

Abstract
Hexavalent chromium (Cr(VI)) is a potent respiratory toxicant and carcinogen. The most carcinogenic forms of Cr(VI) are the particulate salts such as lead chromate, which deposit and persist in the respiratory tract after inhalation. We demonstrate here that particulate chromate induces DNA double strand breaks in human lung cells with 0.1, 0.5, and 1 ug/cm2 lead chromate inducing 1.5, 2 and 5 relative increases in the percent of DNA in the comet tail, respectively. These lesions are repaired within 24 h and require Mre11 expression for their repair. Particulate chromate also caused Mre11 to co-localize with gamma-H2A.X and ATM. Failure to repair these breaks with Mre11 induced neoplastic transformation including loss of cell contact inhibition and anchorage independent growth. A 5-day exposure to lead chromate induced loss of cell contact inhibition in a concentration-dependent manner with 0, 0.1, 0.5 and 1 ug/cm2 lead chromate inducing 1, 78 and 103 foci in 20 dishes, respectively. These data indicate that Mre11 is critical to repairing particulate Cr(VI)-induced double strand breaks and preventing Cr(VI)-induced neoplastic transformation.
Keywords: chromium, lead chromate, Mre11, DNA double strand break, neoplastic transformation
Introduction
Cr(VI) compounds represent both an occupational and environmental hazards of increasing public health concern. Exposure is common, as these compounds are used extensively in occupational settings and are common contaminants in industrial waste sites. Cr(VI)) compounds are well-established as human carcinogens. Studies have shown that solubility plays a key role in the carcinogenicity of Cr(VI), with the most potent carcinogens being the water-insoluble or ‘particulate’ compounds (1, 2). The genotoxic mechanism of these particulates involves chronic extracellular dissolution releasing a soluble Cr(VI) anion that enters cells and induces DNA damage (3). The ultimate carcinogen, however, is not Cr(VI) as once inside cells it is rapidly reduced to Cr(III) via the production of short-lived highly reactive species such as Cr(V) and reactive oxygen species (4). One of these metabolic products or some combination of them then exerts the genotoxic and carcinogenic effect.
Our previous study showed that particulate lead chromate induces DNA double strand breaks in human lung cells. While much is understood about the formation and repair of radiation-induced DNA double strand breaks, the genes involved in the mechanism of repairing Cr(VI)-induced DNA double strand breaks and in preventing Cr(VI)-induced neoplastic transformation are poorly understood. The repair of radiation-induced DNA double strand breaks involves checkpoint signaling pathways that initially sense the DNA double strand break, then amplify this signal and finally transduce it to effectors that produce the appropriate biological responses (5). To detect the DNA damage, repair factors such as the MRN complex (Mre11/Rad 50/NBS1) are recruited to the damaged ends (6). Histone protein H2A.X is phosphorylated forming foci over large chromatin domains surrounding the break (7). Various kinases such as ataxia telangiectasia mutated (ATM), ATM-Rad3-related (ATR) and DNA-dependent protein kinase (DNA-PK) are activated and, in turn, transduce the signal to downstream proteins, leading to the activation of checkpoints that prevent cell division and repair damage (7, 8). Cr(VI) is known to induce H2A.X phosphorylation and ATM expression at low concentrations and ATR expression at high concentrations (9–11) however, the role of MRN after Cr(VI) exposure has not been investigated.
DNA double strand breaks represent a major concern for the maintenance of genomic integrity. If left unrepaired, they can result in permanent cell cycle arrest, induction of apoptosis or mitotic cell death (12), and if repaired incorrectly, they are hypothesized to lead to carcinogenesis through directly induced or delayed chromosomal rearrangements (13). Previous studies indicate that particulate Cr(VI) can induce both morphological and neoplastic transformation in cultured rodent cells, but studies in primary human skin cells show that Cr(VI) cannot induce foci formation in these cells (14–17). We reasoned that if Cr(VI)-induced DNA double strand breaks then the inability to repair these lesions should allow for neoplastic transformation in human cells. Because of its importance as an early sensor of damage, in this study we focused on Mre11 and investigated the consequence of its absence on particulate Cr(VI)-induced DNA double strand break repair and neoplastic transformation.
Materials and Methods
Chemicals and Reagents
Lead chromate (CAS No: 7758-97-6) and Triton X-100 (CAS No: 9002-93-1) were purchased from Sigma/Aldrich (St. Louis, MO). Trypsin/EDTA, sodium pyruvate, penicillin/streptomycin, phosphate buffered saline (PBS), L-glutamine and Gurr’s buffer were purchased from Invitrogen Corporation (Grand Island, NY). Methanol (CAS No: 67-56-1), acetone (CAS No: 67-64-1) and acetic acid (CAS No: 64-19-7) were purchased from J.T. Baker (Phillipsburg, NJ). Dulbecco’s minimum essential medium and Ham’s F-12 (DMEM/F-12) 50:50 mixture was purchased from Mediatech Inc. (Herndon, VA). Cosmic calf serum (CCS) was purchased from Hyclone (Logan, UT). Tissue culture dishes, flasks and plasticware were purchased from Corning Inc. (Acton, MA).
Cells and Cell Culture
WTHBF-6 cells are a clonal cell line derived from primary human bronchial fibroblasts with reconstituted telomerase activity. They exhibit a diploid karyotype, normal growth parameters, and an extended lifespan. These cells have clastogenic and cytotoxic responses to metals similar to their parent cells (17). Mre11-deficient cells (ATLD2) were a gift from Dr. Yossi Shiloh, Tel Aviv University. The cells were created from a patient with ataxia telangiectasia-like disease (ATLD), a disease that results from a genetic deficiency in Mre11 (8). This cell line is human skin fibroblast cell and was immortalized with hTERT. Cells were maintained as subconfluent monolayers in DMEM/F-12 supplemented with 15% CCS, 2 mM L-glutamine, 100 U/ml penicillin/100 ug/ml streptomycin and 0.1 mM sodium pyruvate and incubated in a 5% CO2 humidified environment at 37°C. They were fed three times a week and subcultured at least once a week using 0.25% trypsin/1mM EDTA solution. All experiments were performed on logarithmically growing cells.
Chromium Preparations
Lead chromate was used as a particulate Cr(VI) model. It was prepared and administered to cells as previously described (9, 17).
Neutral Comet Assay
DNA double strand breaks were measured using our published methods (9, 18). Briefly, 80,000 cells were seeded into a 6 well plate and treated with lead chromate. Cells were suspended at 1 × 105/ml in PBS and prepared in microgels for comet analysis. Microgels were prepared with 8.5 ul of cell suspension mixed with 85 ul of 1% low-melting agarose and spread on a CometSlide (Trevigen). Slides were allowed to solidify on ice and then lysed by immersion in pre-chilled lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10, 10% sodium lauryl sarcosinate ) containing additional 1% Triton X-100 at 4 °C for 30 min. After lysis, the slides were immersed in digestion solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10) with 1 mg/ml proteinase-K for 2 h at 37 °C. Next, slides were placed in a horizontal gel electrophoresis apparatus containing TBE buffer for 10 min to allow equilibration of microgels. Electrophoresis was carried out at 30V (1V/cm) for 10 min at 4 °C. All the steps beginning with lysis through electrophoresis were done in low light to avoid background-induced DNA damage. Microgels were then immersed in 70% ethanol for 5 min and air-dried. One slide at a time was stained with 50 ul of SYBR Green I. Slides were viewed by epifluorescence microscopy using an Olympus microscope with the Loats Comet system.
Immunofluorescence for Gamma-H2A.X Foci Formation
Discrete nuclear foci of phosphorylated H2A.X (gamma-H2AX) are visible by immunofluorescence and appear to be true markers of double strand breaks (19). Immunofluorescence was done based on our previous publication (9). Logarithmic growing cells were grown on 8 well chamber slides. Lead chromate-treated cells were incubated with an anti-Mre11 antibody and anti-gamma-H2A.X antibody or anti-p-ATM at 4 °C overnight. Then, cells were incubated with FITC AlexaFluor 484-conjugated goat anti-rabbit IgG secondary antibody and TEXAS RED anti-mouse IgG secondary antibody. Nuclei were counterstained with 4′-6-Diamidino-2-phenylindole (DAPI). The slides were viewed with an Olympus laser scanning confocal microscope (LSCM) using a 60X objective. Images of the same antibody staining were obtained using the same parameters (brightness, contrast, pinhall, etc.).
Western Blot Analyses
Western blot were performed based on our published methods (9). Briefly, logarithmic growing cells were grown were treated with lead chromate. Cells were lysed and equal samples were resolved by 3%–8% SDS-PAGE for ATM and 4–15% gradient SDS-PAGE gels for Mre11. Nitrocellulose membranes were probed with anti-phospho-ATM antibody overnight or anti-ATM antibody and then probed with HRP conjugated secondary antibody for 1 h. Blots visualized by ECL western blotting detection reagents (Amersham Biosciences, Piscataway, NJ).
Small Interfering RNA
siRNAs (ACAGGAGAAGAGAUCAACU) were designed and synthesized for the target sequence of Mre11 based on published methods (20). Human lung cells were transfected with siRNA using Lipofectamine according to the manufacture’s instructions (Invitrogen). Cells were collected 48 h after transfection to isolate protein for immunoblotting and to detect DNA double stand breaks using assay methods described above.
Transformation Assay
Transformation assay was done as foci formation assay based on published method for C3H/10T1/2 cells (2). 5000 to 100,000 cells were seeded in 60 mm dishes to achieve an optimum confluence which allowed the cells grow for about 1 month without splitting. Each concentration of treatment included 20 dishes of cells. Subconfluent cells were treated for 5 days with lead chromate; washed with 1× PBS twice to remove lead chromate particles; and then fed with culture medium without antibiotic after 5 days treatment. Cells were maintained in a 37 °C, humidified incubator with 5% CO2 and the medium was changed every other day. Cell growth was monitored regularly for cell morphology change and foci formation. Foci were ring cloned and then culture terminated.
Anchorage Independent Assay
Anchorage independence assay is considered a stringent assay for detecting transformation of cultured cells. To determine anchorage independent cell growth, control and treated cells were suspended in 0.35% agar, plated onto a 0.6% base layer in a 60 mm dish at a density of 5 × 104 and grown for 4 weeks (21). Cultures were examined microscopically 24 h after plating to confirm an absence of large clumps of cells. Colonies were visualized by 5% 4-nitro-blue-tetrazolium chloride staining. When cultures were established from anchorage independent colonies, soft-agar colonies were plucked under sterile conditions and then dispersed in trypsin and replated into culture dishes.
Results
Lead chromate-induced DNA double strand break repair in human lung cells
We found that lead chromate induced concentration-dependent increases in DNA double strand breaks with 0.1, 0.5 and 1 ug/cm2 lead chromate inducing 1.5, 2 and 5 percent increases in DNA in the comet tail relative to control, respectively (Figure 1A). After a 24 h recovery, breaks were reduced as the percent of DNA in the tail, relative to control, was reduced to 0.3, 0.9 and 0.5, respectively. These data indicate that the breaks were repaired within 24 h after exposure (Figure 1A). We also checked longer term repair and found that double strand breaks remained at background level over a 96 h recovery period (Figure 1B).
Figure 1. Particulate Cr(VI) Induces Formation and Repair of DNA Double Strand Breaks in Human Lung Cells.


This figure shows that lead chromate induces DNA double strand breaks in human lung cells. (A) Increasing concentrations of lead chromate induced more DNA double strand breaks in human lung cells. After a 24 h exposure, these breaks are all repaired within 24 h. * Significant (p<0.05) differences in comparison to vehicle control. # Within the chromium treatment group, value show significant (p<0.05) differences comparing to 24 h treatment only. (B) DNA Double strand breaks remained at the background level over a 96 h recovery period. For each data point the untreated control levels were subtracted. Data are the mean of at least three independent experiments ± standard error of the mean. (C) Mre11 expression was induced after lead chromate exposure. Hydrogen peroxide was used as a positive control. β-Actin antibody staining is shown as a loading control.
Mre11 is required for the repair of lead chromate-induced DNA double strand breaks
Mre11 expression increased after particulate chromate exposure in human lung cells (Figure 1C), suggesting that Mre11 is part of the repair response for Cr(VI)-induced DNA double strand breaks. Knock-down of Mre11 with siRNA inhibited the repair of particulate Cr(VI)-induced DNA double strand breaks in human lung cells further indicating the importance of Mre11 in repair of these lesions (Figure 2). In the Mre11-inhibited cells the amount of breaks continued to accumulate after the end of exposure reflecting that repair was inhibited (Figure 2B). To confirm the finding, we treated Mre11-deficient skin cells with lead chromate. Results showed that after a 24 h treatment, concentrations of 0.1, 0.5 and 1 ug/cm2 lead chromate-induced DNA double strand breaks with 1.7, 3.1 and, 4.1 percent increases in DNA in the comet tail, relative to control (Figure 3A). These breaks did not significantly decline after a 24 h recovery indicating that repair was not inhibited. We again gave cells up to 96 h to recover and found double strand break repair did occur after 48 h, indicating that repair was delayed in Mre11-deficient cells (Figure 3B). Interestingly, only partial activation of ATM was observed when Mre11-deficient cells were exposed to lead chromate (Figure 3C) suggesting an interaction effect between these two proteins.
Figure 2. Reduced Mre11 Expression in Human Lung Cells Inhibits Repair of Particulate Cr(VI)-Induced DNA Double Strand Breaks.
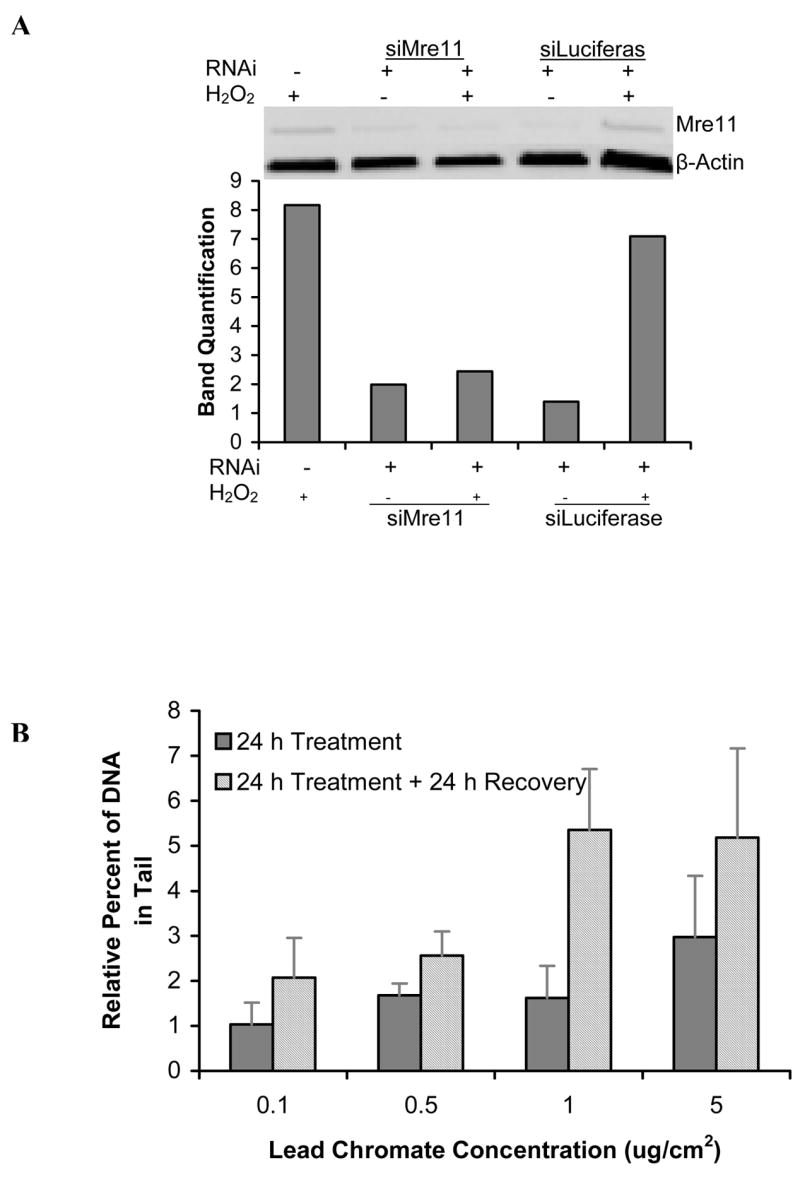
This figure shows that reduced Mre11 expression in human lung cells inhibits repair of lead chromate–induced DNA double strand breaks. (A) siRNA targeted and reduced Mre11 expression in human lung cells. Cells were collected 48 h after being transfected with siRNA. (B) Lead chromate induces DNA double strand breaks in Mre11-suppressed human lung cells in which small interfering RNAs (siRNA) targeted Mre11. After a 24 h exposure, these lesions are not repaired within 24 h. For each data point the background control levels were subtracted. Data are the mean of at least two independent experiments ± standard error of the mean.
Figure 3. Repair of Particulate Cr(VI)-Induced DNA Double Strand Breaks Is Delayed in Mre11-Deficient Cells.


This figure further shows that lead chromate-induced DNA double strand break repair is mediated by Mre11. (A) Increasing concentrations of lead chromate induce more DNA double strand breaks in Mre11-deficient cells. After a 24 h exposure, these breaks are not repaired within 24 h. * Significant (p<0.01) differences in comparison to its vehicle control. (B) Substantial DNA double strand break repair did not occur until 48 h after the end of a 24 h exposure and were not reduced to background levels until 96 h in Mre11-deficient cells. For each data point the background control levels were subtracted. Data are the mean of at least three independent experiments ± standard error of the mean. (C) Lead chromate induced low levels of ATM expression after a 24 h exposure in Mre11-deficient cells. β-Actin antibody staining is shown as a loading control.
Mre11 and ATM are recruited to lead chromate-induced double strand breaks
H2A.X was quickly phosphorylated after lead chromate exposure and formed foci around double strand breaks (9). Mre11 was recruited to and co-localized at these gamma-H2A.X foci, indicating that Mre11 is recruited to the site of double strand breaks (Figure 4A). We also found that phosphorylated ATM foci co-localized to Mre11 foci, further indicating that activated ATM interacts with Mre11 in the repair pathway (Figure 4B).
Figure 4. Mre11 Co-Localize with gamma-H2A.X and ATM in Response to Particulate Cr(VI) Treatment.
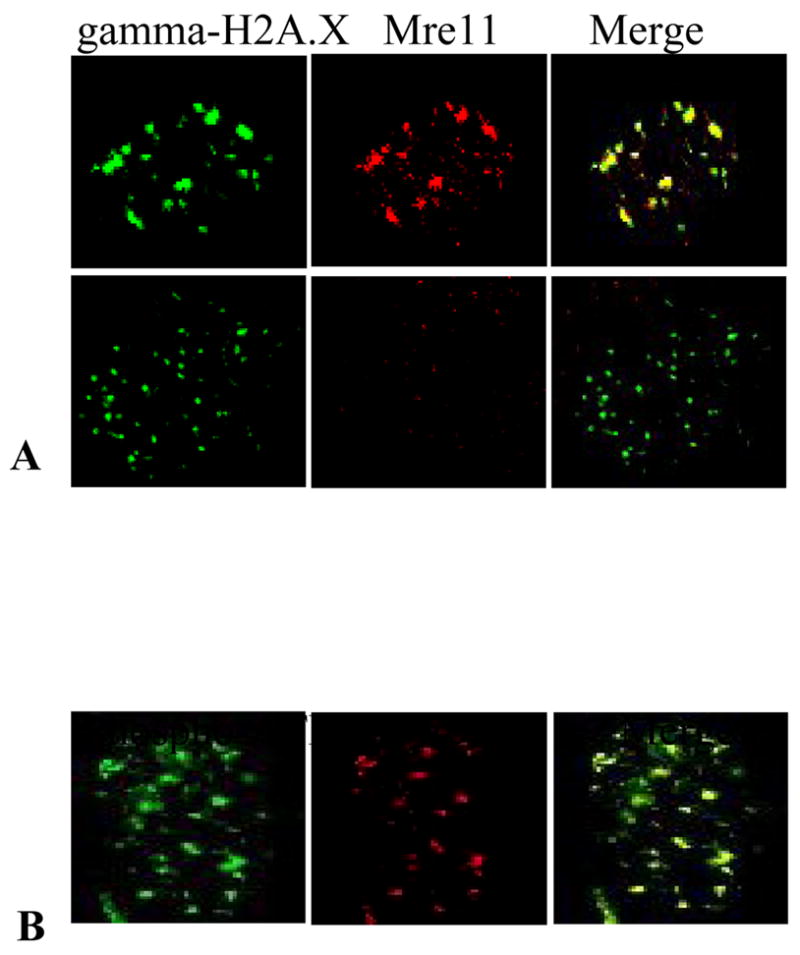
This figure shows that Mre11 is recruited to the double strand break after lead chromate exposure. (A) Mre11 and gamma-H2AX co-localized in human lung cells after 1 ug/cm2 lead chromate treatment (top). No Mre11 foci form in Mre11-deficient cells after 1 ug/cm2 lead chromate treatment (bottom). (B) Mre11 and phosphorylated ATM co-localized in human lung cells after lead chromate treatment.
Lead chromate induces loss of cell contact inhibition and anchorage independence
Adherent cells maintain contact inhibition. They spread out across the culture flask, when two adjacent cells touch; this signals them to stop growing. Cells that lose contact inhibition not only form a monolayer in culture but also pile up on top of one another in foci. A 5-day exposure to lead chromate did not induce foci formation in Mre11-proficient cells indicating an intact repair pathway prevents this growth. In contrast, lead chromate did induce foci formation in Mre11-deficient cells in a concentration-dependent manner with 0, 0.1 and 1 ug/cm2 lead chromate inducing 1, 78, 103 foci in 20 dishes, respectively (Figure 5A, B and Table 1). These foci grew in large masses on top of the monolayer and were grown into cell lines and subcloned.
Figure 5. Particulate Cr(VI) Induces Transformation of Mre11 Deficient Cells.
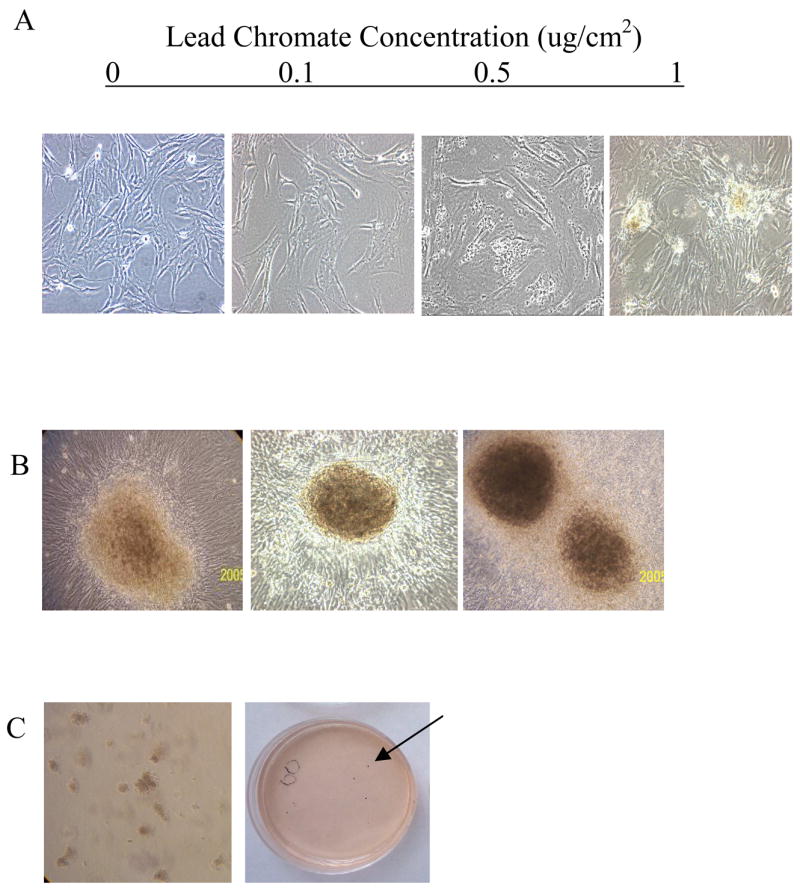
This figure shows that lead chromate induces neoplastic transformation in Mre11-deficient cells. (A) Cell morphology change observed after 5-day treatment with lead chromate in Mre11 deficient cells. Magnification is equal to 100X. (B) Foci produced in Mre11- deficient cells after exposure to 0.1–1 ug/cm2 lead chromate for 5 days. These foci grew in large masses on the top of the monolayer with complex borders. Magnification is equal to 100X. (C) Foci were ring cloned, subcloned, and tested for growth in agar (left). Colonies grow on soft agar as visualized by 5% 4-nitro-blue-tetrazolium chloride staining (arrow, right).
Table 1.
Particulate Cr(VI) Induces Foci Formation in Mre11-Deficient Cells
| Cell Line | Lead Chromate
(ug/cm2) |
Foci frequency
(Total foci/20 dishes) |
Growth in soft agar |
|---|---|---|---|
| BJhTERT | 0 | 0 | − |
| (wildtype) | 0.05 | 0 | − |
| 0.1 | 0 | − | |
| 1* | |||
|
| |||
| ATLD2 | 0 | 1 | − |
| (Mre11-deficient) | 0.1 | 78 | + |
| 1 | 103 | ++ | |
1 ug/cm2 was omitted due to high toxicity.
Normal cells are anchorage dependent and require a surface on which to flatten out and divide. The ability to grow in soft agar is a property acquired by neoplastic cells. We found that foci cells cloned from lead chromate-treated Mre11-deficient cells grew on soft agar in a concentration-dependent manner (Figure 5C, Table 1) indicating these cells were transformed by lead chromate.
Discussion
Recently, we demonstrated that particulate Cr(VI) induces DNA double strand breaks in human bronchial cells (9). In this study, we elucidate some of the key steps in the repair of these lesions and begin to develop a model of DNA double strand break repair for an established human chemical carcinogen in human lung cells. We found that after a 24 h exposure to particulate Cr(VI), DNA double strand breaks were repaired to background levels within 24 h in human lung cells. The repair of these lesions were delayed in Mre-11 depleted lung cells and Mre11-deficient skin cells with no obvious repair occurring within 24 h indicating that Mre11 is essential for proficient repair of these lesions (Figure 2B and 3A). This is the first study to show that Cr(VI)-induced DNA double strand break repair is mediated by Mre11. Our data are the first to consider Mre11 in repair after exposure to an occupational and environmental chemical carcinogen and are consistent with the response of Mre11 to radiation induced DNA double strand breaks (22–24).
Mre11 and gamma-H2A.X were both recruited to the site of DNA double strand breaks after exposure to particulate chromate suggesting that Mre11 serves as a sensor in response to these lesions (Figure 4A). It has previously been shown that gamma-H2A.X is required for MRN foci formation after radiation exposure (2). After particulate chromate treatment, gamma-H2A.X foci were able to form in Mre11-deficient indicating that the presence of functional Mre11 is not required for the formation of gamma-H2A.X foci and that phosphorylation of H2A.X is upstream of Mre11 function. This observation is consistent with radiation exposure which found that gamma-H2A.X recruited MRN complex to sites of DNA damage through a direct interaction with Nbs1 (25).
Previously, we demonstrated that ATM is induced by particulate Cr(VI) (9). In this report we found that ATM colocalized with Mre11 foci linking these two repair events together in the pathway for repairing particulate Cr(VI)-induced DNA double strand breaks (Figure 4B). Recent work with radiation exposure suggests that the MRN complex may be required for full ATM activation, as demonstrated by defective ATM activation and defective phosphorylation of ATM targets such as p53-ser15, Chk2-thr68, SMC1-ser966 and 53BP1 in Mre11 deficient cells after exposure to DNA double strand break-inducing agents (22–25). The MRN complex may act as a double strand break sensor for ATM and recruit ATM to broken DNA molecules (22, 25). Phosphorylated ATM expression was decreased in Mre11-deficient cells after lead chromate exposure as measured by phosphorylation of ATM-ser 1981 extends the hypothesis of the requirement of a functional MRN complex for an optimal activation of the ATM kinase activity to chemical carcinogens.
Having determined that Mre11 plays a role in particulate Cr(VI)-induced DNA double strand break repair, we sought to determine if the inability to repair these lesions could contribute to particulate Cr(VI)-induced carcinogenesis and therefore, establish a direct link between DNA double strand break repair and chemical-induced neoplastic transformation. Previous studies indicate that particulate Cr(VI) cannot induce foci formation in normal human cells (26); and so we reasoned that if Cr(VI)-induced DNA double strand breaks are important in carcinogenesis then the inability to repair these lesions should allow for neoplastic transformation in human cells. We found that particulate Cr(VI) did transform cells deficient in DNA double strand break repair but could not transform normal human cells (Figure 5 and Table 1). Our findings are consistent with the only other previous study to considered particulate Cr(VI) transformation in human cells (27). That study reported lead chromate induced foci formation and anchorage independent growth (27), however, the cells they used were already neoplastic, being derived from an osteosarcoma, and the status of Mre11 and DNA double strand break repair in those cells is unknown.
Our data are consisted with the observations that Mre11 disfunction may be involved in the development of cancer. Defects in Mre11 functions are associated with ATLD, a cancer predisposition syndrome (8) and Mre11 mutations and aberrant transcript, resulting from abnormal splicing event, have been reported in primary tumors (28). Another study showed that Mre11 expression is impaired in gastric cancer with microsatellite instability (29). Gross chromosomal rearrangements, which are often associated with tumor development, were increased by mutations of Mre11 in S. cerevisiae also supports this possibility (30). Fukuda et al. (28) performed a mutation analysis of Mre11 in 159 unselected primary tumors. Three mutations at conserved positions were found in breast and lymphoid tumors suggesting an occasional role for Mre11 alterations in the development of primary tumors.
In this study, we have demonstrated that defective repair of particulate Cr(VI)-induced DNA double strand breaks leads to neoplastic transformation. The accumulated evidence indicates that the mechanism of particulate Cr(VI)-induced carcinogenesis involves extracellular dissolution of the particles leading to the release of soluble Cr(VI) oxyanion which enters the cell and is reduced to Cr(III) and other reactive intermediates (31). One or more of these species induces DNA double strand breaks, which are sensed directly by gamma-H2A.X and the MRN complex, which binds DNA, unwinds the ends and recruits ATM, ultimately leading to repair (5–7). Failure to repair these breaks leads to neoplastic transformation, possibly through the generation of chromosome instability, which has also been observed after particulate Cr(VI) exposure (32, 33).
Acknowledgments
We thank Professor Malcolm Taylor, The University of Birmingham, Cancer Research, UK and Yossi Shiloh, Tel Aviv University for the use of Mre11-deficient cells (ATLD2) and Geron Corporation for the use of the hTERT materials. We thank David Kirstein and Christy Gianios, Jr. for administrative and technical support; Imhoitseme Dako for assistance with cell culture. Grant supported by the National Institute of Environmental Health Sciences grant ES10838 (J.P. Wise) and the Maine Center for Toxicology and Environmental Health at the University of Southern Maine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 49, International Agency for Research on Cancer, Lyons, France (1990).
- 2.Patierno SR, Banh D, Landolph JR. Transformation of C3H/10T1/2 mouse embryo cells to focus formation and anchorage independence by insoluble lead chromate but not soluble calcium chromate: relationship to mutagenesis and internalization of lead chromate particles. Cancer Res. 1988;48:5280–5288. [PubMed] [Google Scholar]
- 3.Xie H, Holmes AL, Wise SS, Gordon N, Wise JP., Sr Lead chromate-induced chromosome damage requires extracellular dissolution to liberate chromium ions but does not require particle internalization or intracellular dissolution. Chem Res Toxicol. 2004;17:1362–7. doi: 10.1021/tx0498509. [DOI] [PubMed] [Google Scholar]
- 4.De Flora S, Serra D, Camoirano A, Zanacchi P. Metabolic reduction of chromium, as related to its carcinogenic properties. Biol Trace Elem Res. 1989;21:179–87. doi: 10.1007/BF02917250. Review. [DOI] [PubMed] [Google Scholar]
- 5.Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13:458–462. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 6.van den Bosch M, Bree RT, Lowndes NF. The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003;4(9):844–9. doi: 10.1038/sj.embor.embor925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nature Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 8.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM. The DNA double-strand break repair gene hMre11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 9.Xie H, Wise SS, Holmes AL, Xu B, Wakeman TP, Pelsue SC, Singh NP, Wise JP., Sr Carcinogenic lead chromate induces DNA double-strand breaks in human lung cells. Mutat Res. 2005;586:160–172. doi: 10.1016/j.mrgentox.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ha L, Ceryak S, Patierno SR. Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis. 2004;25:2265–2274. doi: 10.1093/carcin/bgh242. [DOI] [PubMed] [Google Scholar]
- 11.Wakeman TP, Xu B. ATR regulates hexavalent chromium-induced S-phase checkpoint through phosphorylation of SMC1. Mutat Res. 2006;610:14–20. doi: 10.1016/j.mrgentox.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olive PL. The role of DNA single- and double-strand breaks in cell killing by ionizing radiation. Radiat Res. 1998;150:S42–51. [PubMed] [Google Scholar]
- 13.Morgan WF, Corcoran J, Hartmann A, Kaplan MI, Limoli CL, Ponnaiya B. DNA double-strand breaks, chromosomal rearrangements, and genomic instability. Mutat Res. 1998;404:125–8. doi: 10.1016/s0027-5107(98)00104-3. [DOI] [PubMed] [Google Scholar]
- 14.Fradkin A, Janoff A, Lane BP, Kuschner M. In vitro transformation of BHK21 cells grown in the presence of calcium chromate. Cancer Res. 1975;35:1058–63. [PubMed] [Google Scholar]
- 15.Hansen K, Stern RM. Welding fumes and chromium compounds in cell transformation assays. J Appl Toxicol. 1985;5:306–14. doi: 10.1002/jat.2550050509. [DOI] [PubMed] [Google Scholar]
- 16.Lanfranchi G, Paglialunga S, Levis AG. Mammalian cell transformation induced by chromium(VI) compounds in the presence of nitrilotriacetic acid. J Toxicol Environ Health. 1988;24:251–260. doi: 10.1080/15287398809531158. [DOI] [PubMed] [Google Scholar]
- 17.Elias Z, Poirot O, Baruthio F, Daniere MC. Role of solubilized chromium in the induction of morphological transformation of Syrian hamster embryo (SHE) cells by particulate chromium(VI) compounds. Carcinogenesis. 1991;12:1811–6. doi: 10.1093/carcin/12.10.1811. [DOI] [PubMed] [Google Scholar]
- 18.Singh NP, Stephens RE. X-ray induced DNA double-strand breaks in human sperm. Mutagenesis. 1998;13:75–79. doi: 10.1093/mutage/13.1.75. [DOI] [PubMed] [Google Scholar]
- 19.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 20.Chai W, Sfeir AJ, Hoshiyama H, Shay JW, Wright WE. The involvement of the Mre11/Rad50/Nbs1 complex in the generation of G-overhangs at human telomeres. EMBO Rep. 2006;7:225–30. doi: 10.1038/sj.embor.7400600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wise SS, Elmore LW, Holt SE, Little JE, Antonucci PG, Bryant BH, Wise JP., Sr Telomerase-mediated lifespan extension of human bronchial cells does not affect hexavalent chromium-induced cytotoxicity or genotoxicity. Mol Cell Biochem. 2004;255:103–11. doi: 10.1023/b:mcbi.0000007266.82705.d9. [DOI] [PubMed] [Google Scholar]
- 22.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;15:5612–21. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;15:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, Paull TT. Direct Activation of the ATM Protein Kinase by the Mre11/Rad50/Nbs1 Complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 25.You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 2005;25:5363–79. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biedermann KA, Landolph JR. Induction of anchorage independence in human diploid foreskin fibroblasts by carcinogenic metal salts. Cancer Res. 1987;15:3815–23. [PubMed] [Google Scholar]
- 27.Sidhu MK, Fernandez C, Khan MY, Kumar S. Induction of morphological transformation, anchorage-independent growth and plasminogen activators in non-tumorigenic human osteosarcoma cells by lead chromate. Anticancer Res. 1991;11:1045–1053. [PubMed] [Google Scholar]
- 28.Fukuda T, Sumiyoshi T, Takahashi M, Kataoka T, Asahara T, Inui H, Watatani M, Yasutomi M, Kamada N, Miyagawa K. Alterations of the double-strand break repair gene Mre11 in cancer. Cancer Res. 2001;61:23–26. [PubMed] [Google Scholar]
- 29.Ottini L, Falchetti M, Saieva C, De Marco M, Masala G, Zanna I, Paglierani M, Giannini G, Gulino A, Nesi G, Mariani Costantini R, Palli D. Mre11 expression is impaired in gastric cancer with microsatellite instability. Carcinogenesis. 2004;25:2337–43. doi: 10.1093/carcin/bgh257. [DOI] [PubMed] [Google Scholar]
- 30.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 31.Liu KJ, Shi X. In vivo reduction of chromium (VI) and its related free radical generation. Mol Cell Biochem. 2001;222:41–7. [PubMed] [Google Scholar]
- 32.Holmes AL, Wise SS, Sandwick SJ, Lingle WL, Negron VC, Thompson WD, Wise JP., Sr Chronic exposure to lead chromate causes centrosome abnormalities and aneuploidy in human lung cells. Cancer Res. 2006;15:4041–8. doi: 10.1158/0008-5472.CAN-05-3312. [DOI] [PubMed] [Google Scholar]
- 33.Wise SS, Holmes AL, Xie H, Thompson WD, Wise JP., Sr Chronic exposure to particulate chromate induces spindle assembly checkpoint bypass in human lung cells. Chem Res Toxicol. 2006;19:1492–8. doi: 10.1021/tx0601410. [DOI] [PubMed] [Google Scholar]