

Abstract
κ-conotoxin PVIIA is the first conotoxin known to interact with voltage-gated potassium channels by inhibiting Shaker-mediated currents. We studied the mechanism of inhibition and concluded that PVIIA blocks the ion pore with a 1:1 stoichiometry and that binding to open or closed channels is very different. Open-channel properties are revealed by relaxations of partial block during step depolarizations, whereas double-pulse protocols characterize the slower reequilibration of closed-channel binding. In 2.5 mM-[K+]o, the IC50 rises from a tonic value of ∼50 to ∼200 nM during openings at 0 mV, and it increases e-fold for about every 40-mV increase in voltage. The change involves mainly the voltage dependence and a 20-fold increase at 0 mV of the rate of PVIIA dissociation, but also a fivefold increase of the association rate. PVIIA binding to Shaker Δ6-46 channels lacking N-type inactivation or to wild phenotypes appears similar, but inactivation partially protects the latter from open-channel unblock. Raising [K+]o to 115 mM has little effect on open-channel binding, but increases almost 10-fold the tonic IC50 of PVIIA due to a decrease by the same factor of the toxin rate of association to closed channels. In analogy with charybdotoxin block, we attribute the acceleration of PVIIA dissociation from open channels to the voltage-dependent occupancy by K+ ions of a site at the outer end of the conducting pore. We also argue that the occupancy of this site by external cations antagonizes on binding to closed channels, whereas the apparent competition disappears in open channels if the competing cation can move along the pore. It is concluded that PVIIA can also be a valuable tool for probing the state of ion permeation inside the pore.
Keywords: voltage-gated potassium channels, ion channel pore, potassium binding site, Xenopus expression system
Introduction
Toxins from the carnivorous marine cone snails have been useful tools for the study of voltage-activated calcium and sodium channels (Olivera et al. 1985, Olivera et al. 1990). Recently, we reported the electrophysiological effects of κ-conotoxin PVIIA, the first member of a new family of conotoxins that interact with voltage-gated potassium channels (Terlau et al. 1996). By using the Xenopus oocyte expression system, it was shown that PVIIA inhibits Shaker K+ currents, but not the currents mediated by the rat homologues of the Kv1 family tested so far (Terlau et al. 1996). The same toxin has been shown to act also on one of the two splice variants of the Shaker homologue of lobster (Kim et al. 1997). Like scorpion toxins of the charybdotoxin (CTX)1 family (Miller 1995), PVIIA appears to bind to the extracellular mouth of the ion pore because its interaction with the channel protein is strongly modified by mutations of some of the amino acid residues that are believed to shape the outer pore vestibule (Shon et al. 1998). Nuclear magnetic resonance spectroscopy studies (Scanlon et al. 1997; Savarin et al. 1998) reveal strong similarities between the folding of PVIIA and CTX, but differences in the residues that most strongly affect the binding of the two toxins make PVIIA an interesting tool for additional information on the molecular structure of potassium channels. Most notably, the mutation F425G that increases the CTX-binding affinity of Shaker channels by more than three orders of magnitude (Goldstein et al. 1994) has the opposite effect of making the channels PVIIA insensitive (Shon et al. 1998).
In this paper, we study the mechanism of inhibition by PVIIA of Shaker channels and Shaker Δ6-46 channels lacking fast N-type inactivation. Our analysis shows that the voltage-dependent modification of the partially inhibited currents (Scanlon et al. 1997) cannot be attributed to a change in the gating of toxin-modified channels; e.g., as observed for the binding of the spider venom toxin Hanatoxin (Swartz and MacKinnon 1997). Instead, our findings are consistent with the hypothesis that the toxin blocks the permeation of the channel via a 1:1 binding to the pore, and owes its apparent properties of gating modifier to the dependence of its binding on the state of channel conductance. We shall describe block relaxations during activating pulses showing that the binding of PVIIA to open channels is characterized by a strongly voltage-dependent off rate and a voltage-insensitive on rate, and tonic-block recoveries during resting periods showing that the binding to closed channels has voltage-independent and much slower kinetics. We find that the properties of PVIIA block of Shaker Δ6-46 and Shaker wild-type channels are similar, except that inactivation protects partially the latter from the depolarization-induced unblock, in agreement with the assumption that the toxin senses only the conductive state of the channel. In analogy to CTX block of Ca-activated K+ channels (MacKinnon and Miller 1988) and Shaker channels (Goldstein and Miller 1993), our data are consistent with the idea that the voltage dependence of PVIIA binding to open channels is mainly an indirect effect of the interaction with potassium ions occupying an outer site in the pore. We shall also argue that the binding to closed channels, particularly its sensitivity to external K+, can be described by the same type of toxin–K+ interaction. A consistent molecular model shows that, apart from its use as a structural probe of the outer pore vestibule, PVIIA might also be a valuable tool for investigating more intimate properties of the ion-conduction pathway.
Methods
The solid-phase peptide synthesis of PVIIA was performed as described in Shon et al. 1998. cRNA for Shaker H4 (Kamb et al. 1988) and Shaker–Δ6-46 (Hoshi et al. 1990) was obtained by a standard protocol (Krieg and Melton 1987) using a template with a T7 promoter.
Oocyte Expression System
Oocytes from Xenopus laevis were prepared as described previously (Stühmer 1992). RNA was injected into stage V–VI oocytes and currents were recorded 1–7 d after injection. Whole-cell currents were recorded under two-electrode voltage clamp control using a Turbo-Tec amplifier (npi Electronic). The intracellular electrodes were filled with 2 M KCl and had a resistance between 0.6 and 1.0 MΩ. Current records were low-pass filtered at 1 kHz (−3 dB) and sampled at 4 kHz. For outside-out patch-clamp recordings (Hamill et al. 1981), the aluminum silicate glass pipettes had resistances between 0.8 and 1.2 MΩ. The pipette solution contained (mM): 115 KCl, 1.8 EGTA, 10 HEPES, pH 7.2 with KOH. Currents were measured with an EPC-9 patch clamp amplifier driven by the Pulse+PulseFit software package (HEKA Elektronik). Current records were low-pass filtered at 3 kHz (−3 dB) and sampled at a rate of 10 kHz.
The bath solution in the electrophysiological experiments was either normal frog Ringer's (NFR) containing (mM): 115 NaCl, 2.5 KCl, 1.8 CaCl2, 10 HEPES, pH 7.2, with NaOH, or K+-Ringer containing (mM): 115 KCl, 1.8 CaCl2, 10 HEPES, pH 7.2 with KOH. Leak and capacitive currents were corrected on-line by using a P/n method. In all experiments, the vitelline membranes of the oocytes were removed mechanically with fine forceps. Toxin solution was added to the bath chamber with a Gilson tip pipette. The indicated toxin concentrations correspond to the final concentration in the bath chamber.
Results
Open-Channel Block by PVIIA in Low [K]o (NFR)
As shown by Terlau et al. 1996, the main effect of PVIIA on Shaker-H4 channels is a reversible reduction of the peak voltage-clamp currents with a dose dependence that is consistent with toxin binding to a single site of the channel protein where it blocks ion permeation.
Fig. 1 A shows superimposed current responses to three different step depolarizations, recorded from the same oocyte before and after the addition to the bath of 500 nM PVIIA. All peak amplitudes are reduced roughly 10-fold under toxin, corresponding to an IC50 of PVIIA block of ∼50 nM. A closer inspection reveals, however, that the time course of the currents is sensibly modified. Fig. 1 B shows that the ratio, U (for unblock probability), of toxin to control responses for the same voltage increases after the time to peak approaching a late steady state value that, for 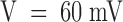 , is about five times larger. Although developing during the onset of inactivation, this effect cannot be attributed to an unblock of inactivated channels because it increases strongly with V in a range where the steady state probability and the time constant of inactivation are fairly constant, ∼0.9 and ∼3.5 ms, respectively. The solid lines in Fig. 1 B are single-exponential fits of U(t), yielding asymptotic values that increase from 0.21 at
, is about five times larger. Although developing during the onset of inactivation, this effect cannot be attributed to an unblock of inactivated channels because it increases strongly with V in a range where the steady state probability and the time constant of inactivation are fairly constant, ∼0.9 and ∼3.5 ms, respectively. The solid lines in Fig. 1 B are single-exponential fits of U(t), yielding asymptotic values that increase from 0.21 at 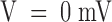 to 0.46 at
to 0.46 at 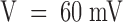 , while the time constants decrease from 27 to 6.3 ms. These data could be related to some average relaxation of toxin binding to open and inactivated channels, but the unfolding of binding and inactivation processes would not be straightforward since their kinetics appear to occur in a similar time range. Nevertheless, the fitted asymptotic values of U(t) can be used to estimate an apparent dissociation constant, K
(O)
app, of PVIIA binding to open (noninactivated) channels. K
(O)
app estimates in the voltage range −20 to +60 mV were fairly well fitted by:
, while the time constants decrease from 27 to 6.3 ms. These data could be related to some average relaxation of toxin binding to open and inactivated channels, but the unfolding of binding and inactivation processes would not be straightforward since their kinetics appear to occur in a similar time range. Nevertheless, the fitted asymptotic values of U(t) can be used to estimate an apparent dissociation constant, K
(O)
app, of PVIIA binding to open (noninactivated) channels. K
(O)
app estimates in the voltage range −20 to +60 mV were fairly well fitted by: 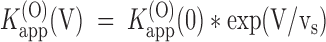 . Data from the experiment in Fig. 1 are shown in Fig. 4 C as □ and fitted with
. Data from the experiment in Fig. 1 are shown in Fig. 4 C as □ and fitted with 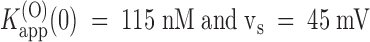 . Mean values of K
(O)
app(0) and vs from four different oocytes tested with toxin concentrations between 100 and 500 nM were 125 nM and 46 mV (Table , columns 2 and 5).
. Mean values of K
(O)
app(0) and vs from four different oocytes tested with toxin concentrations between 100 and 500 nM were 125 nM and 46 mV (Table , columns 2 and 5).
Figure 1.
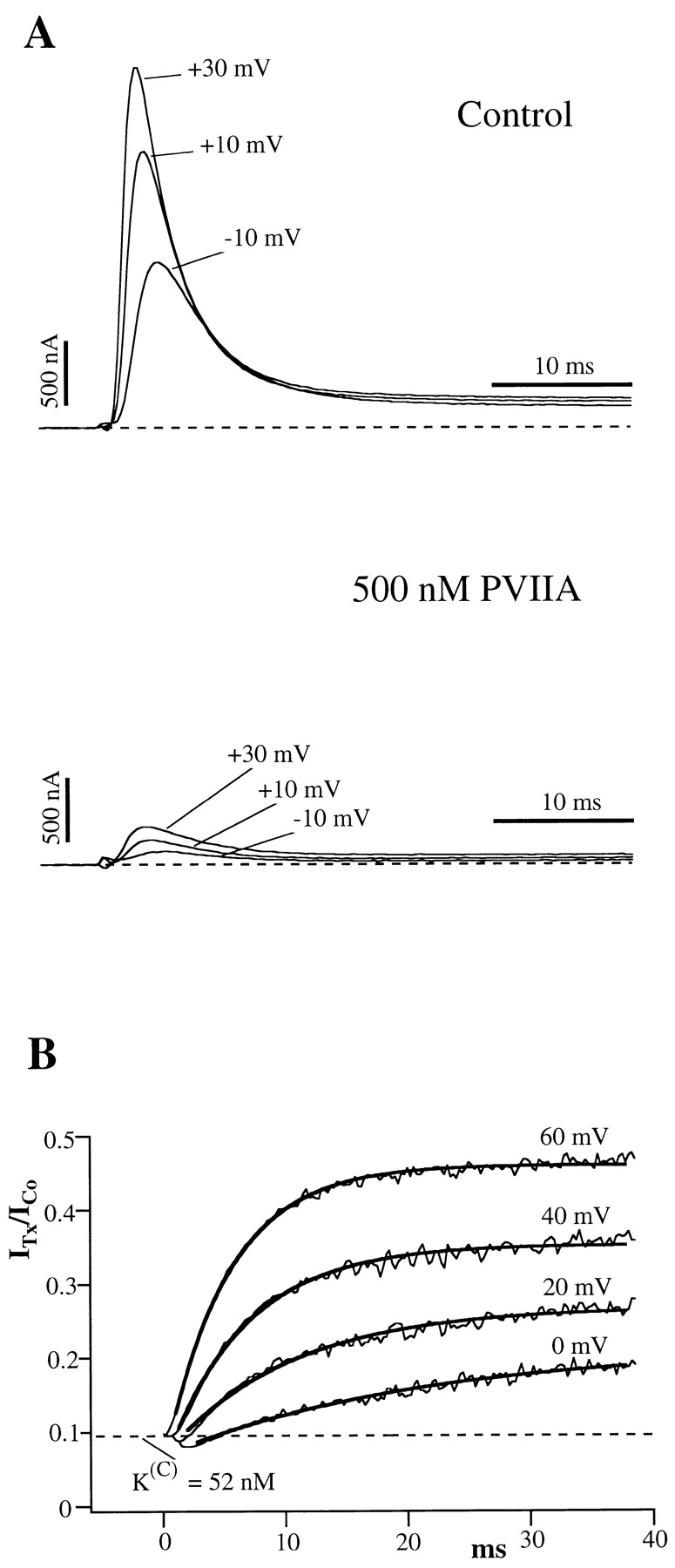
The effect of PVIIA on Shaker-H4 currents in NFR. (A) Superimposed records of voltage-clamp currents from the same oocyte before (top) and after (bottom) the addition of 500 nM PVIIA to the bath solution. All peak currents under toxin are reduced by about the same factor of 10.5, corresponding to a dissociation constant of the toxin from the blocking site of closed channels, 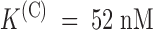 . (B) Toxin to control current ratios, U, for the indicated voltage steps. After the time to peak U(t) is well fitted by a single exponential (solid lines) with a plateau level increasing with voltage. Fitted asymptotic values and time constants (ms): 0.20, 27 (0 mV); 0.27, 16 (20 mV); 0.35, 9.7 (40 mV); and 0.46, 6.3 (60 mV). Interpreted as toxin-free probabilities, the asymptotic values indicate apparent toxin dissociation constants, K
(O)
app, of 125, 185, 270, and 430 nM.
. (B) Toxin to control current ratios, U, for the indicated voltage steps. After the time to peak U(t) is well fitted by a single exponential (solid lines) with a plateau level increasing with voltage. Fitted asymptotic values and time constants (ms): 0.20, 27 (0 mV); 0.27, 16 (20 mV); 0.35, 9.7 (40 mV); and 0.46, 6.3 (60 mV). Interpreted as toxin-free probabilities, the asymptotic values indicate apparent toxin dissociation constants, K
(O)
app, of 125, 185, 270, and 430 nM.
Figure 4.
Voltage dependence of PVIIA-binding to open channels. (A) Semi-logarithmic plot of estimates of the first-order dissociation rate constant, k
(O)
off, derived from U(O) and τ(O) data from the experiment of Fig. 2 (○) and from similar data in high [K]o (•, see Fig. 7). The straight lines are best fits with the equations: 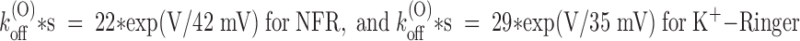 . (B) Linear plot of the second-order association rate constant, k
(O)
on, estimated from the experiments in A. Horizontal lines show the mean values of k
(O)
on*μM*s: 110 for NFR and 130 for K+-Ringer. (C) Semi-logarithmic plot of the dissociation constant, K
(O), estimated from the experiments in A and B (circles) and the apparent dissociation constant measured in the experiment of Fig. 1 for the inactivating Shaker-H4 channels (□). The straight lines are best fits with the equations:
. (B) Linear plot of the second-order association rate constant, k
(O)
on, estimated from the experiments in A. Horizontal lines show the mean values of k
(O)
on*μM*s: 110 for NFR and 130 for K+-Ringer. (C) Semi-logarithmic plot of the dissociation constant, K
(O), estimated from the experiments in A and B (circles) and the apparent dissociation constant measured in the experiment of Fig. 1 for the inactivating Shaker-H4 channels (□). The straight lines are best fits with the equations: 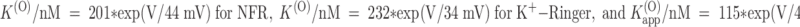 .
.
Table 1.
Parameters of PVIIA binding to Open and Closed States of Shaker Channels
| Type | K (O) (0) | n | k (O) on | n | k (O) off (0) | n | vs | n | K (C) | n | k (C) on | n | k (C) off | n |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nM | mM−1*s−1 | s−1 | mV | nM | μM−1*s−1 | s−1 | ||||||||
| Sh-WT, low K+ | 125 ± 30 | 4 | 46 ± 10 | 4* | 59 ± 16 | 7 | 22 ± 5 | 5 | 1.3 ± 0.4 | 5 | ||||
| Sh-Δ, low K+ | 207 ± 50 | 10 | 132 ± 30 | 10 | 26 ± 5 | 10 | 41 ± 4 | 10 | 48 ± 12 | 8 | 22 ± 4 | 8 | 1.1 ± 0.3 | 8 |
| Sh-Δ, high K+ | 220 ± 40 | 4 | 157 ± 21 | 4 | 31 ± 6 | 3 | 37 ± 6 | 3 | 403 ± 67 | 5 | 1.76 ± 0.07 | 3 | 0.79 ± 0.15 | 3 |
Evidence that the inactivation of Shaker-H4 channels is a damping factor rather than the cause of the above effects is provided by the study of the deletion mutant Shaker–Δ6-46 (Sh-Δ) that lacks fast inactivation (Hoshi et al. 1990). In agreement with Scanlon et al. 1997, we find that the currents mediated by Sh-Δ channels appear even more strongly modified by the presence of partially blocking concentrations of extracellular PVIIA. Fig. 2 A shows voltage-clamp currents for steps to −10, +10, and +30 mV, recorded from an oocyte expressing Sh-Δ channels before and after the bath addition of 200 nM PVIIA. It is seen that the toxin reduces strongly the early phase, but has much less effect on the steady state of the currents. The currents measured at the half-activation time of the normal responses are reduced at all voltages slightly more than fivefold, indicating a PVIIA dissociation constant from the blocking site of ∼50 nM, close to that estimated for Shaker-H4 channels from the reduction of peak currents. However, this early reduction diminishes during the pulse, and the currents approach asymptotic values that are progressively closer to the toxin-free levels at increasing depolarization. As opposed to the case of the gating modifier Hanatoxin from spider venom (Swartz and MacKinnon 1997), we show that these effects can be explained be assuming that PVIIA binding blocks pore conduction without modifying substantially channel gating and that channel opening reduces the toxin-binding affinity entraining a re-equilibration towards a lower block probability. According to this interpretation, the ratio U of toxin to control currents measures the fraction of unblocked channels and is predicted to have several distinctive properties that characterize a simple second-order reaction of channel-toxin association (Fig. 1), where {U} and {B} represent, respectively, an unblocked channel or a channel blocked by the association with the toxin, and where k (O) off and k (O) on are, respectively, the first-order dissociation rate constant and the second-order association rate constant of toxin binding during the step depolarization. A first prediction is that, after a step-like change in the binding parameters, U(t) should follow a single-exponential relaxation from the resting value towards the equilibrium value set by the new binding conditions. Fig. 2 B shows this property for sample voltage steps to 0, 20, 40, and 60 mV. The smooth lines are fits of U(t) with single exponentials rises from the same initial value, U(C) ∼ 0.19, towards asymptotic values, U(O), and with time constants, τ(O), that depend on the step voltage. For V ≥ −20 mV, the open-channel probability is close to its maximum and we find that τ(O) is always much larger than the half-activation time of normal currents. Therefore, these relaxations develop almost exclusively while the channels are fully activated, and this justifies the use of the superscript (O) for the parameters that characterize them.
Figure 2.
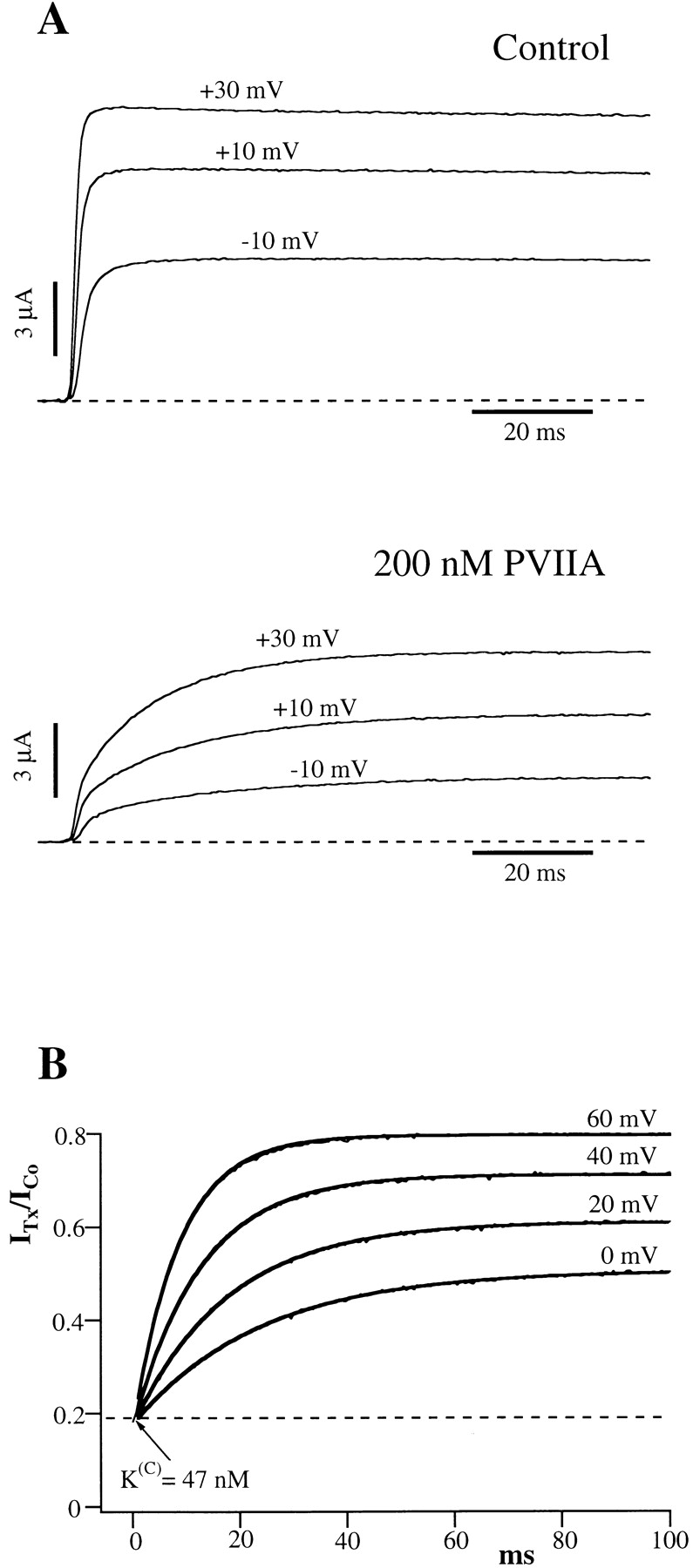
The effect of PVIIA on Shaker–Δ6-46 channels. (A) Voltage-clamp currents recorded from the same oocyte before (top) and after (bottom) the bath-addition of 200 nM PVIIA. Apart from the longer pulse length and PVIIA concentration, the other experimental conditions are as in Fig. 1. The toxin causes a strong reduction of the early currents, but has a much smaller effect on the steady state conductance activated by step depolarizations. (B) Current ratios, or unblock probabilities, U(t), for the indicated voltage steps. At the half-activation time of the control responses, U is ∼0.19 for all voltages and corresponds to a toxin dissociation constant from closed channels, 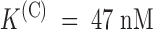 . Thereafter, U(t) is well fitted by a single-exponential relaxation (solid lines). Fitted asymptotic values, U(O), and time constants, τ(O) (ms): 0.51, 23 (0 mV); 0.61, 17 (20 mV); 0.71, 12 (40 mV); and 0.80, 8.5 (60 mV). The estimated open-channel dissociation constants corresponding to the values of U(O) are:
. Thereafter, U(t) is well fitted by a single-exponential relaxation (solid lines). Fitted asymptotic values, U(O), and time constants, τ(O) (ms): 0.51, 23 (0 mV); 0.61, 17 (20 mV); 0.71, 12 (40 mV); and 0.80, 8.5 (60 mV). The estimated open-channel dissociation constants corresponding to the values of U(O) are: 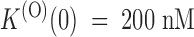 ,
, 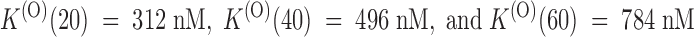 .
.
Scheme S1.

A second expectation is that the dependence of both U(C) and U(O) on the toxin concentration, [T], should follow simple Langmuir isotherms:
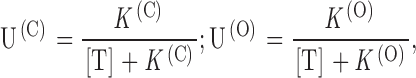 |
where K
(C) and K
(O) are the dissociation constants characterizing, respectively, the equilibrium binding of PVIIA to resting (closed) or activated (open) channels. Fig. 3 illustrates a single experiment in which [T] was changed progressively from 0 to 10, 20, 50, 100, 200, 500 nM, 1 μM, and back to 0. At each [T], a standard series of current–voltage responses to various pulse potentials, Vp, was recorded with 5-s stimulation intervals at a holding potential of −100 mV. Fig. 3 A shows the [T] dependence of the responses to 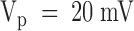 . Notice that the record obtained after washing-out the 1-μM PVIIA solution shows a small “run up” of the preparation and a very small residual unblock that we attribute to the remaining presence of a few nanomolar PVIIA around the oocyte. Ratios of the toxin to the initial control responses of Fig. 3 A are shown in B, together with single-exponential fits that are almost indistinguishable from the data at all times after 1 ms from the half-activation time of the control response. Estimates of U(C) and U(O) from these fits are plotted in Fig. 3 C as a function of [T]. The solid lines show that the data are indeed well fitted by Langmuir isotherms, yielding in this case
. Notice that the record obtained after washing-out the 1-μM PVIIA solution shows a small “run up” of the preparation and a very small residual unblock that we attribute to the remaining presence of a few nanomolar PVIIA around the oocyte. Ratios of the toxin to the initial control responses of Fig. 3 A are shown in B, together with single-exponential fits that are almost indistinguishable from the data at all times after 1 ms from the half-activation time of the control response. Estimates of U(C) and U(O) from these fits are plotted in Fig. 3 C as a function of [T]. The solid lines show that the data are indeed well fitted by Langmuir isotherms, yielding in this case 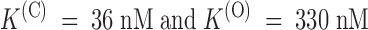 .
.
Figure 3.

Dose response of PVIIA effects on Sh-Δ channels. (A) Voltage-clamp currents for a voltage step to +20 mV from a holding potential of −100 mV recorded from the same oocyte exposed at increasing values of [T] as indicated. The trace marked “ ” was recorded after washing out the 1 μM PVIIA solution; notice a small “run up” and a residual slow unblock indicating that a few nanomolar PVIIA are still present around the oocyte. (B) Ratios, U, of the toxin to the initial control responses in A are plotted vs.
” was recorded after washing out the 1 μM PVIIA solution; notice a small “run up” and a residual slow unblock indicating that a few nanomolar PVIIA are still present around the oocyte. (B) Ratios, U, of the toxin to the initial control responses in A are plotted vs. 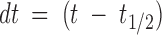 , where t
1/2 is the time of half activation of the control response; single-exponential fits (smooth lines) are indistinguishable from the data for dt > 1 ms; departures at earlier times are likely due to inadequate voltage-step control. (C) [T] dependence of the extrapolated values of the U(t) data in B for
, where t
1/2 is the time of half activation of the control response; single-exponential fits (smooth lines) are indistinguishable from the data for dt > 1 ms; departures at earlier times are likely due to inadequate voltage-step control. (C) [T] dependence of the extrapolated values of the U(t) data in B for 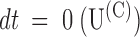 and dt → ∞(U(O)); the solid lines are best fits with Langmuir isotherms:
and dt → ∞(U(O)); the solid lines are best fits with Langmuir isotherms: 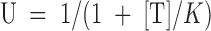 . The fitted K values for U(C) and U(O) data are, respectively,
. The fitted K values for U(C) and U(O) data are, respectively, 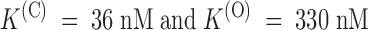 . (D) [T] dependence of the time constant τ(O) of the exponential fits in B; the solid line is the best fit with the expression:
. (D) [T] dependence of the time constant τ(O) of the exponential fits in B; the solid line is the best fit with the expression: 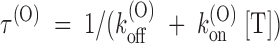 ; the fitted parameters are
; the fitted parameters are 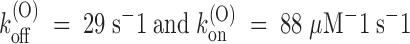 ; the ratio k
(O)
off/k
(O)
on coincides with
; the ratio k
(O)
off/k
(O)
on coincides with 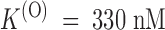 from the fit of U(O) data in C.
from the fit of U(O) data in C.
A third expectation is that τ(O) should depend on [T] according to:
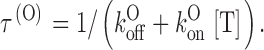 |
Estimates of τ(O) from the single exponential fits of Fig. 3 B are plotted as a function of [T] in D. The solid line is the least-squares fit of the data according to the above relationship, which is clearly well obeyed.
Finally, we expect a simple relationship between the estimates of k
(O)
off and k
(O)
on from the above fit of the [T] dependence of τ(O) and the equilibrium dissociation constant K
(O) estimated from the fit of U(O) data: 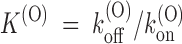 .
.
The values of k (O) off and k (O) on fitting the τ(O) data of Fig. 3 D, respectively, 29 s−1 and 88 μM−1 s−1, yield indeed the same estimate of K (O) ∼ 330 nM as the fit of the U(O) data of Fig. 3 C.
In most experiments, only two toxin concentrations were tested on the same oocyte, or experiments were done for a single [T] value in the range of 100–500 nM. The consistency of the results of these experiments with the bimolecular character of the toxin-block reaction was shown indirectly by the agreement of the estimates of K (O), k (O) off, and k (O) on obtained from single measurements of U(O) and τ(O) in any given condition according to the inverse relationships:
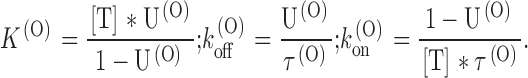 |
1 |
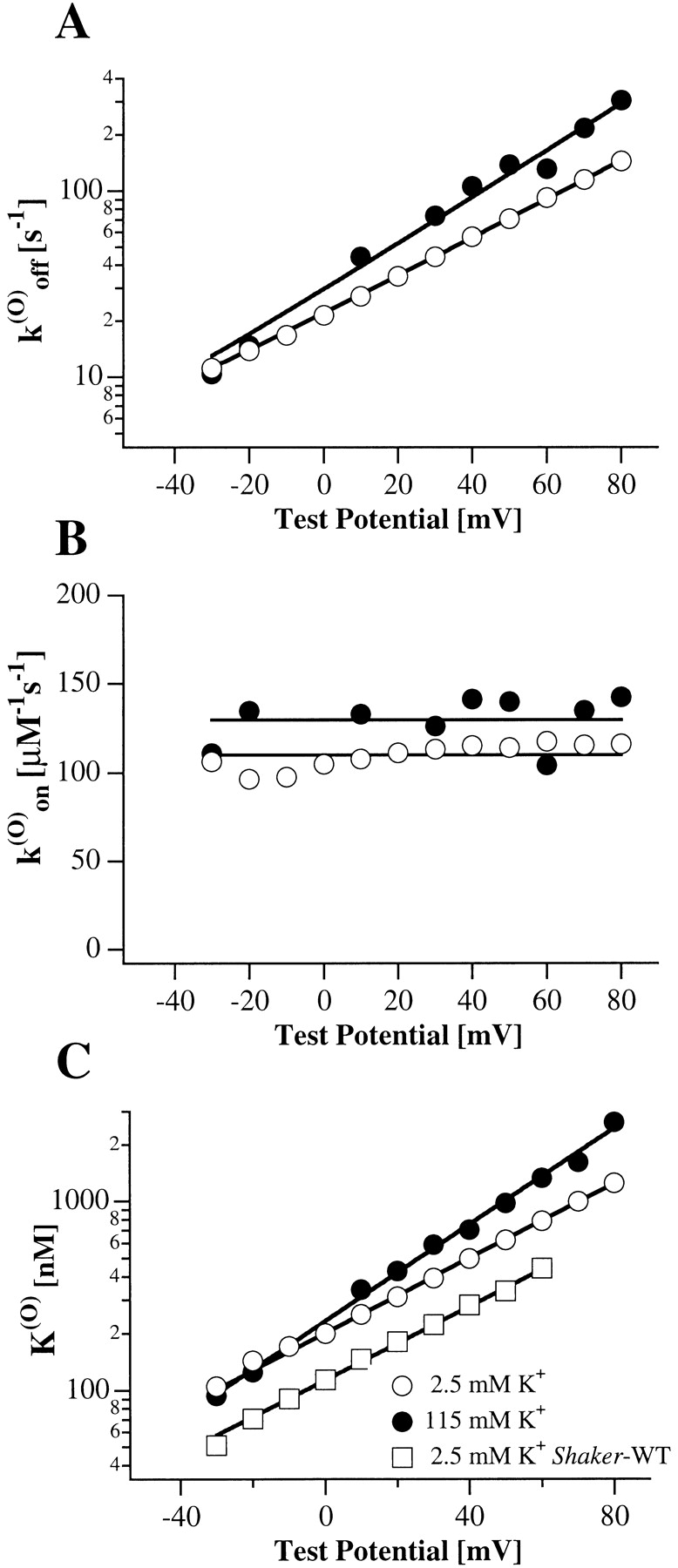
The most interesting feature of PVIIA-block relaxations is their strong voltage dependence. In the experiment of Fig. 2, U(O) increased from 0.51 to 0.8 and τ(O) decreased from 23 to 8.5 ms as V was increased from 0 to 60 mV. Converting these data according to yields the voltage dependencies of the binding parameters shown in Fig. 4 as ○. The figure shows also plots of similar data from a representative experiment in K+-Ringer to be described later (Fig. 4, •, see Fig. 7). Fig. 4A and Fig. C, shows that both k
(O)
off and K
(O) increase with voltage according to a simple exponential law. The straight lines fitting the semilogarithmic plots of k
(O)
off and K
(O) were drawn according to the expressions: 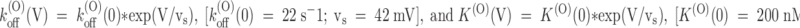 . Consistently, Fig. 4 B shows that k
(O)
on has no systematic trend with a mean value of 110 s−1 μM−1. Mean values of k
(O)
on, k
(O)
off(0), K
(O)(0), and vs
. Consistently, Fig. 4 B shows that k
(O)
on has no systematic trend with a mean value of 110 s−1 μM−1. Mean values of k
(O)
on, k
(O)
off(0), K
(O)(0), and vs
 are given in columns 2–5 of Table .
are given in columns 2–5 of Table .
Figure 7.

Relaxation of PVIIA-block of open Sh-Δ channels in symmetric high-K+ solutions. (A) Currents in response to a standard current–voltage protocol recorded from an outside-out patch without (left) and with (middle) 1 μM PVIIA in the bath K+-Ringer. Plots of the late currents (right) show that most of the inward currents, but not so much the outward currents at large depolarizations, are blocked by 1 μM PVIIA. (B) Direct comparison of three of the control records with the respective records under toxin scaled by a constant factor of 3.3. The records at +20 mV match almost exactly, whereas the others match only during the early rising phase. (C) Current ratios for different voltage steps show a block increase (−30 and +10 mV) or decrease (+50 and +70 mV) that is well fitted by a single exponential (smooth lines). The extrapolated ratio at the half-activation time of the control records is ∼0.3 for all voltages and corresponds to 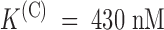 . Fitted time constants and asymptotic values: 6.8 ms, 0.09 (−30 mV); 5.6 ms, 0.25 (10 mV); 4.0 ms, 0.50 (50 mV); and 2.8 ms, 0.62 (70 mV).
. Fitted time constants and asymptotic values: 6.8 ms, 0.09 (−30 mV); 5.6 ms, 0.25 (10 mV); 4.0 ms, 0.50 (50 mV); and 2.8 ms, 0.62 (70 mV).
Fig. 4 C shows also that the apparent dissociation constant, K (O) app, estimated from the unblock of Shaker-H4 channels, is systematically lower than K (O). This is consistent with the idea that the reduction of toxin block occurs in the open state, so that the effect is strongly reduced if the channels visit frequently, and for relatively long periods, the inactivated (closed) state.
Binding of PVIIA to Closed Channels in Low [K]o (NFR)
PVIIA has obviously free access to the site of block also when the channels are closed during resting hyperpolarizations. The dissociation constant of PVIIA binding to closed channels, K (C), can be easily measured by the reduction with [T] of the early responses to pulse stimulations under resting conditions. Our estimates of K (C) in NFR, in the range of 35–80 nM, were not significantly different for Shaker-H4 or Shaker-Δ channels (Table , column 6). These estimates are approximately fourfold lower than those of K (O)(0) and it is important to know what changes in the kinetic parameters of toxin-binding contribute to this difference.
The kinetics of PVIIA binding to closed channels cannot easily be measured from wash-in/wash-out experiments because testing toxin block at any time grossly upsets the block itself and changes the meaning of later tests. A correct wash-in/wash-out experiment should be performed repetitively with a fast-perfusion system testing in each trial PVIIA block at a different time from wash-in. However, the marked change of PVIIA block caused by a pulse depolarization allows us to perform a conceptually identical experiment, by testing at different times the after-pulse re-equilibration of PVIIA binding to closed channels. Fig. 5 shows the results of a double-pulse experiment on an oocyte expressing Sh-Δ channels. Each stimulation consisted of two successive pulses of 40 ms to 40 mV separated by a variable resting period, Ti, at −100 mV. Fig. 5, top, shows the responses for Ti increasing from 5 to 200 ms, recorded before and after the addition of 200 nM PVIIA to the bathing NFR solution. It is seen that the two successive responses in the control experiment are always virtually identical, indicating that the channels recover completely their resting state after 5 ms at −100 mV. With PVIIA, however, the second response reveals a long memory of the effects induced by the first pulse: for short interpulses it is dominated by a fast rising phase much as the toxin-free response, and its “tonic” characteristics are not fully recovered even after 200 ms. Fig. 5, bottom left, plots as a function of Ti the ratio of the early currents elicited by the second and first pulses at the half-time of the toxin-free response. This ratio, which is virtually unity in control measurements at all Ti, with PVIIA is ∼4 for 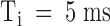 , and is still ∼2 for
, and is still ∼2 for 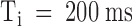 . We interpret this phenomenon as due to a relatively slow reequilibration of PVIIA binding to closed channels, and we take the early amplitude of the second response, normalized to control, as the fraction of toxin-free channels at the time of onset of the second pulse. As shown in Fig. 5, bottom right, this quantity decays with Ti as a single exponential, from
. We interpret this phenomenon as due to a relatively slow reequilibration of PVIIA binding to closed channels, and we take the early amplitude of the second response, normalized to control, as the fraction of toxin-free channels at the time of onset of the second pulse. As shown in Fig. 5, bottom right, this quantity decays with Ti as a single exponential, from 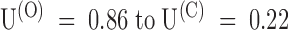 , with a time constant, τ(O), of 190 ms. We interpret U(O) as the toxin-free probability of open channels at the end of the conditioning pulse, U(C) as the equilibrium toxin-free probability of closed channels, and τ(C) as the relaxation time of PVIIA binding to closed channels. Accordingly, the last two quantities yield estimates of the association and dissociation rate constants, k
(C)
on and k
(C)
off, of PVIIA binding to closed channels. From the experiment of Fig. 5, we obtain:
, with a time constant, τ(O), of 190 ms. We interpret U(O) as the toxin-free probability of open channels at the end of the conditioning pulse, U(C) as the equilibrium toxin-free probability of closed channels, and τ(C) as the relaxation time of PVIIA binding to closed channels. Accordingly, the last two quantities yield estimates of the association and dissociation rate constants, k
(C)
on and k
(C)
off, of PVIIA binding to closed channels. From the experiment of Fig. 5, we obtain: 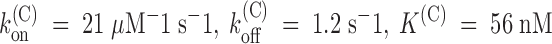 . Mean estimates
. Mean estimates  are given in Table . Compared with open-channel properties, both the on and off rates of PVIIA binding to closed channels are much slower. The approximately fourfold higher value of K
(O)(O) relative to K
(C) results from the combination of an increase of the rate of PVIIA dissociation by a factor of ∼24 and an increase of the association rate by a factor of ∼6. The most important conclusion from these measurements is that, while the low value of k
(C)
off could be thought as an extrapolation to hyperpolarized potentials of the voltage dependence of k
(O)
off (see Fig. 4 A), the sixfold lower value of k
(C)
on is incompatible with the voltage independence of k
(O)
on (see Fig. 4 B) and indicates that closed channels indeed have different toxin-binding properties. In two oocytes, the above double-pulse protocol was applied using variable holding potentials between −60 and −120 mV and we observed no significant change in the estimates of the binding parameters (data not shown). Thus, unlike for open channels, the interaction of PVIIA with closed channels appears to be voltage insensitive and this conclusion is also qualitatively consistent with the above reported observation that even at large positive potentials Shaker-H4 channels that are closed by the inactivation gate appear protected from toxin unblock.
are given in Table . Compared with open-channel properties, both the on and off rates of PVIIA binding to closed channels are much slower. The approximately fourfold higher value of K
(O)(O) relative to K
(C) results from the combination of an increase of the rate of PVIIA dissociation by a factor of ∼24 and an increase of the association rate by a factor of ∼6. The most important conclusion from these measurements is that, while the low value of k
(C)
off could be thought as an extrapolation to hyperpolarized potentials of the voltage dependence of k
(O)
off (see Fig. 4 A), the sixfold lower value of k
(C)
on is incompatible with the voltage independence of k
(O)
on (see Fig. 4 B) and indicates that closed channels indeed have different toxin-binding properties. In two oocytes, the above double-pulse protocol was applied using variable holding potentials between −60 and −120 mV and we observed no significant change in the estimates of the binding parameters (data not shown). Thus, unlike for open channels, the interaction of PVIIA with closed channels appears to be voltage insensitive and this conclusion is also qualitatively consistent with the above reported observation that even at large positive potentials Shaker-H4 channels that are closed by the inactivation gate appear protected from toxin unblock.
Figure 5.

Relaxation of PVIIA binding to closed Shaker-Δ channels after a depolarizing pulse. (Top) Superimposed records of responses to double-pulse stimulations before (Control) and after the addition of 200 nM PVIIA to NFR; each stimulation consisted of a conditioning 40-ms pulse to 40 mV, followed by an identical test pulse with a variable pulse interval, Ti. Successive stimulations were separated by 3-s pauses at −100 mV. The vertical bars represent 3 μA. (Bottom left) The amplitude of the second response at the half-activation time of control is normalized to the first response and plotted as a function of Ti; control data (○) are virtually unity for all Ti. (Bottom right) Plot of the toxin-to-control ratio of the early second response as a function of Ti; the steady state value of 0.22 for 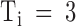 s is the toxin-to-control ratio of the first response; the solid line represents a single-exponential decay from 0.86 to 0.22 with a time constant of 190 ms.
s is the toxin-to-control ratio of the first response; the solid line represents a single-exponential decay from 0.86 to 0.22 with a time constant of 190 ms.
Due to inactivation, the recovery of the tonic binding of PVIIA to Shaker-H4 channels after a conditioning stimulus appears rather peculiar, as illustrated by Fig. 6. The control recordings from a double-pulse protocol follow a classical pattern showing that the second response increases with Ti as more channels recover from the inactivation produced by the conditioning pulse: the ratio of second to first peak current approaches 1 as a single exponential with a time constant of 28 ms (Fig. 6, ○ and solid line in bottom left). The recordings with 100 nM PVIIA added to the bath show instead a marked overshoot of the second response that subsides very slowly. This effect has a simple explanation if we assume that the recovery from the unblock induced by the conditioning pulse is much slower than recovery from inactivation. Consistently, as shown in Fig. 6, bottom right, the fraction of toxin-free channels, estimated from the toxin to control ratio of the second peak response, decreases monotonically with Ti. Fitting this decay towards the steady state ratio of the first peak responses with a single exponential yields 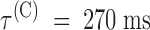 . Combining this estimate with that of the asymptotic toxin-free probability,
. Combining this estimate with that of the asymptotic toxin-free probability, 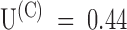 , we estimate in this experiment:
, we estimate in this experiment: 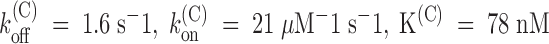 . Mean estimates of the parameters of PVIIA binding to closed Shaker-H4 channels are given in Table . It is seen that they are not significantly different from those of Shaker-Δ channels, supporting the idea that PVIIA does not distinguish the resting state of the two phenotypes.
. Mean estimates of the parameters of PVIIA binding to closed Shaker-H4 channels are given in Table . It is seen that they are not significantly different from those of Shaker-Δ channels, supporting the idea that PVIIA does not distinguish the resting state of the two phenotypes.
Figure 6.

Recovery of tonic PVIIA effects in Shaker-H4 channels. Same experimental protocol as in Fig. 5. (Top) Superimposed double-pulse responses before (Control) and after addition of 100 nM PVIIA to NFR. Successive stimulations were separated by 2-s pauses at −100 mV. (Bottom left) The second peak amplitude is normalized to the first and plotted as a function of Ti; control data (○) show the usual recovery from inactivation, which is well fitted by a single exponential (solid line) with a time constant, 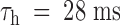 ; the same type of data under toxin (•) shows a marked overshoot that has not subsided after 250 ms. (Bottom right) Plot of the toxin-to-control ratio of the second peak response as a function of Ti; the steady state value of 0.44 for
; the same type of data under toxin (•) shows a marked overshoot that has not subsided after 250 ms. (Bottom right) Plot of the toxin-to-control ratio of the second peak response as a function of Ti; the steady state value of 0.44 for 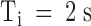 is the mean ratio of the first peak responses; the solid line represents a single-exponential decay from 0.71 to 0.44 with a time constant of 270 ms.
is the mean ratio of the first peak responses; the solid line represents a single-exponential decay from 0.71 to 0.44 with a time constant of 270 ms.
Effect of [K]o on the Binding of PVIIA
The voltage dependence of PVIIA dissociation from open channels is strongly reminiscent of the properties of CTX block of Ca-activated potassium channels (MacKinnon and Miller 1988) or Shaker channels (Goldstein and Miller 1993), which arise from the interaction of CTX with potassium ions occupying an outer site in the channel pore. To investigate the interaction of PVIIA with potassium ions, we studied the effect of PVIIA on Sh-Δ channels using high extracellular concentrations of potassium ions, 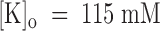 . For more reliable measurements in the voltage range of negative-resistance characteristics of this preparation, few experiments were performed on excised outside-out patches, but very similar results were also obtained outside of this range from experiments on whole oocytes.
. For more reliable measurements in the voltage range of negative-resistance characteristics of this preparation, few experiments were performed on excised outside-out patches, but very similar results were also obtained outside of this range from experiments on whole oocytes.
Fig. 7 illustrates a representative experiment on an outside-out patch exposed to symmetric 115-mM K+ solutions. Fig. 7 A shows superimposed current records from standard current–voltage stimulation protocols applied before (left) and after (middle) the addition to the external bath of 1 μM PVIIA. The right diagram gives plots of the late currents at the end of 100-ms pulses as a function of pulse voltage. It is seen that 1 μM PVIIA blocks most of the inward currents, but has a much smaller effect on the outward currents at large depolarizations. This is consistent with the results obtained with NFR 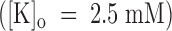 in the external bath. By interpreting the steady state ratios of toxin to control currents as toxin-free probabilities, we obtain for the open-channel dissociation constant, K
(O), similar estimates and the same voltage dependence as in NFR (Fig. 4 C, •, and mean estimates in Table ).
in the external bath. By interpreting the steady state ratios of toxin to control currents as toxin-free probabilities, we obtain for the open-channel dissociation constant, K
(O), similar estimates and the same voltage dependence as in NFR (Fig. 4 C, •, and mean estimates in Table ).
The comparison of the whole time course of test and control responses also reveals that toxin-block relaxations have properties similar to those observed in low K+ solutions, although starting from a different resting-block equilibrium. Fig. 7 B shows three of the control records in A 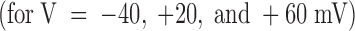 superimposed with the respective records under toxin scaled by a constant factor of 3.3. It is seen that this scaling makes the currents recorded at +20 mV match almost exactly for the whole duration of the pulse, whereas the match for the other records is good only for the initial rising phase. At later times, the inward currents at −40 mV are further depressed approximately fourfold and the outward currents at +60 mV undergo an approximately twofold increase. The most obvious interpretation of these results is that two thirds of the channels are tonically blocked at 1 μM PVIIA according to a toxin dissociation constant from closed channels, K
(C) ∼ 430 nM, which is about equal to K
(O) at +20 mV, whereas depolarizations below or above +20 mV, leading to lower or higher K
(O) values, cause an increase or decrease of PVIIA block. In agreement with this interpretation, the current ratios after almost complete activation follow single-exponential relaxations (Fig. 7 C, smooth lines), as expected from the reequilibration of a toxin-block reaction. The fitting parameters of these relaxations can be used to estimate the rate constants characterizing the binding of PVIIA to open channels. These estimates for the experiment of Fig. 7 are plotted in Fig. 4A and Fig. B, •. The comparison with the data obtained in NFR (Fig. 4, ○) shows that both the association and dissociation rate constants of PVIIA binding to open channels are insensitive to changes of [K]o. Mean estimates of k
(O)
on, k
(O)
off(0), K
(O)(0), and vs from four different experiments are given in columns 2–5 of Table .
superimposed with the respective records under toxin scaled by a constant factor of 3.3. It is seen that this scaling makes the currents recorded at +20 mV match almost exactly for the whole duration of the pulse, whereas the match for the other records is good only for the initial rising phase. At later times, the inward currents at −40 mV are further depressed approximately fourfold and the outward currents at +60 mV undergo an approximately twofold increase. The most obvious interpretation of these results is that two thirds of the channels are tonically blocked at 1 μM PVIIA according to a toxin dissociation constant from closed channels, K
(C) ∼ 430 nM, which is about equal to K
(O) at +20 mV, whereas depolarizations below or above +20 mV, leading to lower or higher K
(O) values, cause an increase or decrease of PVIIA block. In agreement with this interpretation, the current ratios after almost complete activation follow single-exponential relaxations (Fig. 7 C, smooth lines), as expected from the reequilibration of a toxin-block reaction. The fitting parameters of these relaxations can be used to estimate the rate constants characterizing the binding of PVIIA to open channels. These estimates for the experiment of Fig. 7 are plotted in Fig. 4A and Fig. B, •. The comparison with the data obtained in NFR (Fig. 4, ○) shows that both the association and dissociation rate constants of PVIIA binding to open channels are insensitive to changes of [K]o. Mean estimates of k
(O)
on, k
(O)
off(0), K
(O)(0), and vs from four different experiments are given in columns 2–5 of Table .
From the data of Fig. 7 B, we estimate a tonic block of 66% in 1 μM PVIIA, which corresponds to a PVIIA dissociation constant of 430 nM and shows that, at variance with the open channel properties, the binding of PVIIA to closed channels is very sensitive to [K]o. Our mean estimate of K(C) from five oocytes at 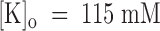 was ∼400 nM, about a factor of 8 higher than at
was ∼400 nM, about a factor of 8 higher than at 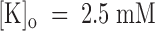 . We can ask whether the lower affinity of PVIIA at
. We can ask whether the lower affinity of PVIIA at 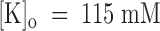 is due to a smaller rate of association or to a larger rate of dissociation by analyzing a double-pulse experiment such as that illustrated in Fig. 8. The experiment was done on a whole oocyte in the presence of 1 μM PVIIA and the protocol consisted of several double stimulations, with pulses to −20 mV separated by a variable interpulse interval (Ti) allowing resting periods of at least 3 s between successive stimulations. Fig. 8 A shows a sample of the responses obtained for Ti values between 30 and 420 ms. In agreement with what we described above, we observe that the conditioning pulse induces an increase of toxin block that appears as a reproducible peak in the first response of each successive double stimulation. However, for small Ti values, the second response has a more normal appearance and no peak, indicating that the fraction of blocked channels is near the equilibrium for open-channel block at −20 mV that was already achieved by the end of the conditioning pulse. We can judge qualitatively the slow recovery of the lower equilibrium probability of PVIIA binding to closed channels by the reappearance of a peak in the second response, which is clearly seen only in the records with
is due to a smaller rate of association or to a larger rate of dissociation by analyzing a double-pulse experiment such as that illustrated in Fig. 8. The experiment was done on a whole oocyte in the presence of 1 μM PVIIA and the protocol consisted of several double stimulations, with pulses to −20 mV separated by a variable interpulse interval (Ti) allowing resting periods of at least 3 s between successive stimulations. Fig. 8 A shows a sample of the responses obtained for Ti values between 30 and 420 ms. In agreement with what we described above, we observe that the conditioning pulse induces an increase of toxin block that appears as a reproducible peak in the first response of each successive double stimulation. However, for small Ti values, the second response has a more normal appearance and no peak, indicating that the fraction of blocked channels is near the equilibrium for open-channel block at −20 mV that was already achieved by the end of the conditioning pulse. We can judge qualitatively the slow recovery of the lower equilibrium probability of PVIIA binding to closed channels by the reappearance of a peak in the second response, which is clearly seen only in the records with 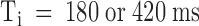 . A more quantitative analysis is shown in Fig. 8 B by plotting, as a function of Ti, the ratio of the early amplitudes of test and conditioning responses taken as means in the time interval of 25–75% rise of the toxin-free response. These data are well fitted by a single exponential increase with Ti with a time constant of 430 ms. Assuming this value as an estimate of τ(C) and using the value of
. A more quantitative analysis is shown in Fig. 8 B by plotting, as a function of Ti, the ratio of the early amplitudes of test and conditioning responses taken as means in the time interval of 25–75% rise of the toxin-free response. These data are well fitted by a single exponential increase with Ti with a time constant of 430 ms. Assuming this value as an estimate of τ(C) and using the value of 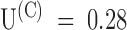 measured from the early fraction of unblocked currents (data not shown), we estimate in this experiment:
measured from the early fraction of unblocked currents (data not shown), we estimate in this experiment: 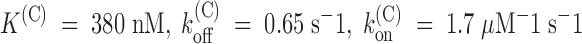 . Mean estimates obtained from three experiments of this type are k
. Mean estimates obtained from three experiments of this type are k
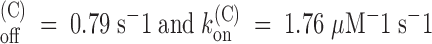 (see Table ). The value of
(see Table ). The value of 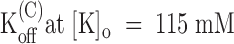 appears equal to that in
appears equal to that in 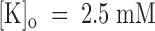 within the experimental error. On the contrary, increasing [K]o from 2.5 to 115 mM decreases by more than one order of magnitude the apparent value of k
(C)
on, which seems to be the only parameter of PVIIA binding to Shaker channels that is very sensitive to [K]o. Notice that we studied the effect of increasing [K]o by substituting Na+ with K+, so that this effect is not comparable to the large decrease of toxin association rates observed at increasing ionic strength for CTX block of Ca2+-activated channels (Anderson et al. 1988) or Shaker channels (Goldstein and Miller 1993).
within the experimental error. On the contrary, increasing [K]o from 2.5 to 115 mM decreases by more than one order of magnitude the apparent value of k
(C)
on, which seems to be the only parameter of PVIIA binding to Shaker channels that is very sensitive to [K]o. Notice that we studied the effect of increasing [K]o by substituting Na+ with K+, so that this effect is not comparable to the large decrease of toxin association rates observed at increasing ionic strength for CTX block of Ca2+-activated channels (Anderson et al. 1988) or Shaker channels (Goldstein and Miller 1993).
Figure 8.

Recovery of tonic PVIIA binding to closed Sh-Δ channels in 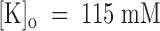 . (A) Superimposed records of responses to double pulses of 40 ms at −20 mV with variable pulse interval, Ti; the stimulation intervals at −100 mV were 3 s; the inset shows a sample of double responses in real time, whereas the main graph shows the same data with the second response translated to the time of onset of the first. The first pulse induces an increase of toxin block seen as a peak in the response; the peak reappears clearly in the second response only for Ti larger than hundreds of milliseconds. (B) The early amplitude of the second response is normalized to the first response and plotted as a function of Ti to follow the recovery of tonic block (the early amplitude was defined as the mean value of the current during the fixed interval in which the toxin free responses rise from 25 to 75% of their maximum); the solid line is the best fit with a single exponential with a time constant of 430 ms.
. (A) Superimposed records of responses to double pulses of 40 ms at −20 mV with variable pulse interval, Ti; the stimulation intervals at −100 mV were 3 s; the inset shows a sample of double responses in real time, whereas the main graph shows the same data with the second response translated to the time of onset of the first. The first pulse induces an increase of toxin block seen as a peak in the response; the peak reappears clearly in the second response only for Ti larger than hundreds of milliseconds. (B) The early amplitude of the second response is normalized to the first response and plotted as a function of Ti to follow the recovery of tonic block (the early amplitude was defined as the mean value of the current during the fixed interval in which the toxin free responses rise from 25 to 75% of their maximum); the solid line is the best fit with a single exponential with a time constant of 430 ms.
Discussion
PVIIA Is a Pore Blocker
We have described in this paper the dose, voltage, and time dependence of PVIIA effects on the currents mediated by Shaker channels. Our results are consistent with the hypothesis that the most prominent effect of toxin-channel association is the block of the channel conductance and rule out a substantial modification of channel gating as observed, e.g., when the channels bind the spider venom toxin Hanatoxin (Swartz and MacKinnon 1997). In fact, there is no way to account for the [T] dependence of the currents shown in Fig. 3 A as a change in the fraction of channels that have a toxin-modified activation, whereas the analysis illustrated in Fig. 3B–D, shows that the toxin-dependent modification of the currents has all the characteristics expected for the relaxation of a bimolecular binding reaction after a step change of the reaction parameters.
PVIIA Block Is State Dependent
An important result of our study for mechanistic interpretations is that the different binding of PVIIA to closed or open channels cannot be attributed solely to the voltage dependence of the reaction rates. First of all, while the low value of k
(C)
off could be thought as the extrapolation of k
(O)
off(V) to hyperpolarizing potentials, the independence of K(O)
on on voltage and [K]o is incompatible with the 6- or 90-fold lower estimates of K(C)
on at 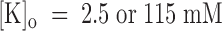 . Secondly, our observation that the relaxations of closed-channel block are independent of the holding potential in the range of −60 to −120 mV shows that, in sharp contrast to the case of open channels, the binding of PVIIA to closed channels is fairly insensitive to the transmembrane voltage. Indeed, the fact that inactivation protects significantly depolarized Shaker-H4 channels from toxin-unbinding is consistent with a similar binding of PVIIA to any nonconducting state of the channels at any voltage.
. Secondly, our observation that the relaxations of closed-channel block are independent of the holding potential in the range of −60 to −120 mV shows that, in sharp contrast to the case of open channels, the binding of PVIIA to closed channels is fairly insensitive to the transmembrane voltage. Indeed, the fact that inactivation protects significantly depolarized Shaker-H4 channels from toxin-unbinding is consistent with a similar binding of PVIIA to any nonconducting state of the channels at any voltage.
An important consideration that justifies a posteriori most of our experimental analysis is that the relaxations of toxin binding to open or closed channels occur on very different time scales. The mean properties that we expect from flickering between open and closed states are weighted for each state in direct proportion to the probability of that state and in inverse proportion to the time constant of the binding relaxation in that state. Since the binding kinetics for closed channels is at least 10× slower than for open channels, the relative weight of the open state in Sh-Δ channels that do not inactivate would be >0.9 for any open probability, P (O) > 0.5. Therefore, it is fair to assume that, during depolarizations of noninactivating channels, we are essentially measuring the binding to open channels. The situation is less favorable for Shaker-H4 channels, where P (O) ∼ 0.1. In this case, we expect that the relative weight of the open state is ∼0.53, and this is indeed fairly consistent with our finding that the K(O) app measured for Shaker-H4 channels is ∼0.6 K(O).
Analogy between PVIIA and CTX
The structural similarity of PVIIA with the scorpion toxins of the charybdotoxin family (Scanlon et al. 1997) and the strong sensitivity of PVIIA efficacy to mutations of some of the amino acid residues that are believed to shape the outer pore vestibule (Shon et al. 1998) indicate that PVIIA acts as CTX by simply blocking the extracellular mouth of the ion pore. The results presented in this paper demonstrate that CTX and PVIIA also share strong similarities in their mechanism of block.
As for the case of CTX block of Ca2+-activated K+ channels (MacKinnon and Miller 1988) or Shaker channels (Goldstein and Miller 1993), we find that PVIIA block is strongly decreased during channel opening with a voltage dependence that is afforded exclusively by the dissociation rate constant and not by the on rate of toxin binding. Also the state dependency of PVIIA binding is qualitatively very similar to that reported for the binding of CTX to Ca2+-activated channels, which occurs with a seven- to eightfold lower rate of association when the channels are fully closed (Anderson et al. 1988).
The only clear difference between CTX and PVIIA appears to be that the latter binds with much faster kinetics, due to a much higher dissociation rate constant. However, several CTX variants also block open Shaker channels with relatively fast kinetics that have been observed by fast perfusion of macropatches during long depolarizing sweeps (Goldstein and Miller 1993; Goldstein et al. 1994). It is interesting to notice that nearly all of the more than 60 variants of CTX characterized by Goldstein et al. 1994 have second-order association rate constants close to the wild type, and in the same range (20–100 μM−1 s−1) of our estimates for PVIIA. None of the CTX variants was studied in a way that would allow a clear separate characterization of closed- and open-channel binding, because slow toxins were studied with multipulse protocols that characterize the binding to closed channels, whereas fast toxins were tested for open-channel block with wash-in/wash-out measurements during long depolarizations. Therefore, we have no direct evidence that CTX block of Shaker channels is state dependent. However, one variant, CTX-R25Q, was studied with both protocols, although with different [K]o conditions (2 vs. 100 mM), and the results of the two measurements were quite different, the fast protocol yielding a 5× larger k on, and 10× larger k off. Although the authors suggest a different explanation related to the different [K]o conditions, we notice that this result is in fact consistent with CTX block having the same state dependency shown here for PVIIA block.
A Model for the State Dependency of PVIIA Block
While we cannot exclude that a PVIIA molecule bound to an open channel senses a significant fraction of the transmembrane electric field, the above discussed analogies with CTX and the antagonizing action of high-K+ solutions on tonic PVIIA block suggest that the unblock observed upon opening the channels in low external K+ may largely arise from the destabilization of toxin binding by internal K+, as in the case of CTX (MacKinnon and Miller 1988). The results reported by Garcia and Naranjo 1999 support to a large extent this hypothesis by showing a significant reduction of the absolute value and voltage dependence of the rate of toxin dissociation upon putatively complete removal of intracellular K+. We discuss below that our findings of a marked difference between the toxin-binding properties of open versus closed channels and of a strong [K]o dependence of closed-channel binding can also have a simple explanation in the context of a model similar to that proposed by MacKinnon and Miller 1988, in which the major factor influencing toxin binding is the state of occupancy of the outermost K+-binding site within the channel permeation pathway.
From now on, we shall refer to this site as if it was the only K+-binding site. The general scheme that we can use to interpret our data is shown in Fig. 2.
Scheme S2.

Fig. 2 assumes that the toxin (Tx) can bind to whatever state of the channels with the same second-order–association rate constant, k 1, whereas its first-order–dissociation rate constant is k (K) −1 or k (0) −1, depending on whether or not the K+-binding site is occupied, and k (K) −1 >> k (0) −1. All the rate constants are otherwise assumed to be voltage independent. The scheme assumes also that K+ binding is in fast equilibrium and is governed by different dissociation constants, K (Tx) d or K (0) d, depending on whether or not there is a bound toxin in the outer pore vestibule. While a bound toxin is blocking the extracellular access, K+ binding to the pore is assumed to be in equilibrium only with the cytoplasmic K+ concentration, [K]i, and the parenthesis in the transition C:Tx ↔ C:K+:Tx indicates that this transition can occur only when the channel is open. A possible molecular picture of the states C:Tx and C:K+:Tx is shown in the cartoon of Fig. 9. When the channel is closed and not blocked by PVIIA [K] stands for the extracellular K+ concentration, whereas when the channel is open [K] is determined by the flux of ions in the pore.
Figure 9.

PVIIA block of the Shaker K+ channel. The cartoon depicts a Shaker channel blocked by PVIIA, occluding a pore that contains three different binding sites for K+ in its narrowest region. When PVIIA is bound to the channel, it experiences a strong or much weaker repulsion, depending on whether the outer-most site is occupied by a K ion (C:K+:Tx) or not (C:Tx). The transition C:Tx → C:K+:Tx involves the concerted movement of two K+, each traversing a small fraction (∼1/3?) of the membrane voltage.
In the case of open channels, it is easily seen that the above scheme reduces to the simple two-state Fig. 1, provided we identify k (O) on with k 1, and k (O) off with an average of k (K) −1 and k (0) −1 weighted according to the relative probabilities of states C:K+:Tx and C:Tx. Assuming for K(Tx) d a voltage dependence associated with the charge translocation occurring along the pore in the transition C:Tx ↔ C:K+:Tx, we can express k (O) off as:
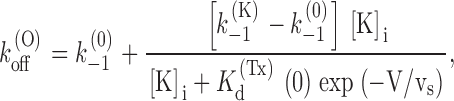 |
2 |
where K
(Tx)
d(0) is the dissociation constant at 0 mV and the voltage dependence is equivalently expressed as if the charge translocation involved the movement of a single K+ across a fraction 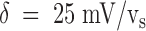 of the transmembrane voltage.
of the transmembrane voltage.
It is important to notice that and the condition 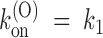 bear no reference whatsoever to the extracellular solution conditions, a strong prediction of Fig. 2 that was verified in our comparative experiments with low- or high-K+ solutions. Notice also that Fig. 2 leads us to conclude that our voltage-independent estimates of k
(O)
on provide a direct measurement of the true rate constant of PVIIA association to open channels.
bear no reference whatsoever to the extracellular solution conditions, a strong prediction of Fig. 2 that was verified in our comparative experiments with low- or high-K+ solutions. Notice also that Fig. 2 leads us to conclude that our voltage-independent estimates of k
(O)
on provide a direct measurement of the true rate constant of PVIIA association to open channels.
The expression of k (O) off according to is the same as that used by MacKinnon and Miller 1988 to describe the voltage and [K]i dependence of the dissociation rate constant of CTX from Ca2+-activated K+ channels and predicts that both dependencies should be sigmoidal. However, it is easily seen that for large values of the ratio k (K) −1/k (0) −1 the voltage dependence of k (O) off is practically exponential (increasing e-fold/vs) in the wide voltage range:
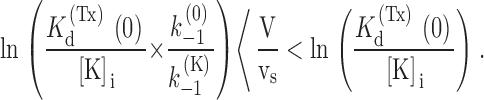 |
In the study of single channels reconstituted in lipid bilayers, MacKinnon and Miller 1988 could show the tendency to saturation of the K+-binding site by using [K]i values up to 700 mM. Likewise, by adjusting internal Ca2+ to maintain channel openings at hyperpolarized potentials, they could also approach the finite lower limit of k
(O)
off. From their data, one estimates K
(Tx)
d(0) values in the range of 1–3 M, k
(K)
−1/k
(0)
−1 ratios (βmax/βmin, in their terminology) in the range of 20–50, and a value of δ close to 1. As discussed below, it is practically impossible in the oocyte preparation to study open-channel block by PVIIA outside the range of the exponential rise of k
(O)
off, so that imposing the consistency of our data with yields only lower estimates for K
(Tx)
d(0) and k
(K)
−1 and a good estimate of vs. Our overall mean estimate of vs ∼ 40 mV, corresponding to 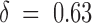 , is in good agreement with the estimates of
, is in good agreement with the estimates of 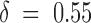 obtained by Scanlon et al. 1997 from the voltage dependence of τ(O) at low [T] values for which τ(O) ∼ 1/k
(O)
off. Low estimates of δ, ranging from 0.31 to 0.62, were also found in the study by Goldstein and Miller 1993 of the voltage dependence of Shaker open-channel block by several CTX variants with relatively fast binding kinetics.
obtained by Scanlon et al. 1997 from the voltage dependence of τ(O) at low [T] values for which τ(O) ∼ 1/k
(O)
off. Low estimates of δ, ranging from 0.31 to 0.62, were also found in the study by Goldstein and Miller 1993 of the voltage dependence of Shaker open-channel block by several CTX variants with relatively fast binding kinetics.
Our measurements of k
(O)
off do not show any obvious deviation from an exponential rise up to 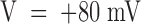 , where k
(O)
off ∼ 200 s−1 (Fig. 4 A). Thus, for consistency with , these data imply k
(K)
−1 >> 200 s−1 and K
(Tx)
d(0) >> [K]i*e2; i.e., K
(Tx)
d(0) >> 0.8 M, in qualitative agreement with the studies of CTX block. The large lower estimate of k
(K)
−1 explains why also the low pedestal predicted by at negative voltages is not seen in our data. According to Fig. 2, k
(0)
−1 is a lower bound for the dissociation rate constant in any channel configuration, so that k
(0)
−1 ≤ k
(C)
off ∼ 1 s−1. Since
, where k
(O)
off ∼ 200 s−1 (Fig. 4 A). Thus, for consistency with , these data imply k
(K)
−1 >> 200 s−1 and K
(Tx)
d(0) >> [K]i*e2; i.e., K
(Tx)
d(0) >> 0.8 M, in qualitative agreement with the studies of CTX block. The large lower estimate of k
(K)
−1 explains why also the low pedestal predicted by at negative voltages is not seen in our data. According to Fig. 2, k
(0)
−1 is a lower bound for the dissociation rate constant in any channel configuration, so that k
(0)
−1 ≤ k
(C)
off ∼ 1 s−1. Since 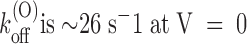 , we expect that k
(O)
off approaches its low pedestal only for voltages more negative than −80 mV, where open channels are impossible to explore.
, we expect that k
(O)
off approaches its low pedestal only for voltages more negative than −80 mV, where open channels are impossible to explore.
In conclusion, the predictions of Fig. 2 are consistent with our data on PVIIA binding to open channels according to the following quantitative estimates:
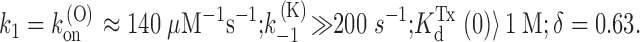 |
We discuss now the predictions of Fig. 2 for the case of closed channels. As already anticipated, the most important difference with respect to the previous case is the absence of C:Tx ↔ C:K+:Tx transitions. An additional important difference is the fact that C: ↔ C:K+ transitions involve only a very fast equilibration with external K+. With these two conditions, Fig. 2 reduces to Fig. 3, where {U} lumps together the unblocked states C: and C:K+, and p is the probability that a U state is a C:K+ state. A further simplification arises from the above consideration that k
(K)
−1 is several hundred times larger than k
(0)
−1, which implies that the probability of state C:K+:Tx is insignificant relative to that of state C:Tx for p < 0.99. This reduces Fig. 3 to a simple two-state scheme (Fig. 4), where 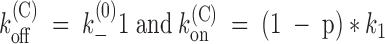 . Both our findings that k
(C)
off is independent of [K]o and k
(C)
on decreases upon increasing [K]o are fully consistent with this interpretation. In principle, the [K]o dependence of k
(C)
on could be used to estimate the dissociation constant of K+ binding to the outer site, K
(0)
d. With the questionable assumption that sodium ions do not compete significantly for the same site, we can tentatively use for p the expression:
. Both our findings that k
(C)
off is independent of [K]o and k
(C)
on decreases upon increasing [K]o are fully consistent with this interpretation. In principle, the [K]o dependence of k
(C)
on could be used to estimate the dissociation constant of K+ binding to the outer site, K
(0)
d. With the questionable assumption that sodium ions do not compete significantly for the same site, we can tentatively use for p the expression:
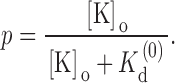 |
Scheme S3.

Scheme S4.

Since we have data only for two values of [K]o, 2.5 and 115 mM, our estimates of K
(0)
d can be only very crude. For 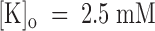 , we estimate k
(C)
on ∼ 1/6 k
(O)
on (i.e., (1 − p) ∼ 1/6) and for
, we estimate k
(C)
on ∼ 1/6 k
(O)
on (i.e., (1 − p) ∼ 1/6) and for 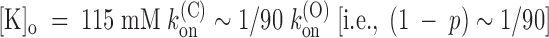 . These two data are roughly consistent, yielding K
(0)
d estimates of 0.5 or 1.3 mM, respectively. Notice that these guesses imply that the ratio K
(Tx)
d(0)/K
(0)
d is in the order of 103, close to our lower estimate for the ratio k
(K)
−1/k
(0)
−1. This is an important verification of the physical consistency of Fig. 2, which requires that the destabilization of PVIIA binding by K+ entrains a reciprocal effect of PVIIA on K+ binding.
. These two data are roughly consistent, yielding K
(0)
d estimates of 0.5 or 1.3 mM, respectively. Notice that these guesses imply that the ratio K
(Tx)
d(0)/K
(0)
d is in the order of 103, close to our lower estimate for the ratio k
(K)
−1/k
(0)
−1. This is an important verification of the physical consistency of Fig. 2, which requires that the destabilization of PVIIA binding by K+ entrains a reciprocal effect of PVIIA on K+ binding.
The low δ value accounting for the voltage-dependent destabilization of PVIIA binding to open Shaker channels may appear in contrast with the model underlying Fig. 2. Finding a value of δ close to 1 was used by MacKinnon and Miller 1988 as evidence that the K+-binding site is located at the extreme outer end of the K+-conduction pore, from where K+ destabilizes CTX binding by speeding up CTX dissociation by a factor of 20. Using the same model, we find that PVIIA dissociation is accelerated by K+ at least 10× more (k
(K)
−1/k
(0)
−1 >> 200), so that we might intuitively expect that the site from which K+ exerts its repulsion on the toxin is even more to the outer extreme of the pore conduction pathway. However, it may be naive to interpret δ as the true electrical distance traversed by a single K+ that leaves the cytoplasmic medium and goes all the way through the pore to reach the outermost binding site in the conduction pathway. A plausible, more realistic interpretation is illustrated by the cartoon in Fig. 9. The conduction pore can be safely assumed to contain three binding sites, in agreement with classical models proposed to explain single-file diffusion properties of potassium channels (Hille and Schwartz, 1978; see Hille 1992 for review) and with the structural study of Doyle et al. 1998. It is also plausible to assume that when the outer pore mouth is occluded by a toxin molecule the pore always has one, but very unlikely all three, of these binding sites occupied. Accordingly, the occupational configurations that most significantly represent the states indicated in Fig. 2 as C:Tx and C:K+:Tx could be those shown in Fig. 9. If this is the case, the transition from C:Tx to C:K+:Tx would involve the concerted movement of two potassium ions, each traversing only a fraction of the electrical distance across the pore; the necessary push/pull of intercalated water molecules might be eased by a water shunt of the toxin plug. Even assuming no voltage drop from the outermost site to the external vestibule, the transition could then involve for each ion a δ value of 1/3, and this would yield an apparent overall 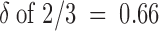 , consistent with our measurements.
, consistent with our measurements.
Conclusions
As a consequence of our study, we propose that the dependence of the PVIIA block on channel conductance does not involve state-dependent changes of the molecular interactions of the toxin with the residues that shape the pore vestibule, but rather the indirect modulation of toxin binding by the interaction with cations occupying nearby sites. For our comprehensive modeling of the voltage, [K]o, and state dependence of PVIIA block, we have used a somewhat natural expansion of the scheme proposed by MacKinnon and Miller (1998) for CTX block. Therefore, we believe that a similar analysis could be applied to CTX variants with relatively fast binding kinetics like PVIIA. In general, our analysis suggests an additional potential value of pore-blocking molecules for the study of ion channels, besides their use as probes of the pore-mouth structure. Indeed, a detailed study of the interaction of these substances with permeant ions, by an appropriate evaluation of the effects of such interaction upon the pore blockade, may also provide important information about more intimate properties of the ion-conduction pathway.
Acknowledgments
We thank Dr. Martin Stocker for providing the Shaker-H4 and Shaker-Δ6-46 RNA. The helpful comments of Dr. Chris Miller on the manuscript are greatly appreciated.
H. Terlau was supported by a grant from the CNR and the SFB “Synaptische Interaktionen in neuronalen Zellmembranen,” and F. Conti by Telethon grant 926.
Footnotes
1used in this paper: CTX, charybdotoxin; NFR, normal frog Ringer; Sh-Δ, Shaker–Δ6-46; Tx, toxin
Portions of this work have been previously published in abstract form (Terlau, H., A. Boccaccio, B.M. Olivera, and F. Conti. 1999. Biophys. J. 76:A150).
References
- Anderson C.S., MacKinnon R., Smith C., Miller C. Charybdotoxin block of single Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J. Gen. Physiol. 1988;91:317–333. doi: 10.1085/jgp.91.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle A.D., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Garcia E., Naranjo D. Blocking mechanism of κ-PVIIA conotoxin on Shaker channels Biophys. J. 76 1999. A332(Abstr.) [Google Scholar]
- Goldstein S.A.N., Miller C. Mechanism of charybdotoxin block of a voltage-gated K+ channel. Biophys. J. 1993;65:1613–1619. doi: 10.1016/S0006-3495(93)81200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S.A.N., Pheasant D.J., Miller C. The charybdotoxin receptor of a Shaker K+ channelpeptide and channel residues mediating molecular recognition. Neuron. 1994;12:1377–1388. doi: 10.1016/0896-6273(94)90452-9. [DOI] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic channels of excitable membranes 2nd ed 1992. Sinauer Associates, Inc; Sunderland, MA: pp. 362–422 [Google Scholar]
- Hille B., Schwarz W. Potassium channels as multi-ion single-file pores. J. Gen. Physiol. 1978;72:409–442. doi: 10.1085/jgp.72.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W., Aldrich R. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Kamb A., Tseng-Crank J., Tanouye M. Multiple products of the Drosophila Shaker gene may contribute to potassium channel diversity. Neuron. 1988;1:421–430. doi: 10.1016/0896-6273(88)90192-4. [DOI] [PubMed] [Google Scholar]
- Kim M., Baro D.J., Lanning C.C., Doshi M., Farnham J., Moskowitz H.S., Peck J.H., Olivera B.M., Harris-Warrick R.M. Alternative splicing in the pore-forming region of Shaker potassium channels. J. Neurosci. 1997;17:8213–8224. doi: 10.1523/JNEUROSCI.17-21-08213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg P.A., Melton D.A. In-vitro RNA-synthesis with SP6 RNA-polymerase. Methods Enzymol. 1987;155:397–441. doi: 10.1016/0076-6879(87)55027-3. [DOI] [PubMed] [Google Scholar]
- MacKinnon R., Miller C. Mechanism of charybdotoxin block of the high-conductance, Ca2+-activated K+ channel. J. Gen. Physiol. 1988;91:335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. The charybdotoxin family of K+ channel-blocking peptides. Neuron. 1995;15:5–10. doi: 10.1016/0896-6273(95)90057-8. [DOI] [PubMed] [Google Scholar]
- Olivera B.M., Gray W.R., Zeikus R., McIntosh J.M., Varga J., Rivier J., de Santos V., Cruz L.J. Peptide neurotoxins from fish-hunting cone snails. Science. 1985;230:1338–1343. doi: 10.1126/science.4071055. [DOI] [PubMed] [Google Scholar]
- Olivera B.M., Rivier J., Clark C., Ramilo C.A., Corpuz G.P., Abogadie F.C., Mena E., Woodward S.R., Hillyard D.R., Cruz L.J. Diversity of conus neuropeptides. Science. 1990;249:257–263. doi: 10.1126/science.2165278. [DOI] [PubMed] [Google Scholar]
- Savarin P., Guenneugues M., Gilquin B., Lamthanh H., Gasparini S., Zinn-Justin S., Ménez A. Three-dimensional structure of κ-conotoxin PVIIA, a novel potassium channel-blocking toxin from cone snails. Biochemistry. 1998;37:5407–5416. doi: 10.1021/bi9730341. [DOI] [PubMed] [Google Scholar]
- Scanlon M.J., Naranjo D., Thomas L., Alewood P.F., Lewis R.J., Craik D.J. Solution structure and proposed binding mechanism of a novel potassium channel toxin κ-conotoxin PVIIA. Structure (Lond.). 1997;5:1585–1597. doi: 10.1016/s0969-2126(97)00307-9. [DOI] [PubMed] [Google Scholar]
- Shon K., Stocker M., Terlau H., Stühmer W., Jacobsen R., Walker C., Grilley M., Watkins M., Hillyard D.R., Gray W.R., Olivera B.M. κ-Conotoxin PVIIA is a peptide inhibiting the Shaker K+-channel. J. Biol. Chem. 1998;77:1–6. doi: 10.1074/jbc.273.1.33. [DOI] [PubMed] [Google Scholar]
- Stühmer W. Electrophysiological recordings from Xenopus oocytes. Methods Enzymol. 1992;207:319–339. doi: 10.1016/0076-6879(92)07021-f. [DOI] [PubMed] [Google Scholar]
- Swartz K.J., MacKinnon R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 1997;18:665–673. doi: 10.1016/s0896-6273(00)80306-2. [DOI] [PubMed] [Google Scholar]
- Terlau H., Shon K., Grilley M., Stocker M., Stühmer W., Olivera B.M. Strategy for rapid immobilization of prey by a fish-hunting marine snail. Nature. 1996;381:148–151. doi: 10.1038/381148a0. [DOI] [PubMed] [Google Scholar]