

Abstract
Anionic phospholipids modulate the activity of inwardly rectifying potassium channels (Fan, Z., and J.C. Makielski. 1997. J. Biol. Chem. 272:5388–5395). The effect of phosphoinositides on adenosine triphosphate (ATP) inhibition of ATP-sensitive potassium channel (KATP) currents was investigated using the inside-out patch clamp technique in cardiac myocytes and in COS-1 cells in which the cardiac isoform of the sulfonylurea receptor, SUR2, was coexpressed with the inwardly rectifying channel Kir6.2. Phosphoinositides (1 mg/ml) increased the open probability of KATP in low [ATP] (1 μM) within 30 s. Phosphoinositides desensitized ATP inhibition with a longer onset period (>3 min), activating channels inhibited by ATP (1 mM). Phosphoinositides treatment for 10 min shifted the half-inhibitory [ATP] (K i) from 35 μM to 16 mM. At the single-channel level, increased [ATP] caused a shorter mean open time and a longer mean closed time. Phosphoinositides prolonged the mean open time, shortened the mean closed time, and weakened the [ATP] dependence of these parameters resulting in a higher open probability at any given [ATP]. The apparent rate constants for ATP binding were estimated to be 0.8 and 0.02 mM−1 ms−1 before and after 5-min treatment with phosphoinositides, which corresponds to a K i of 35 μM and 5.8 mM, respectively. Phosphoinositides failed to desensitize adenosine inhibition of KATP. In the presence of SUR2, phosphoinositides attenuated MgATP antagonism of ATP inhibition. Kir6.2ΔC35, a truncated Kir6.2 that functions without SUR2, also exhibited phosphoinositide desensitization of ATP inhibition. These data suggest that (a) phosphoinositides strongly compete with ATP at a binding site residing on Kir6.2; (b) electrostatic interaction is a characteristic property of this competition; and (c) in conjunction with SUR2, phosphoinositides render additional, complex effects on ATP inhibition. We propose a model of the ATP binding site involving positively charged residues on the COOH-terminus of Kir6.2, with which phosphoinositides interact to desensitize ATP inhibition.
Keywords: phosphatidylinositol, phospholipids, inwardly rectifying potassium channels, sulfonylurea receptor, KATP
Introduction
The ATP-sensitive potassium channel (KATP) plays an important role in the physiology and pathophysiology of many tissues, including pancreas, brain, vascular smooth muscle, and heart muscle. KATP is a member of the family of inwardly rectifying potassium channels and is composed of a pore-forming subunit, Kir6, and a sulfonylurea receptor, SUR. For each subunit, two separate genes encoding two major isoforms have been found and named as Kir6.x and SURx (x = 1 or 2) (for review see by Aguilar-Bryan et al. 1998). Anionic phospholipids, especially the phosphorylated phosphoinositols, modulate the activity of a number of inward rectifier channels by increasing their open probability (Fan and Makielski 1997; Huang et al. 1998), as well as other membrane proteins such as the Na-Ca exchanger (Hilgemann and Ball 1996). A key distinguishing feature of KATP is ATP sensitivity, the inhibition of channel activity by micromolar amounts of ATP. This feature also accounts for the role ATP plays in coupling cellular metabolism to potassium flux and membrane potential. A truncated version of Kir6.2 (Tucker et al. 1997), and also the full-length Kir6.2 (John et al. 1998), were found to exhibit ATP-sensitive currents in the absence of SUR, showing that much of the ATP sensitivity of the channel is conferred by the Kir6 subunit. Moreover, mutation of positively charged residues on Kir6.2 (e.g., K185) reduced ATP inhibition (Tucker et al. 1998). This residue is located near other positively charged residues on Kir6.2 (R176, R177) that we (Fan and Makielski 1997) have previously shown to be involved with the phosphatidylinositol effect on maximal open probability of KATP. In this study, we show that phosphoinositides (PPIs),1 a mixture of several species of phosphatidylinositol, in addition to the previously described activation effect in the absence of ATP inhibition, also desensitize KATP to ATP inhibition. Two recent papers have reported the attenuation effect of phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-4-phosphate (PIP) on ATP-sensitive inhibition primarily in pancreatic KATP (Shyng and Nichols 1998; Baukrowitz et al. 1998). Our results confirm and extend this observation to native cardiac myocytes and for SUR2, the putative cardiac isoform. We also demonstrate and analyze the effect of PPIs on single-channel behaviors of KATP. Our study suggests that the mechanism whereby PPIs desensitize ATP inhibition may involve a multiple-step antagonism between PPIs and ATP binding to Kir6.2, through protein–lipid interaction between Kir6.2 and membrane phospholipids. Some of these results have been presented in abstract form (Fan and Makielski 1999).
methods
Expression of Recombinant cDNAs and Truncation of Kir6.2 cDNA
Mouse SUR2 and mouse Kir6.2 cDNA clones were coexpressed in a COS-1 cell line using a LipofectAMINE transfection kit (GIBCO BRL). The SUR2 we used in these studies was SUR2A, the “cardiac form” with terminal usage of exon 39 (Isomoto et al. 1996) in the full-length variant (Chutkow et al. 1996) including exons 14 and 17. A PCR-based site-directed mutagenesis kit, ExSite (Stratagene, Inc.), was used to generate the COOH-terminal truncation (Kir6.2ΔC35) at amino acid residue 35 of the COOH terminus of mouse Kir6.2. The resulting PCR product was subsequently subcloned into a PCR3.1 vector (T/A cloning kit; Clontech Labs, Inc.), and the PCR product was verified by sequencing.
Native Cardiac KATP Channels
KATP currents were also measured in native cardiac ventricular myocytes isolated from dog and rat ventricles by previously described methods (Wittenberg and Robinson 1981). The source of individual records is noted in the figure legends. No differences in native or cloned (SUR2/Kir6.2) channel activity were noted; therefore, the sources were combined into summary data where indicated.
Electrophysiology Recordings
The patch clamp and data acquisition system was an Axopatch 200B with a 1200 DMA interface using pClamp6.0 software (Axon Instruments) running on a PC computer. Inside-out single- or multiple-channel currents were recorded in an intracellular solution containing (mM) 140 KCl, 2 EGTA, 0.5 MgCl2, 5.5 glucose, and 5 Hepes, pH 7.4, and an extracellular solution containing (mM) 10 KCl, 120 NaCl, 1.8 CaCl2, 0.48 MgCl2, 5.5 glucose, and 5 Hepes, pH 7.2. Phosphoinositides (Sigma Chemical Co.; containing PIP2, PIP, and phosphatidylserine) or phosphatidylcholine (1 mg/ml; Avanti Polar Lipids) were dispersed in solutions during a 10-min ultrasonication on ice. The lipid-containing solutions were used in experiments shortly after the dispersal procedure and were applied to the inner side of the patch membrane. Adenosine hemisulfate salt, ATP potassium salt, and tolbutamide (all from Sigma Chemical Co.) were also used in some experiments. Solution changes in the bath (intracellular membrane side of channel in excised patches) were made within 100 ms by a rapid solution exchange system (DAD-12; ALA Scientific Instruments). All current recordings were filtered at 0.5–2 kHz and digitized at 2–20 kHz. In the figures, a dotted line and a “c–” at the left end of the current recording indicates the level where all channels are closed. Outward currents are shown as upward deviation from this closed level. Unless specified, currents were recorded at membrane potential 0 mV at room temperature.
Data Analysis
Channel activity.
For patches containing ≤5 active channels (which was limited by the software), open activity was assessed by an open probability (P o) using the equation introduced by Spruce et al. 1985. In macropatch recordings with >5 channels, an apparent open probability (NP o) was used. NP o was calculated as the average current in a 5-s time window divided by the single-channel current amplitude determined under the same recording conditions. A 50% threshold criterion was used to detect events, and all events were confirmed visually. Data were reported as mean ± SE. Student's t test was used to compare the significant differences between data sets.
Single-channel kinetics.
Single-channel kinetics was analyzed with methods following those of Davies et al. 1992. Because our experiments were done under conditions similar to those reported by Davies et al., we adopted their analysis parameters. In brief, single-channel recordings showing no obvious overlapped channel activity were used in the analysis. All currents were filtered at 2 kHz and digitally sampled at 20 kHz. Events were detected at a 50% threshold. Distributions of open and closed times were constructed against a logarithmic time scale with event duration log-binned at a resolution of 25 bins per log unit and a minimum resolution of tmin = 150 μs (Davies et al. 1992). Exponential fits to the histograms were performed by a maximum likelihood fitting strategy using the following exponential equation:
 |
1 |
where S f,,n is the scale factor determined by the proportional contribution of the component to the area under the function, τf,n is the time constant of the component, and subscript f denotes the state of the channel, i.e., o for open and c for closed; subscript n denotes the order of the exponential component (n = 1, 2, 3, 4). The appropriateness of the selection of n was decided by an F-test. Mean open times were calculated and corrected for missed closings by:
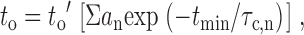 |
2 |
where a n is the area of the exponential component n, and t min is 150 μs as described above. Mean closed times were not corrected if they were reported, because the mean open times were much greater than t min, implying that missed open events were rare. A burst was defined as any series of openings interrupted only by gaps shorter than a specified critical time, t critical (Sakmann and Trube 1984). The following equation of Colquhoun and Sakmann 1983 was iteratively solved to calculate t critical:
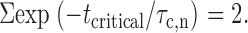 |
3 |
A built-in function of pClamp software to estimate the tcritical that deploys the method of Sigurdson et al. 1987 was also used to help determinate the critical time. The typical value of tcritical was in the range of 3-5 ms.
Simulation of Single-channel Behaviors with Kinetic Models.
A suit of programs for simulation and analysis of single-channel data (Qin et al. 1996) were used to simulate single-channel behaviors. Analysis was carried out according to advice from the authors of the programs. In brief, programs PRE and MIL were used to fit a hypothesized model to the experimental current traces; this process helped the selection of the parameters. Single-channel current events were then simulated by using program SIMU. Programs SKM and MIL, as well as pClamp6.0, were used to analyze the computer-generated events, and the results were compared with the experimental data to evaluate appropriateness of proposed kinetic models.
results
Two Major Effects of PPIs to Activate KATP: Increasing Maximal Po and Desensitizing ATP Inhibition
When applied to the cytoplasmic surface of excised patches, PPIs had two experimentally distinct activating effects: (i) they increased maximal KATP open probability (maximal P o, defined as the P o measured in the absence of inhibitory [ATP]) with an onset period <30 s; and (ii) they desensitized KATP to ATP inhibition with a longer onset period (>3 min). As commonly seen in excised patch experiments, open channel activity of KATP was reduced and eventually lost over several minutes in a process often called run-down. Fig. 1 A shows an example of PPIs applied to a patch containing run-down KATP channels. After the patch was excised, KATP were allowed to run down for ∼5 min in this experiment. With inhibitory concentrations of ATP absent from the solution, PPIs (1 mg/ml) subsequently applied to the inner side of the patch membrane rapidly (within 30 s) reactivated channel activity (similar results observed in 35 patches of dog heart cells, rat heart cells, and SUR2/Kir6.2). The channel activity persisted for many minutes after PPIs were removed from the bathing solution. We further investigated two aspects of this effect of PPIs. First, although run-down is, strictly speaking, an experimental phenomenon, it may have a common mechanism related to physiological regulatory processes involving endogenous phosphatidylinositol. With this in mind, we also examined the effect of PPIs in the absence of notable run-down. Absence of run-down was defined by a value not less than one for the ratio: maximal P o immediately before application of PPIs/maximal P o at the time of patch excision.
Figure 1.

Two effects of PPIs on KATP currents. PPIs (1 mg/ml) and ATP (1 mM) were applied in the bath (internal membrane in these inside-out patches) at the times indicated by the bars. [ATP] was 1 μM unless indicated otherwise. The dashed line represents closed current levels. Currents were recorded from COS-1 cells transfected with Kir6.2/SUR2, and all three examples were multichannel patches. (A) Current recorded after run-down of KATP shows reactivation by PPIs. (B) Current recorded in the presence of ATP to block the channel shows loss of ATP sensitivity after PPIs. (C) Current recorded in the absence of obvious run-down shows that these channels also lost sensitivity to ATP, and this effect persisted after removal of PPIs from the bath.
We selectively applied PPIs to those patches before the appearance of run-down, typically shortly after the patch excision (<5 min). We frequently observed an increased maximal P o after application of PPIs, especially in those patches with maximal P o < 0.5 at the time of patch excision, as we have reported previously (Fan and Makielski 1997). The maximal P o stimulated by PPIs often reached >0.9 at 0 mV, with an average of 0.84 ± 0.11 (n = 15). Such a striking increase provides a large P o range within which endogenous phosphatidylinositol could potentially regulate channel activity. The second aspect of the effect of PPIs on P o in the absence of inhibitory [ATP] was noticed by Furukawa et al. 1996. They observed a time-dependent decrease in the effectiveness of PIP2 to reactivate KATP . They then argued that the effect of PIP2 might require some mediating factors, such as G-actins, that would be depleted or lose activity in an inside-out patch membrane. In our experiments, we particularly reexamined this aspect for PPIs using extraordinarily long patch clamp recording. For three patches that lasted 5–8 h, repeated applications of freshly prepared PPIs (1 mg/ml, prepared within 10 min of use) induced reappearance of KATP activity. Based on the time-dependent decrease in the maximal P o that PPIs could stimulate, we confirmed that the effectiveness of PPIs was reduced over time. For example, after the first treatment with PPIs, one patch contained 11 active channels at a maximal NP o of 9.6. After allowing channel activity to run down, the maximal NP o response to the third subsequent applications of PPIs dropped to 1.8 in 1 h after patch excision. The response to PPIs then became relatively stable at 1.8 in response to additional applications of PPIs in 7 h thereafter. Perhaps the component of KATP activity not reversed by PPIs or other “reactivation” interventions such as MgATP should more appropriately be referred to as run-down. In keeping with former usage, however, we have used the term “reactivatable activity” for that component that could be reactivated.
With longer (>3 min) exposure, PPIs dramatically desensitized KATP to ATP inhibition. When PPIs were applied in the continuous presence of 1 mM ATP (Fig. 1 B), little or no KATP activity was observed within the initial 30 s, demonstrating maintained ATP inhibition at a time when the effect of PPIs on maximal P o was nearly complete. However, after several minutes in PPIs, KATP activity gradually increased in the presence of 1 mM ATP, demonstrating a loss of ATP sensitivity, and activity reached a new stable level after ∼10 min. Similar results were obtained in five patches: one from a dog ventricular cell, three from rat ventricular cells, and one from SUR2/Kir6.2. In the particular experiment shown in Fig. 1 B, KATP had already partially run down before application of PPIs. However, loss of ATP sensitivity with PPIs required neither previous channel run-down nor ATP, nor did it require the continuous presence of PPIs in the bathing solution as was demonstrated in experiments similar to that shown in Fig. 1 C. In a patch (SUR2/Kir6.2) without notable prior run-down (as defined by the ratio noted above), ATP sensitivity was first demonstrated by exposure to 1 mM ATP, then PPIs were applied for 10 min in the absence of inhibitory [ATP]. Desensitization of ATP inhibition was then demonstrated by the failure of 1 mM ATP to suppress KATP currents after removal of the PPIs from the bath solution (Fig. 1 C). In these experiments, slight recovery of ATP sensitivity was observed 7–20 min after PPIs were removed (data not shown). Interestingly, in those patches treated with PPIs where KATP activity was followed for several hours, ATP sensitivity never returned to its initial value, while at the same time the maximal P o gradually decreased to nearly zero. The concentration–response of ATP inhibition before and after exposure to PPIs for 10 min was measured within single patches (SUR2/Kir6.2). [ATP] was stepped between 1 and 1,000 μM in control and between 1 and 10,000 μM after a 10-min exposure to PPIs (for example, see Fig. 2). [ATP] changes in 10-s intervals were stepped in both increasing and decreasing concentrations. As studied in four patches (Fig. 2 C), PPIs desensitized ATP sensitivity by increasing the K i for inhibition nearly 500-fold (before PPIs: K i = 34.9 ± 6.7 μM; after PPIs: K i = 15.6 ± 2.7 mM, P < 0.001) without a significant difference in slope factor (1.03 ± 0.11 versus 0.94 ± 0.17, before versus after PPIs, respectively). For 1 mg/ml phosphatidylcholine, an uncharged phospholipid, a small, variable, statistically insignificant effect (n = 7) was observed on the KATP sensitivity of native rat cardiac myocytes. This result suggested a critical role of the negatively charged head group for the effect on ATP sensitivity. Previously, we had found that negatively charged groups of phosphatidylinositol were needed for reactivation of KATP (Fan and Makielski 1997).
Figure 2.

Concentration-response for ATP inhibition of KATP. Ascending and descending [ATP] response relationships in a single multichannel patch before (A) and after (B) treatment with PPIs (10 min, 1 mg/ml). KATP currents were recorded in inside-out patches from COS-1 cells transfected with SUR2/Kir6.2. Bars and numbers represent ATP concentration except that in B 10K stands for an internal solution with 10 mM [K+] that produced a 0 current level for equimolar [K+] at 0 mV. (C) Summary data for the ATP block concentration–response relationship before and after treatment with PPIs from experiments such as those shown in A and B. Symbols and error bars represent the mean ± SE from four control experiments and three or four experiments after treatment. Summary data for ATP sensitivity was obtained by fitting the dependence of normalized P o on [ATP] using the expression: P = P o,max {1 − 1 /[1 + (K i/[ATP])S]} where P is the normalized P o, in ATP relative to the maximal P o (P o,max) in the absence of ATP or to 1 μM [ATP] that produced little or no inhibition; K i is the half-inhibitory [ATP]; and S is the slope-factor or Hill coefficient. P o,max, K i, and S were free parameters for fitting. Before treatment with PPIs, K i = 34.9 ± 6.7 μM, and after treatment K i = 15.6 ± 2.7 mM. (P < 0.001). The Hill coefficients S were 1.03 ± 0.11 versus 0.94 ± 0.17, respectively. In the inset, PPIs stands for the data collected after the patches was treated for 10 min with PPIs (1 mg/ml) and PPIs were washed out. The same label is used in the subsequent figure legends.
Interference by PPIs of MgATP Antagonism of ATP Inhibition
In the presence of Mg2+, the potency of ATP inhibition of KATP is partially reduced, an effect attributed to MgATP stimulation of KATP through interaction with the SUR subunit (Gribble et al. 1998). Much like PPIs, the effect of the presence of Mg2+ shifts the ATP concentration–inhibition curve to the right. Both Mg2+ and PPIs are potentially cellular regulators. Therefore, from both physiologic and mechanistic points of view, it is important to know whether PPIs and MgATP effects on ATP inhibition are simply additive or interactive (synergistic or antagonistic). ATP inhibition of KATP was reduced in the presence of MgCl2 2.2 mM both before and after treatment with PPIs (Fig. 3 A), but the change in ATP inhibition was much less dramatic after treatment. Summary data (Fig. 3 B) show that before treatment with PPIs, ATP inhibition in the presence of Mg2+ decreased by ∼10-fold, whereas after treatment with PPIs, the same [Mg2+] caused only a 1.5-fold decrease in ATP inhibition. For this experiment, we used a fixed [MgCl2] for all [ATP] to avoid the additional errors introduced by titration of free [Mg2+]. However, this meant that the [Mg2+] was likely to be reduced at the higher [ATP] and could have had a reduced effect. We therefore chose a 5-min exposure to 0.5 mg/ml PPIs, which gave a K i of 2.3 mM, much less than the 16 mM obtained with longer exposures (Fig. 2 C). Under this nonsaturating condition we were able to record the proportional changes of KATP activity over a common [ATP] range, mitigating the possible problem caused by comparing channel activity at different [Mg2+]. In addition, in 2 mM of ATP, the highest [ATP] we used, doubling [MgCl2] to 4.4 mM did not significantly change P o (data not shown). Therefore, we conclude that the Mg2+ effect is not strictly additive to the effect of PPIs and that the two effects interfere each other, and likely share a linked mechanism.
Figure 3.

Effects of PPIs and Mg2+ on ATP inhibition. (A) Current traces recorded in a patch excised from a rat ventricular myocyte before (Control) and after 5-min treatment with PPIs (0.5 mg/ml) (labeled as PPIs). The dotted lines indicate unitary channel current levels. The unitary channel current level in MgCl2 2.2 mM was scaled up to the same level in Mg 0 to assist in making the comparison. c– denotes the closed level. In control, 0.5 mM ATP inhibited KATP (middle trace), but this inhibition was antagonized by MgCl2 2.2 mM (right trace). After treatment with PPIs, more channels were open in low [ATP], 0.5 mM was less effective in suppressing KATP, and MgCl2 2.2 mM slightly antagonized ATP inhibition. (B) Summary data for effects of PPIs and MgCl2 2.2 mM on ATP sensitivity were fitted as described for Fig. 2. The symbols represent the mean ± SE for nine patches. Before treatment with PPIs, K i = 30 ± 2.8 μM in 0 Mg (•) and 333 ± 56 μM in 2.2 mM Mg (○) (P < 0.001). After treatment with PPIs, K i = 2.3 ± 0.4 mM in 0 Mg (▴) and 3.4 ± 0.2 mM in 2.2 mM Mg (▵) (P = 0.03).
Changes of Single-channel Kinetics Underlying Desensitization of ATP Inhibition by PPIs
How do PPIs affect KATP activity and ATP sensitivity at the single-channel level? For short exposure to PPIs, we had previously shown that the single-channel conductance was unaffected and that increased activity was associated with longer mean open time and shorter closed time (Fan and Makielski 1997). For longer exposure to PPIs, we have now investigated single KATP channel patches at different [ATP] to explore possible mechanisms accounting for desensitization of ATP inhibition by PPIs. Single-channel conductance was again unchanged for the longer exposure to PPIs. To measure kinetics, special care was taken to ensure stable channel properties. Each recording at a given [ATP] typically lasted from 2 to 8 min to attain sufficient data for analysis. Because fluctuation in channel kinetics occurred with prolonged exposure to PPIs, we limited the exposure to 5 min (in our experience, this duration did not cause appreciable instability of kinetics). When recording after PPIs, if a reduction in P o was noted, exposure to PPIs was then repeated to restore it. The membrane potential was set to 0 mV to minimize passive noise and background current drift.
Under these experimental conditions, untreated single-channel currents exhibited characteristic KATP kinetics (Fig. 4 A, Control) consistent with those reported in the literature (e.g., Davies et al. 1992). Current events were determined at > tmin = 150 μs resolution (see methods and example of idealized events in the top trace of Fig. 4 B). Current flickering was seen and resolved for all ATP concentrations in untreated and treated channels (Fig. 4 B). Also typical for this channel, the open channel activity occurred in bursts of activity separated by closed intervals longer than the critical time t critical (see methods for determination of t critical, which was generally 3–5 ms) and is best seen in Fig. 4 B. Even longer closures seen in the slower time scale (Fig. 4 A) isolated groups of bursts into clusters. Due to the difficulty in obtaining stable recordings of sufficient length, this cluster behavior was not further analyzed. A typical example of the channel activity histograms for a single-channel patch is shown in Fig. 5, and the parameters of the fits are given in Table .
Figure 4.

Effects of PPIs on ATP sensitivity at the single-channel level. Single-channel KATP current recorded in an inside-out patch at 0 mV from a rat ventricular cell before (left) and after (right) treatment with PPIs (5 min, 1 mg/ml). c– denotes the closed level. Panels show data in compressed (A) and expanded (B) time scales. Concentration of ATP is indicated above the correspondent current trace. See text for details.
Figure 5.

Histograms of open and closed time distributions and kinetic properties of a single KATP channel recorded at 0 mV from a rat cardiac ventricular cell. (A) Closed time distribution analyzed from the same patch as shown in Fig. 4. The solid lines were fitted with a probability density function containing the sum of four exponential functions (). (B) Open time distribution. The solid lines were fitted with the sum of three exponential functions. For both A and B, the left panels are the control and the right panels are after 5-min treatment with PPIs (1 mg/ml). The parameters of probability density functions obtained from the fitting are listed in Table .
Table 1.
Parameters of the Closed- and Open-Time Distribution Functions Fit to the Histograms in Fig. 5
| Control | After PPIs | |||
|---|---|---|---|---|
| [ATP] (μM) | 1 | 100 | 1 | 100 |
| 5 min, 1 mg/ml | ||||
| Closed times | ||||
| τc,1 (ms) | 0.11 | 0.11 | 0.12 | 0.11 |
| Sc,1 | 1690 | 356 | 4918 | 1942 |
| τc,2 (ms) | 1.0 | 1.0 | 0.92 | 1.1 |
| Sc,2 | 92 | 10 | 283 | 94 |
| τc,3 (ms) | 14.2 | 16.5 | 11.4 | 17.5 |
| Sc,3 | 21 | 8 | 104 | 32 |
| τc,4 (ms) | 134.5 | 689.2 | 60.3 | 101 |
| Sc,4 | 29 | 33 | 7 | 10 |
| Open times | ||||
| τo,1 (ms) | 0.28 | 0.24 | 0.27 | 0.25 |
| So,1 | 32 | 13 | 105 | 18 |
| τo,2 (ms) | 3.8 | 1.2 | 6.8 | 6.5 |
| So,2 | 31 | 29 | 192 | 35 |
| τo,3 (ms) | 34.5 | 15.5 | 47.4 | 40.5 |
| So,3 | 175 | 63 | 768 | 210 |
Statistically, the closed time distribution was best fit to four exponential components according to (see methods) at either low or high [ATP] (Fig. 5 A, left). The two components with the faster time constants corresponding to the flicker closures did not change with [ATP] in agreement with earlier studies (Zilberter et al. 1988; Qin et al. 1989; Davies et al. 1992; Alekseev et al. 1998). The other two exponential components had time constants greater than t critical, meaning these components represented closures between bursts. Raising [ATP] increased both the time constant and the fractional amplitude of the slowest exponential component significantly. Open time distributions could be fit by three exponential components (Fig. 5 B, left). The time constant of the slowest component was reduced at higher [ATP] (Table ). These characteristics are again consistent with the analysis of Davies et al. 1992. The only difference is the necessity for a three-exponential expression to describe the open time distribution in our analysis. We found that the third component improved the fitting in a statistically significant manner. There are several notable differences between the kinetics parameters obtained from outward current (Davies et al. 1992; this study), and that from inward currents (Qin et al. 1989; Shyng and Nichols 1998; Alekseev et al. 1998), as follows. (a) Multiple components are necessary to fit the open time distribution for the outward current, whereas a single component is usually sufficient for the inward current. (b) The open time distribution has a major component of a time constant >5 ms for outward current, whereas for the inward current the time constant is much shorter (<1 ms). These differences have been noted previously and attributed to voltage and/or current dependent gating kinetics (Zilberter et al. 1988). We chose outward currents to estimate the rate constant for ATP binding because the longer open events provided an advantage in reducing errors in the measurement of event duration, and also because outward currents are of physiological interest. Current traces on the right side of Fig. 4 are the single-channel currents recorded after treatment with PPIs. The closed time distribution in PPIs was again adequately fit by four exponential components with the two faster components representing the closures within bursts and the two slower components representing the gaps between bursts. The open time distribution was well fit with three exponential components (Fig. 5, right). The corresponding time constants of the two gaps between bursts, however, were smaller than before treatment with PPIs (Table ). Compared with control, the time constant for the slowest component was much less sensitive to [ATP] and the mean open time and the mean burst duration were increased at the same [ATP] after treatment with PPIs (Table ). The prolongation and increased number of the events of the slowest component was most notable and is the major cause of the increase in mean open time. Table summarizes the analysis for P o, corrected mean open time, mean burst duration, and mean closed time for four single-channel patches from rat heart ventricular myocytes. PPIs altered the [ATP] dependence of all of these parameters. Fitting P o with the expression described in the legend to Fig. 2 gave K i = 35 ± 7 μM before PPIs and 5.8 ± 0.6 mM after treatment with PPIs. Compared with the dramatic change in the [ATP] dependence of the mean closed time (t c), the change in the [ATP] dependence in the mean burst duration (t b) for [ATP] 1–1,000 μM was relatively less affected (Table ). For example, treatment with PPIs increased the mean open time, t o, by 5-fold and the mean burst duration, t b, by 8-fold at 1 mM [ATP], whereas the mean closed time, t c, decreased by 94-fold. These findings suggest shortening of interburst gaps contributed mainly to the 70-fold increase in P o caused by PPIs. The lengthening in the slowest component of open times after treatment with PPIs also contributed significantly to the increase in P o.
Table 2.
Relation between Open Probability, Corrected Mean Open Time, Mean Burst Duration, and Mean Closed Time of Single Channel Current Events and ATP Concentration Before and After Treatment with PPIs (5 min, 1 mg/ml)
| Untreated | After 5-min treatment with PPIs | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 mg/ml | |||||||||
| [ATP] mM | 0.001 | 0.01 | 0.1 | 1 | 0.001 | 0.01 | 0.1 | 1 | 10 |
| P o | 0.64 ± 0.02 (4) | 0.56 ± 0.04 (3) | 0.11 ± 0.03 (4) | 0.01 ± 0.002 (3) | 0.93 ± 0.02 (4) | 0.91 ± 0.04 (3) | 0.88 ± 0.04 (3) | 0.7 ± 0.04 (3) | 0.27 (2) |
| t o (ms) | 8.2 ± 1.1 (4) | 6.2 ± 0.6 (3) | 4.1 ± 0.4 (4) | 1.5 ± 0.3 (3) | 11.2 ± 0.7 (4) | 10.9 ± 0.8 (3) | 10.1 ± 0.3 (3) | 6.4 ± 0.6 (3) | 3.1 (2) |
| t b (ms) | 243 ± 51 (4) | 190 ± 48 (3) | 27 ± 3 (4) | 20 ± 5 (3) | 439 ± 20 (4) | 401 ± 32 (3) | 228 ± 21 (3) | 156 ± 36 (3) | 21 (2) |
| t c (ms) | 26 ± 4 (4) | 118 ± 20 (3) | 421 ± 36 (4) | 2350 ± 214 (3) | 3.7 ± 0.5 (4) | 4.5 ± 0.4 (3) | 7.0 ± 2.6 (3) | 25 ± 3 (3) | 83 (2) |
Data recorded from patches of ventricular myocytes of rat heart containing a single active K ATP channel. K i values are 35 ± 7 μM and 5.8 ± 0.6 μM through fitting the data with the expression: P = P o,max {1 − 1/[1 + (K i/[ATP])S]}, where P is the normalized P o, in ATP relative to the maximal P o (P o,max), and S is the slope factor, or Hill coefficient. The slope factors are between 0.9 and 1.3. Filter cutoff frequency for events analysis was fc = 2.0 kHz.
The Rate of ATP Inhibition and Dissociation
Mean open time, t o, varied with [ATP] both with and without PPIs (Table ), indicating that the ATP-inhibited state is an open state(s). Davies et al. 1992 used a method to obtain apparent ATP inhibition rate constants that did not depend upon other details of the underlying kinetic model. If we assume that (a) ATP binds to the open states to inhibit the channel; (b) rates of all ATP-dependent transitions linked to the open states are uniform and independent each other; and (c) ATP does not affect the relative occupancy within the set of open states, then the reciprocal of t o has a linear relation to [ATP] given by:
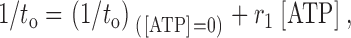 |
4 |
where the slope r 1 will also be the apparent association rate constant for ATP causing inhibition. In the absence of evidence disqualifying these assumptions, we have accepted them for this analysis but use the term apparent affinity and apparent rate constants to acknowledge that state-dependent interactions may also play a role (Shyng and Nichols 1998). Fig. 6 A compares the relationship between [ATP] and 1/t o with and without PPIs in rat ventricular myocytes. The linear fit both cases well, but untreated channels demonstrated a steeper slope (0.8 mM−1 ms−1) than channels treated with PPIs (0.02 mM−1 ms−1). Multiplying these slopes by the K i (35 μM and 5.8 mM, determined earlier) gives dissociation rate constants of 0.03 and 0.12 ms−1 for channels untreated and treated with PPIs, respectively. Considering the proposed tetrameric stoichiometry of KATP, the actual inhibition and dissociation constants should be one-fourth of these values if ATP binds to any of four monomers to induce inhibition. Thus, at the single-channel level, PPIs both decreased the apparent ATP association rate and increased the apparent ATP dissociation rate for KATP.
Figure 6.

Plot of the reciprocal of the corrected mean open time (to) against [ATP] before (•) and after treatment with PPIs (5 min, 1 mg/ml) (○). (A) Data from rat ventricular myocytes (Table ). The lines are fits to with inhibition rate constants of 0.8 and 0.02 mM−1 ms−1 for K is of 35 μM and 5.8 mM for control and after treatment with PPIs, respectively. (B) Data for Kir6.2ΔC35 channels expressed in COS-1 cells converted from Table . The lines are obtained by taking an inhibition rate constant of 0.6 and 0.3 mM−1 ms−1 for K is of 247 μM and 2.1 mM for control and after treatment with PPIs, respectively.
Desensitization of ATP Inhibition by PPIs in Kir6.2ΔC35
Which subunit, SUR2 or Kir6.2, interacts with PPIs to cause the antagonism of ATP inhibition? Previously, using site-directed mutagenesis, we have shown that the reactivation effect of PPIs on KATP was through an interaction on the Kir6.2 subunit (Fan and Makielski 1997). Moreover, phosphatidylinositol is known to have a direct interaction with other inwardly rectifying K+ channels (Fan and Makielski 1997; Huang et al. 1998). Therefore, we explored whether the ATP desensitization effect of PPIs also localized to the channel subunit. When Kir6.2 was expressed alone, either with or without a green florescence protein tag on its COOH terminus (John et al. 1998), or with COOH-terminal truncation (Tucker et al. 1997), Kir6.2 retained ATP sensitivity, albeit reduced. We examined the effect of PPIs on ATP inhibition of the COOH-terminal truncated Kir6.2, Kir6.2ΔC35, that when expressed alone without SUR2 gave KATP currents. In the presence of low [ATP] (1 μM), the gating behavior of Kir6.2ΔC35 was characterized by short openings and less obvious short bursts (Fig. 7 A), consistent with previous observations in a similarly truncated Kir6.2 (Tucker et al. 1997; Drain et al. 1998; Trapp et al. 1998). Kir6.2ΔC35 currents also ran down after patch excision, albeit somewhat slower than when in the presence of SUR2. PPIs restored channel activity after run-down, providing further evidence that this effect is mediated through an interaction on Kir6.2 (Fig. 7B and Fig. C, and Table ). The increase in P o after reactivation by PPIs was reflected in the prolongation of mean open time and mean burst duration, and the profound shortening of mean closed time (Table ).
Figure 7.

PPIs reactivate KATP current in Kir6.2ΔC35. Single-channel currents were recorded from an inside-out patch from a COS-1 cell transfected with Kir6.2ΔC35. c– indicates the closed open level. The bottom trace in each panel represents a detail at a greater time resolution of the record in the top panel. The diagonal lines indicate the part of the top record that was selected for display in the lower record. (A) KATP current recorded immediately after excision of the patch before run-down. (B) Current recorded 10 min after excision of the patch after the current had run down. (C) Current recorded after a 5-min treatment with PPIs (1 mg/ml).
Table 3.
Open Probability, Corrected Mean Open Time, and Mean Closed Time of the Single-Channel Current Events Recorded from COS-1 Cells Transfected with Kir6.2ΔC35 Before and After Run Down, and After 10-min Treatment with PPIs
| Before run down | After run down | After PPIs | |
|---|---|---|---|
| average, 6 min | 1 mg/ml, 3 min | ||
| P o | 0.03 ± 0.01 (4) | 0.004 ± 0.003 (4) | 0.05 ± 0.04 (4) |
| t o (ms) | 1.5 ± 0.03 (3) | 1.9 ± 0.1 (3) | 2.6 ± 0.3 (3) |
| t c (ms) | 49 ± 7 (3) | 568 ± 23 (3) | 47 ± 6 (3) |
Filter cutoff frequency fc = 2.0 kHz. Data from a patch that contained more than one active channel is included in P o.
Representative traces from a patch with a single Kir6.2ΔC35 channel (Fig. 8 A) show that its kinetics are distinctly different from those of native KATP or cloned SUR2/Kir6.2 KATP currents. They also show that Kir6.2ΔC35 retains some sensitivity to ATP inhibition in control and that PPIs also desensitize ATP inhibition for this channel. Summary data for the ATP inhibition of Kir6.2ΔC35 (Fig. 8 B) show that K i in the control was 247 μM for ATP inhibition, compared with 35 μM for SUR2/Kir6.2 (Fig. 2), a sevenfold difference. Thus, the truncated version of the channel is much less sensitive than in the presence of the SUR subunit. These features of COOH-terminal truncated Kir6.2 are consistent with the data published by Tucker et al. 1997 for the truncated Kir6.2 and those by John et al. 1998 for the full length of Kir6.2. As with SUR2/Kir6.2, treatment with PPIs (10 min, 1 mg/ml) significantly (P < 0.001) shifted the concentration–response curve for ATP inhibition to the right, resulting in a larger value of K i of 2.1 mM (Fig. 8 D). However, in SUR2/Kir6.2 the same treatment desensitized K i by nearly 500-fold from 35 μM to 16 mM (Fig. 2). These results suggest that although the essential effect of PPIs on reactivation and desensitization of ATP inhibition are retained in Kir6.2ΔC35, differences do exist. The SUR subunit and/or the alteration to the COOH terminus may modulate the effects of PPIs.
Figure 8.

ATP block of Kir6.2Δ35 before and after treatment with PPIs (1 mg/ml). (A) KATP currents from single-channels recorded at the [ATP] indicated. Traces at left were obtained before treatment with PPIs; at right traces were obtained 10 min after treatment. c– and o– indicate closed and open current levels, respectively. (B) Concentration–response relationship of ATP block before and after treatment of Kir6.2ΔC35 with PPIs. Symbols and error bars represent the mean ± SE (n = 1–4 for each data point) in control and 10 min after treatment. Lines represent fitting of the data to a binding equation as in Fig. 2. K i is the concentration of half inhibition and S is the slope factor or Hill coefficients. K i = 247 ± 188 μm in control, and 2.17 ± 0.93 mM after treatment with PPIs (P < 0.001). S = 1.3 and 1.4 before and after treatment with PPIs, respectively.
We also analyzed the single-channel kinetics for ATP inhibition of Kir6.2ΔC35 current before and after treatment with PPIs (Table ). Burst behavior was more ambiguous in this mutant. In control, ATP increased the mean closure time and reduced the mean open time, consistent with those previously reported for Drain et al. 1998 and Trapp et al. 1998. ATP dependence in the mean closed time was profoundly reduced after treatment with PPIs. On the other hand, the mean open time and burst duration remained dependent upon [ATP]. The change in the ATP dependence of the mean closed time agrees with the similar changes seen in native cardiac myocytes (Fig. 5). The closed time distribution for Kir6.2ΔC35 was adequately fit by two exponential components (τc,1 = 0.59 ± 0.05 ms, and τc,2 = 17.3 ± 2.1 ms, n = 4) rather than the four components required for native myocytes and SUR2/Kir6.2. The slower component (τc,2) varied directly with [ATP], from 17.3 ± 2.1 ms at 10 μM ATP to 132.0 ± 19 at 1 mM ATP (n = 3) (P < 0.001). The open time distribution was adequately fit by two exponential components (τo,1 = 0.15 ± 0.04 ms and τo,2 1.12 ± 0.19 ms, n = 4). The time constant for the slower component of open time, τo,2, decreased at higher [ATP] from 1.12 ± 0.19 ms at ATP 10 μM to 0.81 ± 0.14 ms at ATP 1 mM, although the relationship was weaker than that of SUR2/Kir6.2. After treatment with PPIs, the [ATP] dependence of both time constants for the slower component of closed times and the slower component of open times were reduced, with τc,2 = 41.2 ± 7.1 ms and τo,2 = 2.61 ± 0.51 ms at ATP 10 μM, and τc,2 = 43.5 ± 11.2 ms and τo,2 = 1.69 ± 0.35 at ATP 1 mM (n = 3), respectively. Again, this tendency of the change in single-channel kinetics caused by PPIs is qualitatively in agreement with the observation made in native KATP and SUR2/Kir6.2. With the same method and expression (see ), we estimated ATP inhibition and dissociation rates for Kir6.2ΔC35 untreated and treated with PPIs (Fig. 6 B).
Table 4.
Open Probability, Corrected Mean Open Time, Mean Burst Duration, and Mean Closed Time of the Single-Channel Current Events Recorded from COS-1 Cells Transfected with Kir6.2ΔC35 in Response to ATP Before and After 10-min Treatment with PPIs
| Untreated | After 10-min treatment with PPIs | |||||
|---|---|---|---|---|---|---|
| 1 mg/ml | ||||||
| [ATP] (mM) | 0.01 | 0.1 | 1.0 | 0.01 | 0.1 | 1.0 |
| P o | 0.069 ± 0.012 (4) | 0.047 ± 0.01 | 0.008 ± 0.003 | 0.07 ± 0.01 | 0.067 ± 0.02 | 0.045 ± 0.07 |
| t o (ms) | 1.1 ± 0.2 (4) | 0.9 ± 0.1 | 0.71 ± 0.1 | 2.2 ± 0.2 | 1.9 ± 0.3 | 1.5 ± 0.2 |
| t b (ms) | 23 ± 3.5 | 11 ± 1.8 | 3.5 ± 0.5 | 37 ± 5.8 | 31 ± 3.2 | 16 ± 2.8 |
| t c (ms) | 17 ± 1.8 (4) | 27 ± 2.9 | 125 ± 14 | 42 ± 5.6 | 38 ± 5.4 | 44 ± 9.2 |
Filter cutoff frequency fc = 2.0 kHz. Data were averages from three (without the number) or four (with the number) measurements in two patches.
Adenosine Inhibition of KATP Before and After Treatment with PPIs
To test the hypothesis that charged lipid–protein interactions play a role in the effect of PPIs on ATP sensitivity, we probed the mechanism of inhibition by testing uncharged adenosine to represent the uncharged adenosine moiety of ATP. Fig. 9 A shows an example of the experiments with adenosine before and after a 10-min treatment with PPIs. Note that in this example the control was taken after a 20-s period of perfusion with PPIs. This brief treatment with PPIs induced little or no change in ATP sensitivity; ATP inhibition had a K i (37 μM measured in one patch) not significantly different from the value obtained without any treatment with PPIs (Fig. 2). Adenosine (1 mM) produced a moderate inhibition (∼20% of maximal P o,). After a 10-min treatment with PPIs, ATP sensitivity was profoundly reduced, whereas the inhibitory effect of adenosine was much less affected. We repeated the same experiments in Kir6.2ΔC35. Fig. 9 B a shows current recordings from a typical experiment. This patch contained at least five active channels. Because of the short openings typical of Kir6.2ΔC35, macroscopic currents with this number of channels present appear very noisy. Figure 9 B b better demonstrates the current levels using a histogram transform at a current resolution (bin width) of 0.05 pA. Each peak of the histogram represents an open channel level. The statistical values of P o at different [ATP] and [adenosine] relative to the maximal P o measured at 1 μM ATP in the same preparations are given in Fig. 10. We noticed that adenosine also produced less inhibition in Kir6.2ΔC35 than in KATP of rat ventricular myocytes. The statistical data also demonstrated that treatment with PPIs significantly attenuated ATP inhibition, but PPIs had little effect on adenosine inhibition.
Figure 9.


Effect of adenosine on KATP before and after treatment with PPIs. (A) KATP currents recorded from a ventricular myocyte of rat heart. Current traces on the left were recorded after a 20-s perfusion of PPIs (1 mg/ml). ATP and adenosine were added at the concentrations shown. Traces on the right were recorded after 10-min treatment with PPIs (1 mg/ml). c– indicates the closed current level Traces were recorded from a single patch, and at least six channels were present in this patch. (B) Effect of adenosine on current from a COS-1 cell expressing Kir6.2ΔC35. (a) Current traces were recorded before (left) and after (right) treatment for 10 min with PPIs (1 mg/ml). c– indicates the current level where all channels were closed. Traces were recorded from a single patch, and at least five channels were present in this patch. (b) Amplitude histogram calculated from the corresponding currents in A (see text for details).
Figure 10.

PPIs reduce ATP inhibition, but not adenosine inhibition. Summary data for the effects of ATP and adenosine (Ade) on KATP from rat ventricular myocytes (A) and Kir6.2ΔC35 (B). Normalized P o is a quotient of the NP o measured in ATP or adenosine before and after exposure to PPIs for 10 min, divided by the NP o present at the beginning of the experiment. S.S. indicates statistical significance by a paired t test with P < 0.05; and N.S. represents an insignificant difference by t test with P > 0.05.
Discussion
Multiple Effects of PPIs on KATP Activity
We have shown that PPIs affect KATP activity for native cardiac cells and for the cardiac isoform of recombinant KATP (SUR/Kir6.2) expressed in COS-1 cells. Phosphatidylinositol has several effects on KATP: (a) it increases maximal P o (Fig. 1 and Fan and Makielski 1997); (b) it desensitizes ATP inhibition (Fig. 2 and Shyng and Nichols 1998; Baukrowitz et al. 1998, for SUR1 and mutated Kir6.2); (c) it attenuates MgATP antagonism of ATP inhibition (Fig. 3); and (d) it decreases sulfonylurea block (Fan, Z., unpublished observation). In this study, we provide detailed analysis of the desensitization of ATP inhibition by phosphatidylinositol using a preparation of various PPIs. We also analyzed the role of SUR in the effects of PPIs on ATP inhibition. The nature of the interaction of PPIs with KATP to affect maximal P o has been characterized previously (Fan and Makielski 1997; Huang et al. 1998). In this study we characterized the desensitization of ATP inhibition by PPIs and explored the mechanism of this effect.
Do PPIs Interact with Kir or SUR to Decrease ATP Sensitivity?
Although the characteristic regulation for KATP is inhibition by intracellular ATP, nucleotides regulate KATP in a complex manner. Hydrolysis of ATP is not necessary for inhibition, suggesting that ATP binds directly to one of the subunits of KATP. Tucker et al. 1997 showed that truncated Kir6.2 with either 26 or 36 amino acids truncated from the COOH terminus produced KATP currents that retained ATP sensitivity, and we have confirmed this finding for a Kir6.2 with 35 amino acids truncated from the COOH terminus (Kir6.2ΔC35). John et al. 1998 reported a full-length cDNA of Kir6.2 that, although expressed at extremely low efficiency, produced currents with ATP sensitivity similar to truncated Kir6.2, eliminating the possibility that artifacts were induced by terminal deletion. Thus, at least part of the ATP sensitivity is probably caused by ATP binding to Kir6.2 and not to SUR. Similarly, we isolated the effects of PPIs to Kir6.2 using the same strategy with Kir6.2ΔC35. Comparing the effect of PPIs with (Fig. 2 B) and without (Fig. 8 B) SUR2 reveals that PPIs are effective in desensitizing ATP inhibition for Kir6.2ΔC35 in agreement with Baukrowitz et al. 1998. Even though the kinetics of the Kir6.2ΔC35 and SUR2/Kir6.2 differed considerably, PPIs in the absence and presence of ATP affected kinetics in a similar way, indicating that the mechanism underlying desensitization of ATP inhibition by PPIs probably primarily involves Kir6.2. However, the data also suggest that SUR introduced additional influences on the desensitization effect of PPIs. The SUR2 subunit increased ATP sensitivity of KATP (Fig. 2 and Fig. 8), as previously described for SUR1 (Tucker et al. 1997). However, in the presence of PPIs this increased sensitization was lost and even relatively reversed after treatment with PPIs. The NH2 terminus of Kir6.2 has been suggested to face SUR (Clement et al. 1997), and considering that PPIs are proposed to bind at the COOH terminus (Fan and Makielski 1997), we hypothesized that PPIs may disconnect functional linkage or transduction of conformational changes from SUR to the K+ channel pore, but not dissociate the two subunits physically. Consistent with this hypothesis, we found that PPIs attenuated Mg2+ (via MgATP) stimulation of KATP in the presence of inhibitory [ATP] (Fig. 3), an effect previously shown to require interaction between SUR and Kir subunits (Gribble et al. 1998). In addition to suggesting that SUR and Kir are functionally unlinked in the presence of PPIs, this finding is important per se, showing that under physiological conditions desensitization by PPIs can be at least partly masked by MgATP. Additional evidence that suggests the effect of PPIs on the SUR2/Kir6.2 interaction comes from experiments in which we found that treatment with PPIs significantly attenuated block of KATP by tolbutamide, a sulfonylurea reagent (Fan, Z., unpublished observation). Sulfonylureas are proposed to bind at SUR in order to block the K+ current through the pore (Aguilar-Bryan et al. 1998). Although the data suggest that PPIs interact primarily with Kir6.2 to increase open probability and to decrease ATP sensitivity, the SUR subunit also has a modulatory function, which is not surprising given the complexity of the structure/function of this channel.
Properties of PPI–Channel Interaction Accounting for Desensitization
Based on the activity sequence and effects of cascade products of phospholipids, we previously proposed a structural mechanism for the effect of anionic phospholipids on maximal P o involving the interaction of membrane phospholipids with positively charged amino acids on the COOH terminus of Kir6.2 (Fan and Makielski 1997). Further independent biochemical assays have verified that electrostatic interaction is a key element mediating the binding of phosphatidylinositol to inwardly rectifying K+ channels (Huang et al. 1998). The desensitization effect seems to share a similar charge-dependent property. The observation of a relation between ATP sensitivity and membrane surface negative charge can be traced to the pioneer work by Deutsch et al. 1994, in which they noted that screening of the surface charge sensitized ATP inhibition. Our negative results with the uncharged phosphatidylcholine support the idea that the anionic head group is required for the desensitization of ATP inhibition by PPIs. Two features of the desensitization are (i) the requirement for intact membrane lipids because inositol triphosphate is not effective (Shyng and Nichols 1998); and (ii) the correlation between the charges of the head group and the strength of the desensitization with the order of PIP2 > PIP > PI (Shyng and Nichols 1998; Baukrowitz et al. 1998). Both of these features were also found for the effect on maximal P o (Fan and Makielski 1997). In that study we also hypothesized that a region of the cytoplasmic COOH-terminal segment of Kir6.2 immediately adjacent to the second transmembrane spanning segment, rich in positively charged amino acids, interacts with the head groups of anionic membrane phospholipids. Mutations in either two (176R, 177R) or one (176R) adjacent arginines in this region decreased maximal P o, attenuated the effect of PPIs on P o and reduced the binding of PPIs to the channels (Fan and Makielski 1997; Shyng and Nichols 1998; Huang et al. 1998). ATP sensitivity has not been characterized for these mutants partly because of very low maximal P o and rapid run-down in these channels. However, it has been shown that ATP sensitivity was affected when neutralizing a positively charged K185 on the COOH terminus (Tucker et al. 1998), a residue close to the tentative phosphatidylinositol binding region underlying the effect on maximal P o. We consider that an electrostatic lipid-protein interaction also accounts for the desensitization effect of PPIs.
When interpreting the modest reduction of ATP inhibition in mutation of N160, Shyng et al. 1997 proposed that ATP preferentially bind to and stabilize the closed channel, rather than inhibit the channel directly from an open state. According to this hypothesis, an intervention to increase maximal P o also decreases ATP sensitivity by reducing entry to the closed state. This can be called a collateral effect because the decrease of ATP sensitivity is not caused by a change of the intrinsic ATP binding affinity. According to this hypothesis, the previously described increase in maximal P o by PPIs (Fan and Makielski 1997) would imply a decrease in apparent ATP sensitivity if ATP binds selectively to closed states. However, several aspects of the data show that PPIs do not act solely by this mechanism to desensitize ATP inhibition. First, we have shown that the desensitization had a time course that could be clearly separated experimentally from the effect on maximal P o (Fig. 1). The separation of the effects is in the range of several minutes, but the time constants for ATP binding and dissociation are in the range of several milliseconds to seconds. This separation, therefore, cannot be explained by a kinetic delay. Second, we found that in the patches that lasted for several hours, after treatment with PPIs the maximal P o decreased to nearly zero while ATP sensitivity did not increase. Third, PPIs desensitized Kir6.2ΔC35 (247 μM versus 2.1 mM, before versus after treatment with PPIs, respectively) while the maximal P o was not significantly altered (0.69 versus 0.7) (Fig. 6 and Table ). In the next two sections, with the aid of modeling analysis of single-channel kinetic behaviors, we interpret the desensitization effect of PPIs to be primarily due to a result of change in the intrinsic ATP binding affinity.
Single-Channel Kinetics of ATP Inhibition and Desensitization by PPIs
Our analysis, as well as those reported by other laboratories, has indicated that the kinetics of outward KATP currents are sufficiently complicated to require two or more components to fit each of the open and closed time distributions. Conventional models with three states (Qin et al. 1989) or four states (Gillis et al. 1989; Alekseev et al. 1998) connected linearly are commonly used to account for the single-channel behaviors of KATP. With progress in understanding channel structure, more complex models such as that described by Nichols et al. 1991 were proposed to utilize structural information. A conclusive discrimination between these models will certainly require data beyond that provided in our report. Nonetheless, in the context of comparison between simpler key steps in more complex models, interpreting single-channel behaviors in response to ATP and PPIs can provide insight into the mechanism of desensitization and ATP inhibition.
The effects of PPIs on single-channel kinetics of KATP in the absence of ATP have been analyzed (Fan and Makielski 1997; this paper, Tables I–III). Mean open time was increased and mean closed time was decreased after treatment with PPIs requiring that PPIs affect a transition that is directly connected to open state(s). Given this consideration, a collateral change in ATP sensitivity secondary to change in maximal P o could be caused if ATP binds preferentially to closed states of the channel in a linear scheme (Fig. 1), where the arrows represent the direction toward which ATP or PPIs facilitate transition. Fig. 1 can qualitatively reproduce the burst behaviors we observed. If ATP binds only between the bursts, as when the channel is in C1 or C2, the probability that ATP binds to the channel would be reduced if PPIs increase burst duration and reduce the closed times between bursts. However, this scheme cannot explain the other kinetic changes we observed experimentally. In such a scheme, ATP should not affect open times, but ATP shortened open times, and PPIs altered the ATP effect on open times as well (Table and Table ). Interestingly, open times of Kir6.2ΔC35 had a similar [ATP] dependence (Table IV; and Drain et al. 1998; Trapp et al. 1998). Also, according to Fig. 1, the mean duration of the slowest component of closed intervals between bursts would be predicted to be dominated by [ATP], and PPIs should not affect this duration, but this was not the case (see τc,4 in Table ). Changes in both of these parameters are major contributors to the increase in channel activity in the presence of ATP after treatment with PPIs. Hence, the effect of PPIs on single-channel kinetics reflecting desensitization of ATP inhibition cannot be caused solely by interactions with ATP inhibitory closed states. In a recent study, Babenko et al. 1999a also reported that a model such as Fig. 1 could not account for the SUR influence on ATP inhibition and proposed that a true change in ATP affinity occurred.
Scheme S1.

Further considerations cause us to favor a change in intrinsic ATP affinity as the primary mechanism of desensitization by PPIs. In addition to the reasons already given, we tested this hypothesis using a relatively model-independent method (Davies et al. 1992, and results) to evaluate the single-channel behaviors. This method assumes a transition by which ATP drives the channel directly from an open state to a closed state.
Fig. 2 predicts a linear relationship between the reciprocal of mean open time and [ATP] that provides a reasonable fit to the experimental data (Fig. 6). In this scheme, the action of PPIs could be interpreted as desensitizing ATP inhibition either by competing with ATP directly or by changing the ATP binding affinity allosterically. The analysis gives an ATP association rate constant regardless of how many ATP-dependent transitions are involved, provided that these transitions have the same set of rate constants. To account for our data we expand Fig. 2 to include additional states.
Scheme S2.

In Fig. 3, C1 and C2 represent closed states analogous to the closed states in usual linear sequential models. Multiple open states (O1 and O2) are included to account for the multiple components in the open time distribution. Although the open channel time distribution (Table ) actually consist of three components, indicating that three open states may exist, for simplification purpose only two open states were used in this scheme. Similar consideration was also applied to determination of the closed states. Our simulation (introduced below) proved that this simplification did not affect our conclusion. Transitions to additional closed states, Crs (“r” for “reactivatable”), represent the closed conformation of the channel from which PPIs can reactivate the channel by preventing them from entering these states. This branch entering from the open state is an expansion of Fig. 1. Data in Table indicate that PPIs preferentially prolong the slower components of open times with little effect on the fastest component in the absence of ATP. Unlinking the Cr states from the O1 state that dominates the fast component can reproduce these data. The idea of two pathways, one ATP-sensitive and one insensitive, that can close the open channel, has been recently suggested (Trapp et al. 1998).
Scheme S3.

The model in Fig. 3 accounts for the salient single-channel features of our data with PPIs and ATP. For Fig. 3, 1/τo can be expressed as a linear function of [ATP]:
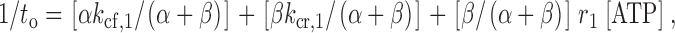 |
5 |
where the transition rates are noted in Fig. 3. The rate constants k cr,1 and r 1 are assumed ∝1/[PPIs]. When [ATP] = 0, Fig. 3 is reduced to the first two terms in , the sum of which is the value of intersection in plot of 1/t o versus [ATP]. Treatment with PPIs decreases k cr,1, predicting a lowering of this intersection. also predicts that the intersection must be >0. These predictions fit the experimental findings (Fig. 6A and Fig. B).
Quantitatively, our analysis showed that after treatment with PPIs a major change occurred in the ATP association rate, suggesting a competitive antagonism. This is in general agreement with rate constants measured by Baukrowitz et al. 1998, who used a fast solution exchange and an analysis method that is also model independent. The dissociation rate also changed, especially for Kir6.2ΔC35 in which the [ATP] dependence of mean open time is relatively weak. The reason for such a deviation from strict competitiveness is unclear, but a more complicated process such as a multiple-step binding (discussed below), or an allosteric effect may be implied. In fact, in Fig. 3 the ATP dissociation rate deviates from the product of measured K i and ATP association rate by a factor of 1.6 (control) or 1.3 (PPIs). In this model, this factor represents a source of collateral influence of the kinetics on the ATP sensitivity. But this influence is very small comparing to the change in intrinsic ATP affinity that is required to reproduce the data. Using Fig. 3 and the measured ATP binding rate constants, we simulated the single-channel current. Other rate constants in the model were obtained by fitting the model to the experimental single-channel data using maximum likelihood as a criterion (Qin et al. 1997). The final rate constants were rounded as simple as possible. Table lists the rate constants and statistical data of the simulated single-channel currents. The model quantitatively reproduced all of the salient features of the single-channel currents observed experimentally and listed in Table . The following features are representative and emphasized. First, the model reproduced the [ATP]-inhibition relationship for both control and treatment with PPIs. The Hill coefficient of the simulation data is close to 1, consistent with the data. Second, the model accurately simulates the shortened mean open time and mean burst duration with increased [ATP] and the prolongation of these parameters with PPIs at increased [ATP]. Third, the prolongation effect of PPIs on mean open time and burst duration in the absence of inhibitory ATP was also reproduced. Fourth, the prolongation of the mean closed time by ATP and the dramatic decreasing effect of PPIs on this parameter are also well reproduced. Several quantitative deviations of the simulation results from the experimental data such as the mean closed times at high [ATP], and the mean burst duration at low [ATP] in PPIs is likely to be caused by the simplification of the model and can be corrected by introducing additional states. In summary, although complex kinetic schemes might improve the fit of the experimental data, our analysis based on Fig. 2 provides a reasonable representation of the kinetic changes with ATP and PPIs observed experimentally.
Table 5.
Model Simulations of the Open Probability, Corrected Mean Open Time, Mean Burst Duration, and Mean Closed Time of Single-Channel Current Events and ATP Concentration Before and After Treatment with PPIs
| Untreated | Treatment with PPIs | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ATP binding rate (s−1 · M−1) | r 1 = 800000 | r 1 = 20000 | |||||||
| Other rates (s−1) | k cf = 10000 | α = 60 | k cr,1 = 3 | k cf, = 10000 | α = 60 | k cr,1 = 0.3 | |||
| k cf,−1 = 3500 | β = 1000 | k cr,−1 = 5 | r −1 = 45 | k cf,−1 = 3500 | β = 1000 | k cr,−1 = 5 | r −1 = 150 | ||
| [ATP] (mM) | 0.001 | 0.01 | 0.1 | 1 | 0.001 | 0.01 | 0.1 | 1 | 10 |
| P o | 0.61 | 0.53 | 0.11 | 0.003 | 0.92 | 0.9 | 0.88 | 0.8 | 0.24 |
| to (ms) | 8.3 | 7.5 | 5.0 | 1.3 | 9.0 | 8.9 | 8.7 | 7.2 | 3.2 |
| tb (ms) | 280 | 119 | 19 | 2.0 | 1650 | 1090 | 460 | 78 | 16 |
| tc (ms) | 26 | 47 | 97 | 651 | 1.8 | 1.9 | 2 | 4.3 | 21 |
The data were measured from single-channel events generated from the listed rate constants and Fig. 3, shown in the text. A 90-s length of simulated current recording was used for each datum. ATP binding rates were from the calculation in Fig. 6. Ki of ATP inhibition was obtained by fitting the data with the expression: P = P o,max {1 − 1/[1 + (K i/[ATP])S]}, where P is the normalized P o in ATP relative to the maximal P o (P o,max) and S is the slope factor, or Hill coefficient. Simulated channel events have K i values of 35 μM and 5.8 mM, corresponding to channels untreated or treated with PPIs, respectively. The slope factors are 1.4 and 1.2 for channels untreated and treated with PPIs, respectively. Channel events had a unitary amplitude of 2 pA plus a noise of 0.5 pA (SD) at closed level and a noise of 0.8 pA (SD) at open channel level. The events were filtered digitally through a low-pass filter at a cutoff frequency fc 5 2.0 kHz.
A Speculative Molecular Mechanism for ATP Binding and Desensitization by PPIs
To explain the change in the intrinsic ATP binding affinity caused by PPIs, we propose a hypothetical molecular model that provides a mechanistic and structural basis for the ATP and phosphatidylinositol interaction at an ATP binding site. The model is adapted from a model first proposed generically by Jencks 1975. We hypothesize that ATP binding to Kir6.2 occurs through two interactions: an electrostatic interaction between the negatively charged phosphates of ATP with positively charged amino acids of Kir6.2, and a hydrophobic interaction between the nucleotide of ATP and Kir6.2. Fig. 11 depicts the proposed electrostatic interaction (K A and K Au) and hydrophobic interaction (K B and K Bu) where u represents interactions leading to the state where both moieties are bound. The channel is considered closed, probably by a conformational change, when the adenosine moiety and the phosphate group from the same ATP molecule (right configuration) occupy both sites. Occupation of either site lowers the binding energy and favors formation of the double-occupancy configuration. Whether occupation of a single site can also induce the conformational change required to close the channel is not clear, but our experiment using adenosine suggests that binding of the adenosine moiety alone may be sufficient to close the channel, although energetically this is less likely to occur.
Figure 11.

A model to account for phosphatidylinositol desensitization of ATP inhibition of KATP. The ATP molecule is shown as a stick diagram having an ∼120° angle between the adenosine group (ring structures) and the phosphate group (crosshatched end). The protein binding site is depicted as a “hook” with positively charged residues, indicated by ⊕, representing the site for electrostatic interaction with the negatively charged phosphates, and the hook representing a hydrophobic binding site for the adenine moiety. KA denotes the association constant of the step in which a complex of ATP and channel is formed through the binding of the hydrophilic phosphate group, and KB denotes the association constant of the step forming the complex through the binding of the hydrophobic adenosine moiety. These steps are bimolecular bindings presumably with the same binding constants of the individual parts binding to their binding site. KAu and KBu are the association constants of the subsequent binding of the second part of the ATP molecule to the binding site, which is a unimolecular (thus the u in the superscript) process. Binding of PPIs to the charged part of the ATP binding site (denoted in the diagram as negatively charged heads with tails pointing downward) causes a decrease rate of ATP association involving phosphate group (denoted by an apostrophe and smaller arrows). The rightmost state represents the state in which both parts of the ATP molecule are bound to the binding site. The states with adenosine binding to the site (bottom and right) are proposed to cause nonconducting channels.
When the head groups of PPIs occupy the charged amino acids (Fig. 11, right), then the electrostatic interaction is not favored and ATP binding is weakened. This model is plausible in that electrostatic interactions have been shown to guide charged ligands to their binding sites in many proteins; our model is based upon these precedents (Jencks 1975), and also fits the activity sequence ATP > ADP > AMP > adenosine (Ashcroft 1988). The model predicts that PPIs desensitize ATP inhibition but not adenosine inhibition, in agreement with our experimental data (Fig. 10).
This model requires that the ATP binding site and the PPIs binding site be at or near the same location on the KATP channel. The precise location of the ATP binding site is not known, but recent studies have suggested that it may be on the COOH terminus of Kir6.2. Mutation and deletion of residues in the NH2 terminus of Kir6.2 reduced ATP inhibition (Takano et al. 1998; Tucker et al. 1998), but these changes were thought to exert their effect primarily by increasing P o (Babenko et al. 1999a; Babenko et al. 1999b; Koster et al. 1999), i.e., invoking the collateral effect, and not by a direct effect at an ATP binding site. Drain et al. 1998 identified two distinct regions (T171 – K185, and G334 – I337) on the COOH terminus of Kir6.2 critical for ATP inhibition. Mutation of residues within these regions decreased ATP inhibition profoundly. The region T171 – I185 includes the putative PPIs binding site identified with the effect on maximal P o. Whether or not this putative region where ATP binding and PPIs interact coincides with or overlaps the binding region accounting for the effect of PPIs on maximal P o has not been determined. Finally, this model, although very speculative, is based upon precedent for lipid–protein interactions and may stimulate the development of testable hypotheses regarding the underlying structural/function mechanisms for ATP inhibition, changes in ATP sensitivity, and the desensitization effect of PPIs.
Physiologic Role of PPI–KATP Interaction
Do phosphatidylinositols have a physiological or pathophysiological role in modulating KATP function? In theory, a change in composition of phosphatidylinositol could affect a change in the ATP sensitivity of KATP. The compositions of the individual phosphatidylinositol species, including PIP3, PIP2, PIP, and PI, are substrates of cellular signaling pathways and subject to tight regulation by specific kinases (Loijens et al. 1996), suggesting that these lipids may be good candidates for regulation of KATP. Wortmannin, an inhibitor of phosphatidylinositol kinases, blocked MgATP reactivation of KATP in native cardiac myocytes, supporting this pathway as possibly a physiologic mechanism (Xie et al. 1999). We have observed that a purified phosphatidylinositol-4-phosphate 5-kinase (Boronenkov and Anderson 1995) enhanced channel activity in the presence of MgATP, mimicking the effects of PPIs (Fan, Z., and J.C. Makielski, unpublished observation). Other highly regulated fixed anion charges, such as those in the actin cytoskeleton (Furukawa et al. 1996), are also potential candidates for this interaction. Such regulation might explain the observations of significant variance in the ATP sensitivity of KATP in the same type of cells (Findlay and Faivre 1991; Takano and Noma 1993) or even in the same patch recorded at a different time (Qin et al. 1989). Perhaps this interaction can cause the opening of KATP in intact cells at levels of ATP that close channels in cell-free patches where PPIs may be quickly degraded.
In conclusion, two groups have reported that phosphorylated PPIs decrease ATP block of SUR1/Kir6.2 and mutations of Kir6.2 expressed in COS cells (Shyng and Nichols 1998) and oocytes (Baukrowitz et al. 1998). Our results confirm that this desensitization also applies to Kir6.2 expressed with the cardiac isoform SUR2 and in native cardiac myocytes. Additional information provided by our study includes a description of the effect at the single-channel level for both SUR2/Kir6.2 and a truncated Kir6.2 expressed alone, suggesting a complex effect of PPIs on both ATP association and dissociation rates. In addition, the interaction of PPIs with MgATP desensitization of ATP inhibition, combined with differences in the details of how PPIs exert their effects on ATP sensitivity when SUR is present (SUR2/Kir6.2 versus Kir6.2ΔC35), suggest that the SUR subunit maintains a modulatory role in the effect. Finally, based upon our results, the effects of PPIs are considered at single-channel and molecular levels. A kinetic model accounting for the channel behaviors in response to ATP affinity change, and a molecular model of two sites accounting for coordinated ATP binding to cytoplasmic domains of Kir6.2 subunit, are offered to explain the effect of PPIs on ATP sensitivity.
Acknowledgments
The technical assistance of Kevin Galles is gratefully acknowledged.
Support for this work was provided by the Oscar Rennebohm Foundation, a Scientist Development Grant of the American Heart Association National Center, and National Institutes of Health grants HL58133 (to Z. Fan) and HL57414 (to J.C. Makielski).
Footnotes
1used in this paper: PIP, phosphatidylinositol-4-phosphate; PIP2, phosphatidylinositol-4,5-bisphosphate; PPI, phosphoinositide
References
- Aguilar-Bryan L., Clement J.P., Gonzalez G., Kunjilwar K., Babenko A., Bryan J. Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 1998;78:227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- Alekseev A.E., Brady P.A., Terzic A. Ligand-insensitive state of cardiac ATP-sensitive K+ channels. Basis for channel opening. J. Gen. Physiol. 1998;111:381–394. doi: 10.1085/jgp.111.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft F.M. Adenosine 5′-triphosphate-sensitive potassium channels. Annu. Rev. Neurosci. 1988;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- Babenko A.P., Gonzalez G., Bryan J. Two regions of sulfonylurea receptor specify the spontaneous bursting and ATP inhibition of KATP channel isoforms J. Biol. Chem. 274 1999. 11587 11592a [DOI] [PubMed] [Google Scholar]
- Babenko A.P., Gonzalez G., Bryan J. The N-terminus of Kir6.2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels Biochem. Biophys. Res. Commun. 255 1999. 231 238b [DOI] [PubMed] [Google Scholar]
- Baukrowitz T., Schulte U., Oliver D., Herlitze S., Krauter T., Tucker S.J., Ruppersberg J.P., Fakler B. PIP2 and PIP as determinants for ATP inhibition of K-ATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- Boronenkov I.V., Anderson R.A. The sequence of phosphatidylinositol-4-phosphate 5-kinase defines a novel family of lipid kinases. J. Biol. Chem. 1995;270:2881–2884. doi: 10.1074/jbc.270.7.2881. [DOI] [PubMed] [Google Scholar]
- Chutkow W.A., Simon M.C., Le Beau M.M., Burant C.F. Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channels. Diabetes. 1996;45:1439–1445. doi: 10.2337/diab.45.10.1439. [DOI] [PubMed] [Google Scholar]
- Clement J.P., IV, Kunjilwar K., Gonzalez G., Schwanstecher M., Panten U., Aguilar-Bryan L., Bryan J. Association and stoichiometry of K(ATP) channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Colquhoun D., Sakmann B. Fitting and statistical analysis of single channel records. In: Sakmann B., Neher E., editors. Single-channel Recording. Plenum Press. ; New York, NY: 1983. [Google Scholar]
- Davies N.W., Standen N.B., Stanfield P.R. The effect of intracellular pH on ATP-dependent potassium channels of frog skeletal muscle. J. Physiol. 1992;445:549–568. doi: 10.1113/jphysiol.1992.sp018939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch N., Matsuoka S., Weiss J.N. Surface charge and properties of cardiac ATP-sensitive K+ channels. J. Gen. Physiol. 1994;104:773–800. doi: 10.1085/jgp.104.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain P., Li L., Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z., Makielski J.C. Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol.Chem. 1997;272:5388–5395. doi: 10.1074/jbc.272.9.5388. [DOI] [PubMed] [Google Scholar]
- Fan Z., Makielski J.C. Phosphoinositides reduce ATP block of ATP-sensitive K+ channels. Biophys. J. 1999;76:A417. [Google Scholar]
- Findlay I., Faivre J.F. ATP-sensitive K channels in heart muscle. Spare channels. FEBS Lett. 1991;279:95–97. doi: 10.1016/0014-5793(91)80259-6. [DOI] [PubMed] [Google Scholar]
- Furukawa T., Yamane T., Terai T., Katayama Y., Hiraoka M. Functional linkage of the cardiac ATP-sensitive K+ channel to the actin cytoskeleton. Pflueg. Arch. Eur. J. Physiol. 1996;431:504–512. doi: 10.1007/BF02191896. [DOI] [PubMed] [Google Scholar]
- Gillis K.D., Gee W.M., Hammond A., McDaniel M.L., Fahlke L.C., Misler S. Effects of sulphonamides on a metabolite regulated ATP-sensitive K+ channel in rat pancreatic β-cells. Am. J. Physiol. 1989;257:C1119–C1127. doi: 10.1152/ajpcell.1989.257.6.C1119. [DOI] [PubMed] [Google Scholar]
- Gribble F.M, Tucker S. J., Haug T., Ashcroft F. M. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc. Natl. Acad. Sci. USA. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D.W., Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2 . Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- Huang C.L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Isomoto S., Kondo C., Yamada M., Matsumoto S., Higashiguchi O., Horio Y., Matsuzawa Y., Kurachi Y. A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. J. Biol. Chem. 1996;271:24321–24324. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- Jencks W.P. Binding energy, specificity, and enzymatic catalysisthe circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- John S.A., Monck J.R., Weiss J.N., Ribalet B. The sulphonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K+ channels linked to the green fluorescent protein in human embryonic kidney cells (HEK 293) J. Physiol. 1998;510:333–345. doi: 10.1111/j.1469-7793.1998.333bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster J.C., Sha Q., Shyng S.-L., Nichols C.G. ATP inhibition of KATP channelscontrol of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J. Physiol. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loijens J.C., Boronenkov I.V., Parker G.J., Anderson R.A. The phosphatidylinositol 4-phosphate 5-kinase family. Adv. Enzyme Reg. 1996;36:115–140. doi: 10.1016/0065-2571(95)00005-4. [DOI] [PubMed] [Google Scholar]
- Nichols C.G., Lederer W.J., Cannell M.B. ATP dependence of KATP channel kinetics in isolated membrane patches from rat ventricle. Biophys. J. 1991;60:1164–1177. doi: 10.1016/S0006-3495(91)82152-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin D.Y., Takano M., Noma A. Kinetics of ATP-sensitive K+ channel revealed with oil-gate concentration jump method. Am. J. Physiol. 1989;257:H1624–H1633. doi: 10.1152/ajpheart.1989.257.5.H1624. [DOI] [PubMed] [Google Scholar]
- Qin F., Auerbach A., Sachs F. Identification of single-channel currents using the segmentation k-means method. Biophys. J. 1996;70:A227. [Google Scholar]
- Qin F., Auerbach A., Sachs F. Maximum likelihood estimation of aggregated Markov processes. Proc. R. Soc. B. 1997;264:375–383. doi: 10.1098/rspb.1997.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann B., Trube G. Voltage-dependent inactivation of inward-rectifying single- channel currents in the guinea-pig heart cell membrane. J. Physiol. 1984;347:659–683. doi: 10.1113/jphysiol.1984.sp015089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S.-L., Nichols C.G. Membrane phospholipid control of nucleotide sensitivity of K-ATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Shyng S.-L., Ferrigni T., Nichols C.G. Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J. Gen. Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdson W.J., Morris C.E., Brezden B.L., Gardener D.R. Stretch activation of a K+ channel in molluscan heart cells. J. Exp. Biol. 1987;127:191–209. [Google Scholar]
- Spruce A.E., Standen N.B., Stanfield P.R. Voltage-dependent ATP-sensitive potassium channels of skeletal muscle membrane. Nature. 1985;316:736–738. doi: 10.1038/316736a0. [DOI] [PubMed] [Google Scholar]
- Takano M., Noma A. The ATP-sensitive K+ channel. Prog. Neurobiol. 1993;41:21–30. doi: 10.1016/0301-0082(93)90039-u. [DOI] [PubMed] [Google Scholar]
- Takano M., Xie L.H., Otani H., Horie M. Cytoplasmic terminus domains of Kir6.x confer different nucleotide-dependent gating on the ATP-sensitive K+ channel. J. Physiol. 1998;512:395–406. doi: 10.1111/j.1469-7793.1998.395be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S., Proks P., Tucker S.J., Ashcroft F.M. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker S.J., Gribble F.M., Proks P., Trapp S., Ryder T.J., Haug T., Reimann F., Ashcroft F.M. Molecular determinants of KATP channel inhibition by ATP. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker S.J., Gribble F.M., Zhao C., Trapp S., Ashcroft F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Wittenberg B.A., Robinson T.F. Oxygen requirements, morphology, cell coat and membrane permeability of calcium-tolerant myocytes from hearts of adult rats. Cell Tissue Res. 1981;216:231–251. doi: 10.1007/BF00233618. [DOI] [PubMed] [Google Scholar]
- Xie L.-H., Takano M., Kakei M., Okamura M., Noma A. Wortmannin, an inhibitor of phosphatidylinositol kinases, blocks the MgATP-dependent recovery of Kir6.2/SUR2A channels. J. Physiol. 1999;514:655–665. doi: 10.1111/j.1469-7793.1999.655ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter Y., Burnashev N., Papin A., Portnov V., Khodorov B. Gating kinetics of ATP-sensitive single potassium channels in myocardial cells depends on electromotive force. Pflueg. Arch. Eur. J. Physiol. 1988;411:584–589. doi: 10.1007/BF00582382. [DOI] [PubMed] [Google Scholar]