

Abstract
The Gram-positive bacterium Group B Streptococcus (GBS) is an important cause of serious neonatal and adult infections. Toll-like receptor 2 (TLR2) recognizes components of the cell wall of Gram-positive bacteria and is critical for defense against certain invasive pathogens. In GBS, penicillin-binding protein 1a (PBP1a), encoded by ponA, is required for virulence. PBPs participate in cell wall synthesis and in previous studies; the absence of PBP1a was shown to result in subtle changes in the cell wall ultrastructure. Here, we examine the role of TLR2 in defense against GBS infection and the impact of mutation of ponA on TLR2-mediated host responses. We demonstrate TLR2-recognition of both WT GBS and the ponA mutant in vitro. TLR2−/− mice were significantly more susceptible than WT mice to infection with either strain of GBS, indicating a crucial role for TLR2 in defense against GBS. Additionally, the ponA mutant was severely attenuated for virulence in both strains of mice. The mutation in ponA did not affect cytokine expression by WT or TLR2−/− mice. These data indicate that TLR2 is required for host defense against GBS and this response is unaffected by the absence of PBP1a and the resultant changes in cell wall ultrastructure.
Keywords: Streptococcus agalactiae, Toll-like receptor 2, Penicillin-binding protein 1A
1. Introduction
The Gram-positive bacterium Group B Streptococcus (GBS, Streptococcus agalactiae) is an important cause of neonatal sepsis, pneumonia, and meningitis and is increasingly implicated in soft-tissue and invasive infections in the elderly and the immunocompromised [1–6]. Neonatal infections result from ascending movement of GBS from the lower maternal genital tract into the amniotic fluid with subsequent aspiration prior to birth, or from aspiration of infected maternal secretions during delivery. GBS then invades across epithelial and endothelial layers in the neonatal lung to cause bacteremia [7]. Accordingly, host defense against invasive GBS infection requires either containment to prevent invasion and subsequent bloodstream infection, or rapid elimination once such bacteremia occurs.
Neonates are relatively deficient in various aspects of adaptive immunity [8]. Therefore, neonates are dependant upon the innate immune system in order to mount a successful immune response to invading pathogens, such as GBS [8]. Activation of the innate immune response, in neonates and in adults, is controlled in large part by the Toll-like receptor (TLR) family of pattern-recognition receptors. TLRs are transmembrane proteins that recognize specific pathogen-associated molecular patterns. TLRs activate immune responses through several intracellular signaling proteins, such as MyD88, Toll-/interleukin-1-receptor (TIR) domain containing adaptor-inducing interferon (TRIF), TRIF-related adapter molecule (TRAM), and TIR domain containing adapter protein (TIRAP) [9, 10]. Signaling through these adapter proteins results in cell activation, induction of co-stimulatory proteins, and production of pro-inflammatory cytokines, such as TNF-α and IL-6 [10, 11], and other proteins important for host defense, such as antimicrobial peptides (AMP) [12].
GBS and other Gram-positive bacteria can activate host immune effector cells through TLR2 [13, 14]. The specific component or components of these bacteria that can initiate intra-cellular signaling cascades through TLR2 has not been conclusively identified. Lipopeptides and peptidoglycan (PG) from Gram-positive and Gram-negative bacteria, and lipoteichoic acid (LTA) from Gram-positive bacteria have all been proposed as the ligand(s) for TLR2 [11, 12, 15–18]. How TLR2 recognizes GBS is not completely elucidated. TLR2, as part of a heterodimer with TLR6, can bind the LTA of GBS [19] and may recognize other components of the cell wall of Gram-positive bacteria. Henneke et al. have demonstrated that TLR2 recognizes soluble secreted factors from GBS, in addition to LTA [20].
We have previously shown that PBP1a, a surface-localized penicillin-binding protein encoded by ponA, plays an important role in the pathogenesis of GBS infection as a mutant lacking PBP1a is attenuated for virulence in a neonatal rat sepsis infection model [21, 22] and cleared more rapidly from the lungs of neonatal rat pups in an aerosol infection model [23]. Additional characterization of the mutant revealed that PBP1a promotes resistance of GBS to killing by host innate immune antimicrobial peptides, which likely contributes to the virulence defect of this mutant [24].
PBP1a is a transmembrane bifunctional PG synthase that catalyzes the oligomerization of nascent PG strands and the cross-linking of these PG chains into the mature cell wall [25]. Orthologs of PBP1a are found in both Gram-positive and Gram-negative bacteria [26–28]. The absence of PBP1a results in subtle changes in the cell wall ultrastructure including a reduced cross-linking of the PG that is detectable by HPLC analysis [24]. As TLR2 recognizes the cell wall of Gram-positive bacteria, in this study we investigate whether the mutation in PBP1a affects recognition of GBS by TLR2 and examine the contribution of TLR2 to innate immune defense against infection by GBS.
2. Results
2.1. TLR2-mediated cell activation by WT GBS and the ponA mutant
Although GBS can activate cells through TLR2, the specific moiety or moieties responsible for this activation have not been identified [19, 20, 29, 30]. We compared the ponA mutant with WT GBS in TLR2 gain-of-function assays to examine whether TLR2-mediated cell activation differed between the two strains. In these experiments, the activation of HeLa cells, transiently transfected with either human or murine TLR2, after stimulation with varying concentrations of WT GBS or the ponA mutant, was measured using a NF-κB luciferase reporter system.
Serial dilutions of either WT GBS or the ponA mutant GBS were added to HeLa cells transfected with either human TLR2 or murine TLR2. A dose-dependent activation of NF-κB mediated by both human and murine TLR2 was observed (Fig. 1). At an inoculum of 5 × 106 CFU, activation of NF-κB by TLR2 was significantly increased by exposure to WT GBS, when compared to non-transfected HeLa cells. The same inoculum of the ponA mutant also activated NF-κB to an equivalent extent to that of WT GBS. This stimulation of NF-κB activation was still evident for both strains of GBS at an inoculum of 2.5 × 106 CFU, but was not detectable at 1.25 × 106 CFU.
Figure 1.

WT GBS and the ponA mutant activate human and murine TLR2. HeLa cells were transiently transfected with human TLR2, murine TLR2, or empty vector. WT GBS (WT) or the ponA mutant (ponA) at a range of concentrations (X equals 106 CFU of bacteria) was added to the HeLa cells and activation of TLR2 measured. No stimulation indicates cells were where only media was added and IL-1 is a positive control that activates NF-κB production in TLR2-independent manner. Data shown are representative of 4 separate experiments performed in duplicate ± SD.
No increase in luminescence was seen without infecting the HeLa cells with GBS (Fig. 1, no stimulation). The responsiveness of the transfected HeLa cells to a TLR2 agonist was confirmed with the use of heat-killed flagellin-deficient Listeria monocytogenes [31] (data not shown). A robust luminescence was seen following exposure of transfected HeLa cells with IL-1, which stimulates NF-κB activation independently of TLR2 (Fig. 1, IL-1) [32, 33].
2.2. Virulence of WT GBS and the ponA mutant in WT adult mice
The ponA mutant was originally identified in a virulence screen in an intraperitoneal sepsis model in neonatal rat pups. In this model, the LD50 of the ponA mutant was 10 to 80-fold higher than WT GBS [22]. To determine whether the ponA mutant was also attenuated for virulence in adult mice, we compared the virulence of the isogenic strains in a mouse sepsis infection model, as described in section 5.4. The results from two independent determinations of the LD50 of WT GBS and the ponA mutant in WT mice were combined, as described in section 5.7. The LD50 for WT GBS was 3.6 × 107 CFU (95% confidence interval 1.6 × 107 − 8.2 × 108 CFU), as compared to a LD50 for the ponA mutant of 3.6 × 108 CFU (95% confidence interval 7.8 × 107 − 1.6 × 109 CFU), a statistically significant difference (p = 0.01) in virulence, similar to our previous studies in neonatal rat pups [22].
2.3. Time to death of WT mice following infection with isogenic GBS strains
To investigate any difference in kinetics of killing between WT GBS and the ponA mutant, we performed time to death assays in WT mice. A range of inocula were used and a representative experiment in which the mice were infected with ~2.0 × 108 CFU of WT or ponA mutant GBS is shown in Fig. 2. More mice succumbed to infection by the WT strain than to the ponA mutant, confirming the results of our LD50 assays; however, no change in the rapidity of killing was noted between the two strains of GBS.
Figure 2.

Time to death assays in WT mice following infection. WT mice were infected with an equivalent dose of either WT GBS (▴; n=9) or ponA mutant (○; n=9). The number of mice in each group is indicated in the figure. Data shown are representative of 3 independent experiments.
2.4. Competitive Index Assays in WT mice
To investigate how the isogenic GBS strains competed in a mixed infection, we performed competitive index assays. Following 18–24 hours of infection, all mice were bacteremic. However, very few to none of the ponA mutant were recovered from WT mice (Table 1). The competitive index was <0.01 ± 0.01 in WT mice, consistent with severe attenuation of the ponA mutant compared to the WT strain that we observed in the LD50 and time to death assays. These data indicated that the presence of the WT strain does not complement the virulence defect of the ponA mutant in a mixed infection.
Table 1.
Competitive Index of WT and ponA mutant GBS in WT and TLR2−/− adult mice
| Strain of Mouse | Total # of bacteria recovereda | # of ponA mutant recovereda | Competitive Index |
|---|---|---|---|
| WT | 28 | <1 | <0.0328 |
| 149 | 1 | 0.00616 | |
| 3.39 × 103 | <17 | <0.00459 | |
| 4.85 × 103 | <12 | <0.0018 | |
| 6.70 × 103 | 201 | 0.0275 | |
| 9.09 × 103 | <45 | <0.00459 | |
| 1.17 × 105 | <293 | <0.0018 | |
| 2.13 × 105 | <1.07 × 103 | <0.00459 | |
| 6.08 × 106 | <1.52 × 104 | <0.0018 | |
|
| |||
| TLR2−/− | 12.3 | 0.9 | 0.039 |
| 71 | 0.7 | 0.00935 | |
| 94 | 1.4 | 0.0084 | |
| 878 | 4 | 0.0042 | |
| 3.78 × 103 | <9 | <0.0021 | |
| 4.80 × 103 | <12 | <0.0021 | |
| 6.68 × 103 | <17 | <0.0021 | |
| 1.1 × 105 | 1.10 × 103 | 0.0056 | |
| 1.43 × 105 | <715 | <0.00467 | |
| 6.37 × 105 | <1.59 × 103 | <0.00234 | |
| 1.40 × 106 | <7.0 × 103 | <0.00467 | |
| 3.80 × 106 | <1.9 × 104 | <0.00467 | |
| 4.26 × 106 | <2.13 × 104 | <0.00467 | |
| 4.84 × 106 | <1.21 × 104 | <0.0021 | |
CFU/10 μl of blood
2.5. Time to death of TLR2−/− mice following infection with the isogenic GBS strains
TLR2 recognizes components of the cell wall of Gram-positive bacteria and is, therefore, an important component of the innate immune response to Gram-positive bacterial infection. We employed TLR2−/− mice to investigate whether TLR2 enables mice to differentially respond to WT GBS, which expresses PBP1a, compared to the ponA mutant, which does not. We first addressed this question by examining the impact of the mutation in ponA in time to death assays using TLR2−/− mice. Similar to the studies described above, the TLR2−/− mice were infected with a range of inocula. A representative experiment in which the mice were infected with ~2.0 × 108 CFU of WT or ponA mutant GBS is shown in Fig. 3. The TLR2−/− mice were more susceptible to infection by WT GBS than to the ponA mutant and the kinetics of killing of the TLR2−/− mice did not appear to differ between the two strains of GBS.
Figure 3.
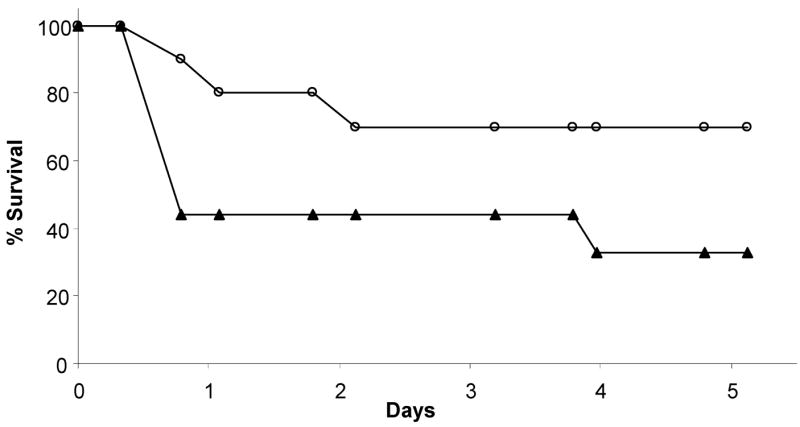
Time to death in TLR2−/− mice following infection. TLR2−/− mice were infected with an equivalent dose of either WT GBS (▴; n=10) or ponA mutant (○; n=9). The number of mice in each group is indicated in the figure. Data shown are representative of 3 independent experiments.
2.6. Competitive Index Assays in TLR2−/− mice
To further investigate the role of TLR2 in the response to GBS, we performed competitive index assays in TLR2−/− mice. As with the WT mice, following inoculation with a 1:1 ratio of WT and ponA mutant GBS, by 18–24 hours following inoculation all of the TLR2−/− mice were bacteremic. However, very few to none of the ponA mutant were recovered from the TLR2−/− mice and the competitive index was calculated to be <0.007 ± 0.01 (Table 1). This result was consistent with severe attenuation of the ponA mutant compared to the WT strain of GBS in TLR2−/− mice in the time to death assays and indicates that the presence of the WT strain does not complement the virulence defect of the ponA in TLR2−/− mice. The competitive index determined in the TLR2−/− mice was not significantly different from that determined in WT mice (p = 0.56).
2.7. Induction of cytokine production by WT GBS and the ponA mutant in WT and TLR2−/ mice
Binding of TLR2 to its ligand(s) activates intracellular signaling cascades, stimulates activation of NF-κB, and results in the production of pro-inflammatory cytokines, such as TNF-α and IL-6 [10, 11]. As TLR2-mediated NF-κB production increased in vitro following infection with both WT GBS and the ponA mutant (see Fig. 1), we were interested to determine whether production of TNF-α and IL-6 in WT mice differed following infection with the two strains of GBS. In addition, we wondered whether the increased mortality seen in TLR2−/− mice following infection with either WT GBS or the ponA mutant might result from inability to mount an effective inflammatory response following infection. To investigate these questions, we measured the amount of TNF-α and IL-6 produced in vivo following infection of WT and TLR2−/− mice by either WT GBS or the ponA mutant.
Blood was collected from WT mice following infection with either WT GBS or the ponA mutant. The bacterial load in the blood, serum TNF-α and IL-6 levels were determined for each mouse. As shown in Fig. 4A, for a given bacterial load, WT mice produced the same amount of TNF-α regardless of the GBS strain that was administered Similar results were obtained using TLR2−/− mice where following infection with an equivalent bacterial load of either strain; we detected the same amount of TNF-α produced (Fig. 4B).
Figure 4.

Cytokine production by WT and TLR2−/− mice following infection with WT and ponA mutant GBS. Mice were infected with either WT GBS (▵, solid line) or the ponA mutant (○, dashed line). The bacterial load in the blood and levels of TNF-α and IL-6 in the serum were quantified. TNF-α (pg/ml) produced by WT mice per CFU (A) and TLR2−/− mice (B). IL-6 (pg/ml) produced by WT mice per CFU (C) and TLR2−/− mice (D).
We also quantified the amount of IL-6 produced in response to infection. The WT mice produced the same amount of IL-6 for the same bacterial load of either of the isogenic GBS strains (Fig. 4C). A similar observation was made for IL-6 production by TLR2−/− mice with equivalent bacterial loads of the WT and ponA mutant strain (Fig. 4D). These data show that both WT and TLR2−/− mice are able to generate an equivalent robust TNF-α and IL-6 response following infection with either WT GBS or the ponA mutant. Linear regression modeling demonstrated that the amount of TNF-α or IL-6 produced was correlated with the level of bacteremia; the amount of TNF-α or IL-6 produced was dependent only upon the CFU of bacteria present in the blood of the infected mice, rather than the strain of GBS that was used.
3. Discussion
GBS is a common commensal organism in the female vaginal tract, where it is held in check by as yet unknown mechanisms [34, 35]. When GBS comes in contact with epithelial or mucosal surfaces in a relatively immunocompromised host, such as a neonate or an elderly or diabetic adult, it has the capacity to mount an invasive infection. Neonates at risk for GBS infection lack effective adaptive immune responses and are dependent upon components of the innate immune system, such as the TLRs, for defense [8]. Characterizing the interaction of GBS with components of innate immunity enhances our understanding of the pathogenesis of neonatal invasive disease.
TLR2 is activated by cell-wall components of Gram-positive bacteria, such as GBS, released by the bacterium or as an integral part of its cell wall. TLR2 plays an important role in host defense against Gram-positive organisms and its role in defense against GBS is the subject of ongoing investigation [19, 20, 36–38]. TLRs such as TLR2, may not bind directly to their target. Instead, they may interact with other pattern recognition receptors which do bind the target and serve as a scaffold for the subsequent intracellular signaling. This would allow a given TLR to be able to potentially recognize and respond to several pathogen-associated molecular patterns [39].
The integrity of the bacterial cell wall is essential for bacterial survival. Biosynthesis of cell wall PG is the function of PBPs. Class A PBPs such as GBS PBP1a are bifunctional proteins with both transglycosylase activity that extends the cell wall glycan chains and a transpeptidase activity that cross-links the PG chains [25]. There appears to be functional redundancy in PBP activity such that inactivation of one PBP can often be compensated for by others [25–27, 40–43]. We have previously shown that PBP1a is important for virulence of GBS in neonatal rat pups, both in an intraperitoneal sepsis model [21, 22] and in lung colonization model [23].
HPLC analysis of PG from the GBS ponA mutant revealed subtle alterations in the cell wall compared to the WT strain. Specifically, a reduction in the degree of cross-linking of the PG was detected [24]. As TLR2 is known to recognize components the cell wall of Gram-positive bacteria, we investigated whether recognition of GBS was affected by the absence of PBP1a and the contribution of TLR2 to innate immune defense against invasive GBS infection.
In an in vitro transfection system, we were able to reconstitute the TLR2 signaling pathway and demonstrate that both WT GBS and the ponA mutant are recognized by TLR2 and activate signaling in a dose-dependent manner. When using equivalent numbers of each strain, we did not detect any difference in the magnitude of activation, suggesting that the absence of PBP1a and the resultant changes in the cell wall do not affect recognition by TLR2 or subsequent intracellular signaling.
We then compared the virulence of the isogenic GBS strains in WT mice in a sepsis infection model. The ponA mutant was significantly attenuated for virulence compared to WT GBS as measured by the LD50 assays; the LD50 for the mutant was 10-fold greater than the WT strain. The ponA mutant also displayed a competition defect in our competitive index assays using WT mice. This suggests that the survival defect of the ponA mutant is not due to lack of production of a secreted factor, such as a toxin as in a mixed infection, as such a deficient factor would be provided by the WT GBS. These data support our hypothesis that the survival defect of the ponA mutant is linked to the changes in the cell wall. In time to death experiments, for a given dose of bacteria, fewer mice succumbed to infection with the ponA mutant than to the WT strain. There did not appear to be a difference in the kinetics of killing between the two strains of GBS. These data suggest that the virulence defect of the ponA mutant is not simply due to a delayed ability of the ponA mutant to mount an invasive infection, as the time course of when deaths occurred was similar between the two strains. Rather, this would imply that WT mice are more likely to clear the ponA mutant, but, if unable to do so, are susceptible to lethal infection.
We then examined whether TLR2-mediated recognition was altered by the absence of PBP1a and investigated the contribution of TLR2 to protecting mice from lethal infection. Our hypothesis was that if PBP1a was required for TLR2-mediated immune recognition and defense, then the attenuation of the ponA mutant that we observed in WT mice would be ameliorated or eliminated in TLR2−/− mice. In our studies, the TLR2−/− mice were more susceptible than WT mice to both the WT and ponA mutant. These observations are consistent with studies demonstrating an increase in susceptibility to WT GBS infection [37] and increased mortality in the absence of TLR2 following infection with other Gram-positive bacteria [44]. Additionally, there was also no difference in the kinetics of killing by the two strains in the TLR2−/− mice. Consistent with our in vitro observations, these data indicated that the absence of PBP1a does affect TLR2-mediated recognition of GBS in vivo.
To further define the role of TLR2 in the response to GBS infection, we investigated the ability of WT and TLR2−/− mice to mount a pro-inflammatory cytokine response. Interestingly, both WT and TLR2−/− mice produced similar amounts of TNF-α and IL-6 when bacteremic with either the WT or ponA mutant GBS strain. This suggests that TLR2−/− mice are able to generate an inflammatory response, as measured by TNF-α and IL-6 production, equivalent to that of WT mice following infection with GBS despite lacking this important component of the innate immune system. Our studies indicate that TLR2 is important for defense against GBS infection as TLR2−/− mice are more susceptible to infection with both the WT strain and ponA mutant. However, TLR2−/− mice mounted a robust cytokine response that was essentially equivalent to that of WT mice suggesting that other mechanisms of cytokine production that are TLR2-independent are operating. Such a mechanism(s) would appear to be less effective without TLR2, based on time to death assays. Further studies are needed to define the mechanisms by which TNF-α and IL-6 are produced in response to GBS infection in a TLR2−/− background.
4. Conclusions
Our data demonstrate an important role for TLR2 in defense against GBS infection. The absence of TLR2 profoundly impaired the ability of mice to survive infection by GBS but did not significantly alter early, pro-inflammatory responses. TLR2 did not appear to mediate the early virulence defect of the ponA mutant. However, the absence of PBP1a clearly results in profound attenuation of GBS virulence, a phenotype we have now extended to healthy, adult WT mice. Additionally, these studies demonstrate that that the absence of PBP1a does not result in any changes to the cell wall of GBS that impact effective recognition by TLR2. This would suggest that TLR2 and the innate immune system is tolerant to minor changes in their pathogen-associated molecular targets such as the cell wall, which might otherwise prevent recognition and the generation of an effective innate immune response.
5. Materials and Methods
5.1 Bacterial strains
The WT GBS strain is A909, a serotype Ia clinical isolate [45, 46]. The ponA mutant, AJ3F6, is derived from A909 and does not produce PBP1a due to a Tn917 transposon insertion in the ponA gene [21]. Tn917 contains an erythromycin resistance gene which allows for selection of transposon-containing GBS. This transposon is stably maintained even in the absence of erythromycin (Em) and does not have polar effects on the expression of adjacent genes [21, 22]. Production of well-studied virulence factors such as capsular polysaccharide and hemolysin are unaffected in AJ3F6 [21, 22]. Bacteria were grown in tryptic soy broth (TSB; Difco, Franklin Lakes, NJ) or on tryptic soy agar (TSA) at 37°C in 5% CO2. Prior to use, the bacteria were grown overnight, sub-cultured to mid-log phase, washed, resuspended in PBS and adjusted to the appropriate concentration.
5.2 Transfection assays
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (GibcoBRL, Rockville, MD) with 10% heat-inactivated fetal calf serum (HyClone, Logan, UT). The NF-κB reporter construct (ELAM-1 firefly luciferase, ELAM-Luc), the β-actin-Renilla luciferase reporter construct (β-actin Renilla-Luc), the modified pDisplay expression vector, and the expression constructs for murine TLR2 (pmuTLR2) and human TLR2 (phuTLR2) have been described previously [47, 48]. HeLa cells (4.0 × 104) were seeded into 96-well plates and transfected using Polyfect reagent (Qiagen, Valencia, CA) with the following amounts of DNA per well: ELAM-Luc, 0.03 μg; β-actin Renilla-Luc, 0.009 μg; and full-length human or murine TLR2 (minus the signal sequence) cloned in frame with the signal sequence in pDisplay (Invitrogen, Carlsbad, CA), 0.03 μg. The total amount of DNA per well was adjusted to 0.30 μg with empty pDisplay vector. Following transfection, the cells were allowed to grow to confluency and, twenty-four hours after transfection, the cells were stimulated with either WT GBS or the ponA mutant for 2 hours. Heat-killed flagellin-deficient Listeria monocytogenes was used as a positive TLR2-ligand control in one experiment. Human IL-1β (10 ng/ml) was used a positive control for production of NF-κB. Transfected HeLa cells were then lysed, and the amounts of Firefly and Renilla luciferase light units in the lysates were quantified with the Dual-Luciferase reporter assay system (Promega, Madison, WI), which allows for correction of any variability in transfection efficiency. Expression of NF-κB (relative light units) is represented as the ratio of Firefly (ELAM-Luc) to Renilla (β-actin Renilla-Luc) luciferase values per well.
5.3 Animal studies
WT mice were C57BL/6 mice obtained from Harlan, Indianapolis, IN. TLR2−/− mice backcrossed for 10 generations to C57BL/6 were generated by Dr. Shizuo Akira [11]. All inoculations of bacteria were intraperitoneal and in a volume of 100 μl in PBS. Mice were humanely sacrificed when moribund. All experiments were approved by the Institutional Animal Care and Use Committee.
5.4 LD50 Assays and time to death following infection with GBS
To determine the dose of bacteria sufficient to kill 50% of infected mice (LD50), WT mice were infected with serial dilutions (from approximately 2 × 107 CFU to 2 × 1010 CFU) of either WT GBS or the ponA mutant. The number of deaths in each group and the number of survivors was used to calculate the LD50 at five days. For time to death experiments, WT mice and TLR2−/− mice were infected with either WT GBS or the ponA mutant. To permit a comparison of the susceptibility of WT and TLR2−/− mice to killing by WT or ponA mutant GBS, all time to death assays were performed in parallel with age-, weight-, and sex-matched WT and TLR2−/− mice using the same preparations of WT GBS and ponA mutant GBS. Each group of mice was then followed for five days and the time of death recorded when the mice were moribund and sacrificed. For both LD50 and time to death experiments, blood was collected from the mice at the time of death when possible and serial dilutions were plated out to confirm that the animals had succumbed to GBS infection. In some cases, the spleens and livers were harvested, homogenized, and aliquots of this homogenate were serially diluted and plated out to ensure that only GBS was present.
5.5 Competitive Index assays
Competitive index assays were performed as described [22]. Briefly, WT mice and TLR2−/− mice were infected with both WT GBS and the ponA mutant in a 1:1 ratio of the two bacterial strains at a combined inoculum of 1 × 108 CFU. Serial dilutions of WT GBS and the ponA mutant were plated on TSA to determine the exact inoculum and the exact ratio of WT to ponA mutant injected. After sacrifice at 18 – 24 hours following infection, blood from each animal was plated on both TSA plates and plates of TSA containing 1 ug/ml Erythromycin (TSA/Em). The total number of bacteria was determined from the TSA plates, while the number of ponA mutant bacteria was determined from the TSA/Em plates. The competitive index is defined as the ratio of mutant to WT bacteria recovered divided by ratio of mutant to WT bacteria that were inoculated [21, 22]. When no ponA mutant was recovered, and the competitive index was calculated assuming that one of the ponA mutant bacteria had been recovered and the competitive index is expressed as less than this number. For these assays, a competitive index less than 0.1 was considered severe attenuation.
5.6 Cytokine assays
WT or TLR2−/− mice were infected with either WT GBS or the ponA mutant. After sacrifice at 18 – 24 hours following infection, serial dilutions of blood were plated out to determine the level of bacteremia. The blood was allowed to clot and the serum concentrations of TNF-α and IL-6 were determined by ELISA (DuoSet® ELISA Development System, R&D Systems, Minneapolis, MN).
5.7 Statistical Analyses
LD50 values were estimated via a logistic regression model using Stata statistical software (Stata Statistical Software Release 9, College Station, TX). Data from two separate experiments were combined in the logistic modeling. A two-tailed unpaired Student’s t-test was used to assess the statistical significance of the difference in competitive indices determined in WT mice from those determined in TLR2−/− mice. For the cytokine assays, linear regression modeling was performed. Analyses evaluated partial correlations of the type of mouse, strain of bacteria, and the interaction between type of mouse and strain of bacteria. Data from three separate experiments were pooled and used in modeling. SPSS statistical software (SPSS for Windows 10.0, Chicago, IL) was used for these analyses. Statistical significance was set at p < 0.05 except where noted.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01AI52299) awarded to A.L.J. and by the Infectious Diseases Society of America career award for vaccine development, the March of Dimes Basil O’Connor award, and a grant from the National Institutes of Health (K08HD51584), all awarded to SS.W. The authors thank Jerry Zimmerman, MD, PhD for critical review of the manuscript.
Abbreviations
- GBS
Group B Streptococcus
- PBP
Penicillin-binding protein
- PG
Peptidoglycan
- WT
Wild-type
- TLR
Toll-like receptor
- TNF
Tumor necrosis factor
- IL-6
Interleukin-6
- AMP
Antimicrobial peptide
- HPLC
High-Performance Liquid Chromatography
- Em
Erythromycin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baker CJ, Edwards MW. Group B Streptococcal Infections. In: Remington JS, Klein JO, Wilson CB, Baker CJ, editors. Infectious Diseases of the Fetus and newborn Infant. 6. Elsevier Sanders; 2005. pp. 1091–1156. [Google Scholar]
- 2.Schuchat A. Group B streptococcal disease: from trials and tribulations to triumph and trepidation. Clin Infect Dis. 2001;33:751–6. doi: 10.1086/322697. [DOI] [PubMed] [Google Scholar]
- 3.Mazade MA, Edwards MS. Impairment of type III group B Streptococcus-stimulated superoxide production and opsonophagocytosis by neutrophils in diabetes. Mol Genet Metab. 2001;73:259–67. doi: 10.1006/mgme.2001.3185. [DOI] [PubMed] [Google Scholar]
- 4.Henning KJ, Hall EL, Dwyer DM, Billmann L, Schuchat A, Johnson JA, Harrison LH. Invasive group B streptococcal disease in Maryland nursing home residents. J Infect Dis. 2001;183:1138–42. doi: 10.1086/319278. [DOI] [PubMed] [Google Scholar]
- 5.Dunne DW, Quagliarello V. Group B streptococcal meningitis in adults. Medicine (Baltimore) 1993;72:1–10. doi: 10.1097/00005792-199301000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Tyrrell GJ, Senzilet LD, Spika JS, Kertesz DA, Alagaratnam M, Lovgren M, Talbot JA. Invasive disease due to group B streptococcal infection in adults: results from a Canadian, population-based, active laboratory surveillance study--1996. Sentinel Health Unit Surveillance System Site Coordinators. J Infect Dis. 2000;182:168–73. doi: 10.1086/315699. [DOI] [PubMed] [Google Scholar]
- 7.Johri AK, Paoletti LC, Glaser P, Dua M, Sharma PK, Grandi G, Rappuoli R. Group B Streptococcus: global incidence and vaccine development. Nat Rev Microbiol. 2006;4:932–942. doi: 10.1038/nrmicro1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenzel S, Henneke P. The innate immune system and its relevance to neonatal sepsis. Curr Opin Infect Dis. 2006;19:264–70. doi: 10.1097/01.qco.0000224821.27482.bd. [DOI] [PubMed] [Google Scholar]
- 9.Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–33. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–9. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–51. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 12.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]
- 14.Yoshimura A, Takada H, Kaneko T, Kato I, Golenbock D, Hara Y. Structural requirements of muramylpeptides for induction of Toll-like receptor 2-mediated NF-kappaB activation in CHO cells. J Endotoxin Res. 2000;6:407–10. [PubMed] [Google Scholar]
- 15.Dziarski R, Gupta D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: a reevaluation. Infect Immun. 2005;73:5212–6. doi: 10.1128/IAI.73.8.5212-5216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaput C, Boneca IG. Peptidoglycan detection by mammals and flies. Microbes Infect. 2007;9:637–47. doi: 10.1016/j.micinf.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Inamura S, Fujimoto Y, Kawasaki A, Shiokawa Z, Woelk E, Heine H, Lindner B, Inohara N, Kusumoto S, Fukase K. Synthesis of peptidoglycan fragments and evaluation of their biological activity. Org Biomol Chem. 2006;4:232–42. doi: 10.1039/b511866b. [DOI] [PubMed] [Google Scholar]
- 18.Travassos LH, Girardin SE, Philpott DJ, Blanot D, Nahori MA, Werts C, Boneca IG. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004;5:1000–6. doi: 10.1038/sj.embor.7400248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henneke P, Morath S, Uematsu S, Weichert S, Pfitzenmaier M, Takeuchi O, Muller A, Poyart C, Akira S, Berner R, Teti G, Geyer A, Hartung T, Trieu-Cuot P, Kasper DL, Golenbock DT. Role of lipoteichoic acid in the phagocyte response to group B streptococcus. J Immunol. 2005;174:6449–55. doi: 10.4049/jimmunol.174.10.6449. [DOI] [PubMed] [Google Scholar]
- 20.Henneke P, Takeuchi O, van Strijp JA, Guttormsen HK, Smith JA, Schromm AB, Espevik TA, Akira S, Nizet V, Kasper DL, Golenbock DT. Novel engagement of CD14 and multiple toll-like receptors by group B streptococci. J Immunol. 2001;167:7069–76. doi: 10.4049/jimmunol.167.12.7069. [DOI] [PubMed] [Google Scholar]
- 21.Jones AL, Knoll KM, Rubens CE. Identification of Streptococcus agalactiae virulence genes in the neonatal rat sepsis model using signature-tagged mutagenesis. Mol Microbiol. 2000;37:1444–55. doi: 10.1046/j.1365-2958.2000.02099.x. [DOI] [PubMed] [Google Scholar]
- 22.Jones AL, Needham RH, Clancy A, Knoll KM, Rubens CE. Penicillin-binding proteins in Streptococcus agalactiae: a novel mechanism for evasion of immune clearance. Mol Microbiol. 2003;47:247–56. doi: 10.1046/j.1365-2958.2003.03297.x. [DOI] [PubMed] [Google Scholar]
- 23.Jones AL, Mertz RH, Carl DJ, Rubens CE. A Streptococcal Penicillin-Binding Protein is Critical for Resisting Innate Airway Defenses in the Neonatal Lung. J Immunol. 2007 doi: 10.4049/jimmunol.179.5.3196. in press. [DOI] [PubMed] [Google Scholar]
- 24.Hamilton A, Popham DL, Carl DJ, Lauth X, Nizet V, Jones AL. Penicillin- binding protein 1a promotes resistance of group B streptococcus to antimicrobial peptides. Infect Immun. 2006;74:6179–87. doi: 10.1128/IAI.00895-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goffin C, Ghuysen JM. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol Mol Biol Rev. 1998;62:1079–93. doi: 10.1128/mmbr.62.4.1079-1093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popham DL, Setlow P. Cloning, nucleotide sequence, and mutagenesis of the Bacillus subtilis ponA operon, which codes for penicillin-binding protein (PBP) 1 and a PBP-related factor. J Bacteriol. 1995;177:326–35. doi: 10.1128/jb.177.2.326-335.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duez C, Hallut S, Rhazi N, Hubert S, Amoroso A, Bouillenne F, Piette A, Coyette J. The ponA gene of Enterococcus faecalis JH2-2 codes for a low-affinity class A penicillin-binding protein. J Bacteriol. 2004;186:4412–6. doi: 10.1128/JB.186.13.4412-4416.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Born P, Breukink E, Vollmer W. In vitro synthesis of cross-linked murein and its attachment to sacculi by PBP1A from Escherichia coli. J Biol Chem. 2006 doi: 10.1074/jbc.M604083200. [DOI] [PubMed] [Google Scholar]
- 29.Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, Mayadas T, Nizet V, Akira S, Kasper DL, Golenbock DT. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol. 2002;169:3970–7. doi: 10.4049/jimmunol.169.7.3970. [DOI] [PubMed] [Google Scholar]
- 30.Henneke P, Berner R. Interaction of neonatal phagocytes with group B streptococcus: recognition and response. Infect Immun. 2006;74:3085–95. doi: 10.1128/IAI.01551-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Way SS, Thompson LJ, Lopes JE, Hajjar AM, Kollmann TR, Freitag NE, Wilson CB. Characterization of flagellin expression and its role in Listeria monocytogenes infection and immunity. Cell Microbiol. 2004;6:235–42. doi: 10.1046/j.1462-5822.2004.00360.x. [DOI] [PubMed] [Google Scholar]
- 32.Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–10. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kutuk O, Basaga H. Aspirin inhibits TNFalpha- and IL-1-induced NF-kappaB activation and sensitizes HeLa cells to apoptosis. Cytokine. 2004;25:229–37. doi: 10.1016/j.cyto.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Dillon HC, Jr, Gray E, Pass MA, Gray BM. Anorectal and vaginal carriage of group B streptococci during pregnancy. J Infect Dis. 1982;145:794–9. doi: 10.1093/infdis/145.6.794. [DOI] [PubMed] [Google Scholar]
- 35.Manning SD, Neighbors K, Tallman PA, Gillespie B, Marrs CF, Borchardt SM, Baker CJ, Pearlman MD, Foxman B. Prevalence of group B streptococcus colonization and potential for transmission by casual contact in healthy young men and women. Clin Infect Dis. 2004;39:380–8. doi: 10.1086/422321. [DOI] [PubMed] [Google Scholar]
- 36.Flo TH, Halaas O, Lien E, Ryan L, Teti G, Golenbock DT, Sundan A, Espevik T. Human toll-like receptor 2 mediates monocyte activation by Listeria monocytogenes, but not by group B streptococci or lipopolysaccharide. J Immunol. 2000;164:2064–9. doi: 10.4049/jimmunol.164.4.2064. [DOI] [PubMed] [Google Scholar]
- 37.Mancuso G, Midiri A, Beninati C, Biondo C, Galbo R, Akira S, Henneke P, Golenbock D, Teti G. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J Immunol. 2004;172:6324–9. doi: 10.4049/jimmunol.172.10.6324. [DOI] [PubMed] [Google Scholar]
- 38.Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–92. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- 39.Ulevitch RJ, Tobias PS. Recognition of gram-negative bacteria and endotoxin by the innate immune system. Curr Opin Immunol. 1999;11:19–22. doi: 10.1016/s0952-7915(99)80004-1. [DOI] [PubMed] [Google Scholar]
- 40.Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol. 1999;181:3981–93. doi: 10.1128/jb.181.13.3981-3993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McPherson DC, Driks A, Popham DL. Two class A high-molecular-weight penicillin-binding proteins of Bacillus subtilis play redundant roles in sporulation. J Bacteriol. 2001;183:6046–53. doi: 10.1128/JB.183.20.6046-6053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McPherson DC, Popham DL. Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilis. J Bacteriol. 2003;185:1423–31. doi: 10.1128/JB.185.4.1423-1431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Popham DL, Setlow P. Phenotypes of Bacillus subtilis mutants lacking multiple class A high-molecular-weight penicillin-binding proteins. J Bacteriol. 1996;178:2079–85. doi: 10.1128/jb.178.7.2079-2085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–6. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 45.Lancefield RC, McCarty M, Everly WN. Multiple mouse-protective antibodies directed against group B streptococci. Special reference to antibodies effective against protein antigens. J Exp Med. 1975;142:165–79. doi: 10.1084/jem.142.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madoff LC, Michel JL, Kasper DL. A monoclonal antibody identifies a protective C-protein alpha-antigen epitope in group B streptococci. Infect Immun. 1991;59:204–10. doi: 10.1128/iai.59.1.204-210.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J Immunol. 2001;166:15–9. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- 48.Hajjar AM, Ernst RK, Tsai JH, Wilson CB, Miller SI. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat Immunol. 2002;3:354–9. doi: 10.1038/ni777. [DOI] [PubMed] [Google Scholar]