

Abstract
Clostridium difficile toxin B glucosylates the Ras-related low molecular mass GTPases of the Rho subfamily thereby inactivating them. In the present report, toxin B was applied as a tool to test whether Rho proteins participate in the carbachol-induced increase in the Ca2+ sensitivity of force and myosin light chain (MLC) phosphorylation in intact intestinal smooth muscle.
Small strips of the longitudinal muscle of guinea-pig small intestine were incubated in toxin B (40 ng ml−1) overnight. Carbachol-induced force and intracellular [Ca2+], and, in a separate series, force and MLC phosphorylation, were determined.
Carbachol induced a biphasic contraction: an initial rapid increase in force (peak 1) followed by a partial relaxation and a second delayed increase in force (peak 2). The peak of the Ca2+ signal measured with fura-2 preceded peak 1 of force and then declined to a lower suprabasal steady-state level. Peak 2 was not associated with a significant increase in [Ca2+]. Toxin B nearly completely inhibited peak 2 while peak 1 was not significantly inhibited. Toxin B had no effect on the Ca2+ transient.
In control strips, MLC phosphorylation at peak 2 was 27.7 %, which was significantly higher than the resting value (18.6 %). The inhibition of the second, delayed, rise in force induced by toxin B was associated with complete inhibition of the increase in MLC phosphorylation. The resting MLC phosphorylation was not significantly different from that of the control strips.
The initial increase in MLC phosphorylation determined 3 s after exposure to carbachol was 54 % in the control strips. Toxin B also inhibited this initial phosphorylation peak despite the fact that the Ca2+ transient and the initial increase in force were not inhibited by toxin B. This suggests that Rho proteins play an important role in setting the balance between MLC phosphorylation and dephosphorylation reactions even at high levels of intracellular Ca2+.
These findings are consistent with the hypothesis that the delayed rise in force elicited by carbachol is due to an increase in the Ca2+ sensitivity of MLC phosphorylation mediated by Rho proteins.
Contraction and relaxation of smooth muscle is primarily regulated by phosphorylation and dephosphorylation of the regulatory light chains (MLC) of myosin catalysed by the Ca2+-calmodulin-dependent MLC kinase (for review, see Stull, Krueger, Kamm, Gao, Zhi & Padre, 1996) and myosin phosphatase (reviewed by Erdödi, Ito & Hartshorne, 1996). The extent of MLC phosphorylation and hence the magnitude of force production depends on the relative activities of these two enzymes. MLC phosphorylation at a fixed submaximal Ca2+ concentration can be increased by inhibition of the MLC phosphatase, e.g. directly by phosphatase inhibitors such as okadaic acid (Takai, Bialojan, Troschka & Rüegg, 1987) or indirectly by a GTP-dependent process (Kitazawa, Masuo & Somlyo, 1991; Kubota, Nomura, Kamm, Mumby & Stull, 1992). This results in an increase in the Ca2+ sensitivity of MLC phosphorylation and consequently of force. The GTP-dependent inhibition of the MLC phosphatase is thought to underly the agonist-induced increase in Ca2+ sensitivity of force in intact smooth muscle (for review, see Somlyo & Somlyo, 1994). It should be noted, however, that the Ca2+ sensitivity of force production is also potentially modulated by the thin filament linked proteins, caldesmon (Katsuyama, Wang & Morgan, 1992; Pfitzer, Zeugner, Troschka & Chalovich, 1993) and calponin (Itoh, Suzuki, Suzuki, Nakamura, Naka & Tanaka, 1994). These effects are not associated with a concomitant change in MLC phosphorylation (Pfitzer et al. 1993; Itoh et al. 1994).
The signalling cascade leading to the GTP-dependent inhibition of MLC phosphatase and the increase in Ca2+ sensitivity of force production has not yet been fully elucidated. The agonist-induced Ca2+ sensitization, which is also observed in receptor-coupled permeabilized preparations, is mimicked by the poorly hydrolysed GTP analogue, GTPγS, and is inhibited by GDPβS (reviewed by Somlyo & Somlyo, 1994), indicating that the activation of at least one GTP-binding protein is involved. In permeabilized smooth muscle, Ca2+ sensitization can be induced by aluminium fluoride indicating the involvement of a heterotrimeric G protein (Kawase & van Breemen, 1992; Fujita, Takeuchi, Nakajima, Nishio & Hata, 1995; Gong et al. 1996). Interestingly, the agonist- and aluminium fluoride-induced Ca2+ sensitization is inhibited by exoenzyme C3 from Clostridium botulinum (Fujita et al. 1995; Itagaki, Komori, Unno, Syuto & Ohashi, 1995; Kokubu, Satoh & Takayanagi, 1995; Gong et al. 1996), which ADP-ribosylates and inactivates GTPases of the Rho subfamily of Ras-related low molecular mass GTPases (Aktories, Mohr & Koch, 1992). Thus, these data suggest that Ca2+ sensitization of force in permeabilized smooth muscle requires the activation of a heterotrimeric G protein as well as of Rho or Rho-like GTPases. The hypothesis that Rho proteins are involved in the agonist-induced Ca2+ sensitization is further supported by the fact that recombinant constitutively active mutants of Rho increase force at constant [Ca2+] in permeabilized smooth muscle (Hirata et al. 1992; Gong et al. 1996) and augment the agonist-induced Ca2+ sensitization (Otto, Steusloff, Just, Aktories & Pfitzer, 1996). In addition, myosin phosphatase may be inhibited by phosphorylation catalysed by Rho-associated kinase (Kimura et al. 1996), which has been shown to also directly phosphorylate MLC in solution (Amano et al. 1996). While these observations suggest the interesting possibility that Rho proteins may be involved in the regulation of pharmacomechanical coupling in smooth muscle, they could also simply reflect the artificial situation introduced by the permeabilization procedure, the use of cultured cells, isolated proteins and/or the use of recombinant, constitutively active Rho proteins. The effects of the mutated proteins may differ from their normal counterparts because they may have higher levels of GTP loading than their normal counterparts, may be active permanently rather than temporarily, and may be abnormally localized permitting them to interact with effectors that they do not normally contact (Vojtek & Cooper, 1995).
The important question of whether, and if so, to what extent Rho proteins participate in the regulation of excitation- contraction coupling of intact smooth muscle can be addressed by using toxin B from C. difficile as a tool, which, unlike C3, is internalized into intact cells (Florin & Thelestam, 1983). Toxin B is a monoglucosyltransferase which specifically catalyses the glucosylation of low molecular mass GTPases of the Rho subfamily, Rho, Rac and Cdc42, in the putative effector domain (Just, Selzer, Wilm, von Eichel-Streiber, Mann & Aktories, 1995). In cultured cells, toxin B disrupts the actin cytoskeleton and causes cell rounding, in much the same way as microinjected C3 (Just et al. 1995), indicating that toxin B acts through inhibition of Rho proteins. We have recently shown that toxin B inhibits the delayed tension response during contractions elicited by the muscarinic agonist carbachol in intact intestinal smooth muscle strips (Otto et al. 1996). There are several possibilities as to how toxin B could cause this inhibition of tension: (i) toxin B could inhibit the signalling cascade leading to an increase in Ca2+ sensitivity of the myofilaments, a process that has been suggested to underly the delayed tension response (Himpens & Somlyo, 1988); (ii) toxin B could lower cytosolic [Ca2+], or (iii) toxin B could lead to depolymerization of actin filaments, an effect that has been suggested to require an increase in cytosolic [Ca2+] (Gilbert, Pothoulakis, LaMont & Yakubovich, 1995). In order to obtain more insight into the mechanism by which toxin B affects smooth muscle contraction, we simultaneously measured cytosolic [Ca2+] and force in the intestinal smooth muscle strips. Moreover, to determine whether the toxin B-induced inhibition of force was mediated through an alteration of the balance between phosphorylation and dephosphorylation reactions in favour of the latter, we also measured MLC phosphorylation.
METHODS
Tissue preparation
Guinea-pigs (300-400 g) were killed by cervical dislocation and exsanguination. The small intestine was rapidly removed and carefully flushed with oxygenated physiological salt solution (PSS) containing (mM): NaCl, 118; KCl, 5; Na2HPO4, 1.2; MgCl2, 1.2; CaCl2, 1.5; Hepes, 24; and glucose, 10. The pH was adjusted to 7.4 at room temperature (20-23°C). Segments from the small intestine of about 1 cm in length were slid onto a glass rod (5 mm in diameter). The longitudinal muscle layer was separated from the circular layer under a dissecting microscope. Small strips (8-10 mm long and about 200-300 μm wide) were mounted on a myograph to measure in the same strip either force and cytosolic [Ca2+] or force and MLC phosphorylation. All experiments were carried out at room temperature.
Fura-2 loading and experimental protocol
For loading with fura-2, the muscle strips were incubated in a cuvette containing 0.5 ml PSS to which 10 μM of the acetoxymethyl ester of fura-2 (fura-2 AM) dissolved in DMSO premixed with Pluronic F127 (final concentrations, 1 and 0.02 %, respectively) was added. Prior to loading, the preparations were incubated at 4°C for 30 min in order to minimize non-cytosolic compartmentalization (Roe, Lemasters & Herman, 1990). Loading was performed in the dark at 22°C for 4-4.5 h with gentle shaking. After loading, the muscle strips were washed in PSS (50 ml) for 5-10 min to remove the extracellular fura-2 AM. The strips were attached to a myograph with aluminium foil clips for measuring isometric tension (DSC-6BE-4-110-force transducer; Eurosensor, London, UK) in a 1 ml cuvette which was mounted on the stage of an inverted Nikon (TMD) microscope. After mounting, the strips were allowed to equilibrate for about 30 min during which period the solution was changed several times. They were then stimulated with carbachol (100 μM) and the fluorescence signal was recorded continuously during the first 100 s, and 5 min after onset of stimulation for another 10 s. The strips were then relaxed in Ca2+-free PSS containing EGTA (2 mM). After 5 min the fluorescence signal was measured again for 10 s. In some of the preparations, the fluorescence signal was now calibrated as described below. The majority of the strips were removed from the myograph and incubated under dark conditions at room temperature in PSS (2 strips per 500 μl) containing 1 % bovine serum albumin and either the appropriate amount of toxin B buffer (toxin B buffer; for composition see ‘Materials’) or toxin B (40 ng ml−1) overnight (11.5-12 h). On the next morning, in the following referred to as day 2, the strips were again mounted on the myograph, allowed to equilibrate for 30 min followed by stimulation with carbachol, and the fluorescence signal was recorded as before, i.e. at day 1. At the end of each experiment, the fura-2 signal was calibrated.
Recording and calibration of the fura-2 signal
UV light of alternating wavelengths (340 and 380 nm) was obtained by placing a rotating wheel with three pairs of the two different interference filters (340 nm: bandwidth, 12 nm; 380 nm: bandwidth, 11 nm; Ditric Optics Inc., Hudson, MA, USA) rotating at a frequency of 16.7 Hz giving one filter change per 10 ms in front of a 100 W mercury lamp (Nikon). The excitation light was passed through liquid light guides (Oriel) and was focused via a focusing beam probe (Oriel) so that the muscle was excited directly through a quartz window of the cuvette. The fura-2 emission at 500 nm using an interference filter (bandwidth, 40 nm) was passed through the objective lens (× 10; numerical aperture, 0.25; Nikon) and collected in a photomultiplier (R 374; Hamamatsu Photonics). The position of the strip was adjusted to the centre of the field by means of the photomultiplier objective. In this way it was possible to obtain the fura-2 signal from about the same position on the next day after incubation with toxin B. The fluorescence and the force signal were recorded with an Analog Devices RTI board and a personal computer. The fluorescence intensities were integrated over time per filter and the values of three measurements at each excitation wavelength were averaged to obtain one ratio value.
The ratio of the fura-2 fluorescence intensities induced by 340 nm (F340) and 380 nm (F380) was calculated after subtraction of the background intensities. The background fluorescence (including the autofluorescence of the strip) was measured at the end of each experiment after ionomycin treatment and determination of Rmin (fluorescence ratio in the absence of Ca2+) and Rmax (fluorescence ratio in the presence of saturating Ca2+) (see below) by incubating the strips in a Hepes-buffered calibration solution (24 mM Hepes, 1.2 mM MgCl2, 1.2 mM Na2HPO4, 140 mM K+ and 2 mM EGTA at pH 8.6) containing 20 mM MnCl2 to quench the fura-2 fluorescence. Under these conditions, the background intensities were not significantly different for the two excitation wavelengths. Moreover, the absolute background values were not significantly different in strips that were incubated overnight from those in which the background was obtained after activation with carbachol at day 1. The background values obtained at day 1 expressed as percentage of the resting values were 21 ± 3 % (F340) and 16 ± 2 % (F380); those obtained at day 2 were 28 ± 3 and 24 ± 3 % (F340), and 25 ± 4 and 18 ± 2 % (F380) in control and toxin-treated strips, respectively. The increase in the relative background intensities is most probably due to the decrease in the fluorescence intensities excited by F340 and F380 suggesting that some of the dye leaked out of the muscle strips (Roe et al. 1990). Despite this decrease in excited fluorescence intensities, the relative maximal changes in the F340 and F380 signals induced by stimulation with carbachol were not significantly different between day 1 and day 2 in the control and the toxin-treated preparations. There was, however, a small increase in the F340/F380 ratio. This is because the decline in the F340 intensity was smaller than that in the F380 intensity (40 % compared with 50 %). On the other hand, when the absolute Ca2+ values were compared in strips in which the Ca2+ signal was calibrated on day 1 with those calibrated on day 2, no signifant difference was observed. Because of this effect of overnight incubation on the ratio of the fura-2 signal, we compared the absolute Ca2+ values rather than the ratios between control and toxin B-treated strips.
Cytosolic Ca2+ concentrations were calculated using an internal calibration procedure (Himpens, Matthijs, Somlyo, Butler & Somlyo, 1988). The minimum value (Rmin) was obtained by incubating the strips in the Hepes-buffered Ca2+-free calibration solution (see above). Five minutes after incubation with this solution, 50 μM ionomycin was added. After 10 min, when changes in the F340/F380 ratio had stabilized, the minimal ratio was determined. To obtain the maximal ratio (Rmax) the strips were incubated with an excess of calcium (20 mM CaCl2). Thereafter MnCl2 was added to record background values. From these values [Ca2+] was calculated using the formula and an apparent dissociation constant of the Ca2+-fura-2 complex of 224 nM given by Grynkiewicz, Poenie & Tsien, 1985 (see also Himpens & Somlyo, 1988). This method of calculating [Ca2+] may not be accurate if fura-2 binds to proteins in the cell which may change its characteristics of fluorescence as well as the Kd value (Konishi, Olson, Hollingworth & Baylor, 1988; Mitsui & Karaki, 1990; Roe et al. 1990). However, the systematic errors in estimating cytosolic [Ca2+], in particular the effect of the overnight incubation, are most probably identical in toxin B-treated and control strips and will therefore not interfere with the determination of whether inhibition of force by toxin B is associated with a change in Ca2+.
Myosin light chain phosphorylation
Longitudinal smooth muscle strips were treated identically to those used for Ca2+ measurements. They were incubated with or without toxin B overnight (11.5-12 h) and mounted horizontally on the myograph to measure isometric force. After a determined period of stimulation, the muscle strips were rapidly frozen (within ≤1 s) with tongs precooled in liquid nitrogen. The muscle strips were processed for two-dimensional gel electrophoresis as described previously (Fischer & Pfitzer, 1989) using a mini-gel system (Biometra, Göttingen, Germany). MLC phosphorylation was assessed by densitometric analysis of the silver-stained gels using the silver-stain kit from Bio-Rad. Light chain phosphorylation is expressed as the percentage of the total MLC (phosphorylated and non-phosphorylated).
Permeabilization with β-escin
In some experiments, the strips were permeabilized with β-escin after completion of the measurements in intact preparations. The permeabilization procedure has been described elsewhere in detail (Otto et al. 1996). Relaxing solution consisted of 20 mM imidazole, 10 mM EGTA, 10 mM magnesium acetate, 7.5 mM ATP, 10 mM creatine phosphate, 5 mM NaN3, 2 mM dithioerythritol, 1 μM leupeptin and 1.5 μM calmodulin. The contracting solution contained in addition 10 mM CaCl2. Alterations of the free Ca2+ concentration were obtained by mixing relaxing and contracting solutions in the appropriate ratio. The pH of the solutions was 7.0 at room temperature. Ionic strength was adjusted to 150 mM with potassium methanesulphonate.
ADP-ribosylation
UDP-glucosylated Rho proteins are no longer substrates for exoenzyme C3 (Just et al. 1994). Therefore, incubation of smooth muscle strips with the glucosyltransferase toxin B should decrease subsequent C3-induced [32P]ADP-ribosylation in homogenates of the strips. After incubation with or without toxin B (40 ng ml−1) overnight, the strips were rapidly frozen in liquid nitrogen and stored at -80°C. In parallel strips, the effect of the toxin on force production was tested. For ADP-ribosylation, the strips (approximately 30 μg protein, determined by the method of Bradford (1976) using IgG as standard) were mechanically homogenized followed by sonication on ice in 50 μl buffer consisting of 50 mM Tris-HCl pH 7.5, 2 mM MgCl2, 1 mM dithiothreitol, 1 μM leupetin, and 0.1 mM phenylmethylsulphonyl fluoride. Lysates (35 μl aliquots) were incubated with 2 μg ml−1 exoenzyme C3 and 6 μM (10 μCi) [32P]NAD (final volume, 45 μl) at 37°C for 30 min. The reaction was stopped by the addition of 100 μl of ice-cold trichloroacetic acid (30 % w/v) for 30 min followed by centrifugation at 14 000 g for 15 min. The pellets were washed twice with acetone and dissolved in sample buffer consisting of 50 mM Tris-HCl pH 6.8, 4 M urea, 1 % (w/v) SDS, 15 % (v/v) glycerol, 10 mM dithioerythritol, and 0.01 % Bromophenol Blue. The proteins were separated by 15 % SDS-PAGE and 32P incorporation was detected by autoradiography. The band corresponding to Rho proteins was cut out from the Coomassie-stained gel and radioactivity was quantified by Cherenkov counting. To obtain an additional measure of protein loading on the gels, the Coomassie-stained gels were evaluated by densitometry scanning and the radioactivity (c.p.m.) of the excised gel slices was divided by the optical density of the actin band.
In a second set of experiments, the ADP-ribosylation reaction was carried out in β-escin-permeabilized smooth muscle strips which had been incubated with or without toxin B prior to permeabilization. Smooth muscle permeabilized with β-escin is permeable for C3, but has the advantage of structural integrity of the cells compared with a homogenate. The smooth muscle strips were permeabilized with β-escin (see above) and incubated in relaxing solution (50 μl) with 2 μg ml−1 C3 and 6 μM (10 μCi) [32P]NAD (final volume, 57 μl) at 37°C for 30 min. The reaction was terminated by transferring the smooth muscle strips into trichloroacetic acid (20 %) on ice for 10 min. The muscle strips were homogenized in sample buffer and the proteins were separated by 12.5 % SDS-PAGE and 32P incorporation was evaluated as above.
Statistics
Values are shown as means ±s.e.m. (n = number of observations). Difference of responsiveness among groups was analysed by ANOVA followed by Student's paired or unpaired t test as appropriate. P values < 0.05 were considered to indicate significant differences.
Materials
Toxin B from C. difficile was purified as described previously (von Eichel-Streiber, Harperath, Bosse & Hadding, 1987), and was stored in aliquots at -80°C in a buffer (toxin B buffer) containing 50 mM Tris-HCl, 500 mM NaCl (pH 7.4) at a concentration of 100 ng μl−1. Individual batches were kept on ice after thawing and could be used for approximately 4 weeks before a decline in the activity was noted. Fura-2 AM and Pluronic F127 were obtained from Molecular Probes, Inc. [32P]NAD was purchased from NEN, and ionomycin from Calbiochem. All other reagents were of the highest grade commercially available.
RESULTS
Effect of toxin B on force and cytosolic [Ca2+]
The first 100 s of typical contractile responses of small strips from the longitudinal intestinal smooth muscle from guinea-pig elicited by carbachol obtained at day 2 (i.e. after overnight incubation at room temperature in toxin B or toxin B buffer, referred to as control), and the corresponding Ca2+ transients are shown in Fig. 1. The contractile response observed at day 1 (original tracing not shown, for summary of results see Fig. 2) was not significantly different from the control response obtained on day 2. Before (day 1) and after incubation with toxin B buffer (day 2), carbachol elicited a biphasic contraction, an initial rapid rise in force to a first maximum (peak 1) followed by a partial relaxation to 54 ± 4.5 % of peak 1 and a delayed rise in force to a second maximum (peak 2), after which force declined to a steady-state value which was close to basal levels (referred to as plateau in Figs 2 and 4). Neither peak 1 nor peak 2 was significantly different at day 2. In contrast, incubation for 11.5-12 h with toxin B (40 ng ml−1) resulted in more than 80 % inhibiton of peak 2 (P < 0.01; Figs 1B and 2A). In some experiments, force was even completely inhibited (see Fig. 1B). Inhibition of peak 1 amounted to about 20 % on average and did not reach the level of significance. However, the inhibitory effect on peak 1 was variable; in a few preparations it amounted to about 50 %. The reason for this variability is not known at present. Steady-state force 5 min after exposure to stimulation was also not significantly inhibited by toxin B (Plateau in Fig. 2A). Heat-denatured toxin B had no effect on force (not shown).
Figure 1. Representative tracings of force and intracellular [Ca2+] of longitudinal muscle layer of the small intestine from guinea-pig.

The strips were incubated in buffer (A) or in 40 ng ml−1 toxin B (B) overnight (11.5 h) at room temperature. Tissues were stimulated with 100 μM carbachol and force and fura-2 fluorescence were monitored simultaneously. Average force of peak 1 was 0.87 ± 0.07 mN in control strips (n = 33), and 0.76 ± 0.07 mN in toxin B-treated strips (n = 33; strips used for phosphorylation determination were included).
Figure 2. Effect of toxin B on force and intracellular [Ca2+] in intestinal smooth muscle strips.

After loading with fura-2, the strips were activated with carbachol (100 μM) and force (A) and [Ca2+] (B) were measured simultaneously (referred to as day 1; n = 12). After incubation in toxin B (n = 6) or in buffer (n = 6) overnight at room temperature, the strips were activated again (referred to as day 2). The plateau value was determined 5 min after exposure to carbachol. PSS, physiological salt solution; EGTA, response in Ca2+-free PSS containing 2 mM EGTA. □, day 1;  , control, day 2;
, control, day 2;  , toxin B, day 2. Columns and bars represent means ±s.e.m.; n.s., not significant; **P < 0.01.
, toxin B, day 2. Columns and bars represent means ±s.e.m.; n.s., not significant; **P < 0.01.
Figure 4. Effect of toxin B on MLC phosphorylation in intact guinea-pig intestinal smooth muscle.

The experimental protocol was as described in Fig. 2. , control, day 2; , toxin B, day 2. The inset shows the force tracing of a control preparation to indicate the times at which the strips were freeze clamped. a, resting phosphorylation before addition of carbachol; b, phosphorylation determined 3 s after exposure to carbachol; c, minimal force level between peak 1 and peak 2; d, peak 2; e, plateau value determined 5 min after exposure to carbachol. Phosphorylation determined at peak 2 (d) in the control strips was significantly higher than the resting value (P < 0.05). Columns and bars represent means ±s.e.m. for 5-11 determinations; ***P < 0.001; **P < 0.01; n.s., not significant.
The fura-2 signal recorded simultaneously with force in the intact preparations (Figs 1 and 2B) revealed that the initial rise in force was always preceded by a rapid increase in intracellular [Ca2+] to a maximum of 621 ± 96 nM reached 2.3 ± 0.3 s after exposure to carbachol (values are given for day 1). While force was still increasing, the Ca2+ signal declined with a half-time of 3.8 ± 0.8 s to a lower sustained level of 279 ± 28 nM, which was higher than the resting level (182 ± 32 nM) before stimulation (Fig. 2B). The delayed contractile response (peak 2) was not associated with a corresponding increase in Ca2+ signal. In nine out of eighteen control preparations, no increase in fluorescence could be detected, whereas in the remaining preparations, the delayed increase in force was preceded by a very small increase in the Ca2+ signal (see Fig. 1A), which was disproportionately smaller than the increase in force. This increase in the apparent Ca2+ sensitivity of force is reflected in a counter-clockwise hysteresis of the force-calcium relationship (Fig. 3). Complete relaxation was induced by switching to a Ca2+-free PSS with 2 mM EGTA in the absence of carbachol. This caused the Ca2+ signal to drop to 101 ± 8 nM.
Figure 3. Dependence of force on intracellular [Ca2+].
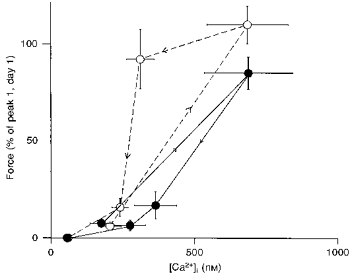
The data are replotted from Fig. 2. ^, control strips evaluated on day 2; •, toxin B-treated strips (day 2). Symbols represent means ±s.e.m. (n = 6).
Neither the shape of the Ca2+ transient nor the intracellular [Ca2+] was significantly affected by incubating the strips in toxin B or buffer (controls) overnight (Fig. 2). In toxin B-treated strips, intracellular [Ca2+] rose to its maximum value (687 ± 156 nM) within 2.8 ± 0.4 s and declined to its plateau value (275 ± 55 nM) with a half-time of 3.6 ± 0.6 s, which was not sigificantly different from the control fibres or from day 1. Thus, the inhibition of force in the presence of toxin B cannot be attributed to a decrease in intracellular [Ca2+]. In some experiments (Fig. 1B), we even observed a complete inhibition of force at suprabasal levels of intracellular Ca2+. In line with this, the force-calcium relationship of toxin B-treated strips did not exhibit a counter-clockwise hysteresis, but rather a small clockwise hysteresis (Fig. 3). The increase in the maximal value of Ca2+ was also observed in the control strips (685 ± 143 nM) and was not significantly different from day 1. In contrast to observations in cultured cells (Gilbert et al. 1995), toxin B did not increase resting [Ca2+] (Fig. 2B).
This result suggests that toxin B inhibits the carbachol-induced increase in the Ca2+ sensitivity of smooth muscle myofilaments. To test this in a more direct manner, we determined whether toxin B inhibits the Ca2+-sensitizing effect of carbachol at a constant submaximal [Ca2+] in β-escin-permeabilized intestinal smooth muscle strips. Under these conditions, toxin B in the presence of its co-substrate UDP-glucose had no effect on force. However, as previously reported, toxin B inhibited the Ca2+-sensitizing effect of carbachol when the strips were incubated with toxin B before permeabilization (Otto et al. 1996). The most likely explanation for the lack of effect when added to β-escin-permeabilized strips is that toxin B, with a molecular mass of 269 kDa, is too large to diffuse into the permeabilized preparations.
Effect of toxin B on MLC phosphorylation
In a separate series of experiments we determined the effect of toxin B on MLC phosphorylation (Fig. 4). The strips were isometrically contracted, the experimental conditions being identical to those for Ca2+ measurements. The strips were freeze clamped before stimulation with carbachol, 3 s after addition of carbachol, which corresponded to the peak of the Ca2+ signal, at the turning point of force between peak 1 and peak 2, at peak 2, and 5 min after addition of carbachol. Resting phosphorylation values determined before stimulation with carbachol and 5 min after exposure to carbachol were not significantly different between control and toxin B-treated strips (Fig. 4). In the control strips, phosphorylation increased to 53.7 ± 1.2 % (n = 5) determined 3 s after exposure to carbachol, and then declined to a significantly lower value at the turning point. Peak 2 was associated with a second increase in phosphorylation (27.7 ± 2.5 %, n = 8), which was significantly higher than the resting value of 18.3 ± 3.3 % (n = 11). Since, in toxin B-treated strips, force at peak 2 was often completely inhibited, the strips were fixed at the same time point during the stimulation with carbachol as were the control strips run in parallel. Phosphorylation determined in this way was significantly lower (16.0 ± 2.9 %, n = 10, P < 0.01) and was similar to the resting value. To our surprise, there was also no significant initial increase in phosphorylation in the toxin B-treated strips (Fig. 4). Since the peak of phosphorylation may have occurred at an earlier or later time point, phosphorylation values were also determined 2 and 4 s after the onset of stimulation. Again, these phosphorylation values were not significantly different from the resting values (data not shown). Thus, our data suggest that toxin B inhibits the increase in MLC phosphorylation without inhibiting the Ca2+ transient (cf. Fig. 2). Maximal force at peak 1 was also not significantly inhibited by toxin B and was 0.87 ± 0.07 mN (n = 33) in control strips and 0.76 ± 0.08 mN (n = 33) in toxin B-treated strips. Phosphorylation determined 5 min after exposure to carbachol (plateau in Fig. 4) was not significantly different between control and toxin B-treated strips (10.6 ± 1.9 and 7.7 ± 2.0 %, respectively).
Effect of toxin B on C3-catalysed ADP-ribosylation of Rho proteins
It has been shown that Rho proteins monoglucosylated by toxin B do not serve as a substrate for exoenzyme C3 (Just et al. 1994). Therefore, C3-catalysed ADP-ribosylation allows us to monitor the toxin B-mediated modification of Rho proteins. Because C3 does not obtain access into intact smooth muscle, the smooth muscle strips were permeabilized with β-escin or homogenized after treatment with toxin B or with buffer. In the control strips as well as in strips which were incubated with heat-denatured toxin B, a significant amount of radioactivity was incorporated into a protein band with a molecular mass of about 22 kDa (Fig. 5). In the toxin B-treated strips, there was still some ADP-ribosylation amounting to 20 % of the control values despite the fact that force (peak 2) was completely inhibited. Since the extent of modification by toxin B or C3 in the intact or semi-intact smooth muscle may depend on structural constraints, we also determined the extent of C3-induced ADP-ribosylation in lysates of toxin B-treated smooth muscle. Interestingly, in this case ADP-ribosylation of toxin B-treated strips amounted to 39 ± 5 % (n = 4) of that in control strips. This result suggests that in the structurally intact smooth muscle strips 20-40 % of the Rho proteins are protected from covalent modification by toxin B or C3.
Figure 5. Effect of treatment with toxin B on exoenzyme C3-induced ADP-ribosylation.

ADP-ribosylation in β-escin-permeabilized strips was determined as described in Methods. A, autoradiogram showing the 22 kDa band: 1, control strip; 2, toxin B-treated strip; 3, strip treated with heat-inactivated toxin B. B, summary of the results of 3 experiments; columns and bars represent means ±s.e.m.
DISCUSSION
Activation of guinea-pig intestinal smooth muscle with the muscarinic agonist carbachol typically elicits a biphasic contraction (Himpens & Somlyo, 1988; Otto et al. 1996). An initial rapid and transient increase in force preceded by a transient rise in intracellular [Ca2+] is followed by a second slower delayed rise in tension. Because this delayed contraction occurred without an appropriate increase in [Ca2+], it was suggested that it was due to an increase in the Ca2+ sensitivity of the myofilaments (Himpens & Somlyo, 1988). This increase in Ca2+ sensitivity is reflected by the counter-clockwise hysteresis of the force-calcium relationship (Fig. 3; see also Himpens, Kitazawa & Somlyo, 1990). The delayed tension response was inhibited by C. difficile toxin B without a detectable change in cytosolic [Ca2+]. This is also reflected by the absence of a counter-clockwise hysteresis of the force-calcium relationship in toxin B-treated strips. If anything, the relationship showed a small clockwise loop indicative of a decrease in Ca2+ sensitivity. The force-calcium relationship is the net result of the activation of sensitizing and desensitizing pathways. A decrease in Ca2+ sensitivity could result from the phosphorylation of MLC kinase by Ca2+-calmodulin-dependent protein kinase II (Word, Tang & Kamm, 1994). This may also account for the low phosphorylation values observed 5 min after exposure to carbachol. Inhibition of the sensitizing pathway with toxin B may therefore unmask the desensitizing pathway reflected in the small clockwise hysteresis.
Toxin B monoglucosylates the small GTPases of the Rho subfamily, Rho, Rac and Cdc42, thereby inactivating them (Just et al. 1995). Incubation of cultured cells with toxin B causes rounding up of these cells, which is associated with disruption of actin filaments (Just et al. 1995). The effect of toxin B on cell morphology is very similar to that of exoenzyme C3 which inactivates Rho proteins by ADP-ribosylation (Just et al. 1995). Microinjection of monoglucosylated Rho proteins into cells causes disassembly of actin stress fibres suggesting that monoglucosylated Rho acts as a dominant inhibitor of endogenous Rho function (Just et al. 1995). The cytopathic effect of toxin B is causally related to the monoglucosyltransferase reaction as indicated by its absence in a mutant, UDP-glucose-deficient cell line (Florin, 1991). These observations suggest that toxin B can be used as a tool to test whether the activation of Rho proteins are involved in smooth muscle contraction. The modification of Rho proteins by toxin B in intact tissues can be monitored by making use of the fact that monoglucosylated Rho proteins are not ADP-ribosylated by C. botulinum exoenzyme C3 (Just et al. 1994). Using this approach, we showed that toxin B-induced inhibition of force was associated with a significant reduction of C3-induced ADP-ribosylation of a protein band with a molecular mass of about 22 kDa, corresponding to that of Rho proteins. In contrast, heat-denatured toxin B neither antagonized the C3-induced ADP-ribosylation nor inhibited force. However, only about 60-80 % of the Rho proteins are accessible to monoglucosylation in intact strips and therefore appear to be required for the sensitization of myofilaments to Ca2+. This is consistent with our recent observation in permeabilized preparations where complete inhibition of carbachol-induced Ca2+ sensitization by C3 was associated with only about 60 % of the Rho proteins being ADP-ribosylated (Otto et al. 1996). Taken together, these results are in support of the idea that the delayed force response of carbachol-induced contraction in intact intestinal smooth muscle is due to an increase in Ca2+ sensitivity mediated by the activation of Rho proteins.
It should be kept in mind that toxin B-induced inhibition of force could be due to mechanisms unrelated to specific intracellular signalling pathways involved in the process of Ca2+ sensitization. The effect of toxin B on the cellular morphology is similar to that of cytochalasins which are known to disrupt the actin cytoskeleton and inhibit tension development in smooth muscle (Adler, Krill, Alberghini & Evans, 1983; Obara & Yabu, 1994). If toxin B-induced inhibition of force was due to a dissociation of actin filaments we would, however, expect that both peak 1 and peak 2 would be inhibited. Moreover, it was proposed that toxin B-induced actin disassembly requires the activation of a calcium influx which occurred within seconds to minutes after addition of toxin B (Gilbert et al. 1995), i.e. on a much faster time scale than the effect on force in our study. We did not measure intracellular [Ca2+] during the initial incubation period. However, we never observed an increase in resting [Ca2+] at the end of the incubation period. We suspect, however, that a rise in intracellular [Ca2+] immediately after addition of toxin B would have resulted in a transient contraction, which we never observed. Therefore, it is unlikely that the inhibitory effect on peak 2 is due to disruption of actin filaments. However, as already reported in our previous study (Otto et al. 1996), the effect on the tension amplitude of peak 1 was variable, i.e. in some preparations we noted an inhibition of force by about 50 %, despite the fact that the Ca2+ transient was never affected by toxin B. The cause of this variability is not known at present. One possibility is that the force-generating capacity is indeed impaired in these preparations. Another possibility is that there are different populations of intestinal smooth muscle cells, some that already exhibit a carbachol-induced sensitization to Ca2+ during the initial phase of the contraction being sensitive to the action of toxin B. Further studies are required to resolve this point.
It has also been reported that toxin B causes a direct membrane depolarization and inhibition of carbachol-induced action potentials in intestinal smooth muscle (Gilbert, Pothoulakis & LaMont, 1989). This was associated with inhibition of the phasic rather than the tonic contractile response and was again of immediate onset. The most obvious difference between our study and that of Gilbert et al. (1989) is that those authors used 100- to 1000-fold higher concentrations of toxin B. At high concentrations, toxin B may have non-specific effects (Shoshan, Florin & Thelestam, 1993). Therefore, we believe that the inhibition of force in our study was not due to inhibition of carbachol-induced action potentials but rather to specific inhibition of the signalling pathway leading to an increase in Ca2+ sensitivity of smooth muscle myofilaments.
This conclusion is also supported by the fact that toxin B inhibited MLC phosphorylation. The agonist-induced increase in Ca2+ sensitivity of smooth muscle contraction is thought to be due to a G protein-mediated inhibition of MLC phosphatase, thereby shifting the balance between phosphorylation and dephosphorylation reactions in favour of the latter leading to an increase in MLC phosphorylation and hence in force at constant [Ca2+] (for review, see Somlyo & Somlyo, 1994). The signalling pathways linking the activation of the membrane-bound receptor to inhibition of myosin phosphatase are not yet fully delineated. An important step appears to be the phosphorylation of the 130 kDa subunit, which decreases the activity of MLC phosphatase in α-toxin-permeabilized smooth muscle (Trinkle-Mulcahy, Ichikawa, Hartshorne, Siegman & Butler, 1995). The enzyme catalysing this reaction could be the Rho-associated protein kinase (Rho-kinase), which has been shown to phosphorylate the myosin-binding subunit of myosin phosphatase in cultured cells thereby decreasing its activity (Kimura et al. 1996). Moreover, Rho-kinase directly phosphorylates the regulatory light chains of myosin in solution (Amano et al. 1996). Both Rho-dependent phosphorylation reactions, therefore, have the potential to increase MLC phosphorylation at constant cytosolic [Ca2+] in smooth muscle. Our results are consistent with this idea. The second peak of the carbachol-induced contraction in the control strips was associated with a small but significant increase in MLC phosphorylation which was completely inhibited by toxin B independent of a change in cytosolic [Ca2+]. We therefore propose that the carbachol-induced delayed contraction (peak 2) is due to an increase in the Ca2+ sensitivity of MLC phosphorylation mediated by Rho or Rho-related proteins.
The surprising result was that toxin B also inhibited the initial increase in MLC phosphorylation, which was only slightly higher than the resting levels before stimulation (see Fig. 4). In other words, in the presence of toxin B, MLC phosphorylation was uncoupled from both the rise in intracellular Ca2+ and tension during the initial phase of the contraction. The degree of MLC phosphorylation is set by the relative activities of the MLC kinase and MLC phosphatase. In theory, the inhibition of MLC phosphorylation could, therefore, be due to either inhibition of MLC kinase or activation of MLC phosphatase. We do not believe that toxin B inhibits MLC kinase activity for the following reason. In strips which were permeabilized with α-toxin after toxin B treatment, we observed an inhibition of force under maximally activating Ca2+ concentrations. Addition of the phosphatase inhibitor okadaic acid (10 μM) increased force about 1.8-fold (n = 4) while in the control preparations okadaic acid had no effect (C. Lucius & G. Pfitzer, unpublished observations). Therefore the most likely explanation is that toxin B inhibits the agonist-induced inhibition of MLC phosphatase. Assuming that, under our experimental conditions, the only action of toxin B is the inhibition of Rho proteins, we propose that the activation of Rho proteins is required for a significant increase in MLC phosphorylation to occur even in the early phase of the carbachol-induced contraction where intracellular [Ca2+] is high.
Since force (peak 1) was not significantly inhibited by toxin B, questions can be raised as to the regulation of force in this preparation. It is tempting to speculate that toxin B unmasks a Ca2+-dependent process which leads to a significant increase in force independent of a rise in MLC phosphorylation. Such a process could be the Ca2+-dependent disinhibition of an inhibitory protein, caldesmon being a putative candidate (for review, see Chalovich, 1992). A more detailed examination of the relationship between force and MLC phosphorylation, however, reveals that there is an apparent dissociation between force and MLC phosphorylation also in the control strips. The initial MLC phosphorylation during the rising phase of the contraction was 54 % while force amounted to about 70 % of maximal force at peak 1. Interestingly, peak 2 amounted to about 80 % of peak 1 but was associated with only 28 % of the MLC phosphorylated. This apparent discrepancy could be explained if the relationship between force and MLC phosphorylation was very steep and non-linear, maximal force being supported by about 30 % phosphorylation as is the case in skinned chicken gizzard fibres (Schmidt, Troschka & Pfitzer, 1995). If this were the case, then the very small increase in MLC phosphorylation in the toxin B-treated strips (see Fig. 4) may be sufficient to account for peak 1. A more detailed time course study of this initial phase of the contraction should reveal whether force and MLC phosphorylation are clearly dissociated in the toxin B-treated strips.
In conclusion, our results are consistent with the idea that the second peak of the carbachol-induced contractions in intact intestinal smooth muscle is due to a transient increase in the Ca2+ sensitivity of MLC phosphorylation mediated by members of the Rho subfamily. The phosphorylation data further suggest that Rho proteins play an important role in setting the balance between MLC phosphorylation and dephosphorylation reactions even at high levels of intracellular Ca2+. Moreover, the activation of Rho proteins appears to occur very early in the course of the contraction. Future studies have to delineate the pathways that link the activation of a membrane-bound, serpentine receptor, in this case a muscarinic receptor, to the activation of the Rho proteins.
Acknowledgments
The excellent technical assistance of A. Richter and the editorial and art work of L. Glahé and D. Metzler are gratefully acknowledged. We also wish to thank L. Moser and H. Ehmke for the assistance with the statistical evaluation of the data. This work was supported by grants from the DFG to G. P., the Swedish Medical Research Council (04x-8268) to A. A., and a scholarship from Humboldt University to C. L.
References
- Adler KB, Krill J, Alberghini TV, Evans JN. Effect of cytochalasin D on smooth muscle contraction. Cell Motility. 1983;3:545–551. doi: 10.1002/cm.970030521. [DOI] [PubMed] [Google Scholar]
- Aktories K, Mohr C, Koch G. Clostridium botulinum C3 ADP-ribosyltransferase. Current Topics in Microbiology and Immunology. 1992;175:115–131. doi: 10.1007/978-3-642-76966-5_6. [DOI] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) Journal of Biological Chemistry. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chalovich JM. Actin mediated regulation of muscle contraction. Pharmacology and Therapeutics. 1992;55:95–148. doi: 10.1016/0163-7258(92)90013-p. [DOI] [PubMed] [Google Scholar]
- Erdödi F, Ito M, Hartshorne DJ. Myosin light chain phosphatase. In: Bárány M, editor. The Biochemistry of Smooth Muscle Contraction. San Diego: Academic Press; 1996. pp. 131–142. [Google Scholar]
- Fischer W, Pfitzer G. Rapid myosin phosphorylation transients in phasic contractions in chicken gizzard smooth muscle. FEBS Letters. 1989;258:59–62. doi: 10.1016/0014-5793(89)81615-1. 10.1016/0014-5793(89)81615-1. [DOI] [PubMed] [Google Scholar]
- Florin I. Isolation of a fibroblast mutant resistant to Clostridium difficile toxin B. Microbial Pathogenesis. 1991;11:337–346. doi: 10.1016/0882-4010(91)90019-7. 10.1016/0882-4010(91)90019-7. [DOI] [PubMed] [Google Scholar]
- Florin I, Thelestam M. Internalization of Clostridium difficile cytotoxin into cultured human lung fibroblasts. Biochimica et Biophysica Acta. 1983;763:383–392. doi: 10.1016/0167-4889(83)90100-3. 10.1016/0167-4889(83)90100-3. [DOI] [PubMed] [Google Scholar]
- Fujita A, Takeuchi T, Nakajima H, Nishio H, Hata F. Involvement of heterotrimeric GTP-binding protein and rho protein, but not protein kinase C, in agonist-induced Ca2+ sensitization of skinned muscle of guinea pig vas deferens. Journal of Pharmacology and Experimental Therapeutics. 1995;274:555–561. [PubMed] [Google Scholar]
- Gilbert RJ, Pothoulakis C, LaMont JT. Effect of purified Clostridium difficile toxins on intestinal smooth muscle. II. Toxin B. American Journal of Physiology. 1989;256:G767–772. doi: 10.1152/ajpgi.1989.256.4.G767. [DOI] [PubMed] [Google Scholar]
- Gilbert RJ, Pothoulakis C, LaMont JT, Yakubovich M. Clostridium difficile toxin B activates calcium influx required for actin disassembly during cytotoxicity. American Journal of Physiology. 1995;268:G487–495. doi: 10.1152/ajpgi.1995.268.3.G487. [DOI] [PubMed] [Google Scholar]
- Gong MC, Iizuka K, Nixon G, Browne JP, Hall A, Eccleston J, Sugai M, Kobayashi S, Somlyo AV, Somlyo AP. Role of guanine nucleotide-binding proteins - Ras-family or trimeric proteins or both - in Ca2+ sensitization of smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1996;93:1340–1345. doi: 10.1073/pnas.93.3.1340. 10.1073/pnas.93.3.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Himpens B, Kitazawa T, Somlyo AP. Agonist-dependent modulation of Ca2+ sensitivity in rabbit pulmonary artery smooth muscle. Pflügers Archiv. 1990;417:21–28. doi: 10.1007/BF00370764. [DOI] [PubMed] [Google Scholar]
- Himpens B, Matthijs G, Somlyo AV, Butler TM, Somlyo AP. Cytoplasmic free calcium, myosin light chain phosphorylation, and force in phasic and tonic smooth muscle. Journal of General Physiology. 1988;92:713–729. doi: 10.1085/jgp.92.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himpens B, Somlyo AP. Free calcium and force transients during depolarization and pharmacomechanical coupling in guinea-pig smooth muscle. Journal of Physiology. 1988;395:507–530. doi: 10.1113/jphysiol.1988.sp016932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata K, Kikuchi A, Sasaki T, Kuroda S, Kaibuchi K, Matsuura Y, Seki H, Saida K, Takai Y. Involvement of rho p21 in the GTP-enhanced calcium ion sensitivity of smooth muscle contraction. Journal of Biological Chemistry. 1992;267:8719–8722. [PubMed] [Google Scholar]
- Itagaki M, Komori S, Unno T, Syuto B, Ohashi H. Possible involvement of a small G-protein sensitive to exoenzyme C3 of Clostridium botulinum in the regulation of myofilament Ca2+ sensitivity in β-escin skinned smooth muscle of guinea pig ileum. Japanese Journal of Pharmacology. 1995;67:1–7. doi: 10.1254/jjp.67.1. [DOI] [PubMed] [Google Scholar]
- Itoh T, Suzuki S, Suzuki A, Nakamura F, Naka M, Tanaka T. Effects of exogenously applied calponin on Ca2+-regulated force in skinned smooth muscle of the rabbit mesenteric artery. Pflügers Archiv. 1994;427:301–308. doi: 10.1007/BF00374538. [DOI] [PubMed] [Google Scholar]
- Just I, Fritz G, Aktories K, Giry M, Popoff MR, Boquet P, Hegenbarth S, von Eichel-Streiber C. Clostridium difficile toxin B acts on the GTP-binding protein Rho. Journal of Biological Chemistry. 1994;269:10706–10712. [PubMed] [Google Scholar]
- Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375:500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- Katsuyama H, Wang CL, Morgan KG. Regulation of vascular smooth muscle tone by caldesmon. Journal of Biological Chemistry. 1992;267:14555–14558. [PubMed] [Google Scholar]
- Kawase T, van Breemen C. Aluminium fluoride induces a reversible Ca2+ sensitization in alpha-toxin-permeabilized vascular smooth muscle. European Journal of Pharmacology. 1992;214:39–44. doi: 10.1016/0014-2999(92)90093-j. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Masuo M, Somlyo AP. G-protein-mediated inhibition of myosin light-chain phosphatase in vascular smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1991;88:9307–9310. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubu N, Satoh M, Takayanagi I. Involvement of botulinum C3-sensitive GTP-binding proteins in α1-adrenoceptor subtypes mediating Ca2+-sensitization. European Journal of Pharmacology. 1995;290:19–27. doi: 10.1016/0922-4106(95)90012-8. [DOI] [PubMed] [Google Scholar]
- Konishi M, Olson A, Hollingworth S, Baylor SM. Myoplasmic binding of fura-2 investigated by steady state fluorescence and absorbance measurements. Biophysical Journal. 1988;54:1089–1104. doi: 10.1016/S0006-3495(88)83045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Nomura M, Kamm KE, Mumby MC, Stull JT. GTP gamma S-dependent regulation of smooth muscle contractile elements. American Journal of Physiology. 1992;262:C405–410. doi: 10.1152/ajpcell.1992.262.2.C405. [DOI] [PubMed] [Google Scholar]
- Mitsui M, Karaki H. Dual effects of carbachol on cytosolic Ca2+ and contraction in intestinal smooth muscle. American Journal of Physiology. 1990;258:C787–793. doi: 10.1152/ajpcell.1990.258.5.C787. [DOI] [PubMed] [Google Scholar]
- Obara K, Yabu H. Effect of cytochalasin B on intestinal smooth muscle cells. European Journal of Pharmacology. 1994;255:139–147. doi: 10.1016/0014-2999(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Otto B, Steusloff A, Just I, Aktories K, Pfitzer G. Role of Rho proteins in carbachol-induced contractions in intact and permeabilized guinea-pig intestinal smooth muscle. Journal of Physiology. 1996;496:317–329. doi: 10.1113/jphysiol.1996.sp021687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfitzer G, Zeugner C, Troschka M, Chalovich JM. Caldesmon and a 20-kDa actin-binding fragment of caldesmon inhibit tension development in skinned gizzard muscle fiber bundles. Proceedings of the National Academy of Sciences of the USA. 1993;90:5904–5908. doi: 10.1073/pnas.90.13.5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe MW, Lemasters JJ, Herman B. Assessment of fura-2 for measurements of cytosolic free calcium. Cell Calcium. 1990;11:63–73. doi: 10.1016/0143-4160(90)90060-8. [DOI] [PubMed] [Google Scholar]
- Schmidt U, Troschka M, Pfitzer G. The variable coupling between force and myosin light chain phosphorylation in triton skinned chicken gizzard fibre bundles: role of myosin light chain phosphatase. Pflügers Archiv. 1995;429:716–721. doi: 10.1007/BF00373992. [DOI] [PubMed] [Google Scholar]
- Shoshan MC, Florin I, Thelestam M. Activation of cellular phospholipase A2 by Clostridium difficile toxin B. Journal of Cellular Biochemistry. 1993;52:116–124. doi: 10.1002/jcb.240520115. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Stull JT, Krueger JK, Kamm KE, Gao Z-H, Zhi G, Padre R. Myosin light chain kinase. In: Bárány M, editor. The Biochemistry of Smooth Muscle Contraction. San Diego: Academic Press; 1996. pp. 119–130. [Google Scholar]
- Takai A, Bialojan C, Troschka M, Rüegg JC. Smooth muscle myosin phosphatase inhibition and force enhancement by black sponge toxin. FEBS Letters. 1987;217:81–84. doi: 10.1016/0014-5793(87)81247-4. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Ichikawa K, Hartshorne DJ, Siegman MJ, Butler TM. Thiophosphorylation of the 130-kDa subunit is associated with a decreased activity of myosin light chain phosphatase in α-toxin-permeabilized smooth muscle. Journal of Biological Chemistry. 1995;270:18191–18194. doi: 10.1074/jbc.270.31.18191. [DOI] [PubMed] [Google Scholar]
- Vojtek AB, Cooper JA. Rho family members: activators of MAP kinase cascades. Cell. 1995;82:527–529. doi: 10.1016/0092-8674(95)90023-3. [DOI] [PubMed] [Google Scholar]
- von Eichel-Streiber C, Harperath U, Bosse D, Hadding U. Purification of two high molecular weight toxins of Clostridium difficile which are antigenically related. Microbial Pathogenesis. 1987;2:307–318. doi: 10.1016/0882-4010(87)90073-8. [DOI] [PubMed] [Google Scholar]
- Word RA, Tang D-C, Kamm KK. Activation properties of myosin light chain kinase during contraction/relaxation cycles of tonic and phasic smooth muscles. Journal of Biological Chemistry. 1994;269:21596–21602. [PubMed] [Google Scholar]