

Abstract
Neurogenesis studies on the adult mouse hippocampal subgranular zone (SGZ) typically report increases or decreases in proliferation. However, key information is lacking about these proliferating SGZ precursors, from the fundamental - what dose of bromodeoxyuridine (BrdU) is appropriate for labeling all S phase cells? - to the detailed - what are the kinetics of BrdU-labeled cells and their progeny? To address these questions, adult C57BL/6J mice were injected with BrdU and BrdU-immunoreactive (IR) cells were quantified. Initial experiments with a range of BrdU doses (25-500 mg/kg) suggested that 150 mg/kg labels all actively dividing precursors in the mouse SGZ. Experiments using a saturating dose of BrdU suggested BrdU bioavailability is less than 15 minutes, notably shorter than in the developing mouse brain. We next explored precursor division and maturation by tracking the number of BrdU-IR cells and colabeling of BrdU with other cell cycle proteins from 15 min to 30 days after BrdU. We found that BrdU and the G2/M phase protein pHisH3 maximally colocalized 8 hr after BrdU, indicating that the mouse SGZ precursor cell cycle length is 14 hr. In addition, triple labeling with BrdU and PCNA and Ki-67 showed that BrdU-IR precursors and/or their progeny express these endogenous cell cycle proteins up to 4 days after BrdU injection. However, the proportion of BrdU/Ki-67-IR cells declined at a greater rate than the proportion of BrdU/PCNA-IR cells. This suggests that PCNA protein is detectable long after cell cycle exit, and that reliance on PCNA may overestimate the length of time a cell remains in the cell cycle. These findings will be critical for future studies examining the regulation of SGZ precursor kinetics in adult mice, and hopefully will encourage the field to move beyond counting BrdU-IR cells to a more mechanistic analysis of adult neurogenesis.
Keywords: BrdU, PCNA, pHisH3, Ki-67, mitosis, neurogenesis
Introduction
Adult hippocampal neurogenesis is increasingly appreciated as a process, not a time point (Kempermann et al., 2004). It begins with precursor proliferation, progresses through neuronal differentiation and survival, and culminates in integration of neurons into hippocampal circuitry. While evidence suggests that each step of this process can be regulated, it is clear that many manipulations that alter adult hippocampal neurogenesis do so by influencing the proliferation of precursors in the subgranular zone (SGZ). This distinction between effects on proliferation and survival underscores the importance of understanding key technical aspects of how cells in the adult mouse SGZ divide. Additional information about how mouse SGZ cells divide and how long they reside in the cell cycle would allow for a more mechanistic exploration of regulation of adult neurogenesis.
The first key piece of information that is needed is the optimal dose of the exogenous marker, bromodeoxyuridine (BrdU), for labeling all cells in S phase of the cell cycle. BrdU doses given to adult mice vary from multiple injections of 50 mg/kg to a single injection of 150 mg/kg (Kempermann et al., 1998, Mandyam et al., 2004), and therefore likely label different cohorts of S phase cells. Identification of the S phase saturating dose of BrdU is a key first step in precise evaluation of the mechanisms underlying regulation of proliferation, and will foster comparison of results across laboratories. An elegant study by Cameron and McKay showed that in the adult rat, a single high dose of BrdU saturated the S phase population without causing overt damage to the labeled cells (see Tables 1 and 2 in (Cameron and McKay, 2001)). However, such information is lacking for the mouse. Given that transgenic mice are increasingly assessed for alterations in proliferation and neurogenesis, and that SGZ precursors appear to be distinct in rat versus mouse (see below), determination of which dose of BrdU is appropriate for mouse studies is needed.
A second piece of information that is needed involves the cell cycle of mouse SGZ precursors. Estimates of the cell cycle and its components in the rat and mouse SGZ are very different (rat vs. mouse: length of cell cycle, 24.7 hr vs. 12-14 hr; length of S phase, 9.5 hr vs. 7.6 hr; percent of cell cycle devoted to S phase, 38% vs. 54-63%; percent of cell cycle devoted to G2/M, 18% vs. 32-38% (Cameron and McKay, 2001,Hayes and Nowakowski, 2002)). This fundamental information has been used to explore key technical details in the adult rat SGZ, such as following the fate and kinetics of several generations of BrdU-labeled cells and their progeny after BrdU injection (Dayer et al., 2003). Such critical information is still needed for the mouse, even considering Hayes and Nowakowski’s groundbreaking work in identifying other cell cycle parameters in the adult mouse SGZ (Hayes and Nowakowski, 2002). Such technical details of the mouse SGZ precursors will help us better utilize transgenic mice that are currently available to mark cells at different stages of cell division (Sawamoto et al., 2001, Overstreet et al., 2004)), therefore allowing us to uncover cellular mechanisms in regulation of adult neurogenesis.
A final piece of information needed is how endogenous cell cycle proteins compare in their ability to provide insight into SGZ precursor proliferation. In studying adult neurogenesis, it is common to label and visualize precursors with exogenous S phase markers, such as BrdU (Miller and Nowakowski, 1988, Cameron and Gould, 1996). Alternatively, expression of endogenous cell cycle markers can be used to detect dividing precursors. Proliferating cell nuclear antigen (PCNA) and Ki-67 have long been used to assess regulation of neurogenesis in tissue where labeling with BrdU is not feasible or untenable, such as in natural populations and human post-mortem tissue (Celis et al., 1986, Bacchi and Gown, 1993, Brown et al., 2003a, Curtis et al., 2003, Dayer et al., 2003, Wharton et al., 2005, Reif et al., 2006). Many studies refer to PCNA or Ki-67 as “endogenous cell cycle markers” and use them almost interchangeably as markers of dividing cells (Kee et al., 2002, Gil et al., 2005, He et al., 2005). However, review of the literature suggests that PCNA and Ki-67 have distinct characteristics that should be considered prior to using these markers for studies of adult hippocampal neurogenesis. For example, while Ki-67 expression indicates proliferation, PCNA expression can indicate proliferation, DNA repair, or cell death (Pandey and Wang, 1995). In addition, biochemical analyses indicate the half-life of PCNA is 20 times longer than the half-life of Ki-67 (Khoshyomn et al., 1993, Karamitopoulou et al., 1994, Lopez-Girona et al., 1995). Therefore, PCNA protein expression may remain detectable either long after cell cycle exit or may be reflective of cell death, thus the use of PCNA as a marker of proliferation may overestimate the number of cells in the cell cycle. In vitro data also suggest that PCNA and Ki-67 are not equally expressed in all cell cycle phases (Gerdes et al., 1984, Celis and Celis, 1985, Takahashi and Caviness, 1993, Kawabe et al., 2002, Eisch and Mandyam, 2004). In sum, while PCNA and Ki-67 are often used to label and study the regulation and cell cycle kinetics of proliferating cells, a detailed comparison of the strengths and limitations of these endogenous markers for adult neurogenesis studies is warranted.
Here we probe for answers to these questions about SGZ precursors by qualitatively and quantitatively examining BrdU-immunoreactive (IR) cells in the adult mouse SGZ at multiple time points after BrdU. We first assess the dose of bromodeoxyuridine (BrdU) sufficient to label all S phase cells in the adult mouse SGZ, and we explore whether a higher dose of BrdU labels more cells merely because it has a longer bioavailability. We explore how long BrdU-labeled daughter cells are added to the cell cycle in the adult mouse SGZ, quantifying BrdU-IR cells and clusters as well as colabeling with endogenous cell cycle proteins. We also specifically evaluate the utility of endogenous cell cycle proteins PCNA and Ki-67 to reveal information about SGZ precursors. Finally, we compare cell cycle kinetic data gleaned from using endogenous cell cycle proteins to previous estimations of cell cycle kinetics in the embryo, in other brain regions, and in other species.
Experimental Procedures
Animals
Adult, male C57BL/6J mice (initial weight 23-27 g, 9-11 weeks old; Jackson Laboratories) were used. Mice were group housed (maximum 5/cage) in a facility approved by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC) at the University of Texas Southwestern Medical Center, with a 12:12 light:dark cycle and with free access to food and water. Mice were acclimated to vivarium conditions for at least one week prior to experimentation. All experiments conformed to guidelines of both the UT Southwestern Institutional Animal Care and Use Committee and AAALAC. The investigators took all steps to minimize the number of mice used for these experiments, and to minimize animal suffering.
BrdU injections and tissue preparation
For the BrdU dose response studies, mice received BrdU (Boehringer Mannheim, dissolved in 0.9% saline and 0.007 N NaOH; one injection of 25-500 mg/kg given in equal volumes i.p.; n = 2-4 per dose) to label dividing cells and were sacrificed 2 hrs post injection. For the BrdU time course study, mice received BrdU (one injection of 150 mg/kg i.p.; n = 4-6 for each time point) to label dividing cells and were sacrificed at various time points post-injection to examine proliferation (0.25, 0.5, 2, 8, 15, and 24 hr), differentiation (48 and 96 hr, or 2 and 4 days), and survival (240 and 720 hr, or 10 and 30 days). All mice were anesthetized with chloral hydrate prior to intracardial perfusion with chilled phophate buffered saline (1XPBS; 5 min, flow rate of 7 ml/min) and 4% paraformaldehyde in PBS (pH 7.4, 15 min). The brains were postfixed overnight at 4 °C with 4% paraformaldehyde and stored in 30% sucrose solution. Brains were cut through the hippocampus (bregma -0.82 to -4.24 (Paxinos and Franklin, 2001)) at 30 μm in the coronal plane on a freezing microtome as described previously (Eisch et al., 2000). Sections were stored at 4 °C in 0.1% NaN3 in PBS until processed for immunohistochemistry (IHC).
Antibodies
The following primary antibodies were used for IHC. Rat monoclonal anti-BrdU (cat# OBT0030; clone BU1/75-ICR1; Accurate, Westbury, NY; 1:100) was raised against BrdU. Staining was not seen in animals that did not receive BrdU, and the pattern of staining was similar to that previously reported (Eisch et al., 2000, Mandyam et al., 2004). Rabbit polyclonal anti-pHisH3 (cat# sc-8656; Santa Cruz Biotechnology, Santa Cruz, CA, 1:50) was raised against amino acid sequence containing phosphorylated Ser 10 of Histone H3 of human origin. This antiserum stains a single 20 kDa band on Western blot (Ng et al., 2004). Via IHC, the pattern of staining was similar to that previously reported (Mandyam et al., 2004), with cells presenting a diverse nuclei morphology reminiscent of pro-, meta-, ana-, and telophase. Mouse monoclonal anti-PCNA (cat# MAB4078; clone 19A2; Chemicon International, Temecula, CA; 1:4000) was raised against whole PCNA of human origin. This antiserum stains a single 36 kDa band on Western blot (Migheli et al., 1999). The pattern of staining was similar to that previously reported (Mandyam et al., 2004). Rabbit polyclonal anti-Ki-67 (cat# NCL-Ki-67p; Novocastra Laboratories, Norwell, MA; 1:500) was raised against prokaryotic recombinant fusion protein corresponding to a 1086bp Ki-67 motif-containing cDNA fragment. The pattern of Ki-67 staining was similar to that previously reported (Dayer et al., 2003) with nuclear morphology reminiscent of prophase. For single labeling colorimetric IHC for BrdU, sections were incubated with biotinylated anti-rat IgG (cat# BA4001; Vector Laboratories, Burlingame, CA; 1:200). For double labeling fluorescent IHC for BrdU/pHisH3, sections were incubated with secondary donkey anti-rat CY5 or donkey anti-rabbit CY2 IgG (Cat# 712-175-150; 711-225-152; Jackson ImmunoResearch, West Grove, PA; 1:200). For triple labeling fluorescent IHC BrdU/Ki-67/PCNA sections were incubated with secondary donkey anti-rat CY2 (Cat# 712-225-150; Jackson ImmunoResearch, West Grove, PA; 1:200), donkey anti-rabbit CY3 IgG (711-165-152; Jackson ImmunoResearch, West Grove, PA; 1:200), and HRP-conjugated anti-mouse IgG (cat# PI-2000; Vector Laboratories, Burlingame, CA; 1:400) followed by CY5 tyramide signal amplication (cat# SAT705A; PerkinElmer life sciences, Boston MA; 1:50).
Immunohistochemistry
Every ninth section through the hippocampus was slide mounted and dried overnight prior to IHC. Slides were coded prior to IHC and the code was not broken until after analysis was complete. Slide mounted sections were subjected to three pretreatment steps as described previously (Mandyam et al., 2004). Slides were incubated with 0.3% H2O2 for 30 min to remove endogenous peroxidase activity. Non-specific binding was blocked with by incubation in 3% serum and 0.3% Triton-X in 1XPBS for 30 min. Sections were then incubated with primary antibody (in 3% serum and 0.3% Tween-20) for 18-20 hr at room temperature. After washing with 1XPBS, the sections were exposed to either fluorescent- and HRP-tagged (triple labeling studies) or biotin-tagged (single labeling studies) secondary antibodies for 60 min. For single labeling, slides subsequently incubated in ABC for 1 hr (cat# PK-6100; Vector Laboratories, Burlingame, CA) and staining was visualized with diaminobenzadine (DAB; cat# 34065; Pierce Laboratories, Rockford, IL). Sections were counterstained with Fast Red (Vector). For triple labeling, the sections were simultaneously incubated with all primary antibodies (BrdU, PCNA, Ki-67 or pHisH3), followed by sequential fluorescent- (for BrdU, pHisH3, Ki-67) or HRP-tagged (for PCNA) secondary antibody staining. Tyramide signal amplification (cat# SAT704A; PerkinElmer Life Sciences, Boston, MA) was used to visualize PCNA staining, and DAPI was used as a fluorescent counterstain (Roche, Basel, Switzerland). Omission or dilution of the primary antibody resulted in lack of specific staining, thus serving as a negative control for IHC.
Microscopic analysis and quantification
After IHC, three types of analysis were performed to localize and quantify immunoreactivity. First, bright field or epifluorescent staining of the coded slides was visualized and quantified with an Olympus BX-51 microscope. Staining was examined and quantified in the subregions of the hippocampal dentate gyrus (bregma -0.82 to -4.24) as previously described (Mandyam et al., 2004, Donovan et al., 2006), including the SGZ, molecular layer (Mol), hilus (Hil) and outer granular cell layer (oGCL; Fig 1h; (Paxinos and Franklin, 2001)). The SGZ was the focus of these studies as it has been shown to be the most proliferative region in the dentate gyrus. However, the other hippocampal regions were quantified to assess the proliferative capacity of the dentate gyrus subregions and to contrast them with the more neurogenic SGZ (Eisch and Mandyam, 2004, Donovan et al., 2006). The cells in the SGZ divide in clusters (Garcia-Verdugo et al., 1998, Cameron and McKay, 2001). To gain insight into how cells divide within each cluster over time, and how clusters of cells separate into daughter clusters as they mature, we quantified BrdU-IR cell and cluster counts at each dose and each time point. DAB-stained tissue was counted for cells and clusters, with a cluster defined as one or more IR cells touching each other at 400X or 1000X magnification (Fig 2a-c). BrdU-IR cells and clusters were quantified in the dentate gyrus using the optical fractionator method in which every ninth section through the hippocampus was examined (bregma -0.82 mm to -4.24 mm; (Eisch et al., 2000, Paxinos and Franklin, 2001)). The total number of BrdU-IR cells and clusters in the layers of the dentate gyrus were multiplied by 9 and are reported as total number of clusters or cells per layer. Raw data for cluster or cell counts were subjected to statistical analysis. On the same slides, cell death was assessed by observing and quantifying BrdU-IR pyknotic cells – chromatin dense cells evident with counterstain – throughout the SGZ and the outer granule cell layer (Cameron and Gould, 1996).
Figure 1. Dose response of BrdU in C57BL/6J mice.
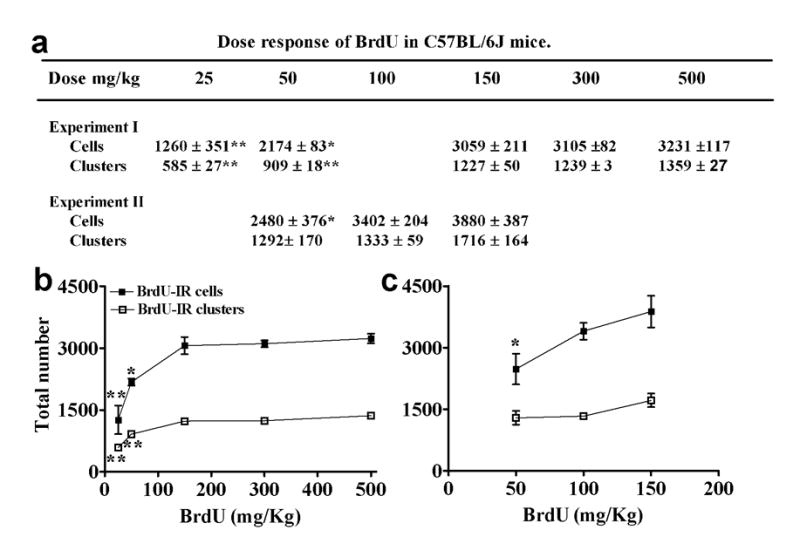
(a) Tabular representation of BrdU dose response from Experiment I (25, 50, 150, 300 and 500 mg/kg) and Experiment II (50, 100, 150 mg/kg). (b-c) Graphical representation of the total number of BrdU-IR cells or clusters in the SGZ after a single injection of BrdU 2 hr earlier from experiment I (b) and experiment II (c). n=3-4 for each BrdU dose, **p<0.01 and *p<0.05 when compared to 150 mg/kg dose, by one-way ANOVA Tukey’s post-hoc analysis.
Figure 2. Morphology and quantitative analysis of BrdU-IR cells and clusters at different time points after a single injection of BrdU.
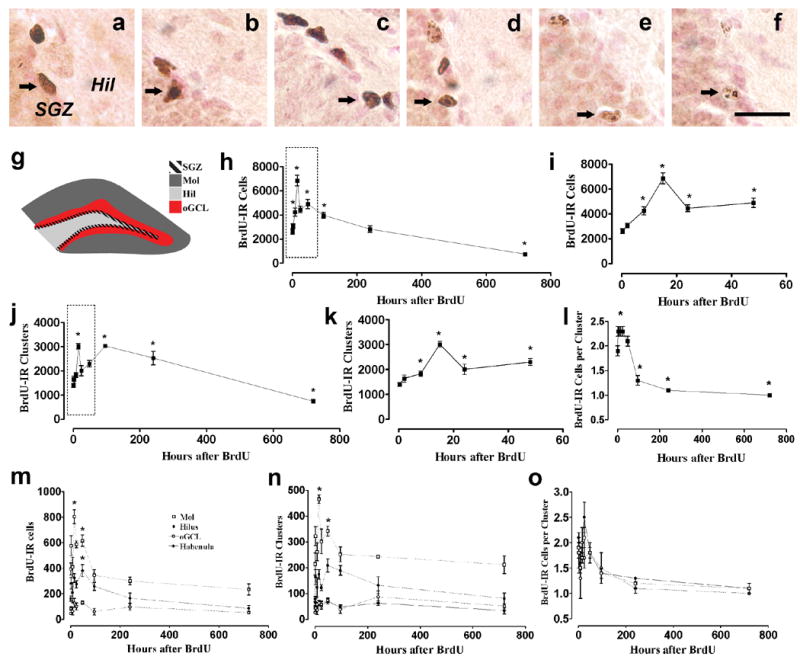
a-f: Dark brown staining of BrdU-IR cells at 2 hr (b), 8 hr (c), 15 hr (d); light brown with punctate staining of BrdU-IR cells at 96 hr (d), 240 hr (f) and 720 hr (g). Clusters of multiple BrdU-IR cells are seen in proliferation time points (b-d); clusters of single BrdU-IR cells are seen in survival time points (e-g). Arrow points to BrdU-IR cells. g: Schematic of one coronal section of mouse dentate gyrus (bregma -2.18) indicates the different regions examined: Subgranular zone (SGZ, hatched), molecular layer (Mol, dark gray), hilus (Hil, light gray) and outer granule cell layer (oGCL, red not hatched). h-l: Quantitative data of BrdU-IR cells (h-i), clusters (j-k) and cells per cluster (l) in the SGZ. All time points are significantly different from 15 hr time point in (h). All other time points except 96 hr and 240 hr are significantly different from 15 hr time point (j). m-o Quantitative data of BrdU-IR cells (m), clusters (n) and cells per cluster (o) in the Mol, Hilus and oGCL. Data is expressed as mean ± SEM. n = 4-8 animals at each time point. *p<0.05 when compared to 0.25 hr time point by one-way ANOVA, Dunnett’s post-hoc analysis. Scale bar = 10 μm in f, applies a-f.
Second, after quantification of BrdU-IR cells and clusters throughout the dentate gyrus by DAB staining, phenotypic analysis was performed on fluorescently labeled BrdU-IR cells. Using a confocal microscope (Zeiss Axiovert 200 and LSM510-META; emission wavelengths 488, 543, and 633), cells were scanned and optically sectioned in the Z plane as described previously (Mandyam et al., 2004). All analysis was done at a magnification of 630X to 1000X. Fluorescently labeled confocal images presented here were taken from one 0.45 μm optical slice and imported into Photoshop (Adobe Systems) for composition purposes. For pHisH3/BrdU analysis, all pHisH3 cells in the SGZ from each mouse (n = 4-6) at each time point post BrdU (0.25, 2, 8, 15, 24, 48, 72, 96, 240, 720 hr) were subjected to phenotypic analysis to determine colocalization of pHisH3 and BrdU. The percent of pHisH3-IR cells that were BrdU-IR were subjected to statistical analysis. For BrdU/Ki-67/PCNA analysis, fifty to sixty BrdU-IR cells in the SGZ from each mouse (n = 4-6) at each time point post BrdU (0.5, 2, 8, 15, 24, 48, 72, 96, 240, 720 hr) were subjected to phenotypic analysis to calculate the proportion of cells that were BrdU/PCNA/Ki-67, BrdU/PCNA-IR, BrdU/Ki-67-IR, or only BrdU-IR at each time point. These ratios were subjected to statistical analysis.
Third, after phenotypic analysis of BrdU-IR with Ki-67, PCNA or pHisH3, further validation of colocalization was achieved by importing stacks of Z images into a 3D reconstruction program, Volocity (Improvision). Rotation, transient modification of contrast and brightness, orthogonal analysis, and 3D rendering were performed as described earlier (Mandyam et al., 2004).
Data analysis
Data are represented as mean ± SEM, and are considered significant if p<0.05. Statistical analysis was performed with either one-way ANOVA followed by Tukey’s post hoc comparisons or Student’s t test using GraphPad Prism version 3.00 for Mac (GraphPad Software).
Results
Dose response of BrdU in adult C57BL/6J mice
To determine the relationship between BrdU dose and BrdU-IR cell counts in the adult mouse SGZ, we performed two separate dose response experiments. In the first experiment (Fig 1a, Experiment I; Fig 1b), we compared 50 and 150 mg/kg doses (Hayes and Nowakowski, 2002, Kronenberg et al., 2003, Mandyam et al., 2004) with lower (25 mg/kg) and higher (300, 500 mg/kg) doses. In the second experiment (Fig 1a, Experiment II; Fig 1c), we directly compared 50, 100 and 150 mg/kg doses. In both dose response experiments, mice were sacrificed 2 hr after a single BrdU injection and BrdU-IR cells were qualitatively examined and quantified in the SGZ.
In the first dose response experiment, qualitative analysis revealed that BrdU-IR cells in mice that received injections of 25 or 50 mg/kg BrdU were significantly lighter in staining, with punctate cells so faint that they were barely visible. No obvious differences were seen in the staining and appearance of BrdU-IR cells in mice that received injections of 150, 300 or 500 mg/kg. Quantitatively, the number of BrdU-IR cells was lowest in the 25 mg/kg mice, significantly greater in the 150 mg/kg mice, but not significantly different between 150, 300, and 500 mg/kg mice (Fig 1a, Experiment I; Fig 1b). We concluded that a dose of 150 mg/kg or higher was needed to label all the cells in – or saturate – S phase, and that less than 150 mg/kg was insufficient to label all the S phase cells in the adult mouse SGZ.
We next tested this conclusion by directly comparing 150 mg/kg BrdU with doses of BrdU commonly used in mouse neurogenesis studies: 50 and 100 mg/kg (Hildebrandt et al., 1999, Kronenberg et al., 2003, Holmberg et al., 2005, Tozuka et al., 2005). We found that 150 mg/kg labeled significantly more cells than 50 mg/kg (Fig 1a, Experiment II; Fig 1c). However, given the trend for 150 mg/kg to label more cells than 100 mg/kg, and our desire to not be on the rising portion of the labeling curve (Fig 1c), we used a BrdU dose of 150 mg/kg for all remaining studies. Taken together, this suggests that 150 mg/kg of BrdU or higher is sufficient to label all cells in S phase in the adult mouse SGZ.
One explanation for the increased number of labeled cells observed with higher doses of BrdU is that higher doses lead to longer bioavailability of BrdU relative to the lower doses. To address this, mice were given 150 mg/kg BrdU and sacrificed 0.25 or 2 hr later. We found no significant difference in the number of BrdU-IR cells between 0.25 or 2 hr (mean ± s.e.m: 0.25 hr, 2616 ± 204; 2 hr, 3069 ± 185; p = 0.15 by unpaired t test). This confirms that 150 mg/kg labels all actively cycling cells in S phase in the adult SGZ, and that this effect is not due to longer bioavailability of this 150 mg/kg dose. Furthermore, the lack of significant difference in BrdU-IR cell counts between 0.25 and 2 hr suggests that BrdU bioavailability is under 0.25 hr (15 min) in adult mouse SGZ.
BrdU-IR cells and clusters in adult mouse SGZ
Proliferating cells in the adult SGZ divide in clusters (Cameron and McKay, 2001, Mandyam et al., 2004, Kahn et al., 2005). To determine the rate at which cells multiply in the adult mouse SGZ, and to determine key aspects of cell and cluster division, we injected adult mice with an S phase-saturating dose of BrdU (150 mg/kg). BrdU-IR cells and clusters in the SGZ and other dentate gyrus regions were qualitatively examined and quantified over a time course from 0.25 hr to 720 hr (15 min to 30 days).
In keeping with previous descriptions of BrdU-IR cells in the adult mouse SGZ (Hayes and Nowakowski, 2002, Mandyam et al., 2004), qualitative analysis at proliferation time points (0.25, 2, 8, and 15 hr) revealed elongated BrdU-IR cells presenting dark staining throughout the nucleus (Fig 2a-c). Cells were often in clusters, with the densest clusters evident at 8-15 hr after BrdU (Fig 2c). After 96 hr, a mixed population of dark and light stained cells was visible, with lighter cells presenting an oval nuclear morphology and often appearing as single cells (Fig 2d). At later survival time points (240 and 720 hr), the intensity of staining decreased and cells uniformly appeared oval or round in shape and as single cells (Fig 2d-f).
Based on this qualitative assessment of how cells divide in clusters and appear to mature into single cells, we next quantified BrdU-IR cells and clusters in the SGZ and dentate gyrus regions to gain more insight into the dynamic nature of precursor division. As stated above, the number of BrdU-IR cells in the SGZ did not increase between 0.25 and 2 hr (Fig 2h-i). BrdU-IR cells between the 0.25 and 2 hr time point at the SGZ have moved from the S phase where they were labeled into the next phases of the cell cycle, Gap 2 (G2) and mitosis (M). Since we saw no no increase in cell number between 0.25 and 2 hr, we can assume that it takes longer than 2 hr for a BrdU-IR cell in the mouse SGZ to reach M. Previously, G2/M phase of the mouse SGZ cell cycle was assumed to be approximately 4.5 hr based on the first appearance of BrdU-IR mitotic figures 4.5 hr after BrdU injection (Hayes and Nowakowski, 2002). Our quantitative data presented here agrees with this previous qualitative observation that the length of G2/M is greater than 2 hr.
Compared to 0.25 hr, the number of BrdU-IR cells increased at 8 hr (the duration of one half of the cell cycle of mouse SGZ precursors (Hayes and Nowakowski, 2002); Fig 2h), suggesting that some of the BrdU-IR cells had moved into and through M phase. The number of BrdU-IR cells doubled at 15 hr (the duration of one cell cycle; Fig 2i), suggesting that each BrdU-IR cell had been through M phase, and therefore a minimum of one cell cycle by 15 hr. After 15 hr, the number of BrdU-IR cells did not increase, and the number of BrdU-IR cells decreased from 24 hr until 720 hr (Fig 2h-i). The first appearance of BrdU-IR pyknotic cells was at 24 hr, suggesting death of a portion of BrdU-IR cells. However, the number of BrdU-IR pyknotic was extremely low at the 24 hr and later timepoints and the numbers were insufficient for statistical analysis. Therefore, our observation of pyknotic/BrdU-IR cells should be considered descriptive, and not quantitative.
In regards to clusters, the increase in the number of BrdU-IR SGZ clusters at 8 hr and the doubling at 15 hr (compare Fig 2h-i to Fig 2j-k) indicate similar kinetics of BrdU-IR clusters and cells during these early time points after BrdU labeling. A second peak of BrdU-IR cluster number occurred at 96 hr. This suggests that at 96 hr after BrdU injection – four to six cell division cycles after BrdU incorporation (Hayes and Nowakowski, 2002) – cells within a cluster are separating into individual BrdU-IR cells. The gradual decrease between 96 and 240 hr suggests that independent clusters start to disappear – likely due to BrdU dilution or cell death - and that one or both of these mechanisms continue until 720 hr.
Combining the cell and cluster data, we found that the number of cells per cluster increased between 0.25 and 8 hr, underscoring our impression of rapid cell division in clusters, and remained level until 48 hr (Fig 2l). There was a 50% decrease in the cells per cluster at 96 hr compared to 48 hr, and single cells (one BrdU-IR cell per cluster) were evident by 240 and 720 hr (Fig 2e, f and l). The cell per cluster data therefore supports the hypothesis that by 96 hr after BrdU incorporation, individual cells begin to break out of the parent cluster, presumably to differentiate and migrate into the granule cell layer.
BrdU-IR cells and clusters in other dentate gyrus regions
In the same tissue in which the SGZ was analyzed, BrdU-IR cells and clusters in other hippocampal regions were quantified to contrast the proliferative capacity of the molecular layer, hilus and outer granule cell layer (see Fig 2g, Fig 2m-o) with the more neurogenic SGZ (Eisch and Mandyam, 2004, Mandyam et al., 2004). The time course of the number of BrdU-IR cells in the molecular layer did not show an initial increase at 8 hr, but approximately doubled at 15 hr, revealing cell division kinetics similar to the SGZ. The number of cells declined after 15 hr until 720 hr, building on our assumption that cells in the molecular layer and SGZ have similar kinetics of cell birth, cell cycle exit and cell death. The time course of the number of BrdU-IR cells in the hilus did not show an initial increase in the number of cells at 8 hr but doubled 48 hr after BrdU (Fig 2m), implying dissimilar hilar cell cycle kinetics from cells in the SGZ and the molecular layer. The number of BrdU-IR cells in the outer granule cell layer did not double (Fig 2m). In addition of BrdU-IR cell analysis, we further quantified BrdU-IR clusters in the molecular layer, hilus, and outer granule cell layer to highlight changes that may be associated with differences in the proliferative capacities of these dentate gyrus subregions. In the molecular layer, the number of clusters approximately doubled at 15 hr (Fig 2n). Surprisingly, the time course of the number of BrdU-IR clusters in the molecular layer did not show a second peak at 96 hr, suggesting that migration of cells within a cluster in the molecular layer occurs at a different pace than in the SGZ. The time course of the number of BrdU-IR clusters in the hilus and the outer granule cell layer did not reveal any migration pattern of cells from clusters in these regions (Fig 2n). Overall we show that cells in the dentate gyrus subregions are born and multiply in clusters, but that the time course of these divisions is distinct from those that occur in the more proproliferative environment of the SGZ.
Double labeling of BrdU-IR cells with pHisH3 in the adult mouse SGZ
We next restricted our focus to SGZ precursors in order to build on previous studies on the cell cycle of these cells (Cameron and McKay, 2001, Hayes and Nowakowski, 2002). To determine the approximate cell cycle phase of BrdU-IR cells throughout the time course (0.25 to 720 hr), sections from the BrdU time course study (Fig 2) were double labeled with antibodies against BrdU and pHisH3, a marker for G2/M of the cell cycle (Fig 3a-i) (Mandyam et al., 2004). The specificity of pHisH3 for G2/M is due to the selective phosphorylation of HisH3 on the serine 10 residue in G2 and M (Hendzel et al., 1997). Every pHisH3-IR cell was analyzed from each animal at every time point (average of 26.8 ± 4.9 pHisH3 cells from each animal; n = 4-6 animals for each time point) for colocalization with BrdU (Fig 3).
Figure 3. BrdU-IR cells express pHisH3 during G2/M phase of the cell division cycle.
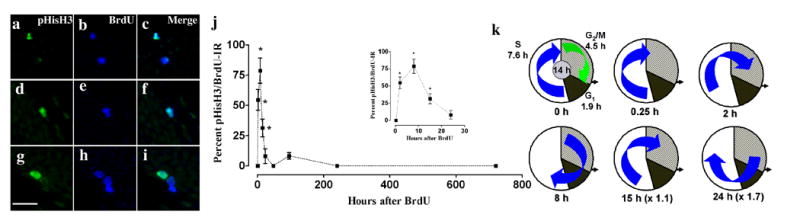
(a-i) Confocal single z plane images of proliferating cells labeled with pHisH3 in green (a, d, g; CY2) and BrdU in blue (b, e, h; CY5). Double labeling of BrdU and pHisH3 (a-c): 2 hr after single BrdU injection, (d-f): 8 hr after single BrdU injection, (g-h): 15 hr after single BrdU injection. j: The proportion of pHisH3-IR cells that are BrdU positive in the SGZ. Every pHisH3-IR cell was analyzed from each animal at every time point (average of 26.8 ± 4.9 pHisH3 cells from each animal; n = 4-6 animals for each time point). Inset in j shows extended x axis between 0 and 30 hr. k: Pie chart indicating the cell cycle of proliferating cells of adult mouse SGZ; S phase (white), G2/M (gray/hatched) and G1 (black). The ratio of the different phases in the pie chart is proportional to the time line of each phase: total cell cycle 14 hr; S 7.6 hr; G2/M 4.5 hr; and G1 1.9 hr (Hayes and Nowakowski, 2002). The position of the blue arrow is indicative of the position of BrdU-IR cells (cells in S phase at time of BrdU injection) at the time point indicated below each pie chart. The percent of BrdU-IR cells in G2/M (blue arrow in pie chart) approximately correlates with the percent of BrdU-IR cells labeled with pHisH3 (see j). The small differences between the actual (x-y graph in j) vs. theoretical (pie chart in k) data may be due to the small number of pHisH3-IR cells available for analysis. The number in parenthesis after the time point below each pie chart suggests the approximate number of cell cycles that have occurred since BrdU incorporation. Data is expressed as mean ± SEM. *p<0.05 when compared to the 0.25 hr time point. Scale bar = 10 μm, applies a-i.
We assessed the percentage of pHisH3-IR SGZ cells that were double-labeled with BrdU (Fig 3j), and we applied this quantitative data to the model of the mouse SGZ precursor cell cycle previously put forth (Hayes and Nowakowski, 2002). We wondered if our data would allow us to fine tune this model in which the length of the cell cycle was estimated to be 12-14 hr, with S phase occupying 7.6 hr and G2/M occupying 4.5 hr (Fig 3k). The percent labeling indicated in Fig 3j approximately correlates with the percent of blue arrow overlapping the G2/M phase at each time point in Fig 3k.
Our analysis revealed that no pHisH3 cells in the SGZ were double-labeled with BrdU at 0.25 hr (Fig 3j ). This suggests that pHisH3 is not expressed in the first 15 minutes of G2/M phase (Fig 3k). The percent of all pHisH3-IR cells that were BrdU-IR was maximal at 8 hr (78.8 ± 10.4%; Fig 3d-f and j). This high percentage is consistent with the large number of S phase cells that will have moved into G2/M phase 8 hr after BrdU injection (Fig 3k). The percent of all pHisH3-IR cells that were BrdU-IR dropped to 8% 24 hr (1 day) after BrdU injection. This lower percentage is expected based on previous models of cell cycle length and the assumption that the some of the initial BrdU-labeled S phase cohort will have exited the cell cycle 24 hr after BrdU injection (Fig 3k). There was a second, non-significant peak at 96 hr compared to the 48 and 240 hr, suggesting that there were even fewer BrdU-IR cells cycling at 96 hr than at 24 hr. Considering Fig 3j and Fig 3k together, two main conclusions can be made. First, S phase is greater than 7 hr, underscoring previous estimations of S phase of the mouse SGZ precursors (Fig 4k; (Hayes and Nowakowski, 2002, Burns and Kuan, 2005)). Second, the length of the cell cycle in SGZ precursors is 14 hr, fine tuning the previous estimation of 12-14 hr (Hayes and Nowakowski, 2002).
Figure 4. Distribution and quantitative analysis of Ki-67 and PCNA-IR clusters in the dentate gyrus of adult mouse brain.
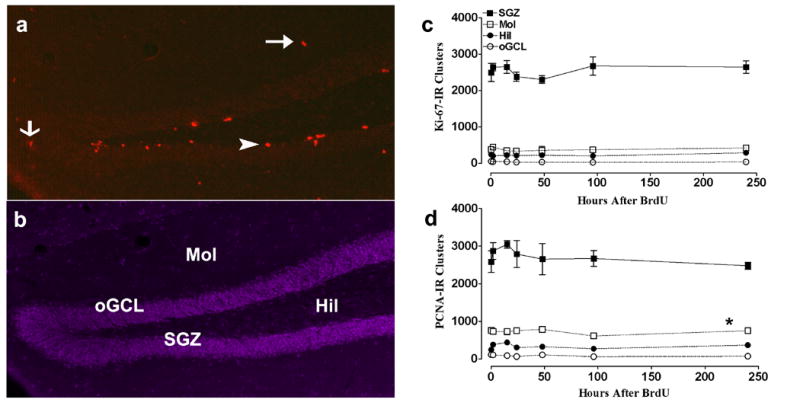
a:Representative coronal section of mouse dentate gyrus stained for PCNA; b: stained for DAPI nuclear staining (200X; bregma - 2.18) indicates the different regions examined: Subgranular zone (SGZ), molecular layer (Mol), hilus (Hil) and outer granule cell layer (oGCL). c: Quantitative data of Ki-67-IR clusters in the dentate gyrus. n = 6-8 animals at each time point. d: Quantitative data of PCNA-IR clusters in the dentate gyrus. n = 4-8 animals at each time point. Arrowhead points to a cell in SGZ, thin arrow points to a cell in oGCL and thick arrow points to a cell in the Mol. Data are expressed as mean ± SEM; *p < 0.05 when compared to Ki-67 Mol clusters by one-way ANOVA Bonferroni’s Multiple Comparison Test.
Comparison of endogenous markers Ki-67 and PCNA in adult mouse SGZ
A steady population of actively dividing precursors exists in the adult SGZ (Cameron and McKay, 2001). However, the steady state of this population has not been addressed with endogenous cell cycle proteins, like Ki-67 or PCNA (Kee et al., 2002, Mandyam et al., 2004), which are both expressed in many phases of the cell cycle in vitro (Celis et al., 1984, Gerdes et al., 1984). In addition, recent work suggests that the circadian cycle can influence hippocampal neurogenesis (Holmes et al., 2004, Kochman et al., 2006). Given that mice for this time course study were injected with BrdU at similar times of day but were sacrificed at different times during the circadian cycle, analysis of the precursor expression of Ki-67 or PCNA at these different sacrifice times would reveal if our results or our interpretation of the results were influenced by the circadian cycle.
We performed Ki-67 and PCNA IHC on hippocampal sections used for the BrdU time course study (Fig 2). This allowed us to quantify the total number of proliferating cells at any given time point. Ki-67 and PCNA-IR cells were seen in the SGZ, Mol, Hil and oGCL as clusters with multiple cells per cluster (Fig 4a-d). Here we report the number of Ki-67 and PCNA-IR clusters across time, but the time course is the same for the number of cells. There were approximately 2700 Ki-67 or PCNA-IR clusters in the SGZ at any given time point, indicating a steady proliferative environment (Fig 4c, d). The number of Ki-67 or PCNA-IR clusters in the subregions of the dentate gyrus (molecular layer, hilus, and outer granule cell layer) were significantly lower than the number of clusters in the SGZ (SGZ: 2673 ± 93; Mol: 731 ± 21; Hil: 335 ± 25; oGCL: 90 ± 8; p<0.01; Fig 4c, d), underscoring that the SGZ is the most proliferative region in the dentate gyrus. Overall there was no significant difference in the number of Ki-67 or PCNA-IR clusters in the SGZ or dentate gyrus subregions over the time course (Fig 4c, d). Interestingly, there were fewer Ki-67-IR clusters in the molecular layer when compared to PCNA (380 ± 14 Ki-67-IR clusters vs. 731 ± 21 PCNA-IR clusters; p<0.05; Fig 4c vs. 4d Mol), suggesting differential labeling pattern of the endogenous markers in the molecular layer subregion of the dentate gyrus. These data indicate that a population of precursors in the SGZ and dentate gyrus subregions can be quantified by endogenous markers, and that the size of this population does not fluctuate over the circadian cycle.
Co-labeling of BrdU-IR cells with PCNA and Ki-67 in the adult mouse SGZ
One way to determine how long after BrdU injection BrdU-labeled progeny continue to cycle is to see how long BrdU-labeled progeny continue to express endogenous cell cycle proteins. To this end, sections from the time course study were triple labeled with antibodies against BrdU and two endogenous cell cycle proteins, PCNA and Ki-67. In vitro data suggests a 20-fold longer half-life of PCNA relative to Ki-67 (Khoshyomn et al., 1993, Karamitopoulou et al., 1994, Lopez-Girona et al., 1995), but this difference has never been shown in situ. We hoped that triple labeling of BrdU/PCNA/Ki-67 would (a) reveal how long daughter cells were added after BrdU incorporation, based on the presence of PCNA or Ki-67 and in comparison with the BrdU cell number data shown in Fig 1 and (b) allow direct comparison of the utility of PCNA and Ki-67 and rough assessment of their in situ half-lives.
PCNA- and Ki-67-IR cells were distributed similarly in the dentate gyrus, with both antibodies frequently labeling the same cell (Fig 5a-l). The portion of cells triple labeled for BrdU/Ki-67/PCNA-IR cells was very high 0.25 hr after BrdU injection, and then decreased from 0.25 to 720 hr, with a corresponding increase in the portion of BrdU-IR cells negative for PCNA and Ki-67 (Fig 5m). This supports that a steady portion of BrdU-IR progeny exit the cell cycle asynchronously over time, presumably to begin the progression towards neuronal maturity (Brown et al., 2003b). Fig 5m also shows that the portion of BrdU-IR progeny that are BrdU/PCNA-IR is still evident 240 hr after BrdU, well after the disappearance of BrdU/Ki-67/PCNA-IR cells. This suggests that Ki-67 and PCNA proteins may have different biological half-lives, with PCNA protein expression persisting long after cell cycle exit. Further insight into this possibility was gained by comparing the time course of BrdU/Ki-67-IR cells vs. BrdU/PCNA-IR cells (Fig 5n). The percent of BrdU-IR SGZ cells that were Ki-67-IR was very high at 0.25 hr and then progressively decreased: 97.9 ± 1.3% at 0.25 hr, 6 ± 3.7% at 96 hr, and 0% at 240 hr. Notably, the percent of BrdU-IR cells that were Ki-67-IR was decreased by 50% at 15 hr (Fig 5n). The percent of BrdU-IR SGZ cells that were PCNA-IR also was very high initially: 100% at 0.25 hr (Fig 5n). However, the rate of decline was sharply different: 100% at 0.25, 2, 8 and 15 hr, gradually decreased to 33.5 ± 8.3% at 96 hr, and did not reach 0% until 720 hr. Emphasizing this significant and dramatic difference in BrdU/Ki-67 vs. BrdU/PCNA populations, the percent of BrdU/PCNA-IR cells declined at statistically different rate than the percent of BrdU/Ki-67-IR cells, with 50% reduction evident at approximately 17.42 hr for Ki-67 and 85.08 hr for PCNA (decay constants via one phase exponential decay: Ki-67 = 0.039 ± 0.005; PCNA = 0.008 ± 0.001; p<0.0001 by unpaired t test; Fig 5n). This rightward shift of PCNA labeling supports its much longer half-life relative to Ki-67, and may indicate that PCNA protein is detectable in BrdU-IR daughter cells long after terminal exit from the cell cycle. In addition to indicating a striking difference in the characteristics of Ki-67 vs. PCNA, these findings suggest that at any given time point, the population of BrdU-IR cells in the SGZ is asynchronously cycling (Ormerod, 2004), with BrdU-IR daughter cells exiting the cell cycle at different times, presumably on their way to their mature phenotype (Dayer et al., 2003).
Figure 5. Comparison of endogenous markers Ki-67 and PCNA with exogenous marker BrdU over a BrdU time course.
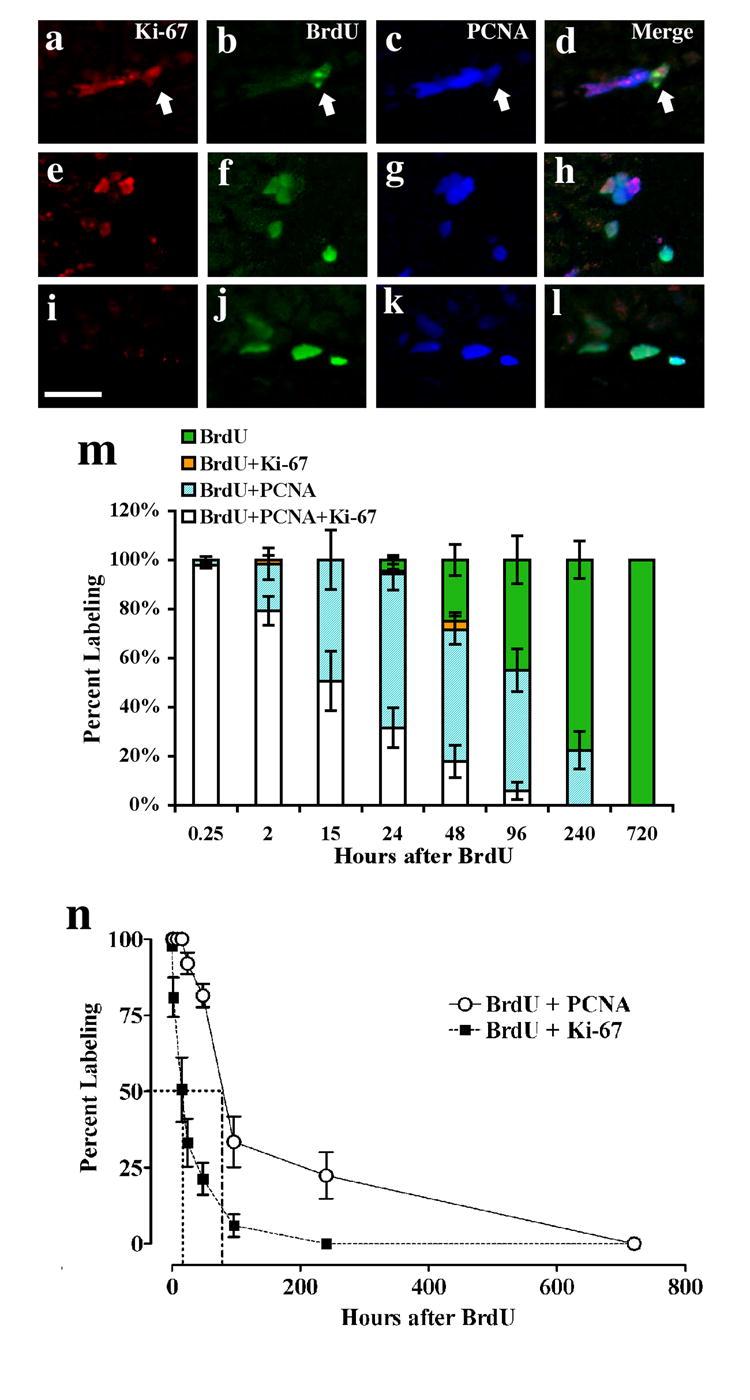
(a-l): Confocal single z plane images of proliferating cells labeled with Ki-67 in red (a, e, i; CY3), BrdU in green (b, f, j; CY3), and PCNA in blue (c, g, k; CY5). Triple labeling of BrdU with Ki-67 and PCNA (a-d): 2 hr after single BrdU injection, (e-h): 15 hr after single BrdU injection, (i-l): 96 hr after single BrdU injection. m: Stacked bar graph indicating ratios of BrdU labeling; white stacked bar- triple labeled BrdU/PCNA/Ki-67-IR cells, orange stacked bar- double labeled BrdU/Ki-67-IR cells, aqua stacked bar- double labeled BrdU/PCNA-IR cells, and green stacked bar- single labeled BrdU-IR cells. 50 to 60 BrdU-IR cells in the SGZ from each mouse were randomly selected for confocal analysis. The ratios of cells are indicated as a percent of 100 and the total sum up to 100% for each group. n: x-y graph of the ratios of BrdU/PCNA and BrdU/Ki-6-IR. The BrdU/PCNA curve is shifted rightward compared to BrdU/Ki-67, the 50% labeling is 15 hr for BrdU/Ki-67 and 80 hr for BrdU/PCNA. n = 4-6 animals for each time point. Data is expressed as mean ± SEM. Scale bar in i = 10 μm, applies a-l.
Discussion
Key aspects of precursor cell division from the adult mouse SGZ are revealed in this study. We identify a dose of BrdU sufficient to saturate the S phase cohort of SGZ precursors in the adult mouse. Using a saturating dose of BrdU, we highlight the tendency of SGZ cells and other proliferative subregions of the dentate gyrus to divide in clusters. Our time course analysis shows the number of cells, clusters, and cells per cluster increasing and then decreasing from 0.25 to 720 hr after BrdU labeling, offering a glimpse into the dynamics of cells dividing in clusters. Our BrdU and pHisH3 analysis confirms that dividing SGZ precursors transiently express pHisH3 during G2/M phase, allows us to fine tune the previous estimate of the cell cycle length to 14 hr, and reveals that that BrdU-IR progeny are detectable up to 96 hr (6-7 divisions) after BrdU labeling. Finally, our BrdU, Ki-67 and PCNA analysis suggests that these two endogenous markers of proliferation have discrete in vivo half-lives, with PCNA likely being detectable long after cell cycle exit.
Below we discuss these data in more detail and underscore the potential of this information to enhance our understanding of the regulation of adult neurogenesis. However, we first must address an assumption underlying this work, and indeed, underlying most adult neurogenesis work that utilizes BrdU: BrdU labels a relatively homogeneous population of SGZ precursors. Initially this assumption may seem incorrect given that multiple populations of proliferating cells exist in the adult hippocampal SGZ (Alvarez-Buylla and Lim, 2004, Kempermann et al., 2004, Abrous et al., 2005), and perhaps each ‘type’ of dividing cell (e.g. stem vs. transient amplifying progenitor) has different cell cycles, or is labeled at different doses of BrdU. However, available data suggests that stem cells (type 1) account for less than 3% of BrdU-IR cells when examined 2 hr after injection (Kronenberg et al., 2003). This emphasizes that a single injection of BrdU primarily labels transient amplifying cells (type 2). Of course, further work is needed to assess if type 2a vs. type 2b cells have similar cell cycles or sensitivities to BrdU labeling. Since the vast majority (85%) of our BrdU-IR cells at 2 hr are presumably type 2a cells (Kronenberg et al., 2003), however, it is reasonable to proceed on the assumption that our data target a relatively homogeneous population of transient amplifying progenitor cells, and therefore are valid to apply to adult mouse SGZ precursors.
Optimal dose of BrdU
Many studies of adult neurogenesis utilize multiple injections of BrdU, spaced by hours or days, in order to label a large number of dividing cells. Certain circumstances, such as cell cycle analysis, require multiple injections of BrdU (Cameron and McKay, 2001, Hayes and Nowakowski, 2002). However, multiple injections of BrdU will result in BrdU incorporation in more regions of the newly synthesized DNA than single injections, and therefore are accompanied by toxicity concerns (Kolb et al., 1999). In addition, data resulting from studies that utilize multiple injections of BrdU are difficult (if not impossible) to interpret due to the mixed “age” of the labeled cells, or how much time has passed since their terminal exit from the cell cycle (Eisch, 2002). A key variability in these experiments is also the dose employed of BrdU. Notably, recent experiments in rat have shown that use of a dose of BrdU that does not label all S phase cells can lead to false conclusions (Eadie et al., 2005). Clearly, identification of a dose of BrdU that saturates, or labels, all S phase cells will facilitate data interpretation, prevent false conclusions, and allow comparison of results across different research groups. While a saturating dose of BrdU is known for the rat (Cameron and McKay, 2001, Eadie et al., 2005), rat and mouse differ in key factors that may influence BrdU incorporation, such as permeability of the blood brain barrier (Yuan et al., 1994). Therefore it is important that the saturating dose of BrdU is specifically determined in the mouse.
We gave adult mice a range of BrdU doses and counted BrdU-IR cells prior to their reaching mitosis ((Hayes and Nowakowski, 2002); Fig 3). This timing was critical in order to examine the S phase cohort without the confounding influence of mitotic cell division presumed to occur approximately 4.5 hr after BrdU labeling. We found the optimal dose to label all S phase cells in the adult mouse SGZ is equal to or greater than 150 mg/kg of BrdU (Fig 1). While this is less than the range of BrdU doses shown to saturate rat SGZ precursors (Cameron and McKay, 2001, Eadie et al., 2005), it is close enough to suggest that rat and mouse require a similar dose of BrdU to saturate S phase cells. In order to prevent the underestimation of S phase cohort in the mouse, especially in experiments involving treatment groups (regulation of proliferation by exercise, drugs of abuse, etc.), we recommend a BrdU dose of 150 mg/kg. While we did not find that 150 mg/kg of BrdU labeled statistically more cells than 100 mg/kg (Fig 1c), we prefer 150 mg/kg since, unlike 100 mg/kg, it does not appear to be on the rising aspect of the dose response curve.
Bioavailability of BrdU
Key aspects of division (e.g. length of S phase, length of cell cycle) are known to change with maturation of the animal (Alexiades and Cepko, 1996) and even between distinct brain regions (Morshead and van der Kooy, 1992, Hayes and Nowakowski, 2002). In addition, other factors that influence BrdU half-life, such as permeability of the blood brain barrier and ability to metabolize BrdU, likely also change with maturation or between brain regions (Sharma et al., 1995, Gould and Gross, 2002). Using a dose of BrdU that labels all S phase cells in the adult mouse SGZ, we find evidence that BrdU availability for incorporation into DNA is less than 15 min. This estimate is less than reported for in the developing mouse brain (Packard et al., 1973, Hayes and Nowakowski, 2000), where 27-38 minutes is needed to label S phase cells. However, this difference is not surprising given characteristics in adult vs. prenatal/early postnatal brain that may influence BrdU metabolism and clearance (Rich and Boobis, 1997, Bauer and Bauer, 2000). While precise evaluation of BrdU bioavailability is outside the scope of this study, our estimate of 15 min may serve as a reasonable starting point for future studies that perform a more directed and thorough analysis of BrdU bioavailability in adult mouse brain (e.g. using brain and serum levels of BrdU along with BrdU-IR cell counts).
Time course of cell division in the adult mouse dentate gyrus
We performed a quantitative analysis of BrdU-IR cells over a thirty-day period to determine the time course of cell division in the adult mouse SGZ. In order to ensure complete labeling of S phase cells at a given time point and to track the cell counts of cells as they mature, mice were given one injection of a saturating dose of BrdU and sacrificed at various time points 0.25 to 720 hr (Fig 2). Cell counts did not change between 0.25 and 2 hr, suggesting that cells had not undergone mitosis after 2 hr, and that G2/M phase is greater than 2 hr. An alternative explanation for a lack of increase between 0.25 and 2 hr that must be considered is cell death (Biebl et al., 2000, Dayer et al., 2003, Heine et al., 2004). This is unlikely for two reasons. First, a length of G2/M greater than 2 hr is consistent with previous estimates in the mouse SGZ (4.5 hr, (Hayes and Nowakowski, 2002)). Second, BrdU-IR cells increase 1.3 fold between 2 to 8 hr and 2.2 fold between 2 and 15 hr, emphasizing that they all passed through mitosis at the expected time (Hayes and Nowakowski, 2002). If cells were dying due to the 150 mg/kg dose, it is unlikely that this doubling from 2 to 15 hr would occur. Indeed, the 2.2 fold increase in the number of cells from 2 hr to 15 hr reflects the completion of one entire cell cycle and mitosis of all cells that were in S phase at the time of injection (for more discussion, see (Cameron and McKay, 2001)). These results fit well with the steady-state cell cycle model previously reported by Hayes and Nowakowski in the adult mouse SGZ (Hayes and Nowakowski, 2002), where the cell cycle is estimated to be 12-14 hr. Notably, the doubling of the cells after the first mitosis strongly suggests that this higher, saturating dose of BrdU is not toxic to dividing cells.
After all BrdU-labeled cells have gone through mitosis, the number of cells decreased 1.5 fold from 15 hr to 24 hr. Since the cells have already gone through mitosis, this loss is unlikely to be due to dilution of BrdU label, but rather to cell death. While the number of BrdU-IR pyknotic (and presumably dying) cells per section were too low to quantify in this study, the earliest time point we found BrdU-IR pyknotic cells was 24 hr after BrdU, similar to in the rat (Dayer et al., 2003). However, while in the rat SGZ the first indirect evidence of cell death is during the first mitosis after BrdU injection (Dayer et al., 2003), in the mouse SGZ the evidence of cell death at 24 hr is between the first and second mitosis. It is possible that use of a saturating dose of BrdU, as used here for the mouse and previously in the rat (Dayer et al., 2003) – while important to gauge cell division prior to mitosis – might artificially induce cell death after mitosis. However, three lines of evidence suggest that this is natural, and not BrdU-induced, cell death. First, BrdU-IR cell counts did not decrease between 24 to 96 hr, as might be expected if BrdU were toxic. Second, combinatorial data with antibodies against several endogenous cell cycle proteins (discussed below) show that precursors were actively dividing at 24, 48 and 96 hr, which would be unexpected if the cells were dying. Third, Dayer et al. saw no enhanced cell loss as they doubled the dose of BrdU (Dayer et al., 2003). Steady loss of cells continued to occur between 96 hr and 720 hr, suggesting that precursor cell death, maturity or exit from the cell cycle contributed to the loss of cells by thirty days after BrdU.
In addition to addressing the time course of cell division in the SGZ of adult mouse, we have quantified precursors over a similar time course in dentate gyrus subregions that have been reported to contribute to hippocampal plasticity and have neurogenic environments (Amaral and Witter, 1995, Wang et al., 2000, Kempermann et al., 2003, Doetsch and Hen, 2005). The dentate gyrus subregions – molecular layer, hilus, and outer granule cell layer – have been examined previously (Mandyam et al., 2004), and have proved useful in solving controversies in regulation of hippocampal neurogenesis (Donovan et al., 2006). Region specific quantitative analysis indicated that precursors in the molecular layer have similar kinetics to those in the SGZ, and that the precursors in hilus and outer granule cell layer are distinct from the cells in SGZ and the molecular layer (Fig 2). While it is unclear what these findings mean at this early stage in the appreciation of dividing cells in other dentate gyrus subregions, in the future the distinct cell cycle kinetics of dentate gyrus subregions may be useful to better understand regulation of hippocampal cytogenesis in these regions, for example, as seen after seizures or in other pathological conditions (Hellsten et al., 2005, Van der Borght et al., 2005, Donovan et al., 2006).
Time course of cluster and cells per cluster counts in the SGZ
Cells are known to divide in clusters, presumably due to their association with the vasculature niche (Palmer, 2002). A few reports have appreciated the difference and relationship between cells and clusters in the rat (Cameron and McKay, 2001, Kahn et al., 2005, Kim et al., 2005) and mouse (Mandyam et al., 2004, Donovan et al., 2006), and one found differential regulation of cells versus clusters (Kahn et al., 2005). Given that the vasculature niche appears critical to the stability and regulation of the clusters of dividing cells (Palmer et al., 2000, Ernst and Christie, 2006), it is surprising that clusters have received relatively little attention. Since clusters may be dynamic in nature, clearly the optimal way to explore how precursors divide in clusters is in thicker sections that maintain the three-dimensional structure of a cluster, and/or with time-lapse microscopy in vitro, as has been used to study developmental neurogenesis (Gal et al., 2006). Technical limitations with such studies preclude immediate application of such an analysis in the mouse SGZ. As a reasonable alternative, we examine the number of cells, clusters, and cells per cluster in the SGZ and adjacent dentate gyrus subregions after a single BrdU injection. This approach has the benefit of low variability between animals (Fig 2), and it has several benefits over comparable in vitro approaches, including the ability to examine cells up to 30 days after a single BrdU injection and to retain the in vivo microenvironment so critical to regulation of adult neurogenesis (Abrous et al., 2005).
Our data suggest that the BrdU time course approach can provide insight into how cells divide in clusters and mature over time. Here we define a BrdU-IR “cluster” as one or more IR cells touching each other at high magnification (Mandyam et al., 2004). At a short, or proliferating, time point after BrdU, BrdU-IR clusters consisted of more than one IR cell, and at survival time points BrdU-IR clusters had only one IR cell. In general, the dynamic pattern of clusters quantified here supports previous qualitatitive data that precursor cells appear as clusters of cells during proliferation and as individual cells at later, survival time points. However, the analysis of the number of BrdU-IR clusters provided additional detail not evident from examining the number of BrdU-IR cells alone. For example, at a time point when the number of BrdU-IR cells was relatively steady (24-96 hr), the number of BrdU-IR clusters increased. One interpretation of these data is that the cells in a given cluster move apart between 24 and 96 hr, an interpretation that is supported when the data are viewed as number of cells per cluster. In fact, since 96 hr appears to be a time point when SGZ precursors no longer appear as clusters, analysis of this time point may be useful in exploring the mechanism underlying regulation of precursor proliferation. For example, additional studies might assay BrdU-IR cells at 96 hr post BrdU for altered levels of key cell-cell adhesion proteins, like EphB2 (Conover et al., 2000) and postmitotic proteins like SOX13 (Wang et al., 2005). In general, the analysis of cells per cluster detailed here for the adult mouse SGZ provides a new tool in the exploration of regulation of adult neurogenesis and mechanisms underlying that regulation.
Endogenous cell cycle markers: stability and half-life of protein
The hippocampal SGZ has a steady output of precursors providing a asynchronous population of cells at any given time point (Seri et al., 2004). If given as a saturating dose, BrdU putatively labels all transient amplifying cells in S phase. Since cells dilute BrdU with each subsequent division, as a result of dilution, BrdU-labeled cells are undetectable by IHC after a few cell divisions (Cameron and McKay, 2001, Dayer et al., 2003). The dilution of exogenous labeling impedes detection of BrdU-IR cells and therefore may hinder precise counts of the precursor population. Utilizing endogenous markers, such as pHisH3, Ki-67, or PCNA, in combination with BrdU to quantify proliferating population partially solves the dilution problem associated with exogenous markers. However, it is unclear how stable endogenous cell cycle proteins are in vivo. This is important given the increasing number of publications that rely on endogenous markers to quantify and assess regulation of cell proliferation.
To determine the stability of endogenous protein labeling in vivo and to find out how long BrdU-IR daughter cells are added to the proliferating population, we labeled BrdU-IR cells with endogenous cell cycle markers for proliferation (pHisH3, Ki-67 and PCNA). The pHisH3/BrdU percent labeling in the SGZ showed transient expression of pHisH3 in BrdU-IR cells at the G2/M phase of the cell cycle. Qualitative analysis of pHisH3/BrdU-IR showed a bell shaped labeling between 0.25 and 24 hr, with none to very little labeling at 0.25 and 24 hr and maximal double labeling at 8 hr. These data fit well with previously reported expression of pHisH3 in the G2/M phase of the cell cycle (Fig 3j and k, pHisH3/BrdU-IR cells in the pie charts; (Hendzel et al., 1997)). Using the expression pattern of BrdU/pHisH3 labeling we were able to estimate the cell cycle length of SGZ precursors. The percent of BrdU-IR cells positive for pHisH3 fits with a 14 hr cell cycle model (Fig 3k, 0 hr), whereas 12, 13 or 15 hr models did not correlate with the expression pattern. This allows us to fine-tune previous estimates of 12-14 hr of the mouse SGZ cell cycle length to 14 hr. Additional studies should find the time course of transient expression of pHisH3 useful to reveal changes in the G2/M phase of the cell cycle. For example, combinatorial analysis of pHisH3/BrdU with Chk1 (G2/M checkpoint protein; (Carr et al., 1995)) will help determine changes in cell cycle progression caused by DNA damage. Importantly, one could use the expression pattern of pHisH3/BrdU to detect induction of cell cycle proteins that prevent programmed cell death. One such example is ErbB2 (Yu, 2001), where co-labeling of pHisH3/BrdU/ErbB2 would determine the anti-apoptotic mechanism of external factors to decrease cell proliferation.
To address the issue of how long cells were putatively dividing after BrdU incorporation, we triple labeled BrdU-IR cells with Ki-67 and PCNA. Ki-67 was chosen because of its expression in most phases of the cell cycle, the short half-life of the protein, and its increasing use to label dividing cells in the adult mammalian SGZ (Gerdes et al., 1984, Karamitopoulou et al., 1994, Kee et al., 2002, Dayer et al., 2003, Eisch and Mandyam, In Press). PCNA was chosen because it is expressed in all phases of the cell cycle including those not expressed by Ki-67 (Gerdes et al., 1984, Celis and Celis, 1985). The disadvantage of PCNA is the long half-life of the protein (Khoshyomn et al., 1993, Lopez-Girona et al., 1995). Nearly all BrdU-IR cells were Ki-67 (97.9%) and PCNA (100%) positive at the 0.25 hr time point, which was expected as both Ki-67 and PCNA are markers of cell proliferation and BrdU-IR cells were mostly in S phase at 0.25 hr (Fig 5m, n). Strikingly, the percent labeling of BrdU with Ki-67 (80.9%) and PCNA (100%) was different at 2 hr. At 2 hr 26% of the BrdU-IR cells have moved into G2/M phase of the cell cycle (based on cell cycle kinetics from (Hayes and Nowakowski, 2002)), suggesting that Ki-67 may not be expressed in all parts of G2/M. The percent labeling of BrdU with Ki-67 (50.6%) and PCNA (100%) at 15 hr is also significantly different. This difference in double labeling can be attributed to the short half-life of Ki-67 and the steady-state model of cell division of mouse SGZ cells (Hayes and Nowakowski, 2002). The decrease in BrdU/Ki-67 labeling to 50% at 15 hr is consistent with our BrdU time course data (compare Fig 2h and 5n). For example, even though there was doubling of BrdU-IR cells between 2 and 15 hr (Fig 2h), only 50% of cells at 15 hr were actively cycling (Ki-67 positive; Fig 5m, n). At 24 hr the 50% of cells that were Ki-67 negative were lost, supporting that some precursors died after the first mitosis. The lack of decrease in BrdU/PCNA labeling at 15 hr suggests that PCNA protein remains detectable in proteins long after the terminal exit of cells from the cell cycle. Therefore, PCNA’s long half-life may lead to overestimation of the proliferating population. The excess labeling with PCNA may hinder the utility of PCNA in future cell cycle kinetic analysis, particularly if a decrease in the number of proliferating cells is expected (Wharton et al., 2005).
The percent labeling of BrdU with Ki-67 (33.2%) and PCNA (92%) at 24 hr reveals evidence of BrdU-IR cells exiting the cell cycle (Fig 5m, n). Cell cycle exit of precursors at 24 hr is further supported by the first appearance of ‘BrdU alone’ cells that are negative for Ki-67 and PCNA. The decrease in percent labeling of BrdU with Ki-67 (6%) to almost 0% at 96 hr suggests that, on average, dividing daughter BrdU-IR cells were added up to 6-7 divisions and were visible up to 4 days after a single injection. Therefore the cell division history of the BrdU-IR cells can be approximately determined up to a period of 4 days (96 hr), this time frame is similar in mouse and rat SGZ precursors (Dayer et al., 2003). It is surprising that 0% of BrdU-IR cells are PCNA-IR 720 hours after labeling. Although there may have been some stem cell precursors with longer cell cycle time in the SGZ labeled by the BrdU pulse, dilution after division may hinder BrdU labeling in such cells. Furthermore, the population of such primary precursors may be so small that some cycling cells may have been excluded by experimental sampling. Importantly, the number of ‘BrdU alone’ cells increased gradually over the time course and at 30 days we can postulate that all the BrdU-IR cells had attained a matured phenotype and exit the cell cycle.
These data with endogenous cell cycle proteins emphasize that Ki-67 and PCNA not only have significant differences in their in vivo half-lives but also that Ki-67 is a more accurate marker for quantitative analysis of cell proliferation of SGZ precursors. Overall, these results stress the utility of endogenous markers in combination with exogenous markers not only to birth date and determine technical details of the cell cycle kinetics of cycling cells, but also to determine important differences in the in vivo half-lives of commonly used endogenous markers to help guide future research in regulation of adult neurogenesis.
Acknowledgments
The authors thank Rebecca Norris and Jessica Yee for outstanding technical assistance. We would like to thank Dr. Giridhar Mandyam for valuable help with staining sections and statistical analysis, and Dr. Diane C. Lagace for critical reading of the manuscript.
This work was supported by a postdoctoral Ruth Kirschstein National Research Service F32 Award from NIDA (DA018017; CDM), a Ruth Kirschstein National Research Service T32 Award from NIDA (DA007290, GCH), and grants from the National Institute on Drug Abuse (DA016765), National Institute of Mental Health (MH66172), National Institute of Aging and National Alliance for Research on Schizophrenia and Depression to AJE.
List of abbreviations
- BrdU
bromodeoxyuridine
- DG
dentate gyrus
- Hil
hilus
- IHC
immunohistochemistry
- IR
immunoreactive
- Mol
molecular layer
- oGCL
outer granule cell layer
- PCNA
proliferating cell nuclear antigen
- pHisH3
phosphorylated histone H3
- SGZ
subgranular zone
Footnotes
Section Editor: Dr. Constantino Sotelo, Cellular Neuroscience
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrous DN, Koehl M, Le Moal M. Adult neurogenesis: from precursors to network and physiology. Physiol Rev. 2005;85:523–569. doi: 10.1152/physrev.00055.2003. [DOI] [PubMed] [Google Scholar]
- Alexiades MR, Cepko C. Quantitative analysis of proliferation and cell cycle length during development of the rat retina. Dev Dyn. 1996;205:293–307. doi: 10.1002/(SICI)1097-0177(199603)205:3<293::AID-AJA9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Lim DA. For the long run: maintaining germinal niches in the adult brain. Neuron. 2004;41:683–686. doi: 10.1016/s0896-6273(04)00111-4. [DOI] [PubMed] [Google Scholar]
- Amaral D, Witter M. The Rat Nervous System San Diego. Academic Press; 1995. Hippocampal Formation; pp. 443–493. [Google Scholar]
- Bacchi CE, Gown AM. Detection of cell proliferation in tissue sections. Braz J Med Biol Res. 1993;26:677–687. [PubMed] [Google Scholar]
- Bauer HC, Bauer H. Neural induction of the blood-brain barrier: still an enigma. Cell Mol Neurobiol. 2000;20:13–28. doi: 10.1023/a:1006939825857. [DOI] [PubMed] [Google Scholar]
- Biebl M, Cooper CM, Winkler J, Kuhn HG. Analysis of neurogenesis and programmed cell death reveals a self- renewing capacity in the adult rat brain. Neurosci Lett. 2000;291:17–20. doi: 10.1016/s0304-3940(00)01368-9. [DOI] [PubMed] [Google Scholar]
- Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci. 2003a;17:2042–2046. doi: 10.1046/j.1460-9568.2003.02647.x. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003b;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Burns KA, Kuan CY. Low doses of bromo- and iododeoxyuridine produce near-saturation labeling of adult proliferative populations in the dentate gyrus. Eur J Neurosci. 2005;21:803–807. doi: 10.1111/j.1460-9568.2005.03907.x. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Gould E. Distinct populations of cells in the adult dentate gyrus undergo mitosis or apoptosis in response to adrenalectomy. J Comp Neurol. 1996;369:56–63. doi: 10.1002/(SICI)1096-9861(19960520)369:1<56::AID-CNE4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay RD. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. Journal of Comparative Neurology. 2001;435:406–417. doi: 10.1002/cne.1040. [DOI] [PubMed] [Google Scholar]
- Carr AM, Moudjou M, Bentley NJ, Hagan IM. The chk1 pathway is required to prevent mitosis following cell-cycle arrest at ‘start’. Curr Biol. 1995;5:1179–1190. doi: 10.1016/s0960-9822(95)00234-x. [DOI] [PubMed] [Google Scholar]
- Celis JE, Celis A. Cell cycle-dependent variations in the distribution of the nuclear protein cyclin proliferating cell nuclear antigen in cultured cells: subdivision of S phase. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3262–3266. doi: 10.1073/pnas.82.10.3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis JE, Fey SJ, Larsen PM, Celis A. Expression of the transformation-sensitive protein “cyclin” in normal human epidermal basal cells and simian virus 40-transformed keratinocytes. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:3128–3132. doi: 10.1073/pnas.81.10.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis JE, Madsen P, Nielsen S, Celis A. Nuclear patterns of cyclin (PCNA) antigen distribution subdivide S- phase in cultured cells--some applications of PCNA antibodies. Leuk Res. 1986;10:237–249. doi: 10.1016/0145-2126(86)90021-4. [DOI] [PubMed] [Google Scholar]
- Conover JC, Doetsch F, Garcia-Verdugo JM, Gale NW, Yancopoulos GD, Alvarez-Buylla A. Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat Neurosci. 2000;3:1091–1097. doi: 10.1038/80606. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson AG, van Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL. Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayer AG, Ford AA, Cleaver KM, Yassaee M, Cameron HA. Short-term and long-term survival of new neurons in the rat dentate gyrus. J Comp Neurol. 2003;460:563–572. doi: 10.1002/cne.10675. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Hen R. Young and excitable: the function of new neurons in the adult mammalian brain. Curr Opin Neurobiol. 2005;15:121–128. doi: 10.1016/j.conb.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J Comp Neurol. 2006;495:70–83. doi: 10.1002/cne.20840. [DOI] [PubMed] [Google Scholar]
- Eadie BD, Redila VA, Christie BR. Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dendritic complexity, and spine density. J Comp Neurol. 2005;486:39–47. doi: 10.1002/cne.20493. [DOI] [PubMed] [Google Scholar]
- Eisch A, Mandyam C. Neural progenitors: Can analysis of endogenous cell cycle markers move us “beyond BrdU”? Current Pharmaceutical Biotechnology. doi: 10.2174/138920107780906540. In Press. [DOI] [PubMed] [Google Scholar]
- Eisch AJ. Adult neurogenesis: implications for psychiatry. Progress in Brain Research. 2002;138:315–342. doi: 10.1016/S0079-6123(02)38085-3. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Barrot M, Schad CA, Self DW, Nestler EJ. Opiates inhibit neurogenesis in the adult rat hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7579–7584. doi: 10.1073/pnas.120552597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisch AJ, Mandyam CD. Progress in stem cell research. Nova Science Publishers Inc; 2004. Beyond BrdU: Basic and clinical implications for analysis of endogenous cell cycle proteins. [Google Scholar]
- Ernst C, Christie BR. Isolectin-IB 4 as a vascular stain for the study of adult neurogenesis. J Neurosci Methods. 2006;150:138–142. doi: 10.1016/j.jneumeth.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Gal JS, Morozov YM, Ayoub AE, Chatterjee M, Rakic P, Haydar TF. Molecular and morphological heterogeneity of neural precursors in the mouse neocortical proliferative zones. J Neurosci. 2006;26:1045–1056. doi: 10.1523/JNEUROSCI.4499-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Verdugo JM, Doetsch F, Wichterle H, Lim DA, Alvarez-Buylla A. Architecture and cell types of the adult subventricular zone: in search of the stem cells. J Neurobiol. 1998;36:234–248. doi: 10.1002/(sici)1097-4695(199808)36:2<234::aid-neu10>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. Journal of Immunology. 1984;133:1710–1715. [PubMed] [Google Scholar]
- Gil JM, Mohapel P, Araujo IM, Popovic N, Li JY, Brundin P, Petersen A. Reduced hippocampal neurogenesis in R6/2 transgenic Huntington’s disease mice. Neurobiol Dis. 2005;20:744–751. doi: 10.1016/j.nbd.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Gould E, Gross CG. Neurogenesis in adult mammals: some progress and problems. Journal of Neuroscience. 2002;22:619–623. doi: 10.1523/JNEUROSCI.22-03-00619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes NL, Nowakowski RS. Exploiting the dynamics of S-phase tracers in developing brain: interkinetic nuclear migration for cells entering versus leaving the S- phase. Dev Neurosci. 2000;22:44–55. doi: 10.1159/000017426. [DOI] [PubMed] [Google Scholar]
- Hayes NL, Nowakowski RS. Dynamics of cell proliferation in the adult dentate gyrus of two inbred strains of mice. Brain Res Dev Brain Res. 2002;134:77–85. doi: 10.1016/s0165-3806(01)00324-8. [DOI] [PubMed] [Google Scholar]
- He J, Nixon K, Shetty AK, Crews FT. Chronic alcohol exposure reduces hippocampal neurogenesis and dendritic growth of newborn neurons. Eur J Neurosci. 2005;21:2711–2720. doi: 10.1111/j.1460-9568.2005.04120.x. [DOI] [PubMed] [Google Scholar]
- Heine VM, Maslam S, Joels M, Lucassen PJ. Prominent decline of newborn cell proliferation, differentiation, and apoptosis in the aging dentate gyrus, in absence of an age-related hypothalamus-pituitary-adrenal axis activation. Neurobiol Aging. 2004;25:361–375. doi: 10.1016/S0197-4580(03)00090-3. [DOI] [PubMed] [Google Scholar]
- Hellsten J, West MJ, Arvidsson A, Ekstrand J, Jansson L, Wennstrom M, Tingstrom A. Electroconvulsive seizures induce angiogenesis in adult rat hippocampus. Biol Psychiatry. 2005;58:871–878. doi: 10.1016/j.biopsych.2005.05.023. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- Hildebrandt K, Teuchert-Noodt G, Dawirs RR. A single neonatal dose of methamphetamine suppresses dentate granule cell proliferation in adult gerbils which is restored to control values by acute doses of haloperidol. J Neural Transm. 1999;106:549–558. doi: 10.1007/s007020050178. [DOI] [PubMed] [Google Scholar]
- Holmberg J, Armulik A, Senti KA, Edoff K, Spalding K, Momma S, Cassidy R, Flanagan JG, Frisen J. Ephrin-A2 reverse signaling negatively regulates neural progenitor proliferation and neurogenesis. Genes Dev. 2005;19:462–471. doi: 10.1101/gad.326905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MM, Galea LA, Mistlberger RE, Kempermann G. Adult hippocampal neurogenesis and voluntary running activity: circadian and dose-dependent effects. J Neurosci Res. 2004;76:216–222. doi: 10.1002/jnr.20039. [DOI] [PubMed] [Google Scholar]
- Kahn L, Alonso G, Normand E, Manzoni OJ. Repeated morphine treatment alters polysialylated neural cell adhesion molecule, glutamate decarboxylase-67 expression and cell proliferation in the adult rat hippocampus. Eur J Neurosci. 2005;21:493–500. doi: 10.1111/j.1460-9568.2005.03883.x. [DOI] [PubMed] [Google Scholar]
- Karamitopoulou E, Perentes E, Diamantis I, Maraziotis T. Ki-67 immunoreactivity in human central nervous system tumors: a study with MIB 1 monoclonal antibody on archival material. Acta Neuropathologica. 1994;87:47–54. doi: 10.1007/BF00386253. [DOI] [PubMed] [Google Scholar]
- Kawabe T, Suganuma M, Ando T, Kimura M, Hori H, Okamoto T. Cdc25C interacts with PCNA at G2/M transition. Oncogene. 2002;21:1717–1726. doi: 10.1038/sj.onc.1205229. [DOI] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, Wojtowicz JM. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods. 2002;115:97–105. doi: 10.1016/s0165-0270(02)00007-9. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Brandon EP, Gage FH. Environmental stimulation of 129/SvJ mice causes increased cell proliferation and neurogenesis in the adult dentate gyrus. Curr Biol. 1998;8:939–942. doi: 10.1016/s0960-9822(07)00377-6. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development. 2003;130:391–399. doi: 10.1242/dev.00203. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Jessberger S, Steiner B, Kronenberg G. Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 2004;27:447–452. doi: 10.1016/j.tins.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Khoshyomn S, Maier H, Morimura T, Kitz K, Budka H. Immunostaining for proliferating cell nuclear antigen: its role in determination of proliferation in routinely processed human brain tumor specimens. Acta Neuropathologica. 1993;86:582–589. doi: 10.1007/BF00294296. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Lee KJ, Shin YC, Choi SH, Do E, Kim S, Chun BG, Lee MS, Shin KH. Stress-induced decrease of granule cell proliferation in adult rat hippocampus: assessment of granule cell proliferation using high doses of bromodeoxyuridine before and after restraint stress. Mol Cells. 2005;19:74–80. [PubMed] [Google Scholar]
- Kochman LJ, Weber ET, Fornal CA, Jacobs BL. Circadian variation in mouse hippocampal cell proliferation. Neurosci Lett. 2006;406:256–259. doi: 10.1016/j.neulet.2006.07.058. [DOI] [PubMed] [Google Scholar]
- Kolb B, Pedersen B, Ballermann M, Gibb R, Whishaw IQ. Embryonic and postnatal injections of bromodeoxyuridine produce age-dependent morphological and behavioral abnormalities. J Neurosci. 1999;19:2337–2346. doi: 10.1523/JNEUROSCI.19-06-02337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg G, Reuter K, Steiner B, Brandt MD, Jessberger S, Yamaguchi M, Kempermann G. Subpopulations of proliferating cells of the adult hippocampus respond differently to physiologic neurogenic stimuli. J Comp Neurol. 2003;467:455–463. doi: 10.1002/cne.10945. [DOI] [PubMed] [Google Scholar]
- Lopez-Girona A, Bosch M, Bachs O, Agell N. Addition of calmodulin antagonists to NRK cells during G1 inhibits proliferating cell nuclear antigen expression. Cell Calcium. 1995;18:30–40. doi: 10.1016/0143-4160(95)90043-8. [DOI] [PubMed] [Google Scholar]
- Mandyam CD, Norris RD, Eisch AJ. Chronic morphine induces premature mitosis of proliferating cells in the adult mouse subgranular zone. Journal of Neuroscience Research. 2004;76:783–794. doi: 10.1002/jnr.20090. [DOI] [PubMed] [Google Scholar]
- Migheli A, Piva R, Casolino S, Atzori C, Dlouhy SR, Ghetti B. A cell cycle alteration precedes apoptosis of granule cell precursors in the weaver mouse cerebellum. Am J Pathol. 1999;155:365–373. doi: 10.1016/S0002-9440(10)65133-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW, Nowakowski RS. Use of bromodeoxyuridine-immunohistochemistry to examine the proliferation, migration and time of origin of cells in the central nervous system. Brain Res. 1988;457:44–52. doi: 10.1016/0006-8993(88)90055-8. [DOI] [PubMed] [Google Scholar]
- Morshead CM, van der Kooy D. Postmitotic death is the fate of constitutively proliferating cells in the subependymal layer of the adult mouse brain. J Neurosci. 1992;12:249–256. doi: 10.1523/JNEUROSCI.12-01-00249.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CP, Lee HC, Ho CW, Arooz T, Siu WY, Lau A, Poon RY. Differential mode of regulation of the checkpoint kinases CHK1 and CHK2 by their regulatory domains. J Biol Chem. 2004;279:8808–8819. doi: 10.1074/jbc.M312215200. [DOI] [PubMed] [Google Scholar]
- Ormerod MG. Cell-cycle analysis of asynchronous populations. Methods Mol Biol. 2004;263:345–354. doi: 10.1385/1-59259-773-4:345. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Hentges ST, Bumaschny VF, de Souza FS, Smart JL, Santangelo AM, Low MJ, Westbrook GL, Rubinstein M. A transgenic marker for newly born granule cells in dentate gyrus. J Neurosci. 2004;24:3251–3259. doi: 10.1523/JNEUROSCI.5173-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard DS, Jr, Menzies RA, Skalko RG. Incorportaiton of thymidine and its analogue, bromodeoxyuridine, into embryos and maternal tissues of the mouse. Differentiation. 1973;1:397–404. doi: 10.1111/j.1432-0436.1973.tb00137.x. [DOI] [PubMed] [Google Scholar]
- Palmer TD. Adult neurogenesis and the vascular Nietzsche. Neuron. 2002;34:856–858. doi: 10.1016/s0896-6273(02)00738-9. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Pandey S, Wang E. Cells en route to apoptosis are characterized by the upregulation of c- fos, c-myc, c-jun, cdc2, and RB phosphorylation, resembling events of early cell-cycle traverse. J Cell Biochem. 1995;58:135–150. doi: 10.1002/jcb.240580203. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Academic Press; San Diego: 2001. [Google Scholar]
- Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, Lesch KP. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006 doi: 10.1038/sj.mp.4001791. [DOI] [PubMed] [Google Scholar]
- Rich KJ, Boobis AR. Expression and inducibility of P450 enzymes during liver ontogeny. Microsc Res Tech. 1997;39:424–435. doi: 10.1002/(SICI)1097-0029(19971201)39:5<424::AID-JEMT5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Yamamoto A, Kawaguchi A, Yamaguchi M, Mori K, Goldman SA, Okano H. Direct isolation of committed neuronal progenitor cells from transgenic mice coexpressing spectrally distinct fluorescent proteins regulated by stage-specific neural promoters. J Neurosci Res. 2001;65:220–227. doi: 10.1002/jnr.1145. [DOI] [PubMed] [Google Scholar]
- Seri B, Garcia-Verdugo JM, Collado-Morente L, McEwen BS, Alvarez-Buylla A. Cell types, lineage, and architecture of the germinal zone in the adult dentate gyrus. J Comp Neurol. 2004;478:359–378. doi: 10.1002/cne.20288. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Westman J, Navarro JC, Dey PK, Nyberg F. Probable involvement of serotonin in the increased permeability of the blood-brain barrier by forced swimming. An experimental study using Evans blue and 131I-sodium tracers in the rat. Behav Brain Res. 1995;72:189–196. doi: 10.1016/0166-4328(96)00170-2. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Caviness VS., Jr PCNA-binding to DNA at the G1/S transition in proliferating cells of the developing cerebral wall. Journal of Neurocytology. 1993;22:1096–1102. doi: 10.1007/BF01235751. [DOI] [PubMed] [Google Scholar]
- Tozuka Y, Fukuda S, Namba T, Seki T, Hisatsune T. GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron. 2005;47:803–815. doi: 10.1016/j.neuron.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Van der Borght K, Wallinga AE, Luiten PG, Eggen BJ, Van der Zee EA. Morris water maze learning in two rat strains increases the expression of the polysialylated form of the neural cell adhesion molecule in the dentate gyrus but has no effect on hippocampal neurogenesis. Behav Neurosci. 2005;119:926–932. doi: 10.1037/0735-7044.119.4.926. [DOI] [PubMed] [Google Scholar]
- Wang S, Scott BW, Wojtowicz JM. Heterogenous properties of dentate granule neurons in the adult rat. J Neurobiol. 2000;42:248–257. [PubMed] [Google Scholar]
- Wang Y, Bagheri-Fam S, Harley VR. SOX13 is up-regulated in the developing mouse neuroepithelium and identifies a sub-population of differentiating neurons. Brain Res Dev Brain Res. 2005;157:201–208. doi: 10.1016/j.devbrainres.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Wharton SB, Williams GH, Stoeber K, Gelsthorpe CH, Baxter L, Johnson AL, Ince PG. Expression of Ki67, PCNA and the chromosome replication licensing protein Mcm2 in glial cells of the ageing human hippocampus increases with the burden of Alzheimer-type pathology. Neurosci Lett. 2005;383:33–38. doi: 10.1016/j.neulet.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Yu D. Mechanisms of ErbB2-mediated paclitaxel resistance and trastuzumab-mediated paclitaxel sensitization in ErbB2-overexpressing breast cancers. Semin Oncol. 2001;28:12–17. doi: 10.1016/s0093-7754(01)90277-5. [DOI] [PubMed] [Google Scholar]
- Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]