

Abstract
Ion transport and regulation of Na+–Ca2+ exchange were examined for two alternatively spliced isoforms of the canine cardiac Na+–Ca2+ exchanger, NCX1.1, to assess the role(s) of the mutually exclusive A and B exons. The exchangers examined, NCX1.3 and NCX1.4, are commonly referred to as the kidney and brain splice variants and differ only in the expression of the BD or AD exons, respectively. Outward Na+–Ca2+ exchange activity was assessed in giant, excised membrane patches from Xenopus laevis oocytes expressing the cloned exchangers, and the characteristics of Na+ i- (i.e., I1) and Ca2+ i- (i.e., I2) dependent regulation of exchange currents were examined using a variety of experimental protocols. No remarkable differences were observed in the current–voltage relationships of NCX1.3 and NCX1.4, whereas these isoforms differed appreciably in terms of their I1 and I2 regulatory properties. Sodium-dependent inactivation of NCX1.3 was considerably more pronounced than that of NCX1.4 and resulted in nearly complete inhibition of steady state currents. This novel feature could be abolished by proteolysis with α-chymotrypsin. It appears that expression of the B exon in NCX1.3 imparts a substantially more stable I1 inactive state of the exchanger than does the A exon of NCX1.4. With respect to I2 regulation, significant differences were also found between NCX1.3 and NCX1.4. While both exchangers were stimulated by low concentrations of regulatory Ca2+ i, NCX1.3 showed a prominent decrease at higher concentrations (>1 μM). This does not appear to be due solely to competition between Ca2+ i and Na+ i at the transport site, as the Ca2+ i affinities of inward currents were nearly identical between the two exchangers. Furthermore, regulatory Ca2+ i had only modest effects on Na+ i-dependent inactivation of NCX1.3, whereas I1 inactivation of NCX1.4 could be completely eliminated by Ca2+ i. Our results establish an important role for the mutually exclusive A and B exons of NCX1 in modulating the characteristics of ionic regulation and provide insight into how alternative splicing tailors the regulatory properties of Na+–Ca2+ exchange to fulfill tissue-specific requirements of Ca2+ homeostasis.
Keywords: sodium–calcium exchange, regulation, alternative splicing
INTRODUCTION
Sodium–calcium exchangers constitute a family of ion counter transporters that play a prominent role in cellular Ca2+ homeostasis (Lederer et al. 1996; Philipson et al. 1996). These integral membrane-spanning proteins are generally thought to serve as Ca2+ efflux mechanisms that are driven by the Na+ electrochemical gradient. Although present in many (and possibly all) tissues of the body, Na+–Ca2+ exchange has been most extensively characterized in cardiac and neuronal tissue. Three unique transporter gene products have been identified in mammals (NCX1, NCX2, and NCX3) and two have been shown to undergo alternative splicing (Quednau et al. 1997). Within the NCX1 subfamily, 12 alternatively spliced isoforms have been identified, whereas a single version of NCX2 has been cloned to date. The existence of three splice variants of NCX3 has been demonstrated (Quednau et al. 1997). The cardiac Na+–Ca2+ exchanger, NCX1.1, remains the most extensively characterized isoform with respect to structure, function, and regulation (Hryshko and Philipson 1997; Lederer et al. 1996; Philipson et al. 1996).
The consequences of alternative splicing of Na+–Ca2+ exchangers are largely unknown. One possibility is that expression of different exon combinations could provide a mechanism for directing Na+–Ca2+ exchangers to appropriate cellular locations. For example, in kidney, the exchanger may reside exclusively in the basolateral membrane of renal cells (Bindels et al. 1992; Reilly et al. 1993; Van Baal et al. 1996). Alternatively, or perhaps coincidentally, splice variants could exhibit altered functional and/or regulatory properties appropriate for different cellular environments and Ca2+ homeostasis requirements. Support for this notion comes from our recent demonstration of significant functional differences in the regulatory phenotypes of alternatively spliced isoforms of the Na+–Ca2+ exchanger from Drosophila (Omelchenko et al. 1998). These splice variants, CALX1.1 and CALX1.2, differ by five amino acids in a region analogous to the alternative splice site of mammalian exchangers (Ruknudin et al. 1997; Schwarz and Benzer 1997). A preliminary report indicates that functional differences exist between Na+–Ca2+ exchangers found in cardiac and renal tissue with respect to modulation by voltage, [Ca2+]i and phosphorylation (Ruknudin et al. 1998). These investigators have also demonstrated preferential expression of splice variants containing the A exon in hippocampal neurons, and the B exon in astrocytes and C6 glioma cells (He et al. 1998). Furthermore, a comparison of exchangers expressed in Xenopus oocytes revealed that the AD, but not the BD splice variant, was regulated by cAMP-dependent protein kinase (He et al. 1998).
In an effort to gain a better understanding of the transport and regulatory consequences of alternative splicing among Na+–Ca2+ exchangers, we selected two versions of NCX1 that differ only in terms of which of its two, mutually exclusive exons (i.e., A and B) is expressed: NCX1.4 (exons AD) and NCX1.3 (exons BD). Since exon A splice variants appear to be expressed predominantly in excitable tissues (e.g., neurons, heart, skeletal muscle), whereas exon B isoforms are more widely distributed (e.g., kidney, liver, lung, astrocytes) (Quednau et al. 1997; He et al. 1998), we reasoned that any observed phenotypic differences might provide insight into how Na+–Ca2+ exchange is tailored to the specific Ca2+ handling requirements of different tissues. Thus, we expressed NCX1.3 and NCX1.4 in Xenopus laevis oocytes and obtained current measurements of Na+–Ca2+ exchange using the giant excised patch-clamp technique (Hilgemann 1990). By using a variety of experimental protocols, we were able to clearly resolve exon-specific influences on the Na+ i- and Ca2+ i-dependent regulatory mechanisms of NCX1.
METHODS
Construction of NCX1.3 and NCX1.4
Kidney and brain samples were collected from adult dogs, rapidly frozen in liquid N2 and stored at −70°C. Total RNA was subsequently isolated by the method of Chomczynski and Sacchi 1987, as modified by Quednau et al. 1997, and splice variants of NCX1 were cloned via reverse transcriptase–PCR (RT-PCR) as described (Quednau et al. 1997). Complementary DNA was amplified in a total volume of 50 μl containing: 1× TaqPlus Precision buffer (Stratagene Inc.), 200 μM dNTP, 5% DMSO, and 25 pM each of reverse phase–purified, canine NCX1.1 forward and reverse primers that span its alternative splicing region (see below). The reaction mixture was heated to 95°C for 5 min and cooled to 80°C before addition of 2.5 U of TaqPlus Precision Polymerase. Amplification was carried out in 35 cycles of denaturation (30 s at 94°C), annealing (60 s at 55°C), and extension (2 min at 72°C), followed by an additional 10 min of extension at 72°C. Forward primer: 5′-TGGAGGTGAAGGTATTGCGA-3′ (corresponding to amino acids 553–559 of canine NCX1.1; Nicoll et al. 1990); reverse primer: 5′-TCTGCATACTGATCCTGGGT-3′ (corresponding to amino acids 861–855). Products from the RT-PCR procedure were purified using GeneClean (Bio101) and subcloned into the pCR-Script vector (Stratagene Inc.) according to manufacturer's instructions. Splice variants of NCX1 were identified by dideoxy sequencing. Clones with exon compositions AD (brain) and BD (kidney) were digested with XhoI and KpnI, and the resulting cassettes repaired into a full-length, canine NCX1.1 clone (Matsuoka et al. 1993) in pBluescript II SK(+) (Stratagene Inc.). The resulting constructs were designated NCX1.4 (exons AD; brain) and NCX1.3 (exons BD; kidney), according to the nomenclature of Quednau et al. 1997.
Synthesis of NCX1.3 and NCX1.4 cRNA
Complementary DNAs encoding NCX1.3 and NCX1.4 were linearized with HindIII (New England Biolabs Inc.) and cRNA synthesized using T3 mMessage mMachine in vitro transcription kits (Ambion Inc.) according to the manufacturer's instructions. After injection with ≈5 ng of cRNA encoding NCX1.3 or NCX1.4, oocytes were maintained at 16°C in solution B minus BSA (see below). Electrophysiological measurements were typically obtained from days 4–6 after injection.
Preparation of Xenopus laevis Oocytes
Xenopus laevis were anaesthetized in 250 mg/liter ethyl p-aminobenzoate (Sigma Chemical Co.) in deionized ice-water for 30 min. Oocytes were then removed and washed in solution A containing (mM): 88 NaCl, 15 HEPES, 2.4 NaHCO3, 1.0 KCl, 0.82 MgSO4; pH 7.6 at room temperature (rt). The follicles were teased apart and the oocytes transferred to 5 ml of solution A containing 80 mg collagenase (Type II; Worthington Biochemical Corp.) and incubated for 45–60 min at rt with gentle agitation. The oocytes were washed several times in solution B containing (mM): 88 NaCl, 15 HEPES, 2.4 NaHCO3, 1.0 KCl, 0.82 MgSO4, 0.41 mM CaCl2, 0.3 mM Ca(NO3)2, 1 mg/ml BSA (Fraction V; Sigma Chemical Co.); pH 7.6 at rt, and then transferred to 5 ml of 100 mM K2HPO4, pH 6.5 at rt, containing 1 mg/ml BSA. After incubation at rt for 11–12 min with gentle agitation, the oocytes were washed several times in solution B at rt. Defolliculated stage V–VI oocytes were selected and incubated at 16°C in solution B minus BSA until injection the following day.
Measurement of Na+–Ca2+ Exchange Activity
Na+–Ca2+ exchange current measurements were obtained using the giant excised patch clamp technique, as described previously (Hilgemann 1990; Hryshko et al. 1996; Trac et al. 1997). Borosilicate glass pipettes were pulled and polished to a final inner diameter of ≈20–25 μm and coated with a Parafilm™:mineral oil mixture to enhance patch stability and reduce electrical noise. To expedite removal of the vitelline layer before membrane patching, oocytes were shrunk slightly by incubation in a solution containing (mM): 100 K-aspartate, 100 KOH, 100 MES, 20 HEPES, 5 EGTA, 5 MgCl2, pH 7.0 at rt, for ≈15 min at rt. After removal of the vitelline layer by dissection, oocytes were placed in a solution containing (mM): 100 KOH, 100 MES, 20 HEPES, 5 EGTA, 5–10 MgCl2, pH 7.0 at rt (with MES), and GΩ seals formed via gentle suction. Membrane patches (inside-out configuration) were excised by progressive movements of the pipette tip. Rapid solution changes (i.e., ≈200 ms) were accomplished using a custom-built, computer-controlled, 20-channel solution switcher. Hardware (Axopatch 200a; Axon Instruments) and software (Axotape) were used for data acquisition and analysis. For outward current measurements, pipette (i.e., extracellular) solutions contained (mM): 100 N-methyl-glucamine–MES, 30 HEPES, 30 tetraethylammonium (TEA)-OH, 16 sulfamic acid, 8.0 CaCO3, 6 KOH, 2.0 Mg(OH)2, 0.25 ouabain, 0.1 niflumic acid, 0.1 flufenamic acid; pH 7.0 at 30°C (with MES). Outward Na+–Ca2+ exchange currents were elicited by switching from Li+ i- to Na+ i-based bath solutions containing (mM): 100 [Na + Li]-aspartate, 20 MOPS, 20 TEA-OH, 20 CsOH, 10 EGTA, 0–9.91 CaCO3, 1.0–1.5 Mg(OH)2, pH 7.0 at 30°C (with MES or LiOH). Magnesium and Ca2+ were adjusted to yield free concentrations of 1.0 mM and 0–30 μM, respectively, using MAXC software (Bers et al. 1994). For inward current measurements, pipettes contained (mM): 100 Na-MES, 10 EGTA, 20 CsOH, 4 Mg(OH)2, 20 TEA-OH, 10 HEPES, 0.25 ouabain, 0.1 niflumic acid, 0.1 flufenamic acid, 0.002 nifedipine, pH 7.0 with MES. Inward currents were activated by switching between Ca2+-free and Ca2+ containing Li+-based bath solutions described above. All experiments were conducted at 30 ± 1°C. All data reported are mean ± SEM, unless indicated otherwise.
RESULTS
Our aim was to determine which aspects of the ionic regulatory profile of NCX1 can be directly attributed to expression of its mutually exclusive exons, A and B. We chose the splice variant pair NCX1.4 (exons AD) and NCX1.3 (exons BD) and characterized their ionic regulatory phenotypes under a variety of conditions.
Inspection of the aligned sequences of exons A (encoding 35 amino acids) and B (encoding 34 amino acids) in Fig. 1 reveals substantial similarity between NCX1.3 and NCX1.4. 13 identities are found and the exons are ≈63% similar with respect to conservative substitutions, with the greatest similarity occurring towards their NH2 termini. Charge reversal occurs at two positions, substitution of charged for neutral residues is observed at six, polar residues coincide with hydrophobics at three, and the overall electric charge of exon A is −2, whereas exon B is +1. Exon B encodes a cysteine residue at position 585 (Nicoll et al. 1990), whereas no cysteines are found in any other exons of the alternatively spliced region of NCX1. Although the specific contribution of dissimilar amino acids between NCX1.3 and NCX1.4 has not been determined, the net effect of interchanging exons A and B is substantially altered ionic regulatory behavior.
Figure 1.

Sequence comparison of the alternatively spliced regions of NCX1.4 and NCX1.3. Single-letter amino acid code is used and identities are boxed. The alternative splicing region of NCX1 is located within the COOH-terminal third of its large intracellular loop. Expression of exons A and B is mutually exclusive.
Na+ i-dependent Regulation of NCX1.3 and NCX1.4
We examined the [Na+]i dependence of peak and steady state outward Na+–Ca2+ exchange currents mediated by NCX1.4 and NCX1.3 to obtain estimates of Na+ transport affinities, as well as the rate and extent of I1 inactivation. Fig. 2 shows representative current traces obtained in response to the rapid (i.e., <200 ms) application of 10–100 mM Na+ i to the cytoplasmic surface of the patches in the continuous presence of 1 μM regulatory Ca2+ i. Transport Ca2+ o in the pipette was constant at 8 mM. After each current activation event, patches were allowed to recover for 32–48 s in Li+ i-containing solution plus 1 μM Ca2+ i before delivery of the next Na+ i pulse. With increasing [Na+]i, the isoforms exhibited similar increases in peak current and in the extent of current inactivation, characteristic of Na+ i-dependent, or I1, inactivation (Hilgemann et al. 1992b). In response to the application of 100 mM Na+ i at 1-μM regulatory Ca2+ i, the rate of inactivation of NCX1.4 was marginally faster than that observed for NCX1.3 (0.29 ± 0.03 s−1, n = 18 vs. 0.22 ± 0.03 s−1, n = 11, respectively, NS). However, the major difference between the splice variants was in steady state current levels produced in response to changes in [Na+]i. Whereas NCX1.4 exhibited a [Na+]i-dependent increase in steady state current levels, similar to that observed with the cardiac Na+–Ca2+ exchanger, NCX1.1 (Matsuoka et al. 1995), steady state currents mediated by NCX1.3 were mainly insensitive to changes in [Na+]i over the concentration range examined (10–100 mM). However, this behavior could be abolished by treatment of the patch with α-chymotrypsin (Fig. 2, bottom), a procedure known to deregulate Na+–Ca2+ exchangers (Hilgemann 1990). After proteolysis, the Na+ i dependence of NCX1.3 became hyperbolic, similar to that observed for peak currents with NCX1.4. Thus, ionic regulation alters the apparent Na+ i affinity of steady state Na+–Ca2+ exchange currents for NCX1.3.
Figure 2.
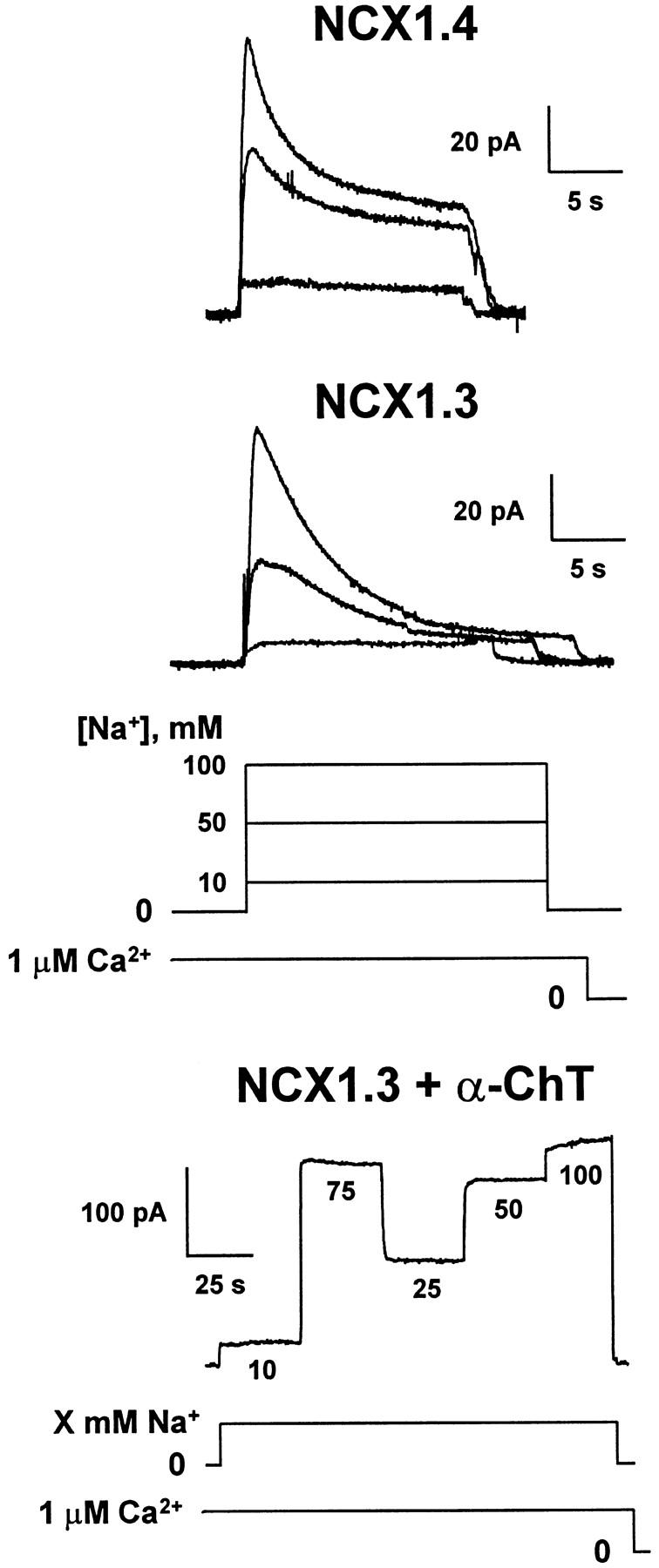
Na+ i dependence of outward Na+–Ca2+ exchange currents of NCX1.4 and NCX1.3. Representative current traces are shown for NCX1.4 and NCX1.3, and for NCX1.3 after proteolysis for ≈60 s with 1 mg/ml α-chymotrypsin. Transport Ca2+ o in the pipette was constant at 8 mM and regulatory Ca2+ i was held at 1 μM for 32–48 s before and during acquisition of the current traces. Outward currents were activated by the rapid (i.e., ≈200 ms) application of the indicated concentrations of Na+ i to the cytoplasmic surface of the patch. After each current activation event, patches were perfused for 32–48 s with 100 mM Li+ i-containing solution plus 1 μM Ca2+ i to permit full recovery from inactivation before delivery of the next Na+ i pulse.
Fig. 3 summarizes the Na+ i dependence of peak and steady state outward Na+–Ca2+ exchange currents derived from pooled data obtained with NCX1.3 and NCX1.4 in the presence of 1 μM regulatory Ca2+ i, as above. Currents were normalized to the values obtained at 100 mM Nai +. For both isoforms, peak currents progressively increased with increasing [Na+]i. Estimates of exchanger affinity for Na+ i based on peak current measurements provided nearly identical values of K d (33 ± 5 mM, n = 4 vs. 31 ± 4 mM, n = 6, for NCX1.3 and NCX1.4, respectively). For comparison, the peak current-derived Na+ i affinity of the cardiac exchanger, NCX1.1, is ≈27 mM (Matsuoka et al. 1995, Matsuoka et al. 1997). With respect to the Na+ i affinity derived from measurements of steady state currents, however, a K d value of 16 ± 1 mM (n = 6) was obtained for NCX1.4, whereas the NCX1.3 isoform appeared to be nearly Na+ i independent over the concentration range examined. Thus, a K d value could not be estimated. However, after treatment with α-chymotrypsin, a K d value of 26 ± 1 mM (three determinations from two patches) was estimated for NCX1.3, similar to that of peak currents for both exchangers.
Figure 3.

Na+ i dependence of peak and steady state outward Na+–Ca2+ exchange current for NCX1.4 and NCX1.3. Outward currents were obtained as described in Fig. 2, and were normalized to the value obtained at 100 mM Na+ i. Data are mean ± SEM of three to six determinations from six patches for NCX1.4, four determinations from four patches for NCX1.3, and three determinations from two patches for NCX1.3 after treatment with 1 mg/ml α-chymotrypsin for ≈60 s.
Fig. 4 illustrates the Na+ i dependence of the NCX1 isoforms in terms of their ratios of steady state to peak currents, or Fss values (Omelchenko et al. 1998). This fraction is a sensitive measure of the extent of I1 inactivation and can provide insight into the stability of the I1 inactive complex. Both splice variants exhibit a decrease in Fss with increasing [Na+]i, typical of I1-inactivated Na+–Ca2+ exchangers, and reflecting entry into the I1 state from the three Na+ i-loaded configuration of the exchanger (Hilgemann et al. 1992b). However, for [Na+]i ≥ 25 mM, the decrease in Fss is more pronounced for NCX1.3 than for NCX1.4. For example, Fss values calculated from currents acquired in response to the application of 100 mM Na+ i were 0.24 ± 0.03 and 0.07 ± 0.01 (n = 20 and 25 determinations from 14 patches) for NCX1.4 and NCX1.3, respectively. Note that for NCX1.3, the extent of I1 inactivation is ≈90% at high [Na+]i, whereas at 10 mM Na+ i it is ≈20%. This substantial inactivation of NCX1.3-mediated transport is likely to be responsible for the peculiar nature of the Na+ i dependence of its steady state currents, as shown in Fig. 3. That is, the increase in steady state current in response to Na+ i appears to be largely offset by the extensive inactivation that occurs as [Na+]i is raised. Consequently, steady state Na+–Ca2+ exchange currents mediated by NCX1.3 do not exhibit a hyperbolic response to rising [Na+]i unless this regulatory mechanism is eliminated by proteolysis with α-chymotrypsin.
Figure 4.

Na+ i dependence of the ratio of steady state to peak current for NCX1.4 and NCX1.3. Currents were obtained as described in Fig. 2. Data are mean ± SEM of 8–20 determinations from 16 patches for NCX1.4, and 4–12 determinations from seven patches for NCX1.3.
Fig. 5 illustrates the current–voltage (IV)1 relationships for NCX1.3 and NCX1.4. Outward currents were activated by switching from 100 mM Li+ i-containing perfusing solution to 100 mM Na+ i, and 1 μM regulatory Ca2+ i was present throughout. The IV relationship was determined before (a) and during (b) exchange current activation, and the former values were subtracted from the latter. From a holding potential of 0 mV, 40-ms voltage steps, in 10-mV increments, were applied from −100 to +100 mV, with a return to the holding potential between each step. Pooled data shown in Fig. 5 (bottom) are from three NCX1.3 and four NCX1.4 patches, with currents normalized to the values obtained at 0 mV. Note that a reversal potential is not observed under these conditions as the pipette solution does not contain Na+ o. The exchanger isoforms exhibited similar IV relationships to that observed for the cardiac exchanger, NCX1.1 (Matsuoka and Hilgemann 1992). We did not observe significant differences in the voltage dependency of the kidney exchanger, in contrast to an earlier report (Ruknudin et al. 1998).
Figure 5.
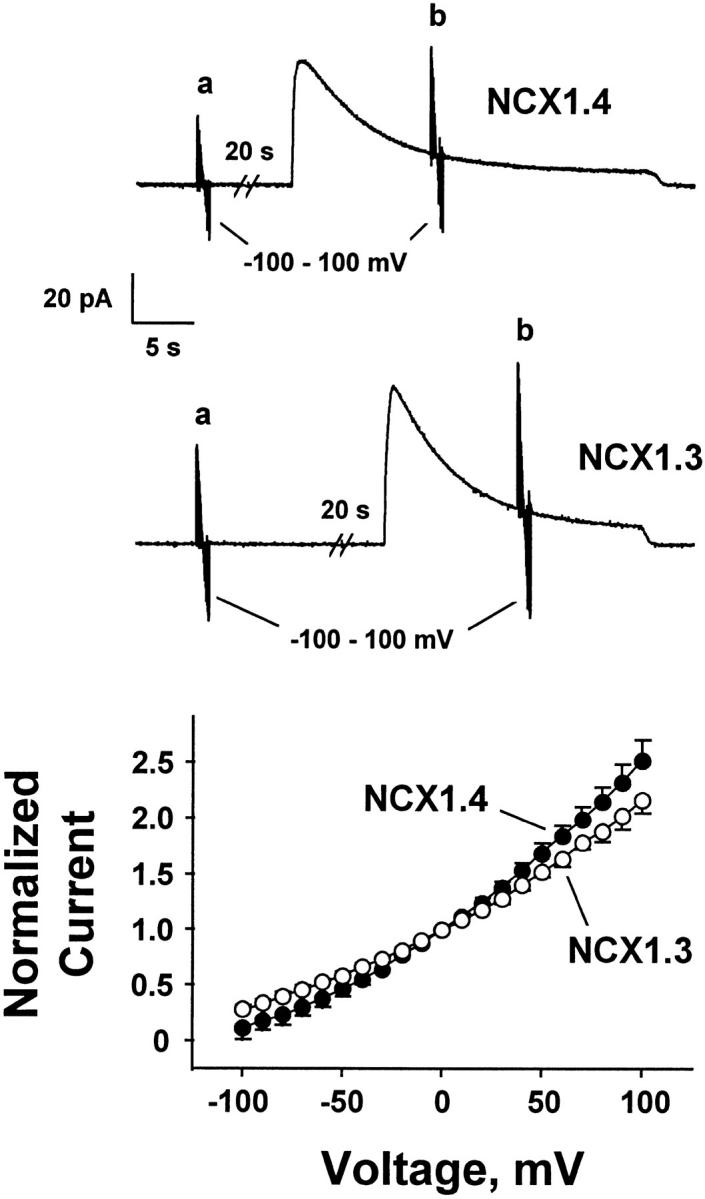
IV relationships for NCX1.4 and NCX1.3. Each voltage-clamp increment (10-mV steps from −100 to 100 mV) was initiated from a holding potential of 0 mV. IV records obtained at a during perfusion with 100 mM Li+ i-containing solution were subtracted from those obtained at b during perfusion with 100 mM Na+ i-containing solution. Regulatory Ca2+ i (1 μM) was present throughout the current recordings. Pooled data shown (bottom) are mean ± SEM from four patches for NCX1.4 and three patches for NCX1.3.
Ca2+ i-dependent Regulation of NCX1.3 and NCX1.4
Fig. 6 illustrates the effects of removal and reapplication of regulatory Ca2+ i on outward Na+–Ca2+ exchange currents mediated by NCX1.3 and NCX1.4. Currents were elicited by applying 100 mM Na+ i, with 1 μM Ca2+ i present before activation. Regulatory Ca2+ i was then removed and reapplied in the middle of the current trace in the continuous presence of 100 mM Na+ i. For NCX1.4, Ca2+ i removal led to rapid and nearly complete inhibition of outward exchange current, with a half-time for current decay of 0.52 ± 0.05 s (n = 5). Similarly, steady state levels were rapidly restored when Ca2+ i was reapplied, with a half-time of 0.49 ± 0.14 s (n = 5). For comparison with NCX1.1, the equivalent protocols yield half-times for loss and reacquisition of steady state current levels of 10.8 and 7.5 s, respectively (Matsuoka et al. 1995). Thus, steady state Na+–Ca2+ exchange currents for NCX1.4 exhibits considerably faster responses to this experimental maneuver than those observed for the cardiac exchanger. In contrast, with NCX1.3, half-times for loss and restoration of steady state current after removal and reapplication of Ca2+ i could not be determined due to the negligible steady state currents generated even in the presence of regulatory Ca2+ i.
Figure 6.

Ca2+ i regulation of outward Na+–Ca2+ exchange currents for NCX1.4 and NCX1.3. Representative records are shown for NCX1.4 and NCX1.3, where currents were activated by applying 100 mM Na+ i in the presence of 1 μM Ca2+ i. Regulatory Ca2+ i was present for 32–48 s before the application of transport Na+ i. Upon approaching steady state current levels, Ca2+ i was removed for 16 s, and then reapplied for a further 16-s interval before deactivating exchange current by returning to a Li+ i-based perfusing solution. The traces shown are typical of five patches for NCX1.4 and three patches for NCX1.3.
Fig. 7 shows representative traces that illustrate the dependence of outward Na+–Ca2+ exchange currents mediated by NCX1.3 and NCX1.4 upon [Ca2+]i. Regulatory Ca2+ i, at the indicated concentrations, was present for 32–48 s before, during, and after current activation with 100 mM Na+ i. For both isoforms, exchange currents are augmented by Ca2+ i. In particular, NCX1.4 behaves similar to the cardiac exchanger, NCX1.1, in that regulatory Ca2+ i not only stimulates exchange activity, but also alleviates I1 inactivation. At 10 μM Ca2+ i, Na+ i-dependent inactivation is nearly eliminated and the current recording adopts a square appearance. With NCX1.3, however, I1 inactivation is still prominent at 10 μM Ca2+ i, in sharp distinction to both NCX1.4 and NCX1.1. That is, regulatory Ca2+ i is not only incapable of alleviating I1 inactivation for NCX1.3, it appears to inhibit outward current generation. These relationships are illustrated graphically in Fig. 8.
Figure 7.
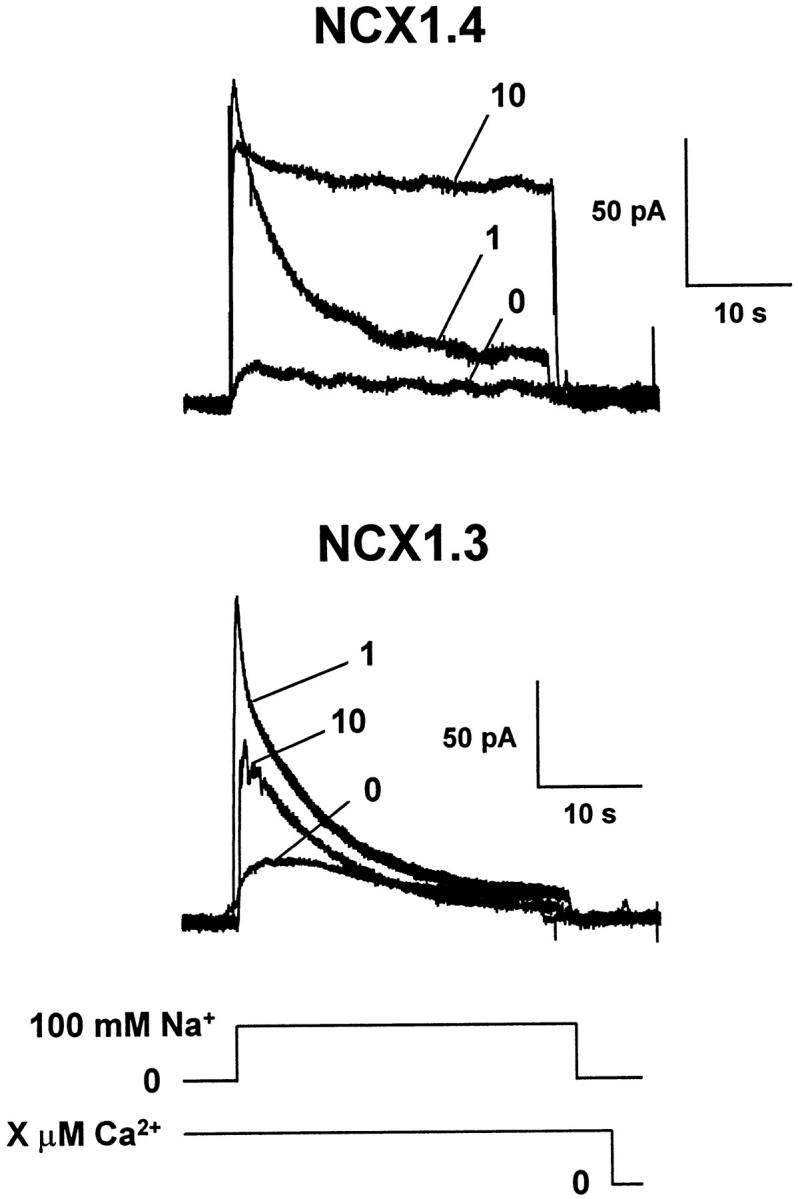
Ca2+ i dependence of outward Na+–Ca2+ exchange currents for NCX1.4 and NCX1.3. Currents were activated by applying 100 mM Na+ i with regulatory Ca2+ i present at the indicated concentrations for 32–48 s before and throughout the current recordings. After a current activation event, patches were perfused with a Li+ i-based perfusing solution containing a new [Ca2+]i for 32–48 s to allow for equilibration and recovery from the previous activation/inactivation event.
Figure 8.
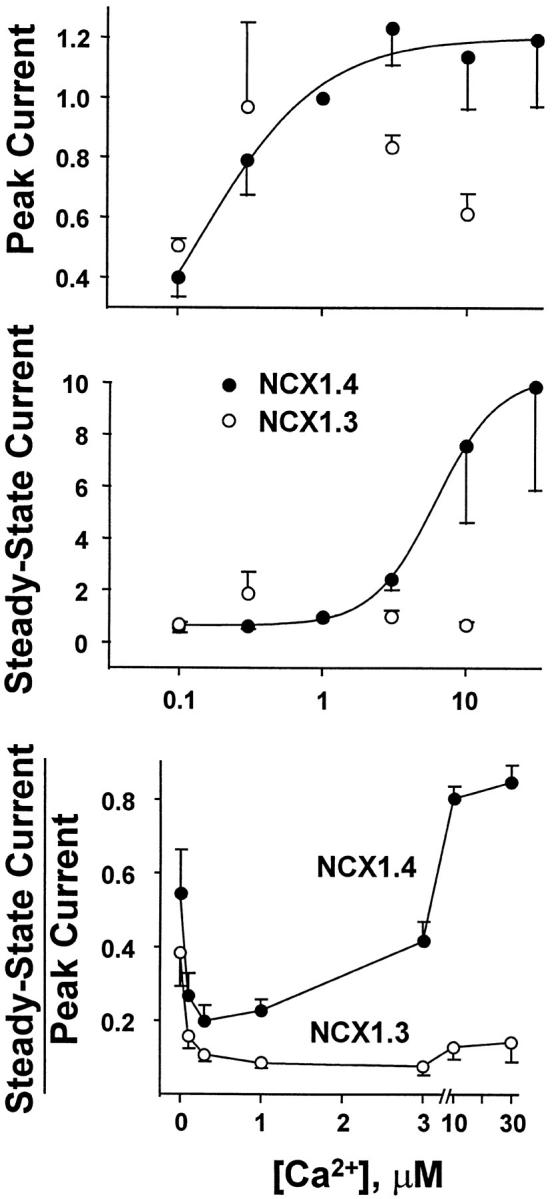
Ca2+ i dependence of peak (top) and steady state (middle) outward Na+–Ca2+ exchange currents for NCX1.4 and NCX1.3. Currents were obtained as described in Fig. 7 and normalized to the value obtained at 1 μM regulatory Ca2+ i. Data are mean ± SEM of three to seven determinations from seven patches for NCX1.4, and three to five determinations from five patches for NCX1.3. The Ca2+ i dependence of the ratio of steady state to peak current for NCX1.4 and NCX1.3 is shown (bottom). Currents were obtained as described in Fig. 7. Data are mean ± SEM of 7–20 determinations from 13 patches for NCX1.4, and 7–19 determinations from 15 patches for NCX1.3.
The pooled data shown in Fig. 8 illustrate the regulatory Ca2+ i dependence of peak and steady state outward currents mediated by NCX1.3 and NCX1.4 in response to the application of 100 mM Na+ i. Currents were normalized to the values obtained at 1 μM regulatory Ca2+ i. Both exchangers initially exhibit an increase in peak outward current as regulatory Ca2+ i is raised (Fig. 8, top). For NCX1.4, data were fit to the Hill equation over the [Ca2+]i range 0–3 μM, providing a K d value of 0.2 ± 0.06 μM. Beyond 3 μM Ca2+ i, however, deviation from simple hyperbolic behavior became evident, and further increases in [Ca2+]i often led to decreased peak outward currents (Fig. 7). This is presumably due to competition between Ca2+ i and Na+ i at the intracellular transport site, a phenomenon previously documented for NCX1.1 and two alternatively spliced isoforms of the Drosophila Na+–Ca2+ exchanger, CALX1 (Matsuoka et al. 1995; Trac et al. 1997; Omelchenko et al. 1998). With NCX1.3, this behavior was more pronounced and regulatory [Ca2+]i > 1 μM mediated a pronounced decrease of exchange current. This reduction in current is also evident in the representative traces shown in Fig. 7. Thus, we could not derive a meaningful estimate of K d for NCX1.3, but visual inspection suggests that at [Ca2+]i < 1 μM, both isoforms are comparably stimulated by regulatory [Ca2+]i. With respect to steady state currents (Fig. 8, middle), NCX1.4 again exhibits a smooth increase of outward current levels with increasing regulatory [Ca2+]i, and similar to that observed for NCX1.1. This reflects the progressive alleviation of I1 inactivation by Ca2+ i. In sharp contrast, the results obtained with NCX1.3 indicate that, at most, I1 inactivation is barely influenced by regulatory Ca2+ i and no obvious Ca2+ i dependence is discernible.
The effects of regulatory Ca2+ i on Fss, the ratio of steady state to peak currents are shown in Fig. 8 (bottom). For NCX1.4, a U-shaped Ca2+ i dependence was obtained. The value of Fss initially declines from 0–0.3 μM Ca2+ i, and then rises as Ca2+ i is increased beyond 0.3 μM. This behavior reflects the observation that, in the absence of Ca2+ i, outward currents are small and show relatively little inactivation. As Ca2+ i increases, however, peak current progressively rises, causing Fss to decrease at intermediate [Ca2+]i. Finally, as [Ca2+ i] is further increased, the extent of I1 inactivation is reduced to near-zero levels and Fss returns to higher values. These characteristics of Fss are typical of the cardiac exchanger, NCX1.1 (Hilgemann et al. 1992a; Matsuoka et al. 1995). In contrast, Na+ i-dependent inactivation of NCX1.3 is essentially insensitive to regulatory Ca2+ i and an L-shaped relationship is obtained. That is, the progressive alleviation of I1 inactivation observed with NCX1.4 and NCX1.1 does not occur for the kidney Na+–Ca2+ exchanger.
The observation that NCX1.3 appears to be inhibited at higher concentrations of regulatory Ca2+ could occur if this exchanger had a higher affinity for Ca2+ at the intracellular transport site. Consequently, lower concentrations of Ca2+ could compete for Na+ i and reduce current magnitude. To test this possibility, we examined inward Na+–Ca2+ exchange currents for both NCX1.3 and NCX1.4. Pipettes contained 100 mM Na+ and inward currents were activated by applying different Ca2+ concentrations (0.1–100 μM) to the cytoplasmic surface of the patch. Typical inward current recordings are shown in Fig. 9. We did not observe any major differences for inward Na+–Ca2+ exchange currents produced by NCX1.3 and NCX1.4. The apparent affinities calculated for Ca2+ activation of inward Na+–Ca2+ exchange currents were 9.0 ± 1.9 μM (mean ± SD, n = 5 patches) for NCX1.3 and 8.1 ± 1.4 μM (mean ± SD, n = 3 patches) for NCX1.4. These values are similar to that reported for NCX1 (e.g., 7 μM; Matsuoka et al. 1995). Furthermore, this indicates that the inhibitory effects of higher regulatory Ca2+ concentrations observed for outward currents from NCX1.3 are unlikely to be due to greater competition at the intracellular transport site.
Figure 9.
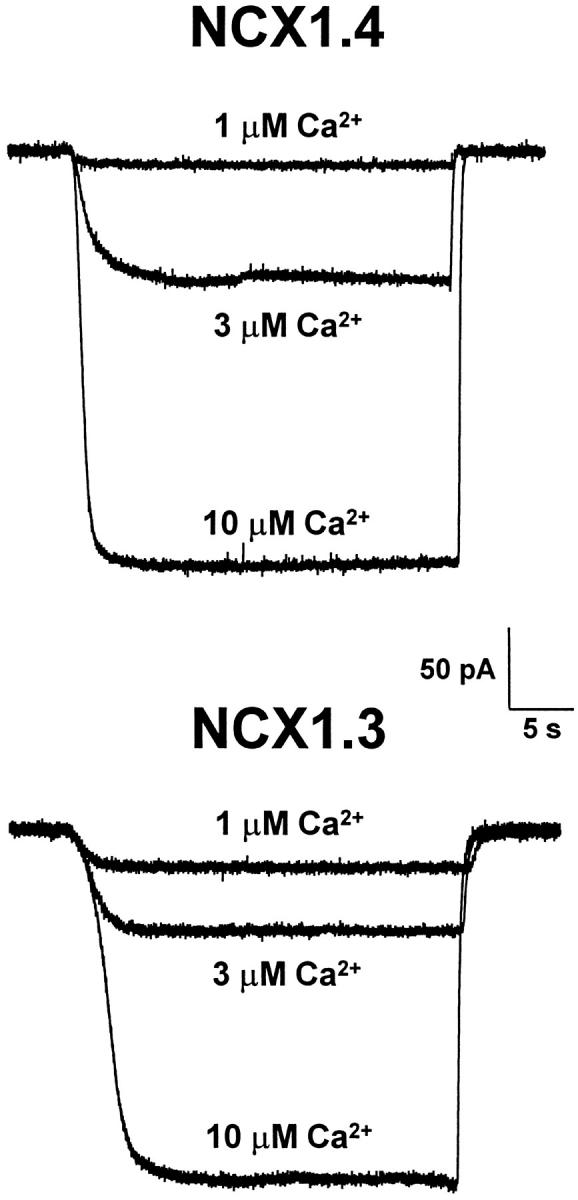
Representative inward Na+–Ca2+ exchange currents produced by NCX1.3 and NCX1.4 are shown in response to the application of three different Ca2+ concentrations (1, 3, and 10 μM). Pipettes contained 100 mM Na+. Similar results were obtained in four additional patches for NCX1.3 and two additional patches for NCX1.4.
Fig. 10 illustrates representative current recordings obtained for paired-pulse experiments conducted at two different regulatory Ca2+ concentrations (0.3 and 10 μM Ca2+ i). In each case, currents were activated by 100 mM Na+ i and transported Ca2+ o in the pipette was constant at 8 mM. Regulatory Ca2+ i, at the indicated concentration, was present throughout the entire paired-pulse trials. For NCX1.4, the second test pulse is substantially reduced after a 4-s interval at 0.3 μM Ca2+ i, whereas the two pulses are nearly identical in magnitude at 10 μM Ca2+ i. This behavior illustrates the ability of Ca2+ i to accelerate exit from, and/or reduce entry into, the I1 inactive state, and is typical of NCX1.1 (Hilgemann et al. 1992a). With NCX1.3, however, I1 inactivation is only weakly affected by regulatory Ca2+ i, and substantial inactivation is observed for paired-pulses even at 10 μM. This difference is shown graphically (Fig. 10, bottom) for representative data over a range of regulatory [Ca2+]i's for a 4-s inter-pulse interval. The parameter chosen to evaluate recovery, (Ipeak − Iss, pulse 2)/(Ipeak − Iss, pulse 1), was used so that recovery values would fall between 0 and 100%. That is, steady state currents are subtracted so that only the portion of current that inactivates is analyzed in terms of its recovery. This behavior was confirmed in a total of four patches each for NCX1.3 and NCX1.4.
Figure 10.
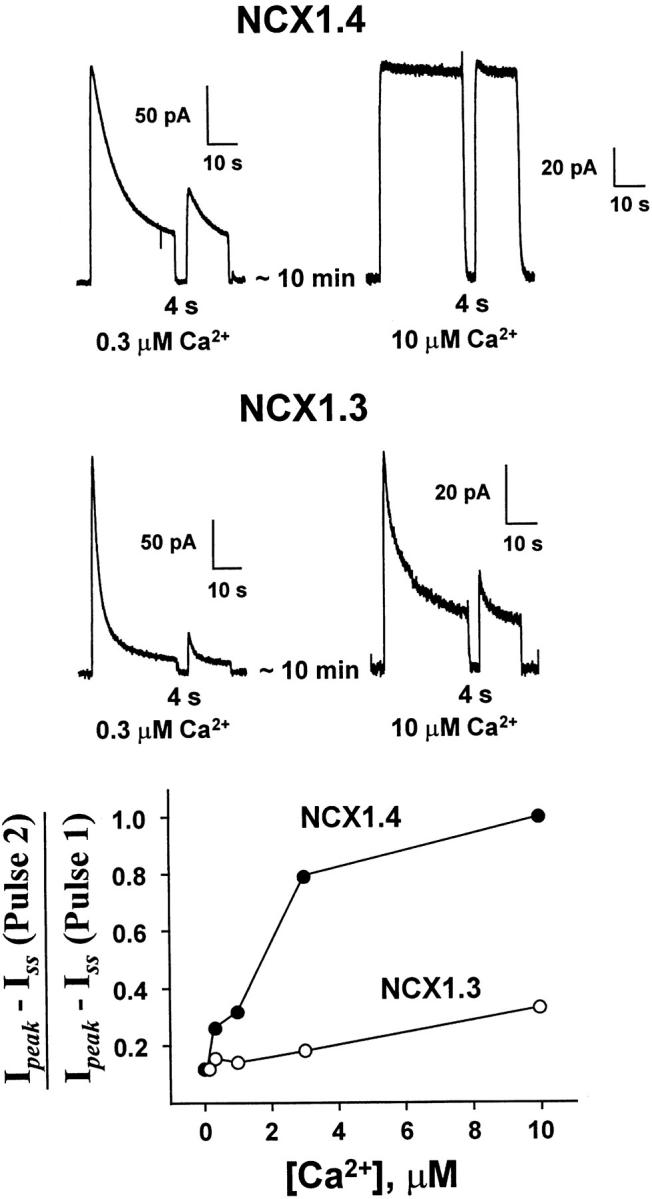
The effect of regulatory Ca2+ i on recovery of NCX1.4 and NCX1.3 from Na+ i-dependent inactivation using paired-pulse stimulation. The indicated concentrations of regulatory Ca2+ i were present throughout the current measurements. The first pulse was activated by applying 100 mM Na+ i for 32 s, followed by a 4-s recovery interval. A second, test pulse was then elicited by reapplication of 100 mM Na+ i. The graph (bottom) shows representative results from four patches each of NCX1.4 and NCX1.3 over a range of regulatory [Ca2+]i.
DISCUSSION
The NCX1 gene encodes a variety of alternatively spliced Na+–Ca2+ exchangers, several with unique tissue distributions. Although the physiological significance of this diversity is unknown, it may reflect different requirements for the maintenance of Ca2+ homeostasis in various cell types. Thus, we examined the ionic regulatory properties of two splice variants of NCX1: NCX1.3 and NCX1.4. These particular exchangers were selected for two reasons. First, NCX1.3 is a prominent splice variant in kidney, whereas NCX1.4 is abundant in brain. Second, these isoforms differ only in terms of expression of the mutually exclusive exons A (NCX1.4) and B (NCX1.3), with expression of the D cassette exon common to both. Therefore, our results provide direct insight into the functional role(s) of the mutually exclusive exons of NCX1. From structure–function considerations, our results point to prominent interactions between the alternative splicing region and other domains subserving the Na+ i- (i.e., I1) and Ca2+ i- (i.e., I2) dependent regulatory processes. These observations also provide a foundation towards understanding the physiological behavior of these transporters in their native environments.
Ionic Regulation of Na+–Ca2+ Exchangers
Sodium-dependent, or I1, inactivation of Na+–Ca2+ exchange current describes the ionic regulatory process that occurs in response to the cytoplasmic application of Na+ i. The phenomenon manifests as a rapid (i.e., ≈200 ms) rise in outward exchange current to a peak value, followed by a relatively slow (i.e., ≈30 s) decay to steady state levels of activity. This mechanism has been characterized extensively in giant excised patch experiments (Hilgemann et al. 1992b), as well as in intact myocytes (Matsuoka and Hilgemann 1994). Experimental and kinetic modeling studies indicate that entry into the I1 state proceeds from the three-Na+ i–loaded configuration of the exchanger, and that exit from I1 is a first-order process (Hilgemann et al. 1992b). Furthermore, structure–function analyses have provided evidence that the XIP region, at the NH2 terminus of the large intracellular loop of the exchanger, plays a pivotal role in this process. Specifically, mutations within the XIP domain of the cardiac exchanger, NCX1.1, and the Drosophila Na+–Ca2+ exchanger, CALX1.1, can accelerate or eliminate the I1 inactivation process (Matsuoka et al. 1997; Dyck et al. 1998).
Calcium-dependent, or I2, regulation describes the stimulatory (e.g., NCX1.1) or inhibitory (e.g., CALX1.1) effect of micromolar [Ca2+]i on both inward and outward currents associated with the forward (i.e., Ca2+ efflux) and reverse (i.e., Ca2+ influx) modes, respectively, of Na+–Ca2+ exchange. Like I1, this process has also been well documented in giant excised patch clamp studies using both cardiomyocytes and Xenopus oocytes expressing NCX1.1 (Hilgemann et al. 1992a; Matsuoka et al. 1995) and CALX1 splice variants (Dyck et al. 1998; Omelchenko et al. 1998). However, the results obtained from intact myocytes pertaining to the operation of the I2 regulatory mechanism have been controversial. For example, stimulation of Na+–Ca2+ exchange current by submicromolar [Ca2+]i has been reported by Kimura et al. 1987, Noda et al. 1988, and Miura and Kimura 1989, whereas a large Ca2+ i-independent component of whole cell Na+–Ca2+ exchange current was observed by Matsuoka and Hilgemann 1994. Studies using Chinese hamster ovary cells expressing NCX1.1 have also demonstrated enhancement of Na+–Ca2+ exchange activity in response to Ca2+ i (Condrescu et al. 1997), an effect that was absent for a deletion mutant shown to lack I2 regulation (Fang et al. 1998). Structure–function studies of NCX1.1 have identified a portion of its large cytoplasmic loop that functions as the regulatory Ca2+ i binding site (Levitsky et al. 1994; Matsuoka et al. 1995). An analogous region is conserved in all cloned Na+–Ca2+ exchangers and, in particular, for CALX1.1, despite the fact that this exchanger is inhibited in response to Ca2+ i (Dyck et al. 1998). Moreover, we have demonstrated limited interconversion of Ca2+ i-dependent regulatory phenotypes between NCX1.1 and CALX1.1 in a chimeric exchanger study (Dyck et al. 1998). However, there is no coherent understanding, at present, of the physiological role of I2 regulation. Virtually all mechanistic information presently available concerning ionic regulation of Na+–Ca2+ exchangers comes from outward current measurements of exchange activity in giant, excised patches (Dyck et al. 1998; Hilgemann 1990; Hilgemann et al. 1992a; Matsuoka et al. 1995). Although this represents the reverse (i.e., Ca2+ influx) mode of transport, it is the most effective means available to allow resolution of mechanistic details of Na+–Ca2+ transport and regulation.
Functional Role of the Alternative Splicing Region
Before recognition of the existence of alternatively spliced isoforms of Na+–Ca2+ exchangers, structure–function studies aimed at delineating the bases of ionic regulation of NCX1 examined the consequences of deleting substantial portions of its large cytoplasmic loop (Matsuoka et al. 1993). In particular, two of these constructs (i.e., Δ240-679 and Δ562-685) eliminated the alternative splicing region, as well as appreciable flanking sequences. With Δ562-685, I1 regulation was ablated, whereas Δ240-679 was associated with loss of both I1 and I2 regulation (Matsuoka et al. 1993). However, given the extent of the structural changes associated with these constructs, it is difficult to evaluate how elimination of the alternative splicing region specifically contributed to the functional consequences. With a third, chimeric exchanger construct (essentially equivalent to NCX1.3), preliminary electrophysiological characterization revealed functional Na+ i- and Ca2+ i-dependent regulation (Matsuoka et al. 1993). We have confirmed these results and extend the characterization of NCX1.3 to demonstrate substantial functional differences between it and the brain- (i.e., NCX1.4) and cardiac- (i.e., NCX1.1) derived exchangers.
Qualitatively, NCX1.4 behaves in a similar fashion to NCX1.1 in terms of its Na+ i and Ca2+ i dependencies and I1 and I2 regulatory profiles. The main difference we observed between NCX1.1 and NCX1.4 lies in the rapidity with which removal and reapplication of regulatory Ca2+ i influences steady state Na+–Ca2+ exchange currents (Fig. 6). The response of NCX1.4 to this maneuver is at least 10× faster than that of NCX1.1 (Matsuoka et al. 1995). Possibly, this attribute of NCX1.4 provides it with the ability to respond appropriately to frequency-encoded signaling in neuronal tissue. In all other regards, NCX1.4 appears similar to the cardiac exchanger. Notably, both NCX1.1 and NCX1.4 share expression of the A exon in their respective alternative splicing regions. Although it is tempting to speculate that exon A alone is responsible for the observed similarities between the cardiac and brain isoforms, the contributions of the cassette exons, alone or in combination, has yet to be evaluated. However, what is clear is that profound differences in regulatory profiles are associated with expression of the A or B exons.
The ionic regulatory behavior of NCX1.3 is considerably different from both NCX1.1 and NCX1.4. Specifically, I1 inactivation appears to be much more prominent for this exchanger. This inactivation process resulted in steady state currents that were inhibited ≈90% compared with peak current values. Furthermore, regulatory Ca2+ i was incapable of alleviating this inhibition, in contrast to the behavior of NCX1.1 and NCX1.4. Finally, we observed a unique behavior of NCX1.3, which suggested that regulatory Ca2+ i exhibits both stimulatory and inhibitory effects (Fig. 7). This is unlikely to reflect differences in competition between Ca2+ and Na+ at the intracellular transport site, as both NCX1.3 and NCX1.4 show similar Ca2+ affinities for inward currents. Consequently, the expression of the B exon adds a novel aspect to the ionic regulatory phenotype of NCX1.3.
Characterization of regulatory phenotypes has only been undertaken for a few members of the Na+–Ca2+ exchanger family (i.e., NCX1.1, NCX1.3, NCX1.4, CALX1.1, and CALX1.2). For example, we have shown that alternatively spliced CALX1 exchangers, which differ by five amino acids, show marked differences in their I1 and I2 regulatory properties (Omelchenko et al. 1998). Although the characterization of regulatory phenotypes and primary structural determinants subserving these processes is advancing, the physiological significance of I1 and I2 regulation is not well understood. It remains the case that the most persuasive evidence suggesting a physiological role for these regulatory mechanisms is the fact that alternative splicing produces changes in their properties. Nevertheless, it is interesting to speculate on the physiological consequences of these regulatory differences. At present, there is no evidence to suggest that these differences exist for the purpose of localizing the exchangers, although this possibility has not been examined. For example, although NCX1.3 is preferentially localized to the basolateral membrane within discrete regions of the nephron (Friedman 1998; Reilly et al. 1993; Van Baal et al. 1996), the role, if any, of alternative splicing in this process is not known. We hypothesize that ionic regulatory mechanisms tailor exchange activity to the Ca2+ homeostatic requirements of individual tissues. For example, Ca2+ transients in cardiac muscle from large animals (e.g., dog, human) occur within the frequency range of ≈0.5–3 Hz. Frequency-encoded signaling within neuronal tissue, however, occurs considerably faster (e.g., 300 Hz), whereas Ca2+ reabsorption in the kidney is likely to be a less dynamic process. As a Ca2+ efflux (and possibly influx) mechanism, the activity of the Na+–Ca2+ exchange system must be able to keep pace with this wide range of Ca2+ flux requirements. If ionic regulation contributes to the ability of Na+–Ca2+ exchangers to accommodate these various cellular Ca2+ flux rates, then the activity of exchangers in a physiological setting may be governed, in part, by the time-averaged consequences of ionic regulation, in addition to the thermodynamic influences of the Na+ and Ca2+ electrochemical gradients.
Acknowledgments
This work was supported by a Medical Research Council of Canada grant (GEC-3) and Heart and Stroke Foundation of Canada grant to L.V. Hryshko, and by the National Institutes of Health grants HL-48509 and HL-49101 (K.D. Philipson). L.V. Hryshko is supported by a Scholarship from the Medical Research Council of Canada.
Footnotes
1used in this paper: IV, current–voltage
References
- Bers D.M., Patton C.W., Nuccitelli R. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- Bindels R.J., Ramakers P.L., Dempster J.A., Hartog A., van Os C.H. Role of Na+/Ca2+ exchange in transcellular Ca2+ transport across primary cultures of rabbit kidney collecting system. Pflügers Arch. 1992;420:566–572. doi: 10.1007/BF00374634. [DOI] [PubMed] [Google Scholar]
- Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Condrescu M., Chernaya G., Lkalaria V., Reeves J.P. Barium influx mediated by the cardiac sodium–calcium exchanger in transfected Chinese hamster ovary cells. J. Gen. Physiol. 1997;109:41–51. doi: 10.1085/jgp.109.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyck C., Maxwell K., Buchko J., Trac M., Omelchenko A., Hnatowich M., Hryshko L.V. Structure–function analysis of CALX1.1, a Na+–Ca2+ exchanger from Drosophilamutagenesis of ionic regulatory sites. J. Biol. Chem. 1998;273:12981–12987. doi: 10.1074/jbc.273.21.12981. [DOI] [PubMed] [Google Scholar]
- Fang Y., Condrescu M., Reeves J.P. Regulation of Na+/Ca2+ exchange activity by cytosolic Ca2+ in transfected Chinese hamster ovary cells. Am. J. Physiol. 1998;275:C50–C55. doi: 10.1152/ajpcell.1998.275.1.C50. [DOI] [PubMed] [Google Scholar]
- Friedman P.A. Codependence of renal calcium and sodium transport. Annu. Rev. Physiol. 1998;60:179–197. doi: 10.1146/annurev.physiol.60.1.179. [DOI] [PubMed] [Google Scholar]
- He S., Ruknudin A., Bambrick L.L., Lederer W. J., Schulze D.H. Isoform-specific regulation of the Na+/Ca2+ exchanger in rat astrocytes and neurons by PKA. J. Neurosci. 1998;18:4833–4841. doi: 10.1523/JNEUROSCI.18-13-04833.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D.W. Regulation and deregulation of cardiac Na+–Ca2+ exchange in giant excised sarcolemmal membrane patches. Nature. 1990;344:242–245. doi: 10.1038/344242a0. [DOI] [PubMed] [Google Scholar]
- Hilgemann D.W., Collins A., Matsuoka S. Steady-state and dynamic properties of cardiac sodium–calcium exchangesecondary modulation by cytoplasmic calcium and ATP J. Gen. Physiol. 100 1992. 933 961a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D.W., Matsuoka S., Nagel G.A., Collins A. Steady-state and dynamic properties of cardiac sodium–calcium exchangesodium-dependent inactivation J. Gen. Physiol. 100 1992. 905 932b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryshko L.V., Matsuoka S., Nicoll D.A., Weiss J.N., Schwarz E.M., Benzer S., Philipson K.D. Anomalous regulation of the Drosophila Na+–Ca2+ exchanger by Ca2+ . J. Gen. Physiol. 1996;108:67–74. doi: 10.1085/jgp.108.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryshko L.V., Philipson K.D. Na+–Ca2+ exchangerecent advances. Basic Res. Cardiol. 1997;92:45–51. doi: 10.1007/BF00794067. [DOI] [PubMed] [Google Scholar]
- Kimura J., Miyamae S., Noma A. Identification of sodium–calcium exchange current in single ventricular cells of guinea-pig. J. Physiol. 1987;384:199–222. doi: 10.1113/jphysiol.1987.sp016450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer W.J., He S., Luo S., DuBell W., Kofuji P., Kieval R., Neubauer C.F., Ruknudin A., Cheng H., Cannel M.B. The molecular biology of the Na+–Ca2+ exchanger and its functional roles in heart, smooth muscle cells, neurons, glia, lymphocytes, and nonexcitable cells. Ann. NY Acad. Sci. 1996;779:7–17. doi: 10.1111/j.1749-6632.1996.tb44764.x. [DOI] [PubMed] [Google Scholar]
- Levitsky D.O., Nicoll D.A., Philipson K.D. Identification of the high affinity Ca2+-binding domain of the cardiac Na+–Ca2+ exchanger. J. Biol. Chem. 1994;269:22847–22852. [PubMed] [Google Scholar]
- Matsuoka S., Hilgemann D.W. Steady-state and dynamic properties of cardiac sodium–calcium exchange. Ion and voltage dependencies of the transport cycle. J. Gen. Physiol. 1992;100:963–1001. doi: 10.1085/jgp.100.6.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S., Hilgemann D.W. Inactivation of outward Na+–Ca2+ exchange current in guinea-pig ventricular myocytes. J. Physiol. 1994;476:443–458. doi: 10.1113/jphysiol.1994.sp020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S., Nicoll D.A., He Z., Philipson K.D. Regulation of the cardiac Na+–Ca2+ exchanger by the endogenous XIP region. J. Gen. Physiol. 1997;109:273–286. doi: 10.1085/jgp.109.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S., Nicoll D.A., Hryshko L.V., Levitsky D.O., Weiss J.N., Philipson K.D. Regulation of the cardiac Na+–Ca2+ exchanger by Ca2+mutational analysis of the Ca2+ binding domain. J. Gen. Physiol. 1995;105:403–420. doi: 10.1085/jgp.105.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S., Nicoll D.A., Reilly R.F., Hilgemann D.W., Philipson K.D. Initial localization of regulatory regions of the cardiac sarcolemmal Na+–Ca2+ exchanger. Proc. Natl. Acad. Sci. USA. 1993;90:3870–3874. doi: 10.1073/pnas.90.9.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura Y., Kimura J. Sodium–calcium exchange current. Dependence on internal Ca and Na and competitive binding of external Na and Ca. J. Gen. Physiol. 1989;93:1129–1145. doi: 10.1085/jgp.93.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll D.A., Longoni S., Philipson K.D. Molecular cloning and functional expression of the cardiac sarcolemmal Na+–Ca2+ exchanger. Science. 1990;250:562–565. doi: 10.1126/science.1700476. [DOI] [PubMed] [Google Scholar]
- Noda M., Shepherd R.N., Gadsby D.C. Activation by [Ca]i and block by 3′4′-dichlorobenzamil of outward Na/Ca exchange current in guinea-pig ventricular myocytes Biophys. J. 53 1988. 342a(Abstr.) [Google Scholar]
- Omelchenko A., Dyck C., Hnatowich M., Buchko J., Nicoll D.A., Philipson K.D., Hryshko L.V. Functional differences in ionic regulation between alternatively spliced isoforms of the Na+–Ca2+ exchanger from Drosophila melanogaster . J. Gen. Physiol. 1998;111:691–702. doi: 10.1085/jgp.111.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipson K.D., Nicoll D.A., Matsuoka S., Hryshko L.V., Levitsky D.O., Weiss J.N. Molecular regulation of the Na+–Ca2+ exchanger. Ann. NY Acad. Sci. 1996;779:20–28. doi: 10.1111/j.1749-6632.1996.tb44766.x. [DOI] [PubMed] [Google Scholar]
- Quednau B.D., Nicoll D.A., Philipson K.D. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am. J. Physiol. 1997;272:C1250–C1261. doi: 10.1152/ajpcell.1997.272.4.C1250. [DOI] [PubMed] [Google Scholar]
- Reilly R.F., Shugrue C.A., Lattanzi D., Biemesderfer D. Immunolocalization of the Na+/Ca2+ exchanger in rabbit kidney. Am. J. Physiol. 1993;265:F327–F332. doi: 10.1152/ajprenal.1993.265.2.F327. [DOI] [PubMed] [Google Scholar]
- Ruknudin A., Lederer W.J., He S., Schulze D.H. Functional differences between heart and kidney isoforms of the Na+/Ca2+ exchanger, NCX1 Biophys. J. 74 1998. 193a(Abstr.) [Google Scholar]
- Ruknudin A., Valdivia C., Kofuji P., Lederer W.J., Schulze D.H. Na+/Ca2+ exchanger in Drosophilacloning, expression, and transport differences. Am. J. Physiol. 1997;273:C257–C265. doi: 10.1152/ajpcell.1997.273.1.C257. [DOI] [PubMed] [Google Scholar]
- Schwarz E.M., Benzer S. Calx, a Na–Ca exchanger gene of Drosophila melanogaster . Proc. Natl. Acad. Sci. USA. 1997;94:10249–10254. doi: 10.1073/pnas.94.19.10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trac M., Dyck C., Hnatowich M., Omelchenko A., Hryshko L.V. Transport and regulation of the cardiac Na+–Ca2+ exchangercomparison between Ca2+ and Ba2+ . J. Gen. Physiol. 1997;109:361–369. doi: 10.1085/jgp.109.3.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Baal J., Yu A., Hartog A., Fransen J.A.M., Willems P.H.G.M., Lytton J., Bindels R.J.M. Localization and regulation by vitamin D of calcium transport proteins in rabbit cortical collecting system. Am. J. Physiol. 1996;271:F985–F993. doi: 10.1152/ajprenal.1996.271.5.F985. [DOI] [PubMed] [Google Scholar]