

Abstract
ADP-glucose (Glc) pyrophosphorylase (AGPase), a key regulatory enzyme in starch biosynthesis, is highly regulated. Transgenic approaches in four plant species showed that alterations in either thermal stability or allosteric modulation increase starch synthesis. Here, we show that the classic regulators 3-phosphoglyceric acid (3-PGA) and inorganic phosphate (Pi) stabilize maize (Zea mays) endosperm AGPase to thermal inactivation. In addition, we show that glycerol phosphate and ribose-5-P increase the catalytic activity of maize AGPase to the same extent as the activator 3-PGA, albeit with higher Ka (activation constant) values. Activation by fructose-6-P and Glc-6-P is comparable to that of 3-PGA. The reactants ATP and ADP-Glc, but not Glc-1-P and pyrophosphate, protect AGPase from thermal inactivation, a result consistent with the ordered kinetic mechanism reported for other AGPases. 3-PGA acts synergistically with both ATP and ADP-Glc in heat protection, decreasing the substrate concentration needed for protection and increasing the extent of protection. Characterization of a series of activators and inhibitors suggests that they all bind at the same site or at mutually exclusive sites. Pi, the classic “inhibitor” of AGPase, binds to the enzyme in the absence of other metabolites, as determined by thermal protections experiments, but does not inhibit activity. Rather, Pi acts by displacing bound activators and returning the enzyme to its activity in their absence. Finally, we show from thermal inactivation studies that the enzyme exists in two forms that have significantly different stabilities and do not interconvert rapidly.
ADP-Glc pyrophosphorylase (AGPase) represents a rate-limiting step in starch synthesis in plants as well as glycogen biosynthesis in bacteria (for review, see Preiss and Romeo, 1994; Preiss and Sivak, 1996; Hannah, 2005). AGPase catalyzes the first committed step in the starch biosynthetic pathway, the conversion of ATP and α-Glc-1-P (G-1-P) to ADP-Glc and pyrophosphate (PPi; for review, see Hannah, 1997). Definitive evidence for the rate-limiting role of AGPase in starch synthesis has been derived through transgenic studies in potato (Solanum tuberosum) tubers (Stark et al., 1992) and seeds of wheat (Triticum aestivum; Smidansky et al., 2002), rice (Oryza sativa; Smidansky et al., 2003; Sakulsingharoja et al., 2004), and maize (Zea mays; Giroux et al., 1996; T.W. Greene and L.C. Hannah, unpublished data; Wang et al., 2007). Variants incorporated into plants giving rise to enhanced starch biosynthesis harbor alterations in allosteric properties and/or alterations in heat stability. Accordingly, the thermal stability and regulatory properties of AGPase have received considerable attention.
While bacterial AGPases are homotetramers, plant and unicellular green algae AGPases are composed of two identical small subunits and two identical large subunits (for review, see Georgelis et al., 2007). Prokaryotic and eukaryotic AGPases have different quaternary structures and regulatory properties (Ballicora et al., 2003), yet their overall kinetic mechanisms appear to be similar (Paule and Preiss, 1971; Kleczkowski et al., 1993). The small and large subunits share much sequence identity, and a gene duplication giving rise to the two subunits apparently occurred early in plant evolution (Bae et al., 1990; Bhave et al., 1990; Georgelis et al., 2007). The present-day small and large subunits are not fully functionally redundant, although bacterial expression of each subunit alone does yield traces of enzyme activity (Iglesias et al., 1993; Burger et al., 2003). Mutations affecting catalytic and allosteric properties of AGPase map to both subunits (Cross et al., 2004, 2005; Hwang et al., 2005). While the small subunit is evolutionarily more conserved compared to the large subunit, this results from fewer genes encoding small subunits and their need to interact with multiple large subunit partners, rather than an inherent inability to successfully accommodate amino acid changes (Georgelis et al., 2007).
At least three mechanisms have been proposed to modulate plant AGPase activity: allosteric regulation, thermal inactivation, and reductive activation. Plant AGPases are activated by several allosteric effectors, with 3-phosphoglyceric acid (3-PGA) activation receiving the greatest attention. Inorganic phosphate (Pi) inhibits activity in many tissues. In the case of the maize endosperm AGPases, Pi negates 3-PGA activation and returns the enzyme's catalytic activity to its basal level observed in the absence of 3-PGA.
The thermal stability of AGPase is also an important characteristic because exposure to high temperatures during plant growth causes reduced grain yield in many internationally important cereal crops, including maize, wheat, and rice (for review, see Singletary et al., 1994). Investigations by others have shown that AGPase is one of the enzymes most affected by elevated temperature (Singletary et al., 1993, 1994). Most plant AGPases, for example, the potato tuber enzyme (Sowokinos and Preiss, 1982; Okita et al., 1990), are heat stable, whereas the cytosolic endosperm isoforms are quite labile. Incubating the maize endosperm AGPase at 57°C for 5 min destroys 96% of the activity (Hannah et al., 1980), whereas the potato tuber enzyme is fully stable at 70°C. Expression of a heat-stable AGPase in the seed of wheat (Smidansky et al., 2002), rice (Smidansky et al., 2003), and maize (Giroux et al., 1996; T.W. Greene and L.C. Hannah, unpublished data) increases grain yield.
A Cys residue near the N terminus of the potato tuber small subunit (Fu et al., 1998) plays an important role in heat stability. While most plant AGPases contain this residue, the heat-labile endosperm enzymes lack this amino acid. Removal of the Cys from the potato tuber enzyme reduces heat stability (Fu et al., 1998), whereas incorporation of this Cys into the maize endosperm enzyme enhances heat stability (Linebarger et al., 2005). The presence of the small subunit Cys also allows for reductive activation of AGPases (Fu et al., 1998; Ballicora et al., 1999; Linebarger et al., 2005). The presence of the Cys can lead to disulfide bridge formation between the two small subunits (Fu et al., 1998; Ballicora et al., 1999; Linebarger et al., 2005). Such activation is impossible for wild-type endosperm AGPases, a phenomenon possibly related to the cytosolic location of the major endosperm AGPases, in contrast to all other AGPases, which are thought to be plastid localized (for review, see Hannah, 2007).
In the studies described below, we show that a diverse series of metabolites affects maize endosperm AGPase in two fundamentally different ways. Metabolites that serve as reactants, activators, or inhibitors of AGPase also stabilize activity to heat inactivation. This includes the reactants ADP-Glc and ATP as well as all known activators and inhibitors. We also describe two previously unknown activators of the maize endosperm AGPase. While the activators and inhibitors differ significantly in structure, all data are consistent with a common binding site (or mutually exclusive sites) for these effector molecules. Finally, we note that the enzyme can apparently exist in two states that differ in their rates of thermal denaturation. This observation has also been noted in apple (Malus domestica) leaf AGPase (Zhou and Cheng, 2005).
RESULTS
Pi Binds Tightly to Maize Endosperm AGPase as Seen from Protection, But Does Not Inhibit Catalytic Activity
Allosteric regulation of AGPase is a physiologically important phenomenon. 3-PGA is the most characterized activator, and its activation is reduced by the presence of Pi. While this is a common feature of virtually all plant AGPases, interesting differences distinguish specific isoforms of the enzyme. In the case of the maize endosperm, Pi does not appreciably reduce activity in the absence of 3-PGA (Fig. 1). Activity was measured in the ADP-Glc synthesis direction either in the presence or absence of 2.5 mm 3-PGA and at various concentrations of Pi. Very little Pi inhibition occurs if 3-PGA is omitted from the reaction mixture. This is in direct contrast to the closely related wheat endosperm AGPase (Gomez-Casati and Iglesias, 2002), whereby Pi interacts with the enzyme to inhibit activity and 3-PGA then acts as a deinhibitor. In contrast to maize and wheat, the barley endosperm AGPase is insensitive to both 3-PGA activation and Pi inhibition (Kleczkowski et al., 1993; Doan et al., 1999).
Figure 1.

Phosphate effects in the presence or absence of 3-PGA. The forward reaction was used to measure the velocity at various concentrations of phosphate using standard reaction conditions. Velocity in the absence (▪) of 3-PGA or in the presence (▴) of 2.5 mm 3-PGA is plotted.
Two mutually exclusive hypotheses explain Pi deactivation of the 3-PGA-stimulated maize endosperm AGPase: (1) Pi does not bind to this isoform in the absence of 3-PGA; or (2) Pi binding to AGPase occurs in the absence of 3-PGA, but binding has no appreciable effect on activity. To distinguish between these possibilities, we tested whether Pi, in the absence of 3-PGA, might affect other properties of maize endosperm AGPase. Heat stability was chosen because previous reports (Gomez-Casati et al., 2000; Zhou and Cheng, 2005) have shown that allosteric effectors and enzyme reactants enhance enzyme stability in the presence of elevated temperatures.
In the first series of experiments, we investigated whether the allosteric activator 3-PGA could reduce or negate heat-induced loss of maize endosperm AGPase activity (Fig. 2). Purified maize endosperm AGPase was mixed with various concentrations of 3-PGA, and one aliquot of each mixture was incubated for 10 min at 42°C and then immediately placed on ice until assay. Heated and unheated aliquots of each mixture were then assayed for AGPase activity, and the percentage of remaining activity is presented in Figure 2. Clearly, 3-PGA reduces the heat lability of maize endosperm AGPase. This is consistent with reports of 3-PGA stabilization of other AGPases (Gomez-Casati et al., 2000; Zhou and Cheng, 2005). It is noteworthy that although 3-PGA protects the enzyme, protection is not complete; in fact, only about 35% of activity is protected at the highest concentration of 3-PGA (50 mm) employed.
Figure 2.

Phosphate and 3-PGA protection from thermal inactivation. Desalted enzymes containing 0.5 mg/mL BSA were heated at 42°C for 10 min in the presence of varying concentrations of Pi (▪) or 3-PGA (▴). Duplicate samples were left on ice and their activity was taken to be 100%. All preparations were assayed for 10 min in the forward direction in the presence of 10 mm 3-PGA. The percentage of activity remaining was then calculated by dividing the heated versus nonheated samples and plotted against the effector concentration.
Pi, in the absence of 3-PGA, also stabilizes maize endosperm AGPase (Fig. 2). In fact, Pi provides substantially more protection than does 3-PGA at the concentrations shown (55% at 50 mm). Hence, the maize endosperm AGPase binds Pi in the absence of 3-PGA; however, this binding does not substantially reduce catalytic activity. To characterize Pi protection of maize endosperm AGPase in more detail, enzyme activity was measured following exposure to temperatures at 42°C for various times in the presence of several Pi concentrations (Fig. 3, A and B). Activity loss follows first order kinetics with respect to time. Values of t1/2 (the time required for loss of one-half of the activity) at each Pi concentration were calculated from the slope of the plot of log activity versus time. t1/2 for inactivation increases with increasing amounts of Pi. A Kd of 65 μm ± 7 μm for Pi was then determined from a replot of the data using the method of Scrutton and Utter (1965).
Figure 3.

A, Thermal stability of AGPase in the presence of varying concentrations of Pi: 0 (▪), 0.25 (▴), 0.5 (▾), 1.0 (♦), 2.5 (•), 5 (▵), and 10 (□). Enzyme preparations were desalted and contained 0.5 mg/mL BSA. Various concentrations of Pi were then added prior to incubation on ice for 10 min. The samples were then heated at 42°C for various times. Duplicate samples were left on ice and their activity was taken to be 100%. All assays and calculations were determined as in Figure 2. Data were plotted as log % activity versus time, and the inactivation constant t1/2 was calculated as described in “Materials and Methods.” B, Determination of phosphate Kd by the method of Scrutton and Utter (1965).
The Activators and Inhibitors of AGPase Are Intimately Intertwined
We next asked whether metabolites known or suspected (Ghosh and Preiss, 1966; Plaxton and Preiss, 1987; Gomez-Casati and Iglesias, 2002) of activating AGPase might also stabilize activity. Prior to these experiments, we monitored activation of AGPase catalytic activity and determined the Ka values in the forward direction (Table I). While activation by 3-PGA has received considerable attention, other metabolites, such as Fru-6-P (F-6-P), Glc-6-P (G-6-P), Rib-5-P, and especially glycerol phosphate, condition activities that are comparable to that produced by 3-PGA. Although the activators F-6-P and G-6-P have activation constants higher than that of 3-PGA, these constants are in the range of their cellular concentration of approximately 0.5 and 3.5 mm, respectively, in the maize endosperm (see Table I; Liu and Shannon, 1981). This is in contrast to the potato tuber AGPase, where 3-PGA is a highly specific activator, with F-6-P stimulating the activity to only 4% of that seen with 3-PGA (Iglesias et al., 1993). Glycerol phosphate activates AGPase to a similar extent as 3-PGA, but the amount needed for one-half activation is approximately 15-fold higher than that of 3-PGA. While many phosphate monoesters activated the maize AGPase, not all did. We found no activation with erythrose-4-P or Rib-1,5-diphosphate. In addition, neither methyl phosphate nor the reducing agent dithiothreitol (DTT) activated or protected the maize endosperm AGPase. These observations from the maize enzyme also stand in contrast to those for the spinach (Spinacia oleracea) leaf chloroplast AGPase, where Rib-5-P and F-6-P activate the enzyme to 15% and 40% of the activity seen with 3-PGA (Ghosh and Preiss, 1966).
Table I.
Ka for activation of various effectors
No activation is seen in the presence of Rib-1,5-diphosphate, erythrose-4-P, or methyl phosphate using concentrations up to 20 mm.
| Activator | Ka | Activitya | Physiological Concentrationb |
|---|---|---|---|
| % | mm | ||
| 3-PGA | 0.22 ± 0.034 | 100 | 1.75 |
| F-6-P | 0.4 ± 0.09 | 70 | 0.5 |
| G-6-P | 2.7 ± 0.79 | 67 | 3.23 |
| Glycerophosphate | 3.6 ± 0.76 | 103 | |
| PEP | 5.0 ± 1.7 | 37 | 0.27 |
| Rib-5-P | 1.0 ± 0.19 | 82 | |
| 6-Phosphogluconic acid | 6.0 ± 2.6 | 14 | |
| Suc-6-P | 11.9 ± 7.1 | 50 | |
| No activator | 10 | ||
| Pi | 6.69 |
Relative activation of AGPase by the indicated metabolite compared to that of activation by 3-PGA.
Data taken from Liu and Shannon (1981).
We then tested the same series of metabolites for their ability to stabilize the enzyme to heat inactivation (Fig. 4). In addition to the activators listed, sulfate was also tested because sulfate inhibits the potato tuber homotetramer (Jin et al., 2005) and saturating levels of sulfate negate the activating effect of 3-PGA. The maize endosperm AGPase is also inhibited by SO4− with a Ki of approximately 20 mm with a [3-PGA] = 2.5 mm (S.K. Boehlein, unpublished data). As shown in Figure 4, all of the metabolites that activate or inhibit activation of the maize endosperm AGPase also stabilize the enzyme against thermal inactivation. While it is evident that all metabolites can stabilize enzyme activity, major differences are seen in terms of the extent of protection and the concentrations of metabolites required for maximal stabilization. For example, 3-PGA and SO4− protect the enzyme to the same extent and also require a similar concentration for maximal protection, while the amount of F-6-P has not reached saturation under these conditions. Pi provides the best protection at saturation against heat inactivation (Fig. 4). The reducing reagent, DTT, and a noneffector ion, KCl, had no effect on the heat stability of the enzyme (data not shown).
Figure 4.

Thermal stability of AGPase in the presence of sulfate, F-6-P, and G-6-P. Desalted enzymes containing 0.5 mg/mL BSA were heated at 42°C for 10 min in the presence of varying concentrations of PO4− (•), 3-PGA (▾), sulfate (♦), F-6-P (▪), or G-6-P (▴). Duplicate samples were left on ice and their activity was taken to be 100%. All preparations were assayed for 10 min in the forward direction in the presence of 10 mm 3-PGA. The percentage of activity remaining was then calculated by dividing the heated versus nonheated samples and plotted against the effector.
We next measured the ability of the substrates to protect AGPase activity. Purified AGPase was mixed with the substrates (5 mm final concentration) listed in Table II and exposed to 42°C for 7.5 min prior to assay in the forward (ADP-Glc synthesis) or back (ADP-Glc degradation) direction. Activities are presented as a percentage of activity remaining from a control maintained on ice prior to assays. Several effectors were also included in these studies. Various kinetic studies have shown that ATP is the first substrate bound to the enzyme and ADP-Glc is the last product released in other AGPase homologs (Paule and Preiss, 1971; Kleczkowski et al., 1993). Hence, it was expected that these kinetically productive complexes formed with reactants might prevent heat-induced activity loss. Under the conditions employed, ADP-Glc provided the greatest amount of protection against thermal inactivation (approximately 60%), regardless of direction. This protection was enhanced by adding either 3-PGA or Pi, and virtually complete protection under these experimental conditions was achieved by the addition of both ADP-Glc and 3-PGA (Fig. 5). It is also interesting that 3-PGA greatly decreased the amount of ADP-Glc required for one-half maximal protection (Fig. 5). The synergistic effect was not surprising because 3-PGA decreases the Km for the substrates as well as increases Vmax (Boehlein et al., 2005).
Table II.
Effect of substrates and effectors on heat denaturation as assayed in both the forward and back directions
Substrates and effectors were added to a final concentration of 5 mm, with the exception of PPi, which was added to a final concentration of 10 mm. All substrate/effector mixes contain 10 mm MgCl2, except where CaCl2 is specified. Enzymes were incubated for 10 min at 42°C, placed on ice, and then assayed in either the forward or reverse direction. ADPG, ADP-Glc; Ca, calcium.
| Effector/Substrate | Forward
|
Back
|
|---|---|---|
| % Activity Remaining | % Activity Remaining | |
| None | 4 | 3 |
| ATP | 14 | 19 |
| ATP + Pi | 58 | 71 |
| ATP + 3-PGA | 62 | 85 |
| ATP + G-1-P + 3-PGA + Ca | 63 | NDa |
| G-1-P | 0 | ND |
| G-1-P + Pi | 51 | ND |
| G-1-P + 3-PGA | 21 | ND |
| ADPG | 59 | 60 |
| ADPG + Ca | ND | 54 |
| ADPG + Pi | 73 | 75 |
| ADPG + 3-PGA | 100 | ND |
| ADPG + PPi + Ca | ND | 47 |
| Pi | 47 | 64 |
| 3-PGA | 30 | 39 |
| Pi + 3-PGA | ND | 67 |
| PPi + 3-PGA | ND | 12 |
| PPi | ND | 0 |
| PPi + Pi | ND | 45 |
ND, Not determined.
Figure 5.

Thermal stability of AGPase in the presence of ADP-Glc and/or 3-PGA. Desalted enzymes containing 0.5 mg/mL BSA were heated at 42°C for 10 min in the presence of varying concentrations of ADP-Glc (▪) or ADP-Glc (▴) with 10 mm 3-PGA. Duplicate samples were left on ice and their activity was taken to be 100%. All preparations were assayed for 10 min in the forward direction in the presence of 10 mm 3-PGA. The percentage of activity remaining was then calculated by dividing the heated versus nonheated samples and plotted against the effector concentration.
Although ATP alone provided some protection (approximately 15%), the addition of Pi or 3-PGA to the ATP/enzyme mixture greatly enhanced enzyme stability at 42°C (Table II). Increasing MgCl2 and ATP concentration to 50 mm allowed 46% of the initial activity to be maintained after heat treatment. PPi did not protect the enzyme against thermal inactivation and actually reduced the amount of stabilization elicited by 3-PGA and Pi. G-1-P also was unable to stabilize the enzyme at concentrations up to 50 mm. Additional experiments were performed in which 3-PGA, ATP, and G-1-P were added to the enzyme in the presence of Ca2+. Ca2+ was used as a cofactor so ATP would bind, but the reaction would not proceed to an appreciable extent during the incubation period (Fu et al., 1998). This set of conditions produced no additional protection compared to experiments containing only ATP and 3-PGA. Also, the addition of ADP-Glc and PPi in the presence of Ca2+ did not condition additional protection compared to addition of only ADP-Glc (Table II).
Taken together, these data are consistent with an ordered kinetic mechanism, with reactants binding productively to free enzyme. In turn, this conditions protection against heat-induced activity loss. 3-PGA and Pi protect against thermal inactivation independently and each acts synergistically with ADP-Glc and ATP to protect the enzyme. Substrates that do not bind productively to free enzyme (PPi and G-1-P) do not enhance thermal stability.
To further characterize binding of these effector molecules to AGPase, the method of Scrutton and Utter (1965) was used to determine the Kd values at 37°C (Table III). The dissociation constant for Pi is the lowest, at 70 μm, while those for 3-PGA, ATP, and ADP-Glc (1.84 mm, 1 mm, and 0.2 mm, respectively) are higher than their corresponding Ka and Km values. However, heat stability experiments were performed on kinetically quiescent enzyme, whereas Ka, Ki, and Km values were determined with catalytically active enzyme. The Kds reported here reflect binding to enzyme in the absence of other ligands. In addition to obtaining the Kd values, the ratios of inactivation constants (k2:k1) were also determined. These reflect the degree of protection provided by the stabilizing metabolites, a smaller ratio indicating a higher degree of protection. All ligands investigated here provide comparable levels of protection except for Rib-5-P, suggesting a common binding mode.
Table III.
Kd for various effector molecules
| Effector Molecule | Kd | k2/k1 |
|---|---|---|
| mm | ||
| Pi | 0.07 | 0.14 |
| 3-PGA | 0.84 | 0.19 |
| ATP | 1.0 | 0.13 |
| ADP-Glc | 0.2 | 0.13 |
| F-6-P | 1.7 | 0.23 |
| Glycerophosphate | 8.7 | 0.17 |
| Rib-5-P | 7.0 | 0.48 |
We then probed the possibility that all the ligands responsible for modulating catalytic activity and thermal stability might bind to the site(s) occupied by 3-PGA. If so, several conditions should be met. First, the addition of a nonsaturating concentration of a second activator should enhance the activation caused by nonsaturating amounts of 3-PGA. This effect was observed when 0.1 mm 3-PGA (one-half Ka value) was added to the enzyme, followed by a second activator equal to one-half its Ka value. Significant additional stimulation was seen for most activators (Table IV). A second prediction of this hypothesis is that a metabolite that activates to a lesser extent than 3-PGA should, at increasing concentrations, inhibit the activation produced by 3-PGA. Phosphoenolpyruvate (PEP) was chosen for this experiment because the activity in the presence of PEP was only about 40% of its activity in the presence of 3-PGA. 3-PGA was used at a concentration of 0.75 mm approximately (3 × Ka) and PEP was varied from 0 to 100 mm (0–20 × Ka). Inhibition by PEP begins at about 25 mm and increases up to about 100 mm (Fig. 6). The decrease in activity caused by the competition of activators indicates that PEP and 3-PGA are binding to the same site or to mutually exclusive sites.
Table IV.
Stimulatory effect of the addition of a secondary activator
All samples contained 0.1 mm 3-PGA (approximately one-half Ka). Metabolites were then added to a concentration of one-half their Ka (see Table I), and fold stimulation was calculated as the amount of activity in the presence of activator divided by amount of activity in the presence of 0.1 mm 3-PGA.
| Activator | Fold Stimulation |
|---|---|
| No secondary activator (0.1 mm 3-PGA) | 1 |
| F-6-P | 1.64 |
| G-6-P | 2.19 |
| Glycerophosphate | 2.15 |
| PEP | 1.55 |
| Rib-5-P | 1.78 |
| 6-Phosphogluconic acid | 1.11 |
| 3-PGA | 1.90 |
Figure 6.

The effect of 3-PGA and PEP on the AGPase activity. The activity of AGPase was measured in the presence of 0.75 mm 3-PGA at varying concentrations of PEP. The percentage of activity was calculated by dividing the amount of activity in the presence of PEP by the amount of activity in the absence of PEP (0.75 mm 3-PGA) and plotted against the PEP concentration.
Several observations suggest that Pi and 3-PGA share a common binding site. Heat protection studies were employed to further investigate this possibility. If 3-PGA and Pi bind to a common site, the presence of one will not have a synergistic effect on the other during thermal protection studies. If, on the other hand, binding of the activator and binding of the inhibitor are independent or coupled, a cumulative or synergistic pattern would be seen. To determine the relationship between 3-PGA and Pi, several activator/inhibitor combinations were used in thermal protection studies (Table II). First, when assayed in the reverse direction, 10 mm Pi conditions 64% protection under the conditions specified. Similarly, 3-PGA conditions 39% protection. Significantly, the combination of 3-PGA and Pi gives 67% protection, a value comparable to the protection observed with only Pi. This suggests that 3-PGA and Pi are binding to the same site or mutually exclusive binding sites. In contrast, we note that the addition of 3-PGA greatly increases the protection caused by the substrates ADP-Glc and ATP (Table II). This is indicative that 3-PGA (and Pi) and the substrates do not share common binding sites.
Thus far, it has been shown that Pi can bind to the enzyme with a fairly high affinity; however, binding does not itself inhibit catalytic activity. A common binding site for Pi and 3-PGA should also be manifest by a Ki value for Pi that is dependent on 3-PGA concentration. This was observed (Fig. 7). As the 3-PGA concentration increases, the amount of Pi needed to inhibit activity rises. Thus, the role of Pi is not to inhibit the enzyme per se, but rather to deactivate the activated state by competing with the activator for binding to the enzyme. To determine whether Pi interacts with the effectors F-6-P and G-6-P in a similar manner, the Ki for Pi in the presence of 3-PGA, F-6-P, and G-6-P was determined. Enzymes were incubated with activators (3-PGA, F-6-P, or G-6-P) at 10× their corresponding Ka values. Pi inhibition studies were then performed to determine the Ki value of Pi in the presence of these activators. If phosphate is able to compete with these activators in a comparable manner, then all three of their phosphate Ki values should be the same. This was the case (Table V).
Figure 7.

A, Inhibition of AGPase activity by phosphate. The activity of AGPase was measured using the forward reaction in the presence of varying concentrations of Pi at several fixed concentrations of 3-PGA: 1 mm (▴), 2.5 mm (•), 5 mm (▾), and 10 mm (♦). The Ki for Pi was determined from the X intercept at each 3-PGA concentration. B, Determination of the binding constant of Pi in the absence of 3-PGA. A plot of Ki versus 3-PGA concentration was used to determine the binding constant of Pi in the absence of 3-PGA.
Table V.
Phosphate Ki in the presence of various activators
| Effectora | Ki |
|---|---|
| mm | |
| 3-PGA | 1.75 ± 0.025 |
| F-6-P | 0.99 ± 0.35 |
| G-6-P | 1.07 ± 0.39 |
| None | >100 mm |
Effectors were used at approximately 10× their corresponding Ka value (see Table I).
Heat Denaturation Curves for AGPase Are Biphasic
While all of the thermal protection experiments described above employed a temperature of 42°C and exposure times ranging up to 10 min, longer incubations at 42°C showed that catalytic activity remaining after the initial exposure to 7 to 10 min exhibited greater heat stability than the activity lost at the beginning of the experiment. Representative data are shown in Figure 8. Catalytic activity was monitored over 40 min in the forward (Fig. 8A) and back (Fig. 8B) directions in the presence of 10 mm Pi. All plots exhibited a clear biphasic pattern of thermal inactivation with a break around 10 min. To determine whether the transition between the two states of the enzyme was a fast or slow process, AGPase plus Pi was placed at 42°C for 10 min and then cooled on ice for 5 min. The sample was then reincubated at 42°C and catalytic activity was monitored over time. If the transition back to the unstable form is a fast process, a rapid loss in enzyme activity should occur during the first 5 to 10 min following re-exposure to heat treatment. Conversely, if the transition is a slow process and the enzyme is locked into a more stable conformation, activity of the preheated enzyme should not exhibit a biphasic decay curve and a lower initial activity should be seen. As can be seen in Figure 8C, the preheated AGPase retains the properties of the more stable configuration of the enzyme. This experiment demonstrates that the enzyme form, which is rapidly heat inactivated, cannot be restored by cooling and that the remaining catalytic activity is due to a more heat-stable form to the enzyme. We then probed whether this interconversion of heat-labile to heat-stable form required the presence of Pi. At 42°C, the wild-type AGPase activity inactivates rapidly, in the absence of Pi, and a clear distinction between enzyme conformations was not obvious. The temperature of inactivation was therefore lowered to 37°C (Fig. 8D) and 30°C (data not shown). These data showed that the biphasic nature of decay is still present, thereby indicating that, although Pi protects the enzyme from heat inactivation, its presence is not obligatory for the biphasic loss of catalytic activity. A similar experiment was performed in the presence of DTT, and no discernable effect on the biphasic nature of inactivation was noted.
Figure 8.

Biphasic nature of heat inactivation. A, AGPase (0.02 mg/mL) was mixed to a final concentration of 10 mm phosphate and incubated on ice for 5 min. Enzymes were then heated at 45°C for the specified amount of time and placed back on ice. When all of the time points were completed, the enzymes were assayed using standard protocols. B, AGPase was prepared as above and assayed in the reverse direction. C, Enzymes were prepared as in A. The entire mixture was then heated at 45°C for 7 min and then cooled on ice. The same experiment was then performed as in A. D, No phosphate was added to the enzyme. Heat treatment was performed at 37°C.
DISCUSSION
Plant transgenic approaches have shown that AGPases harboring alterations in allosteric properties as well as enhanced thermal stabilities can enhance starch synthesis in agriculturally important tissues and organs such as the potato tuber and seeds of maize, wheat, and rice. Accordingly, we have focused our investigations on these two properties. The results reported here show that allostery and heat stability are intimately connected. Metabolites that affect catalytic activity also affect heat stability. Several other features of the maize endosperm AGPase, as outlined below, are noteworthy.
First, while 3-PGA has received the greatest attention as an activator of AGPase activity, we show that the maize endosperm AGPase can also be activated by a variety of other phosphate monoesters. Metabolites ranging from the three-carbon 3-PGA to the 12-carbon Suc-6-P cause comparable levels of activation. Activating metabolites appear to bind at the same or mutually exclusive sites in stimulating maize endosperm AGPase because the addition of metabolites to AGPase partially activated by 3-PGA result in further activity enhancements. This has been directly demonstrated for 3-PGA and PEP, and it is a distinct possibility that all activating metabolites bind to the same site.
While F-6-P and G-6-P both stimulate the activity of several AGPases (Ghosh and Preiss, 1965; Frueauf et al., 2002; Gomez-Casati and Iglesias, 2002), the reported activation levels are less than those observed with the maize endosperm AGPase where F-6-P and G-6-P at physiological concentrations both stimulate the activity to approximately 70% of the maximum observed with 3-PGA (Plaxton and Preiss, 1987; Boehlein et al., 2005). On the other hand, the extent of activation caused by Rib-5-P for the maize endosperm AGPase is much higher, relative to 3-PGA-induced activation, than the level noted for the spinach chloroplast AGPase (Ghosh and Preiss, 1965).
We were surprised to find that glycerol phosphate stimulates the maize endosperm AGPase. This has not been reported for other AGPases, to the best of our knowledge. Whether this provides a mechanism to coordinate starch synthesis and glycolysis and/or lipid synthesis is presently unknown. Alternatively, this activation may simply reflect the structural similarity of glycerol phosphate and 3-PGA.
Metabolites that activate or inhibit AGPase also stabilize the enzyme to thermal inactivation. While Zhou and Cheng (2005) reported that 3-PGA stabilized the apple leaf AGPase and stabilization of the cyanobacterial homolog by 3-PGA and Pi has also been observed (Gomez-Casati et al., 2000), the data reported here show that the range of suitable ligands for the maize endosperm AGPase is much more diverse. Interestingly, while the dissociation constants of ligands that protect against thermal inactivation differ markedly, the degree of protection (as judged by k2:k1 ratios) is quite similar for all metabolites except Rib-5-P. For example, both 3-PGA and glycerol phosphate provide similar levels of protection but differ markedly in their affinity for the enzyme, perhaps as a result of the additional negative charge in 3-PGA. Given the physiological concentrations of these ligands (Table I) and their binding affinities for the enzyme (Table III), maize endosperm AGPase likely enjoys nearly maximal thermal stabilization in its cellular environment.
Protection by ATP and ADP-Glc but not G-1-P and PPi against thermal inactivation is consistent with the well-precedented, ordered kinetic mechanism in which ATP is the first substrate bound and ADP-Glc is the last product released. In this regard, the maize endosperm AGPase exhibits a striking dissimilarity with a cyanobacterial AGPase. This bacterial enzyme is protected from thermal inactivation only by ATP (Gomez-Casati et al., 2000). It is possible that a conformational change occurs after ADP-Glc release in the cyanobacterial enzyme that disfavors rebinding of ADP-Glc. Such an iso-step is unlikely to be present in the maize homolog. The observation that 3-PGA and ADP-Glc acts synergistically to protect the maize endosperm AGPase suggests cooperative binding of these ligands. If a similar situation holds for the cyanobacterial enzyme, ADP-Glc may afford thermal protection if 3-PGA were also present. In addition we would predict that ATP would not protect the potato tuber enzyme in the absence of activator because no apparent activity is seen in the absence of 3-PGA, but, in the presence of 3-PGA, ATP would protect synergistically. Whether these predictions will be borne out awaits experimental testing.
The synergistic interaction of substrates and activators/inhibitors in protecting the maize endosperm AGPase against thermal inactivation suggests that enhanced heat protection occurs when both the active and allosteric sites are occupied. On the other hand, synergy was not observed in studies involving saturating levels of activators and Pi, suggesting that these species bind to the same or mutually exclusive sites. The data obtained from crystallization studies of the potato tuber small subunit homotetramer suggests that Pi has three or four separate binding sites on this particular enzyme and that 3-PGA binds at or near one of these inhibitor binding sites (Jin et al., 2005). This notion is consistent with the work of Morell et al. (1988), who showed that both Pi and 3-PGA blocked binding of pyridoxal phosphate to the spinach leaf AGPase. Comparing the Ka values for the activators with their physiological concentrations (Table II) and the Ki values for Pi (Table V) indicates that the maize endosperm AGPase probably functions at about 50% of its maximal activity, and relatively small changes in [Pi] would have significant impacts on this value. Such data are consistent with our experience in generating mutant forms that increase starch production.
Several explanations can account for the biphasic nature of the thermal stability exhibited by the maize endosperm AGPase. In the simplest case, two forms of enzyme exist at all times. The major form of activity, form 1, denatures rapidly, while the initially minor form 2 inactivates slowly. Therefore, during the first few minutes of heat treatment, large decreases in activity are seen from form 1 inactivation. This is then followed by a second, slow decrease in activity as form 2 is denatured. A second model assumes that before heat treatment, only one form of the enzyme exists. This form is heat labile. Exposure to heat causes the highly active, heat-labile form to convert to a more stable, less active form. Interconversion between the two forms is slow, giving rise to the observed biphasic pattern. A third, hybrid model can also be envisaged. Here, the enzyme exists as one heat-labile form. In the presence of heat, a proportion of molecules are denatured, whereas other molecules are slowly converted into a more heat-stable form. According to this model, both forms have comparable activity. The proposed interconversion of one form to another is reminiscent of the activation process of the potato tuber AGPase (Fu et al., 1998). A several-minute exposure to DTT and ADP-Glc at 37°C gives rise to a 10-fold increase in potato tuber AGPase activity. Subsequent characterization showed that a disulfide bridge between two Cys residues in the N termini of the small subunits must be reduced for activation to occur. ADP-Glc then activates the enzyme. While the conversion of the heat-labile to the heat-stable form of maize endosperm AGPase requires a several-minute exposure to elevated temperatures, the presence of DTT neither blocks nor enhances the conversion process. In addition, the maize endosperm AGPase small subunit lacks the Cys involved in the DTT/ADP-Glc activation process of the potato tuber AGPase.
MATERIALS AND METHODS
Growth and Assay Conditions for Expression of Maize Wild-Type AGPase in Escherichia coli
Escherichia coli AC70R1-504 (Iglesias et al., 1993), which lacks the functional AGPase gene and cannot synthesize glycogen, was transformed with both pMoncSh2 and pMoncBt2 (plasmids containing the large and the small subunit of wild type-AGP, respectively; Giroux et al., 1996), allowed to recover in SOC for 1 h, and directly grown overnight at 37°C in Luria-Bertani media containing 75 μg/mL spectinomycin and 50 μg/mL kanamycin. When the cultures reached an OD600 of 0.7 to 1.0 (16–20 h), they were cooled to room temperature and protein expression was induced by the addition of 0.2 mm isopropyl-β-d-thiogalactoside and 0.02 mg/mL nalidixic acid. Expression continued for 3 h at room temperature with constant shaking. Cells were harvested by centrifugation at 8,000g and the pellets were stored at −80°C.
Purification of AGPase
Maize endosperm AGPase was purified as described elsewhere (Boehlein et al., 2005) with the following modifications. All steps were performed at 4°C and centrifugations were at 30,000g unless otherwise stated. Cell pellets from 4 L of E. coli cultures were extracted in 20 mL of buffer A (50 mm KH2PO4, pH 7.0, 5 mm MgCl2, 0.5 mm EDTA), lysozyme, 50 μg/mL, and protease inhibitors (1 μg/mL pepstatin, 0.1 mm phenylmethylsulfonyl fluoride, 10 μg/mL chymostatin, and 1 mm benzamidine). Cells were thawed on ice and broken in a French press, and the extract was clarified by centrifugation for 20 min. The protein concentration was then adjusted to 30 mg/mL. Three-tenths volume of a 1% protamine sulfate solution was then added, and the mixture was stirred on ice for 20 min, followed by centrifugation for 20 min. The extract was then brought to 45% saturation with solid ammonium sulfate, stirred on ice for 20 min, and centrifuged for 20 min. The resulting pellet was resuspended in buffer A and stored at −80°C. Further purification using column chromatography (ion-exchange and hydroxyapetite) is described in Boehlein et al. (2005). Concentrated, purified proteins were stored at −80°C for many months without appreciable loss of activity.
Prior to kinetic analysis, proteins were desalted using Zeba micro desalt spin columns according to the manufacturer's instructions (Pierce). Proteins were routinely exchanged into 50 mm HEPES, 5 mm MgCl2, 0.5 mm EDTA, assayed for protein concentration, and then bovine serum albumin (BSA; 0.5 mg/mL) was added for stability.
Assay A (Reverse Direction)
A nonradioactive endpoint assay was used to determine the amount of G-1-P produced by coupling its formation to NADH production using phosphoglucomutase and G-6-P dehydrogenase (Sowokinos and Preiss, 1982). Specific details of the reaction are given in Boehlein et al. (2005). Purification of the wild-type enzyme was monitored using assay A.
Assay B (Forward Reaction)
A nonradioactive endpoint assay was used to determine the amount of PPi produced by coupling it to a decrease in NADH concentration. Standard reaction mixtures contained 50 mm HEPES, pH 7.4, 15 mm MgCl2, 1.0 mm ATP, and 2.0 mm G-1-P in a total volume of 200 μL. When activators were added to the reaction, their concentration is specified in tables or figure legends. Reaction tubes were prewarmed to 37°C, and assays were initiated by enzyme addition. Reactions were performed at 37°C and were terminated by boiling for 1.5 min. The reactions were developed by adding 300 μL of coupling reagent (described below) to each tube and the A340 was determined. Blank samples contained complete reaction mixtures without enzyme. The amount of PPi produced was determined from a standard curve using PPi in complete reaction mixtures and omitting enzyme. The change in absorbance between the blank and the reaction was used to calculate the amount of PPi. Reactions were linear with time and enzyme concentration. All kinetic constants were obtained using the forward reaction, unless otherwise stated.
Preparation of Coupling Reagent
The gene encoding the 62-kD PPi-dependent phosphofructokinase from Borrelia burgdorferi was obtained via PCR from genomic DNA purchased from the ATCC. After insertion into a pET duet expression vector, the protein was expressed in BL21DE3 cells and purified according to Deng et al. (1999). The enzyme was concentrated to 0.8 mg/mL and stored in aliquots at −80°C. All other enzymes were purchased from Sigma.
The coupling reagent contained a final concentration of 25 mm imidazole, pH 7.4, 4.0 mm MgCl2, 1.0 mm EDTA, 0.2 mm NADH, 0.725 units of aldolase, 0.4 units of triose phosphate isomerase, 0.6 units of glycerophosphate dehydrogenase, 1.0 mm F-6-P, and 0.8 μg of purified PPi-dependent phosphofructokinase per reaction. The coupling reagent was made fresh daily.
Determination of the Kinetic Constants
Activation constants (Ka) for various AGPase effectors were determined by incubating purified AGPase (0.3 μg) in reaction mixtures with the following components: 50 mm HEPES, pH 7.4, 15 mm MgCl2, 1 mm ATP, and 2 mm G-1-P and varying effector concentrations (0.05–25 mm). Assays were initiated with purified enzyme, incubated 10 min at 37°C, and inactivated by boiling for 1.5 min. Kinetic constants were obtained by nonlinear regression using equations derived from the full kinetic expression using the software program Prism (Graph Pad).
Heat Stability of Purified Maize AGPase
Heat stability was determined using desalted enzyme at a concentration of 2.6 μg protein/mL with or without stabilizing molecules (5 mm). Preparations were placed in a water bath at either 42°C or 37°C for varying times and then cooled on ice. All preparations were subsequently assayed at 37°C for 10 min in the forward direction in the presence of 10 mm 3-PGA. Reactions were started with 1.3 μg of enzyme. Data were plotted as log % activity versus time, and the inactivation constant t1/2 was calculated as follows: slope = −k/(2.3). t1/2 is calculated from the equation k = 0.693/t1/2. Kd was determined by the method of Scrutton and Utter (1965) using the following equations:
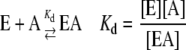 |
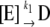 |
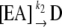 |
where E = free enzyme, D = denatured enzyme, A = stabilizing molecule, Kd = dissociation constant of EA, k1 = rate constant for denaturation of E, and k2 = rate constant for denaturation or EA. If the equilibrium between E, A, and EA is rapid compared with denaturation, then the following relationship applies:
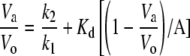 |
Va and Vo are the rates of inactivation of E and the presence and absence of stabilizing molecule A.
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers AF334959 and P55241.
This work was supported by the National Science Foundation (grant nos. IBN–9982626 and 0444031 to L.C.H.) and by the U.S. Department of Agriculture Competitive Grants Program (grant nos. 2000–01488, 2006–03034, and 2007–03575 to L.C.H.).
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: L. Curtis Hannah (hannah@mail.ifas.ufl.edu).
Open Access articles can be viewed online without a subscription.
References
- Bae JM, Giroux MJ, Hannah LC (1990) Cloning and molecular characterization of the brittle-2 gene of maize. Maydica 35 317–322 [Google Scholar]
- Ballicora MA, Fu Y, Frueauf JB, Preiss J (1999) Heat stability of the potato tuber ADP-glucose pyrophosphorylase: role of Cys residue 12 in the small subunit. Biochem Biophys Res Commun 257 782–786 [DOI] [PubMed] [Google Scholar]
- Ballicora MA, Iglesias AA, Preiss J (2003) ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol Mol Biol Rev 67 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave MR, Lawrence S, Barton C, Hannah LC (1990) Identification and molecular characterization of shrunken-2 cDNA clones of maize. Plant Cell 2 581–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehlein SK, Sewell AK, Cross J, Stewart JD, Hannah LC (2005) Purification and characterization of adenosine diphosphate glucose pyrophosphorylase from maize/potato mosaics. Plant Physiol 138 1552–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger BT, Cross JM, Shaw JR, Caren JR, Greene TW, Okita TW, Hannah LC (2003) Relative turnover numbers of maize endosperm and potato tuber ADP-glucose pyrophosphorylases in the absence and presence of 3-phosphoglyceric acid. Planta 217 449–456 [DOI] [PubMed] [Google Scholar]
- Cross JM, Clancy M, Shaw J, Boehlein SK, Greene T, Schmidt R, Okita T, Hannah LC (2005) A polymorphic motif in the small subunit of ADP-glucose pyrophosphorylase modulates interactions between the small and large subunits. Plant J 41 501–511 [DOI] [PubMed] [Google Scholar]
- Cross JM, Clancy M, Shaw JR, Greene TW, Schmidt RR, Okita TW, Hannah LC (2004) Both subunits of ADP-glucose pyrophosphorylase are regulatory. Plant Physiol 135 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Roberts D, Wang X, Kemp RG (1999) Expression, characterization, and crystallization of the pyrophosphate-dependent phosphofructo-1-kinase of Borrelia burgdorferi. Arch Biochem Biophys 371 326–331 [DOI] [PubMed] [Google Scholar]
- Doan DN, Rudi H, Olsen OA (1999) The allosterically unregulated isoform of ADP-glucose pyrophosphorylase from barley endosperm is the most likely source of ADP-glucose incorporated into endosperm starch. Plant Physiol 121 965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frueauf JB, Ballicora MA, Preiss J (2002) Alteration of inhibitor selectivity by site-directed mutagenesis of Arg(294) in the ADP-glucose pyrophosphorylase from Anabaena PCC 7120. Arch Biochem Biophys 15 208–214 [DOI] [PubMed] [Google Scholar]
- Fu Y, Ballicora MA, Leykam JF, Preiss J (1998) Mechanism of reductive activation of potato tuber ADP-glucose pyrophosphorylase. J Biol Chem 273 25045–25052 [DOI] [PubMed] [Google Scholar]
- Georgelis N, Braun EL, Shaw JR, Hannah LC (2007) The two AGPase subunits evolve at different rates in angiosperms, yet they are equally sensitive to activity-altering amino acid changes when expressed in bacteria. Plant Cell 19 1458–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh HP, Preiss J (1965) The biosynthesis of starch in spinach chloroplasts. J Biol Chem 240 960–962 [PubMed] [Google Scholar]
- Ghosh HP, Preiss J (1966) Adenosine diphosphate glucose pyrophosphorylase. A regulatory enzyme in the biosynthesis of starch in spinach leaf chloroplasts. J Biol Chem 241 4491–4504 [PubMed] [Google Scholar]
- Giroux MJ, Shaw J, Barry G, Cobb BG, Greene T, Okita T, Hannah LC (1996) A single mutation that increases maize seed weight. Proc Natl Acad Sci USA 93 5824–5829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Casati DF, Iglesias AA (2002) ADP-glucose pyrophosphorylase from wheat endosperm. Purification and characterization of an enzyme with novel regulatory properties. Planta 214 428–434 [DOI] [PubMed] [Google Scholar]
- Gomez-Casati DF, Preiss J, Iglesias AA (2000) Studies on the effect of temperature on the activity and stability of cyanobacterial ADP-glucose pyrophosphorylase. Arch Biochem Biophys 384 319–326 [DOI] [PubMed] [Google Scholar]
- Hannah LC (1997) Starch synthesis in the maize endosperm. In BA Larkins, IK Vasil, eds, Advances in Cellular and Molecular Biology of Plants. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 375–405
- Hannah LC (2005) Starch synthesis in the maize endosperm. Maydica 50 497–506 [Google Scholar]
- Hannah LC (2007) Starch formation in the maize endosperm. In O-A Olsen, ed, Endosperm, Developmental and Molecular Biology. Springer Books, Berlin, pp 179–194
- Hannah L, Tuschall D, Mans R (1980) Multiple forms of maize endosperm ADP-glucose pyrophosphorylase and their control by Shrunken-2 and Brittle-2. Genetics 95 961–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SK, Salamone PR, Okita TW (2005) Allosteric regulation of the higher plant ADP-glucose pyrophosphorylase is a product of synergy between the two subunits. FEBS Lett 579 983–990 [DOI] [PubMed] [Google Scholar]
- Iglesias AA, Barry GF, Meyer C, Bloksberg L, Nakata PA, Greene T, Laughlin MJ, Okita TW, Kishore GM, Preiss J (1993) Expression of the potato tuber ADP-glucose pyrophosphorylase in Escherichia coli. J Biol Chem 268 1081–1086 [PubMed] [Google Scholar]
- Jin X, Ballicora MA, Preiss J, Geiger JH (2005) Crystal structure of potato tuber ADP-glucose pyrophosphorylase. EMBO J 24 694–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleczkowski LA, Villand P, Preiss J, Olsen OA (1993) Kinetic mechanism and regulation of ADP-glucose pyrophosphorylase from barley (Hordeum vulgare) leaves. J Biol Chem 268 6228–6233 [PubMed] [Google Scholar]
- Linebarger CR, Boehlein SK, Sewell AK, Shaw J, Hannah LC (2005) Heat stability of maize endosperm ADP-glucose pyrophosphorylase is enhanced by insertion of a cysteine in the N terminus of the small subunit. Plant Physiol 139 1625–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TT, Shannon JC (1981) Measurement of metabolites associated with nonaqueously isolated starch granules from immature Zea mays L. endosperm. Plant Physiol 67 525–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell M, Bloom M, Preiss J (1988) Affinity labeling of the allosteric activator site(s) of spinach leaf ADP-glucose pyrophosphorylase. J Biol Chem 263 633–637 [PubMed] [Google Scholar]
- Okita T, Nakata P, Anderson J, Sowokinos J, Morell M, Preiss J (1990) The subunit structure of potato tuber ADPglucose pyrophosphorylase. Plant Physiol 93 785–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paule MR, Preiss J (1971) Biosynthesis of bacterial glycogen 10. Kinetic mechanism of adenosine diphosphoglucose pyrophosphorylase from Rhodospirillum rubrum. J Biol Chem 246 4602–4609 [Google Scholar]
- Plaxton WC, Preiss J (1987) Purification and properties of nonproteolytic degraded ADPglucose pyrophosphorylase from maize endosperm. Plant Physiol 83 105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss J, Romeo T (1994) Molecular biology and regulatory aspects of glycogen biosynthesis in bacteria. Prog Nucleic Acid Res Mol Biol 47 299–329 [DOI] [PubMed] [Google Scholar]
- Preiss J, Sivak M (1996) Starch synthesis in sinks and sources. In E Zamski, ed, Photoassimilate Distribution in Plants and Crops: Source-Sink Relationships. Marcel Dekker, New York, pp 139–168
- Sakulsingharoja C, Choi SB, Hwang SK, Edwards GE, Bork J, Meyer CR, Preiss J, Okita TW (2004) Engineering starch biosynthesis for increasing rice seed weight: the role of the cytoplasmic ADP-glucose pyrophosphorylase. Plant Sci 167 1323–1333 [Google Scholar]
- Scrutton MC, Utter MF (1965) Pyruvate carboxylase. V. Interaction of the enzyme with adenosine triphosphate. J Biol Chem 240 3714–3723 [PubMed] [Google Scholar]
- Singletary GW, Banisadr R, Keeling PL (1993) Decreased starch synthesis in heat stressed maize kernels results from reduced ADPG-pyrophosphorylase and starch synthase activities. Plant Physiol Suppl (Bethesda) 102 6 [Google Scholar]
- Singletary GW, Banisadr R, Keeling PL (1994) Heat stress during grain filling in maize: effects of carbohydrate storage and metabolism. Aust J Plant Physiol 21 829–841 [Google Scholar]
- Smidansky ED, Clancy M, Meyer FD, Lanning SP, Blake NK, Talbert LE, Giroux MJ (2002) Enhanced ADP-glucose pyrophosphorylase activity in wheat endosperm increases seed yield. Proc Natl Acad Sci USA 99 1724–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidansky ED, Martin JM, Hannah LC, Fischer AM, Giroux MJ (2003) Seed yield and plant biomass increases in rice are conferred by deregulation of endosperm ADP-glucose pyrophosphorylase. Planta 216 656–664 [DOI] [PubMed] [Google Scholar]
- Sowokinos J, Preiss J (1982) Pyrophosphorylases in Solanum tuberosum. III. Purification, physical, and catalytic properties of ADPglucose pyrophosphorylase in potatoes. Plant Physiol 69 1459–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark D, Timmerman K, Barry G, Preiss J, Kishore G (1992) Regulation of the amount of starch in plant tissues by ADP glucose pyrophosphorylase. Science 258 287–291 [DOI] [PubMed] [Google Scholar]
- Wang Z, Chen X, Wang J, Liu T, Liu Y, Zhao L, Wang G (2007) Increasing maize seed weight by enhancing the cytoplasmic ADP-glucose pyrophosphorylase activity in transgenic plants. Plant Cell Tissue Organ Cult 88 83–92 [Google Scholar]
- Zhou R, Cheng L (2005) Binding of 3-phosphoglycerate leads to both activation and stabilization of ADP-glucose pyrophosphorylase from apple leaves. Funct Plant Biol 32 839–848 [DOI] [PubMed] [Google Scholar]