIIA on K channel is physically blocking the ion conduction pathway (Bezanilla and Armstrong 1972; MacKinnon and Miller 1988). These results also suggest that, as in CTX, the side chain of a basic residue interacts with permeant ions residing in the narrowest region of the pore.
Figure 8.

Effect of internal K+ removal on the dissociation rate of κ-PVIIA. (A) Pulse application of 1 mM TEA+/500 nM κ-PVIIA (shaded area) to an outside-out patch. External solution contained 100 mM K+ and the pipette was filled with 100 mM NMG+ (0-K
in//100-K
ex; see methods). Pipette potential was maintained at −90 mV and 400-ms pulses were applied from −70 to +50 in 10-mV increments. These records represent the average of four identical traces. The dotted line is the zero current level. (B) Voltage dependence of the dissociation rate in the absence of internal K+. A single exponential function was fitted to each time course of current recovery after toxin removal. Dissociation rate constants were calculated from the reciprocal of the resulting time constants. The solid line was traced with the following parameters: 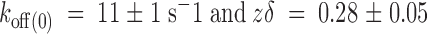 . Each data point represents mean ± SEM for four different patches. For comparison, the dashed line represents the voltage dependence of the dissociation rate measured in whole oocyte bathed in 100 mM external K+
. Each data point represents mean ± SEM for four different patches. For comparison, the dashed line represents the voltage dependence of the dissociation rate measured in whole oocyte bathed in 100 mM external K+
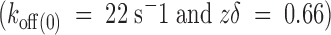 .
.
κ-PVIIA Protects the Shaker Pore from Collapsing
As with CTX (Goldstein and Miller 1993), the complete removal of the internal K+ does not abolish completely the voltage dependence of κ-PVIIA binding to Shaker (Fig. 8). The most economical interpretation for this residual voltage dependence is that a positive charge located in the pore-occluding side chain of the toxin interacts intimately with the narrowest part of the K channel pore, getting located inside the electric field. An electrical distance of 0.2–0.25 coincides with an externally located cationic binding site that is present in the pore of Shaker. A binding site detected at approximately the same electrical distance can accommodate mono- and divalent cations. NH4 +, Cs+, Rb+, and K+ bind to this site with low millimolar or high micromolar affinity (López-Barneo et al. 1993; Hurst et al. 1995; Gómez-Lagunas 1997; Harris et al. 1998). Occupation of this site impeded or delayed C- or P-type inactivation, pore collapsing conformational changes proposed to occur near the external entrance of Shaker pore (López-Barneo et al. 1993; Liu et al. 1996; Harris et al. 1998). On the other hand, Gómez-Lagunas 1997 recently found that the occupation of an external site by the permeant ions or by TEA+ protects Shaker K channels from visiting a long-lived nonconducting state christened “defunct” (Melishuck et al. 1998). This state arises when the channels are opened in the absence or presence of submillimolar K+ concentration; then they are forced to close after its pore occupancy has diminished (Gómez-Lagunas 1997). Closing the channel after the occupying ions are allowed to escape away from the pore promotes a collapse of the pore that traps the gating machinery in a altered set of states (Melishuck et al. 1998). Thus, both types of nonconducting states depend largely on pore occupancy. We tested whether we could promote any of these nonconducting states just by blocking the channel with κ-PVIIA in the absence of internal permeant ions (Fig. 9). Outside-out patches were recorded in 0-K in//100-K ex condition. From a holding potential of −90 mV, the channels were opened by a pulse to −40 mV to ensure a strong driving force for K+ to flow inwardly. First, we determined that an application of nominally zero K+ solution (100 mM NMG+) to the patch pipette that exposed 5–8 ms before the voltage pulse ended was necessary to observe any measurable amount of current reduction between consecutive pulses (shaded area in Fig. 9 A). For an interpulse interval of 5 s, this reduction indicates the population of channels visiting any of the occupancy-dependent nonconducting states. Because Shaker K channels show little rectification between +40 and −40 mV, we expected the external binding site to be empty in <500 μs (Baukrowitz and Yellen, 1996). Thus, the amount of time needed to observe current reduction might represent incomplete wash out of the high K+ solution. In any case, this figure represents an upper limit to the time needed to empty the pore, and is 20–30-fold shorter than the residence time of the bound toxin at −40 mV in this experimental condition (∼150 ms, see Fig. 8 B). After the 40th pulse, <40% of the current remains (Fig. 9 C, ○). On the other hand, Fig. 9 B shows an experiment in which, instead of exposing the open channel to zero potassium, we applied κ-PVIIA to the patch with the intention of occluding the external exit of the pore. In such a case, the K channel pore should be emptied into the internal solution. At 150 ms after channel opening, a 150-ms pulse of 1 mM TEA+/500 nM κ-PVIIA that blocked ∼60% of the channels was applied (Fig. 9, shaded area). While the toxin was still being applied, the channels were forced to close by stepping the voltage back to −90 mV. Thus, given that the toxin blocking time is more than two orders of magnitude longer than the time needed to empty the pore (Table ), closing the channels while they are still blocked should drive a measurable fraction into any occupancy-dependent nonconducting state. Fig. 9 B shows a set of 10 traces representing one of every four acquisitions from one of such an experiment. If the toxin does not protect the channel, ∼40% of the current is expected to have disappeared at the 40th pulse (Fig. 9 C and legend). However, in four experiments like this, no reduction of the current was detected at even the 60th pulse, suggesting that the channel pore cannot collapse while the toxin is bound. Because both types of nonconducting states require or are facilitated by pore vacancy, while κ-PVIIA is bound to the channel, the pore behaves as if occupancy were preserved. The simplest interpretation to this result is that a part of the toxin impedes pore collapse. Thus, we suggest that a component of κ-PVIIA occupies the most externally located K+ binding site of Shaker pore, resulting in a protection of the channel. In normal physiological conditions, such occupancy should reduce the average number of permeant ions residing in the pore.
Figure 9.
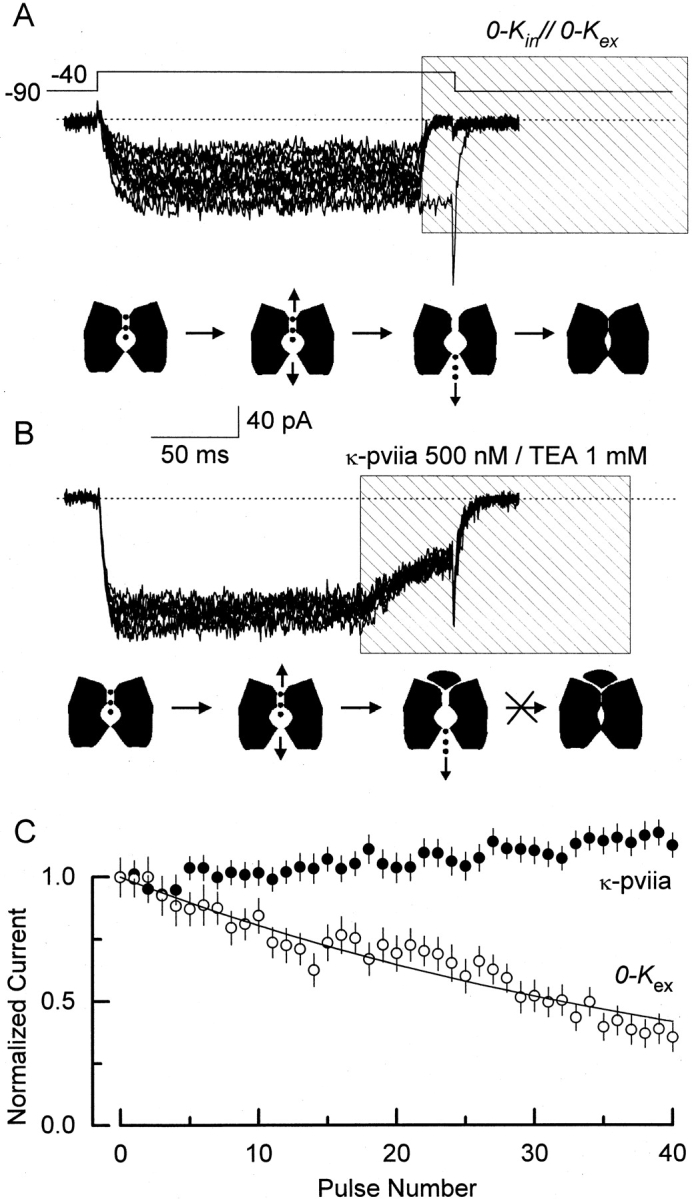
κ-PVIIA protects the Shaker pore from collapsing. Traces show 10 current records from outside-out patches with 100 mM external K+ and 100 NMG+ in the pipette (0-K in//100-K ex, see methods). Except for the initial control in A, traces shown correspond to one in every four acquired. The dotted line indicates the zero current level. The lower cartoon under each set of traces depicts the kinetic states that the Shaker K channels are putatively visiting during the experimental protocol: closed, open-conducting, open-empty, and pore-collapsed. (A) Exposing channels to zero potassium produces a cumulative reduction of the current. Every 5 s, the pipette voltage was stepped from −90 to −40 mV for 200 ms to open channels. After an initial control trace, distinguished by the inward tail current, ∼15 ms near the end of the depolarization, the pulse of a zero K+ solution is given (0-K in//0-K ex). We estimated that the channels are exposed for 5–8 ms to this 0-K in//0-K ex experimental condition. During this time, K+ residing in the pore are allowed to escape, reducing the channel pore occupancy. The fraction of channels not entering nonconductive state(s) promoted by this experimental manipulation is plotted (C, ○). (B) Exposing channels to toxin does not produce reduction in the current. Similar to A, every 5 s, the pipette voltage was stepped from −90 to −40 mV for 200 ms to open channels. At ∼150 ms after the beginning of the voltage pulse, a 150-ms long TEA+/toxin pulse was applied to block the channels (shaded area). During this period, ions residing in the pore of the open/blocked channel are expected to escape away into the intracellular side of the channel. The activating voltage pulse ends when ∼60% of the channels are blocked and near equilibrium. The toxin residence time at this voltage is three orders of magnitude larger than that of possibly the last occupying ion (150 μs vs. 150 ms, see Fig. 8 B; Baukrowitz and Yellen, 1996). Also, this residence time is 20–30-fold larger than the 5–8 ms of exposure to zero K+ needed to observe reduction in the current as shown in A. If the toxin does not protect, and if the 60% toxin binding at the end of the pulse is in equilibrium for more than 5–8 ms, at least a 40% reduction in the current is expected to be produced at the 40th pulse. This is a very conservative low limit that was obtained from the exponential fitting to the 0-K ex data in C. (C) Fractional current averages of non–leak-subtracted records. Averages were measured in the 50–100-ms interval after the beginning of the voltage pulse for both experimental conditions and the error bars represent SD in each interval. A solid line was drawn according to a single exponential with a decay constant of 44 pulses.
Discussion
Here we present evidence for a mechanism of inhibition of a peptide component of the venom of the predator marine snail Conus on K channels. The 27-residue κ-PVIIA inhibits K+ currents by binding to the external vestibule of the Shaker K channel pore with a 1:1 stoichiometry at a rate consistent with a diffusion-limited manner (Miller 1990). It competes with TEA+ and unbinds the channel vestibule in a voltage-dependent manner, probably due to electrostatic interaction with permeant ions inside the pore. Such a mode of action is remarkably similar to that proposed for scorpion toxins of the family α-KTx, the best characterized of all peptide toxin specific for K-channels (Miller 1995).
Effect of Internal K+ on Toxin Unbinding
As with α-KTx, κ-PVIIA voltage dependence of the binding equilibrium to the channel resides in the dissociation rate (see Fig. 3 and Fig. 6). For CTX bound to the external vestibule of the MaxiK-channel, the effective valence of the dissociation rate is 1.0 (Anderson et al. 1988). In the absence of internally present permeant cations, zδ vanishes. Thus, the voltage dependence seems to arise exclusively from the electrostatic repulsion between K+ residing inside the pore and the positively charged toxin (MacKinnon and Miller 1988). By performing extensive mutagenesis of positively charged residues in recombinant CTX, Park and Miller 1992a,Park and Miller 1992b found that neutralization of a single residue, Lys27, also abolished voltage dependence of the dissociation rate, a property not shared with any other residue on the toxin surface. Thus, removal of either the permeant ions or the positive charge of residue 27 produced an equivalent cancellation of zδ, indicating that the ∈-amino group of Lys27 interacts electrostatically with K+ residing in the pore (see Miller 1995). Qualitatively similar, but quantitatively different to the MaxiK-channels, CTX binding to Shaker K channels is less sensitive to the internal K+ concentration. Goldstein and Miller 1993 found that, although neutralization of residue 27 eliminates voltage dependence, after replacing internal K+ with a nonpermeant ion as Li+, there is still some voltage dependence left for CTX unbinding. These authors interpreted the residual valence of 0.2 as the partial electrical distance that the amino group of Lys27 moves into the pore upon toxin binding; as if it goes deeper into the Shaker pore than into the MaxiK one. Our results with κ-PVIIA/Shaker agree qualitatively and quantitatively with those of CTX/Shaker. Replacement of the internal potassium with the nonpermeant NMG+ reduces, but not eliminate, the effective valence of the κ-PVIIA unbinding. For κ-PVIIA, the value zδ is reduced from ∼0.55 in 100 mM internal K+ to 0.29 in nominally zero internal K+ (Fig. 8). This trans-pore effect suggests that, as in CTX, a fraction of the voltage dependence of κ-PVIIA unbinding arises from electrostatic interaction with permeant ions in the conduction pathway. Thus, we postulate that, as with α-KTx, a positively charged residue in the surface κ-PVIIA interacts with permeant ions inside the Shaker K channel pore.
At pH 7.6, κ-PVIIA net charge is ∼4. It contains two Asp, three Lys, and three Arg residues, and it is not amidated as many conotoxins are (Shon et al. 1998). In the absence of experimental data regarding the specific nature of the positively charged amino acid of κ-PVIIA that occludes the pore, we favor a Lys instead of an Arg residue to be the best candidate. First, based on the structural mimicry between CTX and κ-PVIIA on the space position of a Lys associated with a key aromatic residue to conform a functional dyad present at all K channel pore peptide blockers known (Dauplais et al. 1997; Savarin et al. 1998). Second, for all functionally studied K channel–specific families of peptide toxins, independently of the animal origin, substitution of a single Lys, not an Arg, promotes the most dramatic destabilization effects on binding to K channels (Goldstein et al. 1994; Pennington et al. 1996; Dauplais et al. 1997). Third, the agreement with CTX in the magnitude of the change in zδ promoted by the internal K+ replacement by nonpermeant cations suggests a similar electrostatic interaction.
Electrostatic Repulsion of the Toxin and K+ Permeation
In the MaxiK-channel, the internally accessible binding site that destabilizes CTX binding has an affinity for K+ in the 1–3-M range (MacKinnon and Miller 1988; Park and Miller 1992b). A simple interpretation for the high effective valence of the toxin destabilization process  is that a K+ would have to travel across the whole transmembrane electric field to interact with CTX. In other words, the internal cation has to permeate all the way across the pore to bind a site near the external end to electrostatically repel CTX. In agreement with the idea that equates permeation with occupancy of an external site, half-maximal K+ concentration for K conductance of the MaxiK-channel is near 300 mM (Moczydlowsky et al. 1985; Eisenmann et al. 1986; MacKinnon et al. 1989). However, this K channel can host multiple ions in single-file occupancy (Yellen 1984; Eisenmann et al. 1986; Cecchi et al. 1987; Neyton and Miller 1988b), a key feature of ion permeation through K channels (Hodgkin and Keynes 1955; Hille and Swartz, 1978; Doyle et al. 1998). With K+ in the millimolar range, three or four binding sites are already occupied (Neyton and Miller 1988b). Most probably, in the multiply occupied pore, and with the ionic conduction blocked from the external end by the toxin, a K+ cannot permeate through the single-file of resident cations inside the ionic pathway whose occupancies are in equilibrium with the internal side of the channel. Instead, the occupancy of the externally located binding site depends on the ability of the dwelling ions to “transmit” the information that the internally accessible site is occupied by a new K+. This telegraphic effect, via occupied binding sites, would be ultimately responsible for the strong electrostatic repulsion of the bound toxin. A simple mechanism of transference would be that the K+ binding to the internally accessible site promotes a rearrangement of the ions residing in the pore in such a way that results in an increased probability of occupancy of the most external binding site. This rearrangement requires of ion translocation among neighboring binding sites in analogous way to permeation. In this scheme, a zδ of 1.0 for the toxin destabilization observed in the MaxiK-channel implies a net translocation of one charge across the permeation pathway. Also, it accounts for the close agreement between half-maximal K+ concentration for K conductance and toxin destabilization effect. Thus, in this view, the magnitude zδ is determined by coupling between the binding of K+ to the internal site and occupancy of the most external one.
is that a K+ would have to travel across the whole transmembrane electric field to interact with CTX. In other words, the internal cation has to permeate all the way across the pore to bind a site near the external end to electrostatically repel CTX. In agreement with the idea that equates permeation with occupancy of an external site, half-maximal K+ concentration for K conductance of the MaxiK-channel is near 300 mM (Moczydlowsky et al. 1985; Eisenmann et al. 1986; MacKinnon et al. 1989). However, this K channel can host multiple ions in single-file occupancy (Yellen 1984; Eisenmann et al. 1986; Cecchi et al. 1987; Neyton and Miller 1988b), a key feature of ion permeation through K channels (Hodgkin and Keynes 1955; Hille and Swartz, 1978; Doyle et al. 1998). With K+ in the millimolar range, three or four binding sites are already occupied (Neyton and Miller 1988b). Most probably, in the multiply occupied pore, and with the ionic conduction blocked from the external end by the toxin, a K+ cannot permeate through the single-file of resident cations inside the ionic pathway whose occupancies are in equilibrium with the internal side of the channel. Instead, the occupancy of the externally located binding site depends on the ability of the dwelling ions to “transmit” the information that the internally accessible site is occupied by a new K+. This telegraphic effect, via occupied binding sites, would be ultimately responsible for the strong electrostatic repulsion of the bound toxin. A simple mechanism of transference would be that the K+ binding to the internally accessible site promotes a rearrangement of the ions residing in the pore in such a way that results in an increased probability of occupancy of the most external binding site. This rearrangement requires of ion translocation among neighboring binding sites in analogous way to permeation. In this scheme, a zδ of 1.0 for the toxin destabilization observed in the MaxiK-channel implies a net translocation of one charge across the permeation pathway. Also, it accounts for the close agreement between half-maximal K+ concentration for K conductance and toxin destabilization effect. Thus, in this view, the magnitude zδ is determined by coupling between the binding of K+ to the internal site and occupancy of the most external one.
Although MaxiK-channel's conductance is ∼20-fold higher than that of Shaker K channels, in addition to their high K+ selectivity, they share many ionic conduction properties. Shaker K channel is also a multiple-occupancy channel (Heginbotham and MacKinnon 1993; Pérez-Cornejo and Begenesich 1994). As with the MaxiK-channel, at physiological conditions Shaker can simultaneously host up to four K+ in single file (Stampe and Begenesich 1996). Also, its half-maximal K+ concentration for conductance is near 300 mM (Heginbotham and MacKinnon 1993). Based on these similarities, we expected κ-PVIIA off rate to have a similar dependence on the internal K+, as CTX does in the MaxiK-channel. By reducing the internal K+ from 100 to 15 mM, we anticipated a fivefold increase in the κ-PVIIA residency time. To our surprise, we found that the toxin dissociation rate was nearly identical to that of 100 K+ (Table ). In contrast to the CTX/MaxiK-channel system, in κ-PVIIA/Shaker, the toxin-unbinding rate seems to be rather insensitive to the occupation of a internally accessed binding site. To observe any trans-pore effect on κ-PVIIA unbinding rate, we had to replace K+ completely with the nonpermeant NMG+. Although the contaminating K+ in our internal NMG+ solutions may be enough to keep busy a high affinity binding site in the Shaker pore, thus accounting for the residual zδ, the overall voltage dependence of the toxin unbinding is smaller than in the MaxiK-channel. In such an experimental condition, instead of 1.0 as in the MaxiK-channel, only half of the total effective valence of the dissociation rate is accounted for in the replacement of the internal K+. This low voltage sensitivity attributable to the interaction with internal K+ may not correspond specifically to charge delocalization in the κ-PVIIA basic residue that occludes the pore. Instead, because in Shaker the interaction with K+ also accounts for a small fraction of the total voltage dependence of CTX, this low zδ (together with the low sensitivity to internal K+) may be a reflection of the manner in which the toxin interacts with permeant ions inside the pore of Shaker. The net charge translocation associated with the occupancy of the externally located binding site is 0.3–0.4 of an electronic charge. Thus, it seems that in the MaxiK-channel the electrostatic repulsion coupling between the occupancy of the most internal and external binding sites is three- to fivefold stronger than in Shaker. This difference could represent an average greater charge separation of the ions inside the Shaker pore. Although in physiological conditions both types of channels seem to be occupied simultaneously by four cations (Neyton and Miller 1988b; Stampe and Begenesich 1996), this greater charge separation could arise from either lower occupancy in the toxin-blocked Shaker or greater electrical separation among neighboring binding sites. This suggestion is also in agreement with Terlau et al. 1999(in this issue), who, with different arguments, have made the same suggestion. For us, the most economical explanation to the difference in the MaxiK-channel would be that, in Shaker, the blocking ∈-amino group of the toxins occupies the most external site, at an electrical distance of 0.2–0.3, effectively reducing the number of available sites, and then K+ occupancy. The experiments outlined in Fig. 9 agree with this interpretation, suggesting that κ-PVIIA occupies the most external binding site in Shaker pore. Thus, the electrostatic repulsion can occur only from a more internally located site in the pore. Consequently, the net charge translocation associated with electrostatic interaction with the toxin is less than in the MaxiK-channel because the toxins, κ-PVIIA or CTX, reduce the net K+ occupancy of the pore. In this scheme, the elements for a mechanism of destabilization of a toxin bound to the external vestibule seems to be the same as that for ionic permeation: the exit rate depends on electrostatic repulsion and single-file multiple occupancy.
Fast Prey Immobilization
Conus purpurascens preys on teleost fish. It has been proposed that, upon injection of the Conus venom into the prey, κ-PVIIA is a central player in the excitotoxic cabal, which causes a massive depolarization of excitable tissue that quickly immobilizes the prey (Terlau et al. 1996; Olivera 1997). In this scheme, activation of Na channels and blockade of K channels by the venom produces a nerve depolarization in the site of the injury that is propagated orthodromically by sensory nerves and antidromically by motor fibers. In the following part of the discussion, we review the suggestion that κ-PVIIA behaves as part of an optimized prescription to promote the excitotoxic paralysis of C. purpurascens prey. We have found that the mode of action of κ-PVIIA is nearly identical to the mechanism of action of distantly related scorpion peptide toxins of the α-KTx family: first, both bind with 1:1 stoichiometry to the pore of K-channels and, second, the only action that the peptide toxin performs is to occlude the permeation pathway with a tethered substrate analogue that interrupts K+ transport. The high association rate of κ-PVIIA to Shaker is consistent with a diffusion-limited, but aided by through-space electrostatic effects, protein–protein interaction (Miller 1990; Schreiber and Fersht 1995). A mechanism of such nature seems to be the fastest possible. On the other hand, there are many hints that it is shared with other K channel toxins from distantly related animals as the α-KTx from scorpions and BgK from sea anemone (Gómez-Lagunas et al. 1996; Blanc et al. 1997; Dauplais et al. 1997; Savarin et al. 1998).
In the classical example of structural and functional convergence, the catalytic site of the bacterial subtilisin and the mammalian serine proteinase share a similar space topology. Although dissimilar in their overall construction, the side-chain atoms forming the functional catalytic triad, Ser/His/Asp, are in almost identical relative space positions. The breakdown of the peptide bond proceeds by the same basic mechanism (Fersht 1984; Branden and Tooze 1991). Three structural characteristics of K channel–specific peptide toxins suggest a similarly convergent mechanism of action. First, as noted by Dauplais et al. 1997, toxin affinity for its receptor is disrupted mostly by mutations in a lysine and in an aromatic residue that are usually ∼7 Å apart (see also Goldstein et al. 1994; Stampe et al. 1994). Dauplais et al. 1997 suggested that the Lys-Aromatic residue forms a functional dyad specific for K channels. Second, it has an excess of positive charges that may play an important role in the overall probability of colliding with a negatively charged vestibule (Miller 1990). Third, the excess of charge in the surface of the toxin is unevenly distributed in such a manner that it generates a dipole moment oriented perpendicular to the interaction surface of the toxin (Ben-Tal et al. 1997; Blanc et al. 1997; D. Naranjo, unpublished observations).
Thus, some peptide toxins seem to have had a convergence to a common mechanism of action on K channels. They bind with 1:1 stoichiometry to the most conserved structural locus of the channel protein: the pore. Such a precise mechanism appears to constrain the possible ways in which K channel–specific peptide toxin can be constructed.
Acknowledgments
We thank Miguel Hernandez for technical help and frog care and other members of the laboratory IFC 204-O for support. We are grateful to A. Hernández-Cruz, F. Cifuentes, and F. Gómez-Lagunas for insightful discussions, and to Ramón Latorre, Osvaldo Alvarez, and Christopher Miller for critical reading of the manuscript. Our recognition to Paul Brehm, Irwin Levitan, and Christopher Miller for providing equipment that was essential to develop this project.
This work was partially funded by DGAPA-IN200397 and CONACYT-25247N to D. Naranjo, and by the Juan García Ramos Fund and FOMES-SEP 1997 to E. García.
Footnotes
1used in this paper: CTX, charybdotoxin; κ-PVIIA, κ-conotoxin-PVIIA; MaxiK-channel, large conductance Ca-activated K channel; NMG+, N-methyl-d-glucamine in ionic form; TEA+, tetraethylammonium; TEVC, two-electrode voltage clamp; TFA, trifluoroacetic acid
References
- Anderson C., MacKinnon R., Smith C., Miller C. Charybdotoxin inhibition of Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J. Gen. Physiol. 1988;91:317–333. doi: 10.1085/jgp.91.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C.M. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T., Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a potassium channel. Science. 1995;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Ben-Tal N., Honig B., Miller C., McLaughlin S. Electrostatic binding of proteins to membranes. Theoretical predictions and experimental results with charybdotoxin and phospholipid vesicles. Biophys. J. 1997;73:1717–1727. doi: 10.1016/S0006-3495(97)78203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F., Armstrong C.M. Negative conductance caused by entry of sodium and cesium ions into the potassium channels of squid axons. J. Gen. Physiol. 1972;60:588–608. doi: 10.1085/jgp.60.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc E., Sabatier J.M., Kharrat R., Meunier S., El Ayeb M., Van Rietschoten J., Darbon H. Solution structure of maurotoxin, a scorpion toxin from Scorpio maurus, with high affinity for voltage gated potassium channels. Proteins. 1997;29:321–333. [PubMed] [Google Scholar]
- Branden C., Tooze J. Introduction to protein structure Garland Publishing, Inc., New York. 302pp1991. [Google Scholar]
- Cecchi X., Wolff D., Alvarez O., Latorre R. Mechanism of Cs+ blockade in a Ca2+-activated K+ channel from smooth muscle. Biophys. J. 1987;52:707–716. doi: 10.1016/S0006-3495(87)83265-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestèle S., Qu Y., Rogers J.C., Rochat H., Scheuer T. Voltage sensor-trappingenhanced activation of sodium channels by β-scorpion toxin bound to the S3-S4 loop in domain II. Neuron. 1998;21:919–931. doi: 10.1016/s0896-6273(00)80606-6. [DOI] [PubMed] [Google Scholar]
- Dauplais M., Lecoq A., Song J., Cotton J., Jamin N., Gilquin B., Roumestand C., Vita C., de Medeiros C.L.C., Rowan E.G. On the convergent evolution of animal toxins conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 1997;272:4302–4309. doi: 10.1074/jbc.272.7.4302. [DOI] [PubMed] [Google Scholar]
- Doyle D.A., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Eisenmann G., Latorre R., Miller C. Multi-ion conduction and selectivity in the high-conductance Ca2+-activated K+ channel from skeletal muscle. Biophys. J. 1986;50:1025–1034. doi: 10.1016/S0006-3495(86)83546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A. Enzyme structure and function 1984. W.H. Freeman and Co; New York: pp. 475 pp [Google Scholar]
- Goldstein S.A.N., Miller C. A point mutation in a Shaker K+ channel changes its charybdotoxin binding site from low to high affinity. Biophys. J. 1992;62:5–7. doi: 10.1016/S0006-3495(92)81760-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S.A.N., Miller C. Mechanism of charybdotoxin block of a voltage-gated K+ channel. Biophys. J. 1993;5:1613–1619. doi: 10.1016/S0006-3495(93)81200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S.A.N., Pheasant D.J., Miller C. The charybdotoxin receptor of a Shaker K+ channelpeptide and channel residues mediating molecular recognition. Neuron. 1994;12:1377–1388. doi: 10.1016/0896-6273(94)90452-9. [DOI] [PubMed] [Google Scholar]
- Gómez-Lagunas F. Shaker B K+ conductance in Na+ solutionsa remarkably stable non-conducting state produced by membrane depolarization. J. Physiol. 1997;499:3–15. doi: 10.1113/jphysiol.1997.sp021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Lagunas F., Olamendi-Portugal T., Zamudio F.Z., Possani L.D. Two novel toxins from venom of the scorpion Pandinus imperator show that the N-terminal aminoacid sequence is important for their affinities towards Shaker B K+ channels. J. Membr. Biol. 1996;152:49–56. doi: 10.1007/s002329900084. [DOI] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Harris R.E., Larsson H.P., Isacoff E.Y. A permeant ion binding site located between two gates of the Shaker K+ channel. Biophys. J. 1998;74:1808–1820. doi: 10.1016/s0006-3495(98)77891-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham L., MacKinnon R. Conduction properties of the cloned Shaker K-channel. Biophys. J. 1993;65:2089–2096. doi: 10.1016/S0006-3495(93)81244-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B., Schwarz W. Potassium channels as multi-ion single-file pores. J. Gen. Physiol. 1978;72:409–442. doi: 10.1085/jgp.72.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin A.L., Keynes R. The potassium permeability of a giant nerve fibre. J. Physiol. 1955;128:61–88. doi: 10.1113/jphysiol.1955.sp005291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hurst R.S., Latorre R., Toro L., Stefani E. External barium block of ShH4 IR; evidence for two binding sites. J. Gen. Physiol. 1995;106:1069–1087. doi: 10.1085/jgp.106.6.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Baro D.J., Lanning C.C., Doshi M., Farnham J., Moskowitz H.S., Peck J.H., Olivera B.M., Harris-Warrick R.M. Alternative splicing in the pore-forming region of Shaker potassium channels. J. Neurosci. 1997;17:8213–8224. doi: 10.1523/JNEUROSCI.17-21-08213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Dilger J. Opening rate of acetylcholine receptor channels. Biophys. J. 1991;60:424–432. doi: 10.1016/S0006-3495(91)82068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Jurman M., Yellen G. Dynamic rearrangement of the outer mouth of a potassium channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- López-Barneo J., Hoshi T., Heinemann S., Aldrich R. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- MacKinnon R., Miller C. Mechanism of charybdotoxin block of Ca2+-activated K+ channels. J. Gen. Physiol. 1988;91:335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R., Latorre R., Miller C. Role of surface electrostatics on the operation of a high-conductance Ca2+-activated K+ channel. Biochemistry. 1989;28:8092–8099. doi: 10.1021/bi00446a020. [DOI] [PubMed] [Google Scholar]
- Melishuck A., Loboda A., Armstrong C.M. Loss of Shaker channel conductance in 0K+ solutionsrole of the voltage sensor. Biophys. J. 1998;75:1828–1835. doi: 10.1016/S0006-3495(98)77624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. Competition for block of a Ca2+ activated K+ channel by charybdotoxin and tetraethylammonium. Neuron. 1988;1:1003–1006. doi: 10.1016/0896-6273(88)90157-2. [DOI] [PubMed] [Google Scholar]
- Miller C. Diffusion-controlled binding of a peptide neurotoxin to its K+ channel receptor. Biochemistry. 1990;29:5320–5324. doi: 10.1021/bi00474a016. [DOI] [PubMed] [Google Scholar]
- Miller C. The charybdotoxin family of K+ channel-blocking peptides. Neuron. 1995;15:5–10. doi: 10.1016/0896-6273(95)90057-8. [DOI] [PubMed] [Google Scholar]
- Myers R.A., Cruz L.J., Rivier J.E., Olivera B.M. Conus peptides as chemical probes for receptors and ion channels. Chem. Rev. 1993;93:1923–1936. [Google Scholar]
- Moczydlowsky E., Alvarez O., Vergara C., Latorre R. Effect of phospholipid surface charge on the conductance and gating of a Ca2+ activated K+ channel in planar lipid bilayers. J. Membr. Biol. 1985;83:273–282. doi: 10.1007/BF01868701. [DOI] [PubMed] [Google Scholar]
- Naranjo D., Miller C. A strongly interacting pair of residues on the contact surface of charybdotoxin and a Shaker K+ channel. Neuron. 1996;16:123–130. doi: 10.1016/s0896-6273(00)80029-x. [DOI] [PubMed] [Google Scholar]
- Naranjo D., Brehm P. Modal shifts in acetylcholine receptor channel gating confer subunit-dependent desensitization. Science. 1993;260:1811–1814. doi: 10.1126/science.8511590. [DOI] [PubMed] [Google Scholar]
- Neyton J., Miller C. Potassium blocks barium permeation through a calcium activated potassium channel J. Gen. Physiol. 92 1988. 549 567a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyton J., Miller C. Discrete Ba2+ block as a probe of ion occupancy and pore structure of the high conductance Ca2+ activated K+ channel J. Gen. Physiol. 92 1988. 569 586b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera B.M., Rivier J., Ramilo C.A., Corpuz G.P., Abogadie F.C., Mena E.E., Woodward S.R., Hillyard D.R., Cruz L.J. Diversity of Conus neuropeptides. Science. 1990;249:257–263. doi: 10.1126/science.2165278. [DOI] [PubMed] [Google Scholar]
- Olivera B.M. Conus venom peptide, receptor and ion channel targets, and drug design50 million years of neuropharmacology. Mol. Biol. Cell. 1997;8:2101–2109. doi: 10.1091/mbc.8.11.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprian D., Molday R.S., Kaufman R.J., Khorana G. Expression of a synthetic bovine rhodopsin gene in monkey kidney cells. Proc. Natl. Acad. Sci. USA. 1989;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallaghy P.K., Duggan B.M., Pennington M.W., Norton R.S. Three-dimensional structure in solution of the calcium channel blocker ω-conotoxin. J. Mol. Biol. 1993;234:405–420. doi: 10.1006/jmbi.1993.1595. [DOI] [PubMed] [Google Scholar]
- Park C.S., Miller C. Mapping function to structure in a channel-blocking peptideelectrostatic mutants of charybdotoxin Biochemistry. 31 1992. 7749 7755a [DOI] [PubMed] [Google Scholar]
- Park C.S., Miller C. Interaction of charybdotoxin with permeant ions inside the pore of a K+ channel Neuron. 9 1992. 307 313b [DOI] [PubMed] [Google Scholar]
- Pennington M.W., Mahnir V.M., Khaytin I., Zaydenburg I., Byrnes M.E., Kem W.R. An essential binding surface for ShK toxin interaction with rat brain potassium channels. Biochemistry. 1996;35:16407–16411. doi: 10.1021/bi962463g. [DOI] [PubMed] [Google Scholar]
- Pérez-Cornejo P., Begenesich T. The multi-ion nature of the pore in Shaker K+ channels. Biophys. J. 1994;66:1929–1938. doi: 10.1016/S0006-3495(94)80986-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin V., Kent S.B.H., Tan J.P., Merrifield R.B. Quantitative monitoring of solid phase peptide synthesis by the ninhydrin reaction. Anal. Biochem. 1981;117:147–157. doi: 10.1016/0003-2697(81)90704-1. [DOI] [PubMed] [Google Scholar]
- Savarin P., Guenneugues M., Gilquin B., Lamthanh H., Gasparini S., Zinn-Justin S., Ménez A. Three-dimensional structure of κ-conotoxin PVIIA, a novel potassium channel-blocking toxin from cone snails. Biochemistry. 1998;37:5407–5416. doi: 10.1021/bi9730341. [DOI] [PubMed] [Google Scholar]
- Scanlon M., Naranjo D., Thomas L., Alewood P., Lewis R., Craik D. Solution structure and proposed binding mechanism of a novel potassium channel toxin κ-conotoxin PVIIA. Structure (Lond.). 1997;5:1585–1597. doi: 10.1016/s0969-2126(97)00307-9. [DOI] [PubMed] [Google Scholar]
- Schreiber G., Fersht A. Energetics of protein–protein interactionsanalysis of barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Schnolzer M., Alewood P.F., Jones A., Alewood D., Kent S.B.H. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Int. J. Pept. Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- Shon K.-J., Stocker M., Terlau H., Stühmer W., Jacobsen R., Walker C., Grilley M., Watkins M., Hillyard D.R., Gray W.R., Olivera B.M. κ-Conotoxin pviia is a peptide inhibiting the Shaker K-channel. J. Biol. Chem. 1998;273:33–38. doi: 10.1074/jbc.273.1.33. [DOI] [PubMed] [Google Scholar]
- Stampe P., Kolmakova-Partensky L., Miller C. Intimations of K+ channel structure from a complete functional map of the molecular surface of charybdotoxin. Biochemistry. 1994;33:443–450. doi: 10.1021/bi00168a008. [DOI] [PubMed] [Google Scholar]
- Stampe P., Begenesich T. Unidirectional K+ fluxes through recombinant Shaker potassium channels expressed in single Xenopus oocytes. J. Gen. Physiol. 1996;107:449–457. doi: 10.1085/jgp.107.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz K.J., MacKinnon R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 1997;18:665–673. doi: 10.1016/s0896-6273(00)80306-2. [DOI] [PubMed] [Google Scholar]
- Terlau H., Shon K., Grilley M., Stocker M., Stühmer W., Olivera B.M. Strategy for rapid immobilisation of prey by a fish-hunting marine snail. Nature. 1996;381:148–151. doi: 10.1038/381148a0. [DOI] [PubMed] [Google Scholar]
- Terlau H., Boccaccio A., Olivera B.M., Conti F. The block of Shaker K+ channels by κ-conotoxin PVIIA is state dependent. J. Gen. Physiol. 1999;114:125–140. doi: 10.1085/jgp.114.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. Relief of Na+ block of Ca2+-activated K+ channels by external cations. J. Gen. Physiol. 1984;84:187–199. doi: 10.1085/jgp.84.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]