

Abstract
The substituted cysteine accessibility approach, combined with chemical modification using membrane-impermeant alkylating reagents, was used to identify functionally important structural elements of the rat type IIa Na+/Pi cotransporter protein. Single point mutants with different amino acids replaced by cysteines were made and the constructs expressed in Xenopus oocytes were tested for function by electrophysiology. Of the 15 mutants with substituted cysteines located at or near predicted membrane-spanning domains and associated linker regions, 6 displayed measurable transport function comparable to wild-type (WT) protein. Transport function of oocytes expressing WT protein was unchanged after exposure to the alkylating reagent 2-aminoethyl methanethiosulfonate hydrobromide (MTSEA, 100 μM), which indicated that native cysteines were inaccessible. However, for one of the mutants (S460C) that showed kinetic properties comparable with the WT, alkylation led to a complete suppression of Pi transport. Alkylation in 100 mM Na+ by either cationic {[2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET), MTSEA} or anionic [sodium(2-sulfonatoethyl)methanethiosulfonate (MTSES)] reagents suppressed the Pi response equally well, whereas exposure to methanethiosulfonate (MTS) reagents in 0 mM Na+ resulted in protection from the MTS effect at depolarized potentials. This indicated that accessibility to site 460 was dependent on the conformational state of the empty carrier. The slippage current remained after alkylation. Moreover, after alkylation, phosphonoformic acid and saturating Pi suppressed the slippage current equally, which indicated that Pi binding could occur without cotransport. Pre–steady state relaxations were partially suppressed and their kinetics were significantly faster after alkylation; nevertheless, the remaining charge movement was Na+ dependent, consistent with an intact slippage pathway. Based on an alternating access model for type IIa Na+/Pi cotransport, these results suggest that site 460 is located in a region involved in conformational changes of the empty carrier.
Keywords: mutagenesis, phosphate transport, electrophysiology, Xenopus laevis oocyte
INTRODUCTION
Type II sodium/phosphate (NaPi-II)1 cotransporters belong to a unique class of Na+-coupled cotransport proteins that show no amino acid homology to other known cotransport proteins (Murer and Biber 1997; Murer et al. 1998). They can be further subdivided into two subgroups, type IIa and IIb, based on specific motifs in the COOH-terminal region. Type IIa cotransporters are only expressed in the proximal tubule of the kidney, whereas type IIb are expressed in several tissues, such as lung, small intestine, and testis (Hilfiker et al. 1998). Members of these subgroups share an overall amino acid homology of ∼57% (Hilfiker et al. 1998). Nevertheless, the currently predicted secondary structure indicates that membrane spanning regions and some interdomain linking regions have considerably higher homology (up to 99%; Hilfiker et al. 1998) between the two subgroups. This suggests that these regions are responsible for the general functional characteristics that all type II Na+/Pi cotransporters share, such as high affinity for Pi, Na+ dependence, and electrogenicity. Although the transport kinetics of both subgroups are well characterized (Forster et al. 1997, Forster et al. 1998, Forster et al. 1999a; Hilfiker et al. 1998), nothing is known about functionally important sites and domains within the molecule itself that determine the kinetic characteristics of the type II system, such as voltage dependence, substrate specificity, and substrate affinity.
One technique that offers considerable potential for identifying functionally important residues and/or domains is the substituted-cysteine-accessibility method (SCAM; Akabas et al. 1994) combined with the application of methanethiosulfonate (MTS) derivatives that react specifically with cysteine residues (Smith et al. 1975; Kenyon and Bruice 1977). In particular, charged, and therefore membrane impermeant, MTS derivatives have been used extensively to investigate the topology and structure of membrane-spanning proteins (Akabas et al. 1992; Stauffer and Karlin 1994). For example, the topology of the serotonin transporter and the K+-pore Kv2.1 have been studied by substituting several amino acids with cysteines (Pascual et al. 1995; Chen et al. 1998). This technique has also enabled identification of functionally important regions of the glutamate carrier EAAT1 (Seal and Amara 1998) and the sodium/glucose cotransporter (SGLT-1; Lo and Silverman 1998a,Lo and Silverman 1998b). Moreover, the recent findings by Loo et al. 1998 that the accessibility of a substituted cysteine (Q457C) in the putative sugar-translocation domain of SGLT-1 to MTS reagents is dependent on substrate and membrane potential, and their finding of a correlation between pre–steady state charge movements and fluorescence changes of the labeled cysteine strongly supports the notion that ligand- and voltage-induced conformational transitions are responsible for coupling Na+ and glucose transport.
In the present study, we have adopted a cysteine replacement strategy and substituted 15 selected amino acids with cysteine residues with the aim of identifying sites where the reaction with MTS reagents would lead to a detectable change in transport function. Having no precedent for the selection of residues, we based our choice on the following criteria: (a) residues located between hydrophobic and hydrophilic regions were chosen because these intervening regions could be likely candidates for substrate binding or conformational changes during the transport process (Lo and Silverman 1998a; Loo et al. 1998) (see Fig. 1), (b) serine or alanine residues, where present, were selected for cysteine substitution to minimize changes in the protein, and (c) residues in the amino or carboxy termini were not mutated because these are most likely located intracellularly. Furthermore, based on the evolutionary tree of Na+-coupled Pi cotransporters, both termini are located in quite variable regions (Biber et al. 1996, Biber et al. 1998; Murer and Biber 1997), which suggested they are not involved in basic substrate binding or transport processes.
Figure 1.

Topological representation of the rat type IIa sodium phosphate cotransporter (rat NaPi-IIa). Scheme is based on current hydropathy data and a recent topology study (Lambert et al. 1999) in which eight membrane spanning regions (TM1–TM8) are predicted with extracellular loops between TM1–TM2, TM3–TM4, TM5–TM6, and TM7–TM8, and both the carboxy and amino termini are intracellular. (•) Positions of 15 residues that were individually mutated to cysteines (see methods and materials).
We have used the Xenopus oocyte expression system and electrophysiological methods to characterize the results of the mutagenesis. We report that of the 15 mutants assayed, only one (S460C) was sensitive to MTS reagents. We show that this mutant, under normal conditions, behaved essentially the same as the wild-type (WT) protein insofar as its kinetic characteristics were concerned. However, after exposure to membrane-impermeant MTS reagents, the kinetic properties of the chemically modified protein suggested that the native Ser-460 lies in a region involved in voltage-dependent conformational changes during the cotransport process. Moreover, Ser-460 appeared to be neither involved directly in the binding of the first Na+ ion, nor the subsequent Pi binding, but alkylation of the substituted cysteine at this site led to an inhibition of the final cotransport transition.
MATERIALS AND METHODS
Molecular Biology
Mutations were introduced following the Quickchange Site-Directed Mutagenesis Kit manual (Stratagene Inc.). In brief, 10 ng of the plasmid containing the rat NaPi-IIa cDNA were amplified with 2.5 U PfuTurbo® (Strategene Inc.) DNA polymerase in the presence of 250 nM of primers. PCR amplification was performed with 20 cycles of 95°C (30 s), 55°C (1min), and 68°C (12 min). Next, 10 U of Dpn I were added directly to the amplification reaction and the sample was incubated for 1 h at 37°C to digest the parental, methylated DNA. XL1-blue supercompetent cells were transformed with 1 μl reaction mixture and plated onto LB-ampicillin-methicillin plates. The sequence was verified by sequencing. All constructs were cloned in pSport1 (GIBCO BRL). In vitro synthesis and capping of cRNAs were done by incubating the rat NaPi IIa constructs, previously linearized by NotI digestion, in the presence of 40 U of T7 RNA polymerase (Promega) and Cap Analogue (New England Biolabs Inc.) (Werner et al. 1990).
Immunoblotting of Oocyte Homogenates
Yolk-free homogenates were prepared 3 d after injection (H2O or cRNA). Pools of five oocytes were lysed together with 100 μl of homogenization buffer [1% Elugent (Calbiochem) in 100 mM NaCl, 20 mM Tris/HCl, pH 7.6], by pipetting the oocytes up and down (Turk et al. 1996). To pellet the yolk proteins, samples were centrifuged at 16,000 g for 3 min at 22°C. 10 μl of the supernatant in 2× loading buffer [4% sodium-dodecyl sulfate (SDS), 2 mM EDTA, 20% glycerol, 0.19 M Tris/HCl, pH 6.8, 2 mg/ml bromphenol blue] were separated on an SDS-PAGE gel, and separated proteins were transferred to a nitro-cellulose membrane (Schleicher & Schuell, Inc.). The membrane was then processed according to standard procedures (Sambrook et al. 1989) using a rabbit polyclonal antibody raised against an NH2-terminal synthetic peptide of the rat NaPi-IIa cotransporter. The specificity of the antibody has been demonstrated previously (Custer et al. 1994). Immunoreactive proteins were detected with a chemiluminescence system (Pierce).
Streptavidin Precipitation of Biotinylated Protein
Groups of five oocytes expressing C460S or the WT protein were incubated for 5 min in 100 μM 2-aminoethyl MTS hydrobromide (MTSEA)–Biotin. Biotin-streptavidin precipitation was performed as described previously (Hayes et al. 1994): briefly, after taking a sample for Western blotting, the oocyte homogenate was incubated for 2 h with streptavidin beads and precipitated proteins were eluted with 2× loading buffer at 95oC for 5 min. Samples were loaded on an SDS-gel and immunoblotted after protein separation.
Oocyte Preparation and Injection
Stage V–VI oocytes were prepared as previously described (Magagnin et al. 1992). Oocytes were incubated in modified Barth's solution (see below). Typically, 10 ng of cRNA in 50 nl of water were injected per oocyte and experiments performed 4–6 d after injection.
Solutions and Reagents
All standard chemicals and reagents were obtained from either Sigma Chemical Co. or Fluka AG. The MTS reagents, MTSEA, [2-(triethylammonium)ethyl] MTS bromide (MTSET), and sodium(2-sulfonatoethyl) MTS (MTSES), were obtained form Toronto Research Biochemicals and freshly prepared in DMSO. The concentration of DMSO did not exceed 1% and control experiments indicated no effect on transport function by DMSO at this concentration.
The solution compositions (mM) were as follows. (a) Oocyte incubation (modified Barth's solution): 88 NaCl, 1 KCl, 0.41 CaCl2, 0.82 MgSO4, 2.5 NaHCO3, 2 Ca(NO3)2, 7.5 Tris, pH 7.6, and supplemented with antibiotics (10 mg/liter penicillin, streptomycin). (b) Control superfusate (ND100): 100 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, titrated to pH 7.4 with KOH. For pre–steady state recording, where necessary, isomolar BaCl2 was substituted for CaCl2 to reduce contamination from endogenous Ca2+-activated Cl− currents that were observed for V > −10 mV, except for experiments involving phosphonoformic acid (PFA), which otherwise complexes with Ba2+. (c) Control superfusate (ND0): as for ND100, but with N-methyl-d-glucamine or choline chloride replacing Na+ to maintain iso-osmolar external solutions. Solutions were titrated with HCl and KOH to pH 7.4. (d) Substrate test solutions: inorganic phosphate (Pi) was added to ND100 from a K2HPO4/KH2PO4 stock preadjusted to pH 7.4. For PFA-containing solutions, to take account of this being a trisodium salt, the Na+ concentration of the control solution was increased by 9 mM to maintain the same transmembrane Na+ gradient.
Functional Assays
Tracer uptake.
The procedure used for the 32P-uptake assay has been described in detail elsewhere (Werner et al. 1990). 32P-uptake was measured 3 d after injection of both water- and cRNA-injected oocytes (n = 5).
Electrophysiology.
The standard two-electrode voltage clamp technique was used as previously described (Forster et al. 1997, Forster et al. 1998). Oocytes were mounted in a small recording chamber (100 μl vol) and continuously superfused (5 ml/min) with test solutions precooled to 20°C. Freshly prepared MTS reagents were applied to the oocyte chamber via the common superfusion manifold or using a 0.5-mm diameter cannula positioned near the cell and fed by gravity. Unless otherwise indicated, the steady state response of an oocyte to Pi was always measured at a holding potential (Vh) = −50 mV in the presence of 100 mM Na+. Data were acquired online using DATAC software and compatible hardware (Bertrand and Bader 1986) and sampled at more than twice the recording bandwidth. Recorded currents were prefiltered using an eight-pole low pass Bessel filter (Frequency Devices, Inc.) that was set to 20 Hz for the steady state and 500 Hz or 2 kHz for the pre–steady state measurements. To increase the signal resolution for pre–steady state measurements, we also employed capacitive transient subtraction.
Kinetic Characterization
The modified Hill equation was fit to the dose-response data:
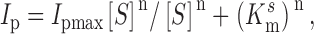 |
1 |
where [S] is the substrate concentration, I pmax is the extrapolated maximum current, K m s is the concentration of substrate S, which gives a half maximum response or apparent affinity constant, and n is the Hill coefficient.
Pre–steady state charge movements were quantified by first subtracting records obtained in 3 mM PFA to eliminate endogenous currents. An exponential fitting routine, based on the Chebychev transform (e.g., Axon Instruments 1998) was used to estimate relaxation time constants. Difference records were integrated to obtain the charge (Q) as a function of transmembrane voltage (V). Q–V data were then characterized by fitting a single Boltzmann function:
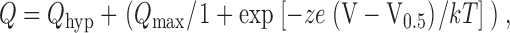 |
2 |
where Q max is the maximum charge translocated, Q hyp is the steady state charge at the hyperpolarizing limit and depends on the holding potential, V0.5 is the voltage at which the charge is distributed equally between the two states, z is the apparent valency per cotransporter, e is the electronic charge, k is Boltzmann's constant, and T is the absolute temperature.
RESULTS
Cysteine Mutagenesis and Identification of One Methanethiosulfonate-sensitive Construct
Fig. 1 shows the location of the residues within the rat isoform of the Na/Pi-IIa protein that we mutated individually to cysteines according to the above criteria. All mutations were confirmed by DNA sequencing and were identical to the WT except for the appropriate base changes. Each of the 15 mutants was expressed in Xenopus oocytes and tested for electrogenic transport activity under whole cell voltage-clamp conditions. For this initial functional assay, oocytes were challenged with a nearly saturating concentration of Pi (1 mM) in the presence of 100 mM Na+, pH 7.4. The Pi-activated current, measured at a holding potential of −50 mV, was compared with that of oocytes expressing the WT protein, obtained from the same donor frog. As shown in Table , six mutants were found to be still active and gave comparable electrogenic responses to the wild type.
Table 1.
Electrogenic Activity of Mutant Constructs
| cRNA | I p | cRNA | I p |
|---|---|---|---|
| nA | nA | ||
| WT | −78.7 ± 10.4 | A393C | −33.3 ± 5.8 |
| N140C | 0 | S418C | 0 |
| S166C | 0 | S434C | 0 |
| S190C | 0 | A460C | −41.3 ± 7.4 |
| L234C | 0 | A513C | 0 |
| A239C | 0 | S532C | −34.7 ± 3.5 |
| S318C | −26.4 ± 5.2 | A538C | −41.3 ± 5.8 |
| S373C | −25.3 ± 4.8 | S583C | 0 |
Cotransport activity of mutants expressed in Xenopus oocytes, assayed as the current induced by 1 mM Pi (I p) in 100 mM Na+, pH 7.4, with the cell voltage clamped to −50 mV. I p values are mean ± SEM (n = 5). Wild-type response is for oocytes from the same donor frog used to express the mutant protein. All mutants that showed expression were exposed to 100 μM MTSEA (2 min) and the response to Pi was retested. Only the response of mutant S460C was suppressed by this procedure.
To test if alkylation of the cysteine residues by the methanethiosulfonate derivative MTSEA would affect the basic transport function, as indicated by the above electrophysiological assay, we incubated oocytes expressing these six active mutants, as well as the WT protein, in 100 μM MTSEA, and then retested for activity under the same conditions as before. The Pi-induced change in holding current, exhibited by the WT (Fig. 2 A) as well as five of the active mutants (S318C, S373C, A393C, S532C, and S538C; data not shown), was unaffected by MTSEA (Table ). In contrast, after incubation in MTSEA, the electrogenic response of mutant S460C showed a significant inhibition (Fig. 2 A) during application of 1 mM Pi. Moreover, prolonged incubation (up to 30 min) in the standard bath medium did not lead to a restoration of function (data not shown). We also found that after alkylation, 32P uptake of S460C was completely suppressed (data not shown), which confirmed that Pi transport was fully inhibited. To demonstrate that the suppression of electrogenic response was an effect of the alkylation and not simply due to the addition of charge in this region (MTSEA is positively charged), we repeated the experiment with the negatively charged MTS reagent, MTSES. Like MTSEA, incubation in 100 μM MTSES, also induced a positive shift in the baseline current during Pi application; i.e., the normal inward current induced by Pi was fully suppressed (data not shown). We also incubated oocytes in 100 μM MTSET, which has been reported to be less permeant than MTSEA (Holmgren et al. 1996) and we obtained the same suppression of Pi response (n = 3). A representative record is shown in Fig. 2 A. This result suggested that both MTSEA and MTSET were acting extracellularly. Moreover, even at high concentrations (1 mM), all three reagents (MTSEA, MTSET, MTSES) had no effect on the electrogenic WT response (data not shown). This was also confirmed in uptake experiments in which there was no statistical difference in 32P uptake between WT-expressing oocytes exposed to 1 mM MTSEA, MTSET, or MTSES and control oocytes. In each case, the 32P uptake was >20-fold higher than that of water-injected oocytes from the same donor frog (results not shown). In contrast to the lack of effect of MTSEA, MTSET, and MTSES on the WT, we did observe a dose dependency of 32P uptake in WT-expressing oocytes exposed to the membrane-permeant reagent methyl-MTS (Lambert, G., J. Biber, and H. Murer, manuscript submitted for publication). This further supported our conclusion that MTSEA, MTSES, and MTSET were only acting extracellularly. All remaining experiments were performed with MTSEA unless otherwise indicated.
Figure 2.
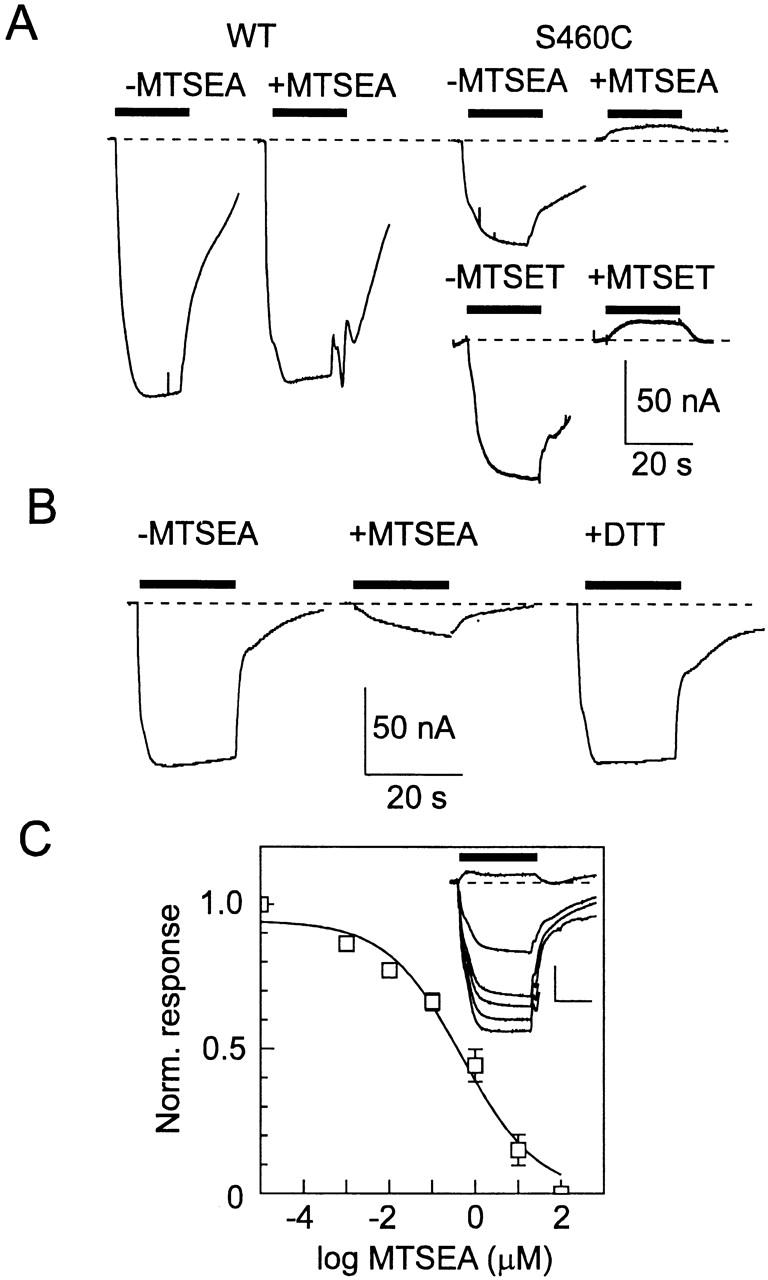
Alkylation using MTSEA leads to a suppression of the electrogenic response in oocytes expressing mutant S460C. (A) Comparison of the Pi-induced current for an oocyte expressing the WT NaPi-IIa before and after exposure to 100 μM MTSEA (left) and two oocytes expressing S460C before and after exposure to 100 μM MTSEA or MTSET for 2 min (right). Bars represent time of application of 1 mM Pi. Dashed line indicates baseline holding current level. Note that Pi induces an upward deflection in the holding current after alkylation. (B) Restoration of Pi response by 15-min incubation in 10 mM DTT (right) for a cell expressing S460C. Incubation in 1.5 μM MTSEA exposure (middle) inhibited the initial Pi response (left) by 80%. (C) Dependency of the Pi response on MTSEA concentration. Inset gives a set of original records showing Pi response (applied during bar) for an oocyte expressing S460C after exposure to successive 10-fold increasing doses of MTSEA from 0.001 to 10 μM. Scale: vertical 50 nA, horizontal 20 s. For this cell, after exposure at 10 μM MTSEA, Pi induced an upward deflection of the baseline current. There was no change in the response after exposure to 100 μM MTSEA. Dose response is pooled from five oocytes. The Pi-induced response was calculated relative to the Pi-induced current after alkylation in 100 μM MTSEA, and was then normalized to the initial response for each cell. Curve is a fit of to the data, which gave an apparent half-maximal concentration = 0.5 μM.
As a further confirmation that chemical modification (alkylation) of Cys-460 was involved, we incubated oocytes that had been previously exposed to either MTSEA (n = 3) or MTSES (n = 3) in the reducing reagent dithiothreitol (DTT, 10 mM, 15 min) and retested for functional activity. As shown in Fig. 2 B for a representative oocyte expressing S460C, exposure to 1.5 μM MTSEA suppressed the Pi-induced response to ∼30% of the initial magnitude, and subsequent DTT incubation restored the Pi-activated response almost to the original level. This finding was consistent with dealkylation occurring in Cys-460, which would thereby restore the original Cys residue and cotransporter function.
The effect of MTSEA on Pi-induced response for S460C was both time and dose dependent and short MTSEA exposures (≤30 s) resulted in large variations (up to 50%) in the amount of suppression of the Pi-activated response for oocytes from the same batch. Although the speed of recovery from Pi application prevented repeated testing of the Pi response after exposure to MTSEA for times shorter than 1 min, application of 100 μM MTSEA, together with continuous application of Pi, showed that the suppression of Pi-induced inward current was complete within 2 min (data not shown). The optimal concentration range was determined from dose-response data (Fig. 2 C) whereby the Pi response (1 mM) was tested after successive 2-min applications of increasing concentrations of MTSEA. These data gave an apparent half-maximal concentration of MTSEA = 0.5 μM (n = 5). MTSEA concentrations up to 100 μM did not result in a further change in the Pi response. In all subsequent experiments, therefore, we routinely applied MTSEA at ≥10 μM for 2–3 min. The response to 1 mM Pi recorded from oocytes expressing the WT NaPi-IIa under the same conditions with repeated application of increasing MTSEA concentrations decreased by ∼20%. This was within the normally observed rundown limits for NaPi-IIa when superfused for periods exceeding 30 min (Forster et al. 1999b) and was therefore not attributable to MTSEA exposure.
Finally, to establish that the loss of transport function by S460C was due to a specific reaction of MTSEA with the Cys-460, we incubated oocytes expressing mutant S460C, as well as the WT protein, in biotin-labeled MTSEA (biotin-MTSEA) and precipitated the protein with immobilized streptavidin (see materials and methods). Expression of both proteins was confirmed by Western blot of the lysate before streptavidin precipitation. This showed that both were expressed at comparable levels (Fig. 3 A). However, as indicated by the immunoprecipitation shown in Fig. 3 B, only the mutant protein and not the WT could be precipitated after incubation with the biotin labeled MTSEA.
Figure 3.
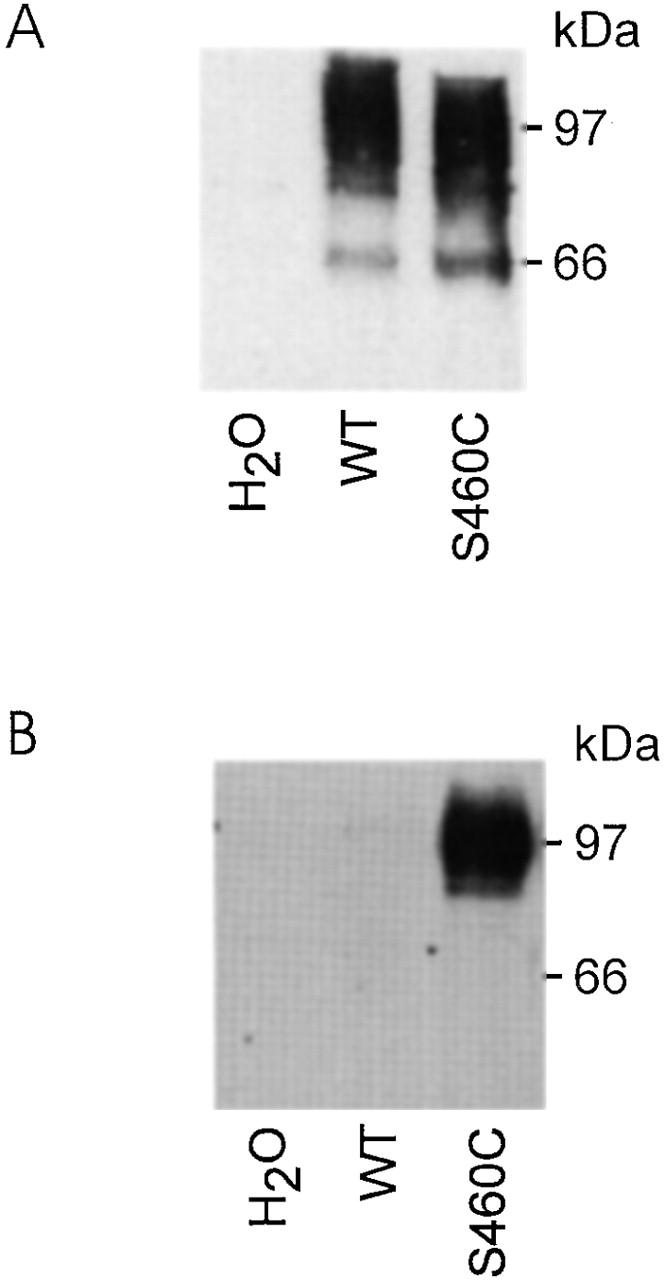
Immunodetection of WT and S460C protein. (A) Western blot obtained from a pool of five oocytes injected either with water, WT, or S460C cRNA. 10 μl of the lysates was separated on a 9% SDS gel and, after blotting, immunoreactive proteins were visualized by incubation with an antibody against the rat NaPi IIa NH2 terminus. This blot confirms that lysate from oocytes expressing S460C presents a similar band (97 kD) to the WT. (B) Streptavidin precipitation of oocyte lysate obtained from pools of five oocytes injected either with water, wild type, or S460C cRNA and incubated with 100 μM MTSEA-Biotin. Cells were then homogenized in 100 μl buffer (see materials and methods) and 90 μl of each lysate was incubated with Streptavidin beads. After washing, bound proteins were eluted with loading buffer (see materials and methods) and the elute was then treated as for A. Single band at 97 kD confirms that the alkylated protein was S460C.
Mutant S460C Displays Electrogenic Characteristics Typical for Type II Na+/Pi Cotransporters
The electrophysiological assay used above provided only a basic confirmation that mutant S460C behaved like the WT. Before making a detailed characterization of the effect of MTS reagents on S460C, we examined whether the replacement of Ser-460 with a cysteine altered any of the specific properties of the cotransporter that have been previously identified from steady state and pre–steady state measurements of the WT (Forster et al. 1998).
We first confirmed that S460C exhibited a dose dependency for the respective substrates (Pi, Na+) that was consistent with the WT. These findings are shown in Fig. 4 A for the Pi-activated dose response and B for the Na+-activated dose response, pooled from representative oocytes expressing the mutant S460C. In each case, a set of original records at the substrate test concentrations is given for a representative oocyte. These were indistinguishable from the typical WT responses under the same conditions (data not shown). For both substrate activation data sets, the steady state currents at the test concentration were normalized to the maximum current predicted from a fit to the whole data set for each cell using the modified Hill equation (). The Pi-activated response was determined at 100 mM Na+ and fits to the data gave a Hill coefficient, nPi = 1.04 ± 0.1 mM and an apparent affinity for Pi (K m Pi) of 0.08 ± 0.01 mM. The Na+ dose response was determined at 1 mM Pi and fits to the data gave a Hill coefficient, nNa = 2.4 ± 0 2 mM and an apparent Na+ affinity (K m Na) of 56 ± 4 mM. These parameters were sufficiently close to the previously reported values for the WT under the same measurement conditions (e.g., nPi = 0.96, K m Pi = 0.057, nNa = 2.9, K m Na = 52 mM; Forster et al. 1998), as well as control oocytes expressing the WT tested in the present study (data not shown), for us to conclude that neither the apparent substrate affinities nor the inferred stoichiometry was affected by the Cys mutation. One further steady state property, which characterizes type IIa Na+/Pi cotransport, namely pH sensitivity, was also found to be unchanged in the mutant S460C when compared with the WT expressed in oocytes from the same batch. In 100 mM Na+, a reduction in superfusate pH from 7.4 to 6.2 gave a 55 ± 4% (n = 5) suppression of the Pi-induced response (1 mM total Pi) compared with 58 ± 4% (n = 5) for the WT.
Figure 4.
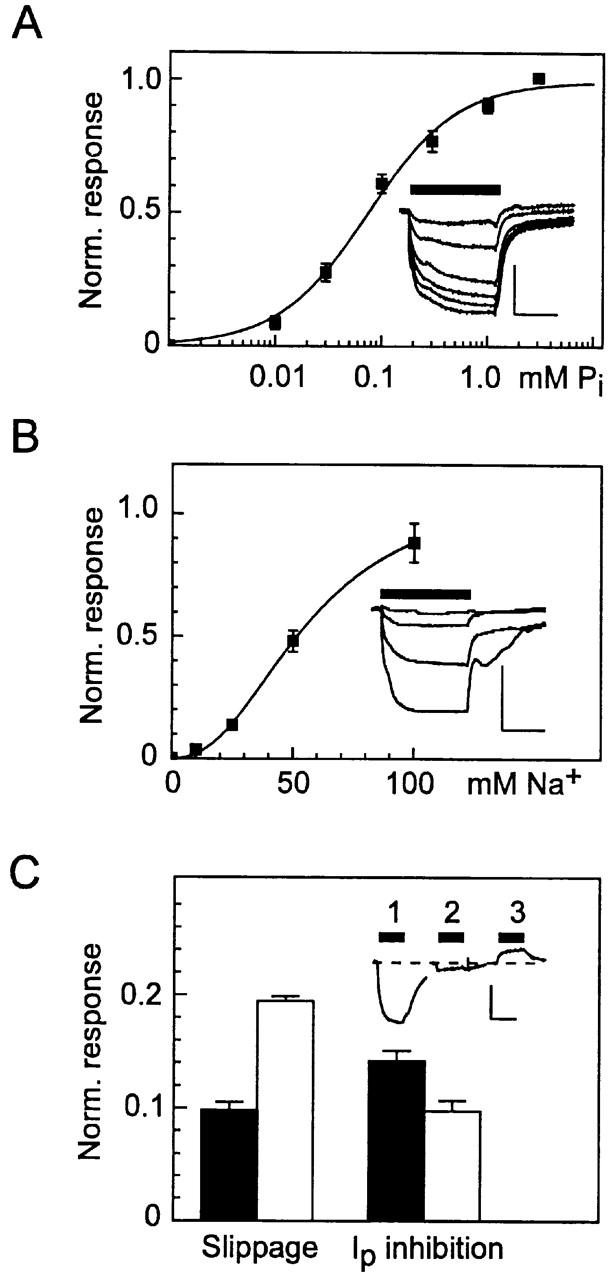
Steady state kinetics of oocytes expressing S460C. (A) Pi dose response determined from original records such as shown in the inset (scale: vertical, 50 nA; horizontal, 10 s) at 100 mM Na+. Data points are pooled from four oocytes from the same batch. was fit to the dose–response data for each cell and the data points were normalized to the predicted maximum current. Continuous line is refit of to the pooled data, giving a Hill coefficient, nPi = 1.04 ± 0.07 and apparent Pi affinity, K m Pi = 0.081 ± 0.01 mM. (B) Na+ dose response determined from original records such as shown in the inset (scale: vertical, 50 nA; horizontal, 10 s), at 1 mM Pi. Data points are pooled from three oocytes from the same batch. Data were treated as in A. Fit of (continuous line) gave a Hill coefficient nNa = 2.35 ± 0.21 and apparent Na+ affinity K m Na = 56.3 ± 4.0 mM. (C) Effect of PFA on the slippage mode (left) and cotransport mode (right) for WT (filled bars) (n = 9) and S460C (open bars) (n = 5). Inset shows an original recording from a cell expressing S460C: (1) response to 0.3 mM Pi, (2) response to 0.3 mM Pi and 3 mM PFA, (3) response to 3 mM PFA. Traces have been aligned to the baseline current in the absence of substrate (dashed line). For the slippage mode assay, bars represent the ratio of trace 3 response to trace 1 response. For cotransport mode assay, bars represent the ratio of trace 2 response to trace 1 response, both relative to the level in the presence of PFA alone (trace 3).
In the absence of Pi and presence of Na+ in the external medium, type IIa cotransporters exhibit a Na+-dependent slippage (or leak) current. For the WT expressed in oocytes, this is inhibited by the Pi analogue and competitive inhibitor for Na+/Pi cotransport, PFA (Forster et al. 1998). As shown in Fig. 4 C (inset) for a representative oocyte expressing S460C, when challenged with both 0.3 mM Pi and 3 mM PFA, a significant suppression of the Pi-activated response occurred (trace 2). Moreover, when oocytes were challenged with PFA alone (trace 3), the holding current at −50 mV was reduced. Pooled data from oocytes expressing the WT or S460C from two donor frogs confirmed this behavior (Fig. 4 C). The data were normalized with respect to the near saturating Pi response to take account of different expression levels. Whereas, the relative inhibition of the Pi response by PFA was similar for both WT and S460C, the latter showed a twofold larger PFA-sensitive current relative to the respective Pi-induced response. This finding suggested that the mutation had altered the kinetics of the cotransporter in the slippage mode.
Voltage dependence was the final property of type IIa Na+/Pi cotransport investigated for the S460C mutant, normally characterized in terms of steady state and pre–steady state behavior. Fig. 5 A shows the steady state voltage dependence of the Pi-induced current (left current–voltage plot), which was obtained by subtracting the holding current in the absence of Pi from that under saturating Pi (1 mM) and 100 mM Na+ (pH 7.4). These data indicate that the voltage dependence of the WT and S460C were indistinguishable for V < 0 mV. Moreover, the voltage dependence of the normalized slippage current (right current–voltage plot) for the WT and S460C, using 3 mM PFA as the blocking agent, was essentially unchanged.
Figure 5.
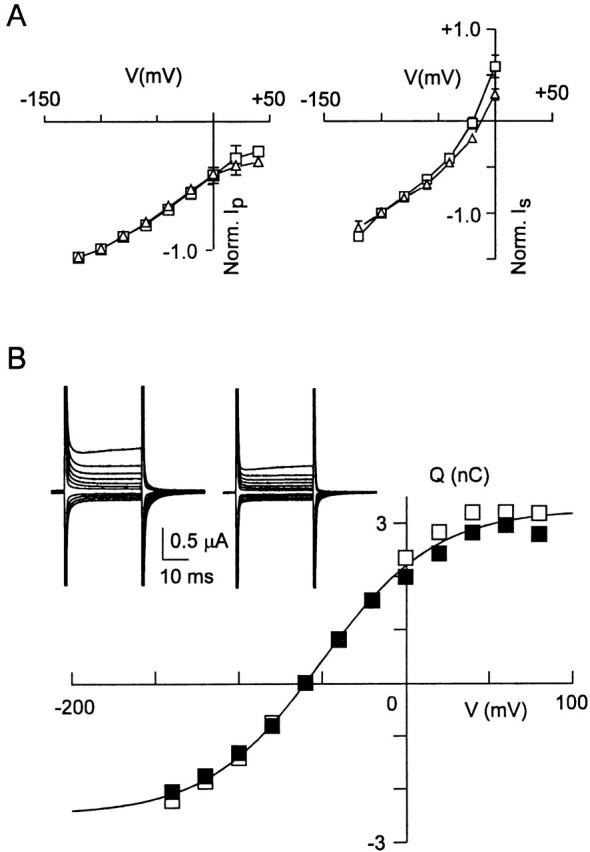
Mutant S460C and wild type show comparable voltage dependence. (A) Steady state current–voltage curves for representative cells expressing wild type (□) and S460C (▵) for the cotransport (left) and slippage (right) modes. For cotransport mode, the current (I p) represents difference between current induced by 1 mM Pi and current in absence of Pi normalized to the value at −100 mV (n = 4). For slippage mode, the current (I s) represents difference between the holding current and current induced by 3 mM PFA, normalized to the value at −100 mV (n = 4). SEMs smaller than symbol size are not shown. (B) Pre–steady state relaxations induced by voltage steps from −60 mV holding potential to voltages in the range −140 to +80 mV in ND100 solution. Inset shows original records before (left) and after (right) application of 3 mM PFA. Q–V curve found by integrating the transient current after subtraction of the PFA response. (▪) ON charge movement, (□) OFF charge movement. Continuous line is fit of to mean of ON and OFF charges, which gave fit parameters: Q max = 5.7 nC, z = 0.7; V0.5 = −51 mV.
Pre–steady state charge movements result from voltage-dependent steps in the type II Na+/Pi cotransporter kinetics (Forster et al. 1997, Forster et al. 1998). Fig. 5 B (inset) shows typical pre–steady state relaxations recorded from a representative oocyte expressing S460C. These relaxations were also blocked by 3 mM PFA, as previously reported for the WT (Forster et al. 1998). The voltage dependence of the apparent charge movement was determined by integrating the current relaxations that remained after subtraction of the PFA response. This procedure was used to eliminate any charge movements not specifically related to S460C that could result from upregulation of other membrane proteins stimulated by injection of S460C cRNA. As illustrated in Fig. 5 B, the Q–V curve for a representative oocyte expressing S460C saturated at extreme potentials for both ON and OFF charge movements. Boltzmann fits to the mean of the ON and OFF charge () gave a midpoint voltage V0.5 = −54 ± 5 mV and apparent valency, z = 0.7 ± 0.4 (n = 5). A representative oocyte expressing the WT protein under the same measurement conditions gave V0.5 = −50 mV and z = 0.6. The turnover φ at −50 mV of the cotransporter can be estimated from :
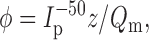 |
3 |
where I p −50 is the Pi-induced current at Vh = −50 mV. For S460C, substitution of the Boltzmann fit data gave φ = 13.5 ± 2.3 s−1 (n = 5), compared with φ = 14 s−1 for the WT.
These data indicate that neither the voltage dependence of the steady state charge distribution nor the apparent valency of the cotransporter substantially changed after mutagenesis. Moreover, the transport turnover in the cotransport mode was unchanged.
Sensitivity of Mutant S460C to Methanethiosulfonate Reagents
A noteworthy result of incubation in MTSEA on the Pi response was the reduction of holding current during Pi application (Fig. 2 A). We investigated this further by testing the response to 3 mM Pi or 3 mM PFA before and after 100 μM MTSEA incubation as shown for a representative oocyte in Fig. 6. These substrate concentrations were chosen to ensure saturation of the responses. After alkylation, the response to PFA remained unchanged, whereas the Pi response was now identical to the PFA response. Moreover, this behavior was found to be consistent for all potentials in the range −80 to 0 mV (data not shown). This result suggested that: (a) alkylation of Cys-460 did not affect the slippage mode, (b) Na+, the cation responsible for slippage current, was still able to bind to the carrier after alkylation, and (c) Pi can still bind to the carrier after alkylation, but subsequent cotransport was suppressed. Noninjected oocytes from the same batch showed small changes in holding current with either Pi or PFA application, under the same conditions, but which were <10% of the responses recorded from S460C-expressing oocytes.
Figure 6.

Effect of alkylation by MTSEA on Pi and PFA response for cells expressing S460C. Substrate was applied during the period indicated by the bar. Cell was voltage clamped to −50 mV. Note that after alkylation, the 3 mM Pi and 3 mM PFA responses superimpose and are identical to the PFA response before alkylation. Vh = −50 mV.
It has been recently reported for SGLT-1 that membrane voltage and substrates can confer an apparent protection to cysteine residues from externally applied MTS reagents (Loo et al. 1998). We therefore investigated whether similar effects could be observed with mutant S460C. We found that 10 μM MTSEA applied together with (a) 1 mM Pi, (b) 3 mM PFA, or (c) Na+ replaced by 100 mM N-methyl-d-glucamine offered no protection since subsequent application of 1 mM Pi resulted in full suppression of the Pi-activated inward current (data not shown). In each case, the membrane potential was held at −50 mV during the entire protocol and MTSEA was applied only after a steady state holding current had been reached under the specific superfusion conditions.
The currently proposed kinetic scheme for type II Na+/Pi cotransport (see Fig. 9) predicts that a voltage step will induce pre–steady state relaxations, contributed by the empty carrier and Na+ binding/debinding, before reaching the final steady state in the slippage mode. Since this mode appeared to be unchanged by alkylation, we would still expect pre–steady state charge movements to be detectable after alkylation. Fig. 7 shows pre–steady state relaxations recorded from a representative oocyte for voltage steps to five test potentials in the presence (A) and absence (B) of external Na+. As before, the endogenous capacitive charging transient was removed by subtracting the response to 3 mM PFA in ND100. Although MTSEA treatment resulted in a significant apparent suppression of relaxations, there was still a charge movement detectable after the endogenous membrane charging was complete (typically after 1–1.5 ms). Moreover, the relaxations were significantly faster in ND0 solution, which indicated that alkylation had altered the kinetics of the empty carrier. The available signal resolution and low expression levels (steady state currents induced by 1 mM Pi were typically 100–150 nA) prevented a full analysis of these relaxations. Nevertheless, single exponential fitting of relaxations induced by large voltage steps indicated that, in 0 mM Na+, alkylation led to an approximately eightfold faster time constant, as shown in Table for three test potentials. Since the relaxations recorded from S460C-expressing oocytes after MTSEA treatment were comparable with the speed of membrane charging, we also confirmed that, under the same perfusion conditions, significant charge movements could not be detected from noninjected oocytes from the same batch once the main capacitive charging was complete.
Figure 9.

Kinetic Scheme for Type II Na+/Pi Cotransport. States occupied in the cis (outward facing) orientation are 1–5. States occupied in the trans (inward facing) orientation are 6–10. The three modes of operation that involve transmembrane reorientation, as revealed by steady state and pre–steady state studies of WT, are indicated by dark shading: empty carrier (10 ⇔ 1), slippage (2 ⇔ 9), and cotransport (5 ⇔ 6). At least two voltage-dependent transitions have been identified: empty carrier (10 ⇔ 1) and first external Na+ binding/release step (1 ⇔ 2). The voltage dependence of the last Na+ binding/release step on the trans side (9 ⇔ 10) has not been characterized. PFA binding places the system in state 2*, which when occupied prevents the slippage and cotransport modes. The lightly shaded region indicates those transitions and associated states that have been shown to remain intact after alkylation. In addition, the zero voltage rate constants for the empty carrier mode increase after alkylation (see text).
Figure 7.
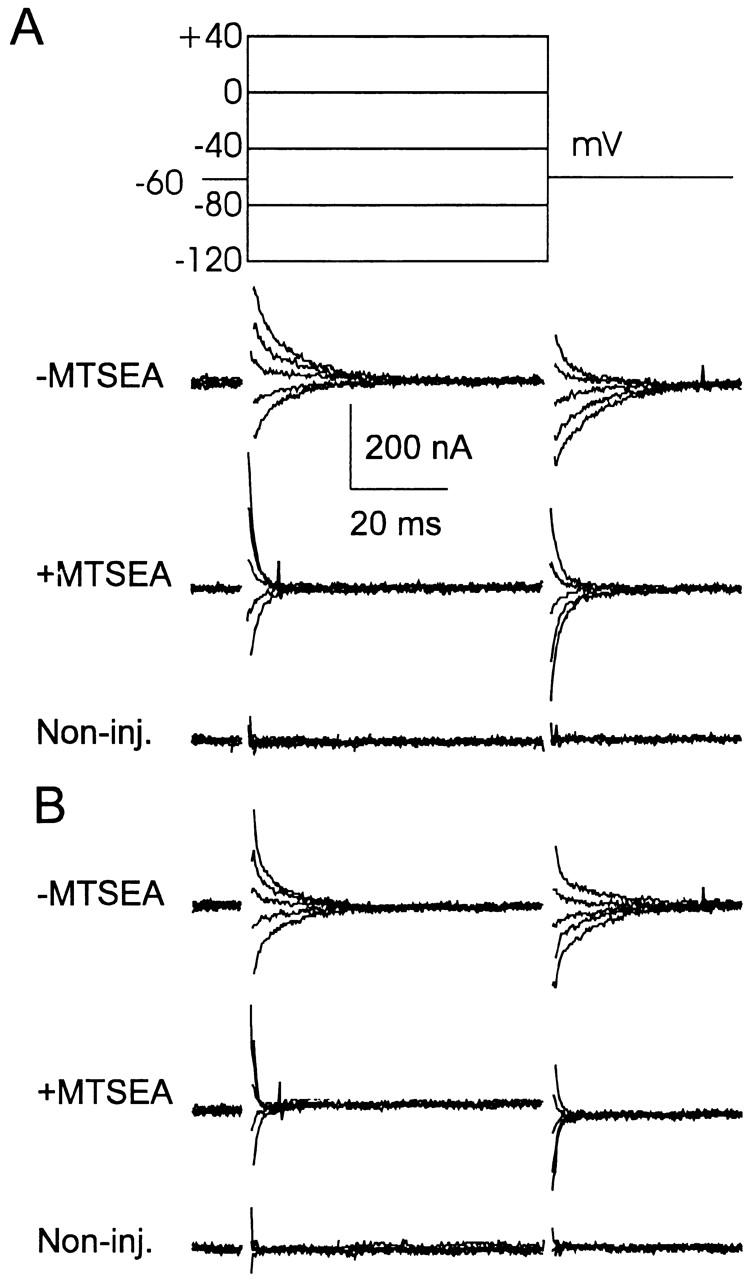
Pre–steady state charge kinetics are faster after MTSEA incubation. Recordings of pre–steady state relaxations from a representative oocyte expressing S460C, superfused in ND100 solution (A) and ND0 solution (B) before MTSEA application (100 μM, 2 min, top) and after MTSEA application (middle). Bottom traces were recorded from a noninjected oocyte from the same batch under the same superfusion conditions (without MTSEA). In each case, oocytes were voltage clamped at −60 mV holding potential and records are shown for voltage steps according to the protocol in A. Each record is the average of eight sweeps with records obtained in 3 mM PFA, 100 mM Na+ was subtracted to eliminate capacitive charging transient. Records were low-pass filtered at 2 kHz and sampled at 50 μs/point. All records were blanked for the first 1.5 ms during the charging period of the oocyte. For this cell, the response to 3 mM Pi before MTSEA was −100 nA, and after MTSEA it was +15 nA relative to the holding current at −50 mV.
Table 2.
The Effect of Alkylation by MTSEA on Pre-Steady State Kinetics
| Test condition | τ (ms) at voltage (mV) | |||
|---|---|---|---|---|
| MTSEA | Na+ | −120 | 0 | +60 |
| mM | ||||
| − | 100 | 5.24 ± 0.2 | 6.8 ± 0.9 | 2.4 ± 0.4 |
| + | 100 | 2.48 ± 0.4 | 1.8 ± 0.3 | 1.2 ± 0.1 |
| − | 0 | 7.7 ± 0.2 | 7.9 ± 0.7 | 5.4 ± 0.7 |
| + | 0 | 1.0 ± 0.3 | 0.9 ± 0.2 | 0.7 ± 0.1 |
Time constants (t) measured by single exponential curve fitting to main component of the pre–steady state relaxations in response to voltage steps from a −60-mV holding potential to the indicated voltage. The endogenous charging component was eliminated by subtracting the response to PFA (see text). Data is pooled from five oocytes expressing S460C before (−) and after MTSEA (+) treatment.
In a final set of experiments, we investigated whether holding potential (Vh) during MTSEA application would also protect against alkylation as reported in the case of SGLT1 (Loo et al. 1998). As illustrated by the original recordings from representative oocytes expressing S460C (Fig. 8 A), when MTSEA was applied during a depolarization to +20 mV in ND100 solution, the subsequent response to Pi at −50 mV was identical to that obtained when MTSEA was applied at −50 mV (Fig. 6). This indicated that in ND100 depolarization to +20 mV did not protect Cys-460. However, when MTSEA was applied in ND0 solution, the magnitude of the Pi response was now dependent on Vh, whereby less inhibition was observed at Vh = +20 mV compared with the response after alkylation at −50 mV. We repeated this protocol for individual oocytes voltage clamped to Vh = +20, 0, and −20 mV during MTSEA exposure, and the pooled results (Fig. 8 B) indicate a clear voltage dependence of inhibition of response by the MTS reagent in 0 mM Na+. For each oocyte tested, we confirmed that exposure to MTSEA at Vh = −50 mV in 100 mM Na+ gave complete inhibition (i.e., the same response during PFA exposure; Fig. 6). We were unable to determine whether at more depolarized Vh further protection of the MTS action was possible because continuous voltage clamping of oocytes at potentials exceeding +20 mV during the MTSEA application period resulted in an irreversible and progressive increase in endogenous leak current. This protocol was also repeated with the anionic MTSES with similar results (data not shown). This confirmed that accessibility to Cys-460 was independent of the charge of the alkylating reagent.
Figure 8.
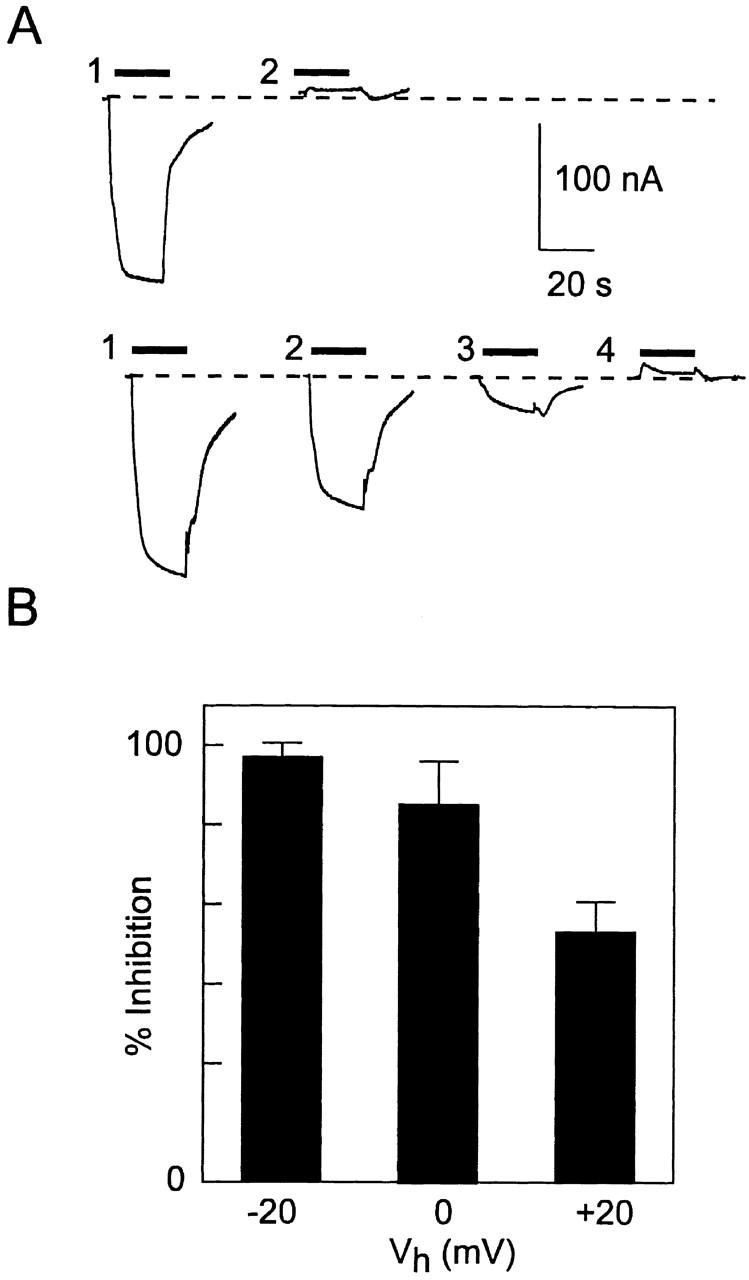
Membrane potential protects against MTSEA suppression of Pi response only in the absence of external Na+. (A) Original recordings from two representative oocytes (top and bottom) from the same donor frog expressing S460C before and after a 2-min exposure to 10 μM MTSEA at different holding potentials. After alkylation, the response was tested each time with 1 mM Pi at −50 mV in ND100. (Top, 1) Initial response, (2) response after alkylation at +20 mV holding potential in ND100. (Bottom, 1) Initial response, (2) response after alkylation at +20 mV in ND0, (3) response after alkylation at −50 mV in ND0, (4) response after alkylation at −50 mV in ND100. The dashed line represents the initial holding current level before Pi application. (B) Pooled data of the inhibition of the Pi response after alkylation in 0 mM Na+, at three holding potentials. n = 3 (−20 mV, 0 mV); n = 5 (+20 mV). For all cells, after reexposure to MTSEA at −50 mV in ND100 solution, the Pi response was the same as the PFA response. The percent change in the Pi-induced electrogenic response was expressed as: 100 · [1 − (I p + + I s)/(I p − + I s)], where I p − and I p + are the Pi-induced current before and after MTSEA exposure, respectively, I s is the PFA-induced change in holding current (slippage current), with all currents expressed as magnitudes. It was assumed that the slippage was fully suppressed by 3 mM PFA so that the true Pi-induced response for saturating Pi was given by the change in current relative to the holding current during PFA exposure.
DISCUSSION
In this study, we investigated the effect of membrane impermeant alkylating reagents on mutant constructs of the type IIa Na+/Pi cotransporter (rat NaPi-IIa). Amino acid residues, hypothesized to be in functionally sensitive regions according to specific criteria based on the topological scheme for NaPi-IIa (Fig. 1), were replaced with cysteine residues. Six of the sites were located intracellularly according to this scheme and might be considered inaccessible to externally applied MTS reagents. Nevertheless, we included them in this study since we could neither fully exclude other candidate topologies (Paquin et al. 1999), nor exclude the possibility that conformational changes might render these sites accessible. In the present study, we have restricted our investigation to the effects of externally applied MTS reagents since it has been shown that the WT response is also inhibited when exposed to membrane-permeant MTS reagents (Lambert et al. 1999). Therefore, our finding that external MTSEA did not affect two functional mutants with cysteines predicted to be on the trans side (S373C and A393C) does not exclude the possibility that they are located in functionally important areas. Further investigations using internally applied MTS reagents and cysteine deletions would be necessary to confirm whether or not these residues are functionally relevant.
Cys-460 Is Alkylated from the cis Side
Of the six functional mutants, only Pi-induced electrogenic response of construct S460C was suppressed by MTSEA. Since this was reversible by DTT, and the WT protein was insensitive to the externally applied MTS reagents MTSEA, MTSES, and MTSET, these findings suggested that only Cys-460 was strongly implicated as the site of alkylation. Moreover, tracer flux studies confirmed that alkylation had indeed suppressed 32P uptake. The immunodetection experiments confirmed that alkylation had left the protein intact and furthermore provided additional evidence that Cys-460 was located extracellularly (predicted to be in the third extracellular loop) (Lambert et al. 1999) since S460C, but not the WT, could be streptavidin-precipitated after incubation with biotin labeled MTSEA. An alternative hypothesis to explain the action of MTS reagents on S460C could be that mutagenesis has exposed another cysteine that is accessible for alkylation. Although we cannot fully exclude this possibility based on our present data, this would appear unlikely since, with the exception of augmented slippage current (see below), the basic kinetic properties of S460C are identical to the WT. We expect that the conformational changes that accompany exposure of another cysteine in a functionally sensitive region also result in significant changes in the cotransport kinetics.
It has been reported that MTSEA is slightly membrane permeant (Holmgren et al. 1996) and this could lead to difficulties in distinguishing between an effect exclusively related to external cysteines and one also involving internal cysteines. In our hands, we observed no change in the WT response after incubation in either of the more charged reagents, MTSET, MTSES, or MTSEA, even up to 1 mM concentrations. In contrast, the membrane-permeant reagent methyl-MTS has been shown to cause a suppression of the Pi-induced response of oocytes expressing the WT protein (our unpublished results). This indicated that alkylation of internal cysteines can interfere with transport function. In the present study, the insensitivity of the WT to all three nominally impermeant reagents, together with the specificity of their effect on S460C, led us to conclude that these reagents were only acting from the cis side.
S460C Behaves According to the Ordered, Alternating Access Model for Type II Na+/Pi Transport
Based on steady state and pre–steady state kinetics of the WT rat NaPi-IIa and flounder NaPi-IIb isoforms, we have proposed a kinetic model for type II Na+/Pi cotransport (Forster et al. 1997, Forster et al. 1998) depicted in Fig. 9. This model belongs to the alternating access or gated channel class of cotransporter models (e.g., Läuger 1991) proposed for other Na+-dependent cotransport systems, including the sodium–glucose cotransporter (Kessler and Semenza 1983; Parent et al. 1992; Loo et al. 1993,Loo et al. 1998). It predicts that accessibility of substrates to their respective binding sites on the cis and trans sides of the membrane relies on a reorientation of the protein, the favored state of which (cis or trans facing) is determined by substrate availability on the respective sides and transmembrane potential. Charge movements (measured as pre–steady state relaxations) induced by voltage steps are predicted to arise from the movement of charges intrinsic to the carrier, as well as the translocation of charged substrates to their binding sites within the transmembrane field. This scheme contrasts with the channel-like substrate hopping model proposed recently (Su et al. 1996) in which major molecular conformational changes do not occur and charge movements arise exclusively from movement of charged substrates within a channel-like pore, assumed to contain multiple binding sites. The recent findings of Loo et al. 1998 support an alternating access type model for SGLT-1, whereby conformational changes are responsible for the coupling of Na+ and sugar transport. Changes in fluorescence of a rhodamine-labeled cysteine residue and pre–steady state relaxations were correlated, as well as the accessibility of this cysteine to MTS reagents and the steady state charge distribution.
In Fig. 9, at least three modes of operation are possible. In the empty carrier mode, the orientation of the charged empty carrier is favored towards the cis side (state 1) when the membrane is depolarized, which increases the accessibility of Na+ ions to a binding site. After the binding of one Na+ ion (transition 1 ⇔ 2), the system operates in the slippage mode, whereby one Na+ ion forms a neutral complex with the empty carrier and slippage occurs through the protein (transition 2 ⇔ 9) in the absence of Pi. Occupancy of state 2 increases the affinity of the protein for Pi, which then binds (transition 2 ⇔ 3) in its divalent form (Forster et al. 1998), together with two additional Na+ ions (transitions 3 ⇔ 4 and 4 ⇔ 5). In this cotransport mode (transition 4 ⇔ 5), reorientation of the fully loaded, neutral carrier to the trans side (transition 4 ⇔ 5) is now favored, and release of the substrates can occur as a result of the low internal Na+ concentration. The cycle is completed by a reorientation of the empty carrier (transition 10 ⇔ 1).
The Pi analogue PFA, which inhibits both slippage and cotransport modes, is assumed to place the system in state 2* when bound. This is consistent with the findings of Busch et al. 1994, who demonstrated that PFA acts as a competitive inhibitor for Pi by shifting the apparent K m for Pi without changing the apparent maximum electrogenic transport rate, Vmax. Since Vmax is determined by the final Na+ binding steps (Forster et al. 1997, Forster et al. 1998). PFA would be expected not to interact directly with these steps, but rather to compete for occupancy of the Pi binding site. Details of kinetics in the trans conformation (states 5–10) are as yet unknown; however, the validity of this model, under zero trans conditions, has been confirmed by simulations in which the main features of steady state and pre–steady state kinetics are adequately predicted (Forster et al. 1998). Comparison of the kinetics of S460C and the WT rat NaPi-IIa indicated that the cysteine substitution did not alter the properties of the cotransport mode. The apparent affinities for both substrates, pH dependence, and voltage dependence were all similar to the WT. Moreover, oocytes expressing S460C gave pre–steady state relaxations both in the presence and absence of external Na+, as predicted from the kinetic scheme (Fig. 9). Of significance was a 50% increase slippage current for S460C relative to the Pi response compared with the WT. Since the turnover in the cotransport mode appeared to be unaffected by mutagenesis, the increased relative slippage current would most likely result from increased turnover in the slippage mode. Furthermore, since the voltage dependence of the slippage mode was the same for WT and S460C, this suggested that mutagenesis had altered the kinetics of transition 2 ⇔ 9 (see , ). Despite this departure from WT behavior, the general similarity of S460C and the WT indicated that the model scheme (Fig. 9) was also valid for this construct.
Effect of Alkylation on the Kinetics of S460C: Structure–Function Implications
Two questions arise with respect to mutant S460C: (a) Can the behavior of S460C after alkylation be explained in terms of the above kinetic scheme? and (b) Which kinetic transitions are associated with the alkylated Cys-460?
The change in the steady state characteristics after alkylation, whereby saturating Pi induced a reduction in holding current that exactly matched that of PFA both before and after alkylation, suggested that site 460 was located in a functionally sensitive region of the molecule. In terms of Fig. 9, an intact slippage pathway after alkylation indicates that the protein can still cycle around the loop 1 ⇔ 2 ⇔ 9 ⇔ 10 ⇔ 1, as well as occupy state 2* when PFA is bound. Moreover, the identical responses to Pi and PFA after alkylation suggest that state 3 (with Pi bound) is also intact, but alkylation of Cys-460 prevents one or more of the subsequent transitions leading to the cotransport mode. This behavior would also strongly suggest that Pi and PFA bind to the same site.
Further support for this interpretation comes from our finding that pre–steady state charge movements, albeit with significantly faster relaxation time constants, were still detectable after alkylation for both the empty carrier and slippage modes. From our recordings, it appeared that alkylation also caused a suppression of the charge movement, even though the magnitude of the slippage current and its steady state voltage dependence remained unchanged. However, we were unable to resolve charge movements at times earlier than 1.5 ms after the voltage step onset and, therefore, one explanation for this apparent discrepancy between the pre–steady state and the steady state data might be that part of the charge movement simply remained undetected.
To investigate this further, we modeled the behavior of a four-state scheme comprising states 1, 2, 9, and 10 (see ).The model predicts pre–steady state relaxations similar to those observed before and after alkylation. Analysis of the model indicated that the steady state slippage current remains constant, as observed experimentally, if the ratio of the zero voltage rate constants for the empty carrier (transition 1 ⇔ 10) was held constant (see , ). We simulated effect of alkylation by arbitrarily increasing both zero voltage rate constants for this transition 10-fold, to accord with our finding of faster pre–steady state relaxations in 0 mM Na+. In terms of an Eyring–Boltzmann transition rate model, this would imply that alkylation reduces the height of the apparent energy barrier of this step. Since the apparent valencies for the voltage-dependent steps are assumed to remain the same, the increased rates for the empty carrier conformational change after alkylation would not change the overall steady state charge distribution.
Our finding of no difference between anionic and cationic MTS reagents in suppressing the Pi response would further suggest that the charge of the alkylated Cys-460 is not in the electric field, and therefore cannot alter the voltage dependence of the empty carrier. This is also consistent with an invariant steady state charge distribution after alkylation. Loo et al. 1998 have reported similar behavior for the Q457C mutant of SGLT-1, whereby the voltage dependence of accessibility of the MTS reagents was also found to be independent of the valency of the reagent.
That Cys-460 is associated with the conformational state of the empty carrier is also suggested from our finding that in 0 mM Na+ the inhibition of the Pi response was dependent on the holding potential during MTS application; i.e., accessibility to Cys-460 was voltage dependent. In the empty carrier mode, membrane potential determines the probability of occupancy of state 1 or state 10. Since such a voltage-dependent change of state implies the movement of charged species within the membrane, the associated conformational changes are hypothesized to alter the accessibility of Cys-460. In 100 mM Na+, we found no protection with holding potential (between −50 and +20 mV) and, similarly, no protection was observed in the presence of either Pi or PFA (together with 100 mM Na+). These findings suggest that once the protein is in state 2 (Na+ bound), state 2* (PFA bound), or state 3 (Pi bound), Cys-460 is readily accessible by externally applied MTS reagents. Interestingly, in the study by Loo et al. 1998 of the SGLT-1 mutant Q457C, alkylation also resulted in inhibition of the cotransport mode, although glucose binding still occurred, just as our data suggest that Pi can still bind to S460C. However, in contrast to S460C, the behavior of this SGLT-1 mutant after alkylation indicated that MTS reagents could only access the cysteine residue in the equivalent of state 2 (Na+ bound).
Conclusions
Our findings indicate that site 460 is located in a functionally sensitive region of the NaPi-IIa molecule, most likely associated with conformational changes of the empty carrier. As indicated in Fig. 9, states 10, 1, 2, and 3 remain intact after alkylation. The subsequent transition or transitions, which are altered by alkylation and thereby inhibit the cotransport mode, remain to be identified. Our findings complement those of Loo et al. 1998 for SGLT-1 and provide further support for the notion that conformational changes accompany substrate binding in Na+-coupled cotransporters.
Acknowledgments
The work was supported by grants to H. Murer from the Swiss National Science Foundation (31-46523), the Hartmann Müller-Stiftung (Zurich), the Olgar Mayenfisch-Stiftung (Zurich), and the Schweizerischer Bankgesellschaft (Zurich) (Bu 704/7-1).
Simulation of a Four-State Scheme to Predict Behavior in Slippage Mode
The slippage mode was modeled as the four-state “iso uni uni” system depicted in 1 A (Segel 1975) and corresponding to states 1, 2, 9, and 10 of the full model in 9. Transitions between states were modeled according to the Eyring–Boltzmann transition rate theory (Adrian 1978). We assumed that the empty carrier has a valency, z c = −1, that moves an electrical distance δ through the membrane between the external and internal Na+ binding sites. A single Na+ ion moves an equivalent electrical distance α′ on the cis side and α″ on the trans side to the respective binding sites (Läuger 1991; Parent et al. 1992) so that α′ + δ + α″ = 1. The translocation (step 2 ⇔ 3) with Na+ bound is electroneutral. The eight pseudo first order rate constants, assuming symmetrical energy barriers for each transition, are then given by:
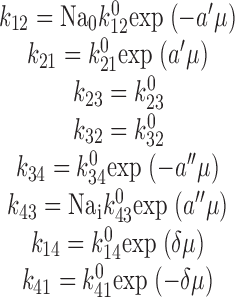 |
and μ = eV/2kT, where, Nao and Nai are the external and internal Na+ concentrations (M), respectively, V is the transmembrane voltage, e is the electronic charge, k is Boltzmann's constant, T is the absolute temperature, and k 0 12, k 0 21, etc., are the rate constants at V = 0.
Steady State and Pre–Steady State Simulations
The time-dependent, net transmembrane current in response to a voltage step is given by the sum of all charge movements that occur within the transmembrane field. Simulations were performed by solving the differential equations describing the transitions using the matrix method to find the eigen values and eigen vectors (Press et al. 1992). Zero voltage rate constants were assigned to each transition based on previous simulations of the WT cotransporter (Forster et al. 1998; see 1, legend).
Figure A1.
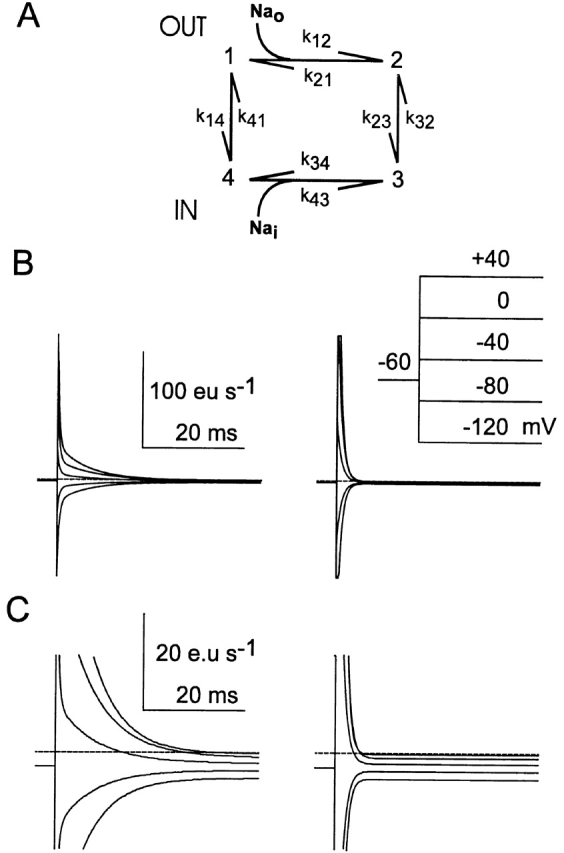
Simulations of slippage mode behavior. (A) Four state kinetic scheme used to simulate the slippage mode. (B) Simulated currents in response to voltage steps indicated in the inset. The parameters for the simulations on the left were: k 0 12 = 8, 000 M−1 s−1, k 0 21 = 2,000 s−1, k 0 23 = 5 s−1, k 0 14 = 120 s−1, k 0 41 = 60 s−1, k 0 34 = 100 s−1, k 0 43 = 1,000 M−1 s−1, α′ = 0.3, α″ = 0.3, δ = 0.4, Nao = 0.1 M, Nai = 0.01 M, T = 20°C. The effect of alkylation (right) was simulated by a 10-fold increase in k 0 14 and k 0 41. (C) Expanded view of the traces in A, which also shows the steady state current levels under the two conditions. The broken line indicates zero baseline current. Note that the current scale (eu, electronic units) gives the apparent charge movement per cotransporter.
The rate constant k 32 was defined according to the condition for microscopic reversibility so that k 32 = k 0 12 k 0 23 k 0 34 k 0 41/(k 0 14 k 0 21 k 0 43). The rate constant k 0 23 was increased twofold to account for the higher slippage observed for S460C. Na+ binding on the trans side was assumed to be voltage dependent, but at low Nai this contributes little to the overall voltage dependence.
1 B shows simulated pre–steady state ON relaxations in response to ideal voltage steps, corresponding to those obtained in 7, before (left) and after (right) alkylation. The effect of alkylation was modeled by assuming that only the zero-voltage rate constants for the empty carrier (k 0 14, k 0 41) were increased 10-fold with no changes to other parameters to account for the faster relaxation time constants measured in 0 mM external Na+ (). Note that for a general four-state system, three nonzero eigen values exist, which would give three time constants in the relaxation. The limited signal resolution available for our data meant that we could resolve a single component that corresponds to the slower component seen in the simulations. The steady state levels preceding the voltage step and after the relaxation is complete are the steady state slippage current at holding potential and target potential, respectively. For the parameters chosen, the expanded traces in 1 C indicate that the slippage current is unaffected by the change in relaxation kinetics, as our data show. Note that the effect of finite voltage-clamp speed and signal filtering on the relaxation time course have not been included in the simulations. Moreover, blanking the first 1.5 ms of the recordings (7) would significantly reduce the amount of detectable charge after alkylation.
A Steady State Approximation
Here we consider the dependence of the steady state current on the rate constants under rate-limiting conditions. The only transmembrane transition involving net charge transfer is 1 ⇔ 4, therefore the steady state current, I s, is given by FDA1 :
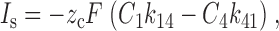 |
FDA1 |
where C 1 and C 4 are the relative occupancies of states 1 and 4, respectively, and F is the Faraday constant. C 1 and C 4 can be found using the King-Altman method (Segel 1975) and, setting z c = −1:
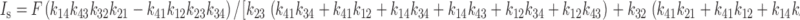 |
A2 |
If transition 2 ⇔ 3 is rate limiting, further simplifies to:
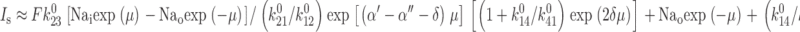 |
A3 |
shows that for fixed Na+ concentrations and apparent valencies, I s is a function of k 0 23 and the ratios of zero voltage rate constants for the other transitions.
Footnotes
1used in this paper: DTT, dithiothreitol; MTS, methanethiosulfonate; MTSEA, 2-aminoethyl MTS hydrobromide; MTSES, sodium(2-sulfonatoethyl)MTS; MTSET, [2-(trimethylammonium) ethyl] MTS bromide; NaPi-IIa, type IIa sodium-phosphate cotransporter; PFA, phosphonoformic acid; SDS, sodium-dodecyl sulfate; SGLT-1, sodium/glucose cotransporter; WT, wild-type
Georg Lambert and Ian C. Forster contributed equally to this work and should be considered co-first authors.
References
- Adrian R.H. Charge movement in the membrane of striated muscle. Annu. Rev. Biophys. Bioeng. 1978;7:85–112. doi: 10.1146/annurev.bb.07.060178.000505. [DOI] [PubMed] [Google Scholar]
- Akabas M.H., Kaufmann C., Archdeacon P., Karlin A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit. Neuron. 1994;13:919–927. doi: 10.1016/0896-6273(94)90257-7. [DOI] [PubMed] [Google Scholar]
- Akabas M.H., Stauffer D.A., Xu M., Karlin A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- Axon Instruments pClamp User's Guide to Clampex and Clampfit 1998. Axon Instruments; Foster City, CA: pp. 217–236 [Google Scholar]
- Bertrand D., Bader C.R. DATACa multipurpose biological data analysis program based on a mathematical interpreter. Int. J. Biomed. Comput. 1986;18:193–202. doi: 10.1016/0020-7101(86)90016-4. [DOI] [PubMed] [Google Scholar]
- Biber J., Custer M., Magagnin S., Hayes G., Werner A., Lotscher M., Kaissling B., Murer H. Renal Na/Pi-cotransporters. Kidney Int. 1996;49:981–985. doi: 10.1038/ki.1996.139. [DOI] [PubMed] [Google Scholar]
- Biber J., Murer H., Forster I. The renal type II Na+/phosphate cotransporter. J. Bioenerg. Biomembr. 1998;30:187–194. doi: 10.1023/a:1020525409894. [DOI] [PubMed] [Google Scholar]
- Busch A., Waldegger S., Herzer T., Biber J., Markovich D., Hayes G., Murer H., Lang F. Electrophysiological analysis of Na+/Pi cotransport mediated by a transporter cloned from rat kidney and expressed in Xenopus oocytes. Proc. Natl. Acad. Sci. USA. 1994;91:8205–8208. doi: 10.1073/pnas.91.17.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.G., Liu-Chen S., Rudnick G. Determination of external loop topology in the serotonin transporter by site-directed chemical labeling. J. Biol. Chem. 1998;273:12675–12681. doi: 10.1074/jbc.273.20.12675. [DOI] [PubMed] [Google Scholar]
- Custer M., Lotscher M., Biber J., Murer H., Kaissling B. Expression of Na-Pi cotransport in rat kidneylocalization by RT-PCR and immunohistochemistry. Am. J. Physiol. 1994;266:F767–F774. doi: 10.1152/ajprenal.1994.266.5.F767. [DOI] [PubMed] [Google Scholar]
- Forster I., Hernando N., Biber J., Murer H. The voltage dependence of a cloned mammalian renal type II Na+/Pi cotransporter (NaPi-2) J. Gen. Physiol. 1998;112:1–18. doi: 10.1085/jgp.112.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster I., Loo D.D.F., Eskandari S. Stroichiometry and Na+ binding cooperativity of rat and flounder renal type II Na+-Pi cotransporters Am. J. Physiol. 276 1999. F644 F649a [DOI] [PubMed] [Google Scholar]
- Forster I., Traebert M., Jankowski M., Stange G., Biber J., Murer H. Protein kinase C activators induce membrane retrieval of type II Na-phosphate cotransporters expressed in Xenopus oocytes J. Physiol. 517 1999. 327 340b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster I.C., Wagner C.A., Busch A.E., Lang F., Biber J., Hernando N., Murer H., Werner A. Electrophysiological characterization of the flounder type II Na+/Pi cotransporter (NaPi-5) expressed in Xenopus laevis oocytes. J. Membr. Biol. 1997;160:9–25. doi: 10.1007/s002329900291. [DOI] [PubMed] [Google Scholar]
- Hayes G., Busch A., Lotscher M., Waldegger S., Lang F., Verrey F., Biber J., Murer H. Role of N-linked glycosylation in rat renal Na/Pi-cotransport. J. Biol. Chem. 1994;269:24143–24149. [PubMed] [Google Scholar]
- Hilfiker H., Hattenhauer O., Traebert M., Forster I., Murer H., Biber J. Characterization of a murine type II sodium-phosphate cotransporter expressed in mammalian small intestine. Proc. Natl Acad. Sci. USA. 1998;95:14564–14569. doi: 10.1073/pnas.95.24.14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren M., Liu Y., Xu Y., Yellen G. On the use of thiol-modifying agents to determine channel topology. Neuropharmacology. 1996;35:797–804. doi: 10.1016/0028-3908(96)00129-3. [DOI] [PubMed] [Google Scholar]
- Kenyon G.L., Bruice T.W. Novel sulfhydryl reagents. Methods Enzymol. 1977;47:407–430. doi: 10.1016/0076-6879(77)47042-3. [DOI] [PubMed] [Google Scholar]
- Kessler M., Semenza G. The small-intestinal Na+, d-glucose cotransporteran asymmetric gated channel (or pore) responsive to ΔΨ. J. Membr. Biol. 1983;76:27–56. doi: 10.1007/BF01871452. [DOI] [PubMed] [Google Scholar]
- Lambert G., Traebert M., Hernando N., Biber J., Murer H. Studies on the topology of the renal type II NaPi-cotransporter. Pflügers Arch. 1999;437:972–978. doi: 10.1007/s004240050869. [DOI] [PubMed] [Google Scholar]
- Läuger P. Electrogenic Ion Pumps 1991. Sinauer Associates Inc; Sunderland, MA: pp. 74–77 [Google Scholar]
- Lo B., Silverman M. Cysteine scanning mutagenesis of the segment between putative transmembrane helices IV and V of the high affinity Na+/glucose cotransporter SGLT1. Evidence that this region participates in the Na+ and voltage dependence of the transporter J. Biol. Chem. 273 1998. 29341 29351a [DOI] [PubMed] [Google Scholar]
- Lo B., Silverman M. Replacement of Ala-166 with cysteine in the high affinity rabbit sodium/glucose transporter alters transport kinetics and allows methanethiosulfonate ethylamine to inhibit transporter function J. Biol. Chem. 273 1998. 903 909b [DOI] [PubMed] [Google Scholar]
- Loo D.D., Hazama A., Supplisson S., Turk E., Wright E.M. Relaxation kinetics of the Na+/glucose cotransporter. Proc. Natl. Acad. Sci. USA. 1993;90:5767–5771. doi: 10.1073/pnas.90.12.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo D.D., Hirayama B.A., Gallardo E.M., Lam J.T., Turk E., Wright E.M. Conformational changes couple Na+ and glucose transport. Proc. Natl. Acad. Sci. USA. 1998;95:7789–7794. doi: 10.1073/pnas.95.13.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magagnin S., Bertran J., Werner A., Markovich D., Biber J., Palacin M., Murer H. Poly(A)+ RNA from rabbit intestinal mucosa induces b0,+ and y+ amino acid transport activities in Xenopus laevis oocytes. J. Biol. Chem. 1992;267:15384–15390. [PubMed] [Google Scholar]
- Murer H., Biber J. A molecular view of proximal tubular inorganic phosphate (Pi) reabsorption and of its regulation. Pflügers Arch. 1997;433:379–389. doi: 10.1007/s004240050292. [DOI] [PubMed] [Google Scholar]
- Murer H., Forster I., Hilfiker H., Pfister M., Kaissling B., Lötscher M., Biber J. Cellular/molecular control of renal Na/Pi-cotransport. Kidney Int. 1998;S2–S10 [PubMed] [Google Scholar]
- Parent L., Supplisson S., Loo D.D.F., Wright E.M. Electrogenic properties of the cloned Na+/glucose cotransporterII. A transport model under nonrapid equilibrium conditions. J. Membr. Biol. 1992;125:63–79. doi: 10.1007/BF00235798. [DOI] [PubMed] [Google Scholar]
- Paquin J., Vincent E., Dugre A., Xiao Y., Boyer J.C., Béliveau R. Membrane Topology of the Renal Phosphate Carrier NaPi-2limited proteolysis studies. Biophys. Biochem. Acta. 1999;1431:315–328. doi: 10.1016/s0167-4838(99)00060-6. [DOI] [PubMed] [Google Scholar]
- Pascual J.M., Shieh C.C., Kirsch G.E., Brown A.M. K+ pore structure revealed by reporter cysteines at inner and outer surfaces. Neuron. 1995;14:1055–1063. doi: 10.1016/0896-6273(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Press W.H., Flannery F.P., Teukolsky S.A., Vetterling W.T. Numerical Recipes in C 1992. Cambridge University Press; Cambridge, UK: pp. 382–397 [Google Scholar]
- Sambrook, J., E.F. Fritsch, and A.M. Maniatis. 1989. Molecular Cloning—A Laboratory Manual. C. Nolan, editor. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 18.66–18.75.
- Seal R.P., Amara S.G. A reentrant loop domain in the glutamate carrier EAAT1 participates in substrate binding and translocation. Neuron. 1998;21:1487–1498. doi: 10.1016/s0896-6273(00)80666-2. [DOI] [PubMed] [Google Scholar]
- Segel, I.H. 1975. Enzyme Kinetics. John Wiley & Sons, New York, NY. 534–535.
- Smith D.J., Maggio E.T., Kenyon G.L. Simple alkanethiol groups for temporary blocking of sulfhydryl groups of enzymes. Biochemistry. 1975;14:766–771. doi: 10.1021/bi00675a019. [DOI] [PubMed] [Google Scholar]
- Stauffer D.A., Karlin A. Electrostatic potential of the acetylcholine binding sites in the nicotinic receptor probed by reactions of binding-site cysteines with charged methanethiosulfonates. Biochemistry. 1994;33:6840–6849. doi: 10.1021/bi00188a013. [DOI] [PubMed] [Google Scholar]
- Su A., Mager S., Mayo S.L., Lester H.A. A multi-substrate single-file model for ion-coupled transporters. Biophys. J. 1996;70:762–777. doi: 10.1016/S0006-3495(96)79616-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk E., Kerner C.J., Lostao M.P., Wright E.M. Membrane topology of the human Na+/glucose cotransporter SGLT1. J. Biol. Chem. 1996;271:1925–1934. doi: 10.1074/jbc.271.4.1925. [DOI] [PubMed] [Google Scholar]
- Werner A., Biber J., Forgo J., Palacin M., Murer H. Expression of renal transport systems for inorganic phosphate and sulfate in Xenopus laevis oocytes. J. Biol. Chem. 1990;265:12331–12336. [PubMed] [Google Scholar]