

Abstract
The effects of human insulin and elevated D-glucose on L-arginine transport and synthesis of nitric oxide (NO) and prostacyclin (PGI2) have been investigated in human umbilical vein endothelial cells isolated from gestational diabetic pregnancies.
The increase in the Vmax for L-arginine transport (9.0 ± 1.1 pmol (μg protein)−1 min−1) in diabetic endothelial cells cultured in 5 mm D-glucose was unaffected following 24 h exposure to 25 mm D-glucose.
Gestational diabetes-induced increases in basal intracellular cGMP and L-citrulline levels (inhibitable by L-NAME) and [Ca2+]i were unaffected by elevated D-glucose. In contrast, PGI2 release was inhibited in diabetic cells exposed to either 5 or 25 mm D-glucose.
Elevated D-glucose attenuated histamine (10 μM, 5 min)-stimulated accumulation of cGMP and L-citrulline in endothelial cells isolated from gestational diabetic pregnancies.
The membrane hyperpolarization (-79 ± 0.9 mV) sustained in diabetic endothelial cells in culture was insensitive to elevated D-glucose.
Elevated D-glucose abolished the stimulatory effect of human insulin (1 nM, 8 h) on L-[3H]leucine incorporation in diabetic endothelial cells cultured in 5 mm D-glucose.
Human insulin reduced the elevated rates of L-arginine transport and cGMP accumulation in diabetic cells cultured in 5 mm D-glucose but failed to reduce increased rates of transport or NO production in cells exposed to 25 mm D-glucose or cycloheximide.
Our findings demonstrate that hyperglycaemia impairs the actions of human insulin on umbilical vein endothelial cells isolated from gestational diabetic pregnancies. Changes in insulin sensitivity and/or its signalling cascade may be affected by hyperglycaemia associated with gestational diabetes, resulting in insulin resistance in endothelial cells derived from the fetal vasculature.
Insulin resistance is a common feature of non-insulin-dependent diabetes mellitus, and insulin-mediated vasodilatation is impaired in patients with insulin resistance caused by obesity or diabetes mellitus (see review, Baron, 1996). The vasodilator actions of insulin in vivo have been attributed to endothelium-derived nitric oxide (NO), since inhibitors of NO synthase abolish insulin-induced increases in leg blood flow in man (Steinberg, Brechtel, Johnson, Fineberg & Baron, 1994; Scherrer, Randin, Vollenweider, Vollenweider & Nicod, 1994) and attenuate insulin-induced potentiation of acetylcholine-mediated increases in forearm blood flow (Taddei, Virdis, Mattei, Natali, Ferrannini & Salvetti, 1995; van Veen & Chang, 1997).
We have recently reported the first cellular evidence that human insulin activates transport of L-arginine (the substrate for NO synthase) and NO release in human umbilical vein endothelial cells and demonstrated that hyperglycaemia reverses the stimulatory action of insulin on the L-arginine-NO signalling pathway (Sobrevia, Nadal, Yudilevich & Mann, 1996). However, the cellular mechanisms underlying insulin-mediated stimulation of endothelium-derived NO synthesis are not well understood (Sobrevia & Mann, 1997). Our previous studies also revealed that gestational diabetes induced phenotypic changes in umbilical vein endothelial cells, which result in a membrane hyperpolarization, activation of L-arginine transport (system y+/hCAT-1) and elevation of basal NO synthesis (Sobrevia, Cesare, Yudilevich & Mann, 1995a). Although these changes in fetal endothelial cell function were maintained in cells cultured for three to five passages in 5 mm D-glucose, this concentration of D-glucose may not necessarily reflect the situation in vivo. To gain further insight into the potential mechanisms linking insulin signal transduction and NO synthesis in gestational diabetes, we have investigated the effects of insulin and elevated D-glucose on L-arginine transport and synthesis of NO and prostacyclin (PGI2) in human umbilical vein endothelial cells isolated from gestational diabetic pregnancies controlled by diet. Basal and agonist-induced release of endothelium-derived NO and PGI2 were correlated with intracellular [Ca2+]i levels and whole-cell patch clamp measurements of membrane potential. Insulin downregulated the elevated rates of L-arginine transport and NO synthesis in gestational diabetic cells cultured in normal D-glucose, but failed to inhibit elevated rates of transport or NO synthesis in diabetic cells exposed to elevated D-glucose. Our findings suggest that hyperglycaemia induces insulin insensitivity in endothelial cells isolated from gestational diabetic pregnancies. An abstract of this work has been published (Sobrevia, Yudilevich & Mann, 1995b).
METHODS
Endothelial cell culture
Umbilical cords were obtained from normal and gestational diabetic pregnancies. Diabetic patients were normotensive, controlled by diet and had a HbA1c plasma level of 8.1 ± 0.3 % (mean ±s.e.m., n = 8). Newborns from gestational diabetic patients had no symptoms of asphyxia or malformation and after 37.5 ± 2 weeks of gestation the mean umbilical vein blood glucose concentration was 2.4 ± 0.3 mm (see review, Dornhorst & Beard, 1993).
Umbilical vein endothelial cells were isolated by collagenase (0.5 mg ml−1) digestion and cultured initially in medium 199 (M199) containing 5 mm D-glucose, 10 % fetal calf serum, 10 % newborn calf serum, 5 mm L-glutamine, 100 i.u. ml−1 penicillin-streptomycin and 0.03 mg ml−1 gentamicin at 37°C in a 5 % CO2 atmosphere (Sobrevia et al. 1995a). Confluent third passage diabetic endothelial cell monolayers were then cultured for 0-96 h in M199 containing 20 % serum and either 5 or 25 mm D-glucose or 5 mm D-glucose + 20 mm D-mannitol (osmotic control). Cell protein, DNA, volume and number were determined in confluent monolayers, whilst L-[3H]leucine and [3H]thymidine incorporation was measured over 24 h in actively replicating cells in the log phase of growth (Sobrevia et al. 1995a).
To investigate modulation of endothelial cell function by insulin, cells were cultured in M199 containing 20 % serum and either 5 or 25 mm D-glucose and increasing concentrations of human insulin (0.1-10 nm), added during the last 8 h of a 24 h incubation period. In some experiments, cycloheximide (17 μM, 24 h) was added to the culture medium to establish whether the effects of elevated D-glucose and/or insulin were dependent on de novo protein synthesis.
Measurements of tetra[3H]phenylphosphonium (TPP+) influx, resting membrane potential and intracellular calcium
Diabetic endothelial cells were cultured for 24 h in M199 containing 20 % serum and either 5 or 25 mm D-glucose in the absence or presence of 1 nM insulin (1 nM, 8 h) (Sobrevia et al. 1996). Influx of the membrane potential sensitive probe [3H]TPP+ (11 nM, 0.12 μCi ml−1) was then measured over various time intervals (0-120 s) in confluent cell monolayers incubated in Krebs solution (37°C) containing either 5 or 25 mm D-glucose in the absence or presence of 1 nM insulin. As changes in membrane potential are known to affect L-arginine transport (see Bogle, Baydoun, Pearson & Mann, 1996), resting membrane potential was measured in subconfluent third passage endothelial cells using the whole-cell patch clamp technique, as described previously (Sobrevia et al. 1995a, 1996). Recordings were made using an EPC-7 amplifier (List Medical, Darmstadt, Germany), and patch pipettes were filled with the following solution (mm): KCl, 135; CaCl2, 0.2; MgCl2, 1.6; Hepes, 10; EGTA, 2; K-ATP, 2.5; Li-GTP, 0.2 (pH adjusted to 7.3 with KOH) and had a resistance of 4-6 MΩ. The membrane potential was measured for at least 1 min, and only stable recordings were included in the analysis.
Diabetic endothelial cells were cultured on glass coverslips in M199 containing 20 % serum and either 5 or 25 mm D-glucose. Endothelial cells pre-exposed to D-glucose for 24 h were then loaded for 15 min at 37°C with 5 μM of the acetoxymethyl derivative of fluo-3 AM (Molecular Probes) in the continued presence of either 5 or 25 mm D-glucose. Intracellular Ca2+ was imaged using a BioRad MRC-600 confocal microscope (Nikon × 60 oil-immersion lens, numerical aperture of 1.4) (Sobrevia et al. 1996).
Endothelial cell transport of amino acids and 2-deoxyglucose
Confluent diabetic endothelial cell monolayers (∼104 cells in a 96-well plate), pre-exposed for 24 h to either 5 or 25 mm D-glucose in the absence or presence of insulin (1 nM, 8 h), were rinsed with warmed (37°C) Krebs solution, composition (mm): NaCl, 131; KCl, 5.6; NaHCO3, 25; NaH2PO4, 1; CaCl2, 2.5; MgCl2, 1; D-glucose, 5; Hepes, 20 (pH 7.4), and then preincubated for 60 min at 37°C in Krebs solution containing 100 μM L-arginine and 5 or 25 mm D-glucose or 5 mm D-glucose + 20 mm D-mannitol. Unidirectional transport of 100 μM radiolabelled L-arginine, L-lysine, L-serine, L-citrulline, L-leucine, L-cystine and 2-deoxy-D-glucose was measured over 30 s or 1 min in cells incubated in Krebs solution containing Na+ and specified concentrations of D-glucose and/or D-mannitol.
Kinetics of L-arginine transport were measured under similar conditions in endothelial cells incubated with increasing concentrations of L-arginine (0.015-1 mm). Kinetic data were analysed using the computer programmes Enzfitter and Ultra Fit (Biosoft, Cambridge, UK) and fitted best by a Michaelis-Menten equation plus a non-saturable component. In all experiments tracer uptake was terminated by removal of the uptake medium 1 s before rinsing monolayers three times with 200 μl ice-cold stop solution (Krebs buffer solution containing 10 mm unlabelled substrate). Radioactivity in formic acid cell digests was determined by liquid scintillation counting, and uptake was corrected for D-[14C or 3H]mannitol disintegrations per minute (d.p.m.) in the extracellular space.
Effect of elevated D-glucose on selectivity of L-arginine transport
Inhibition of L-arginine transport (1 min) by putative competitor amino acids and L-arginine analogues was tested by incubating third passage diabetic endothelial cell monolayers with Krebs solution containing 5 or 25 mm D-glucose + 100 μM L-[3H]arginine in the absence or presence of 1 mm L-lysine, L-serine, L-cystine, L-leucine, L-homoarginine, 2-methylaminoisobutyric acid (MeAIB) and the NO synthase inhibitors NG-monomethyl-L-arginine (L-NMMA), NG-nitro-L-arginine (L-NNA) and NG-nitro-L-arginine methyl ester (L-NAME). L-NNA and L-NAME have previously been reported not to interact with the endothelial cell L-arginine transporter (see Sobrevia et al. 1995a).
cGMP accumulation and prostacyclin release
Confluent third passage diabetic endothelial cells were cultured (24 h) in 24-well plates in M199 containing 20 % serum and either 5 or 25 mm D-glucose. Cells were then washed and preincubated for 15 min with Krebs buffer (37°C) containing either 5 or 25 mm D-glucose, 100 μM L-arginine and the phosphodiesterase inhibitor 3-isobutyl-methylxanthine (IBMX, 0.5 mm). Preincubation media were removed and 500 μl Krebs solution containing either 5 or 25 mm D-glucose and 0.5 mm IBMX added to the wells for a further 5 min (37°C). In some experiments cells were challenged acutely with 10 μM histamine (5 min) or 1 nM human insulin (5 min), although cells were pretreated with insulin for 8 h. The supernatant was removed for analysis of prostacyclin (PGI2) release by radioimmunoassay of its stable metabolite 6-keto-PGF1α. Cells were then placed on ice and incubated with 0.1 n HCl (1 ml per well, 60 min) and 800 μl HCl cell extract and stored at -20°C for radioimmunoassay of cGMP accumulation (index of NO production) following acetylation (see Sobrevia et al. 1995a). cGMP accumulation was also determined in cells preincubated for 15 min with Krebs solution containing 100 μM L-NAME.
Conversion of L-[3H]arginine into L-[3H]citrulline
Confluent diabetic endothelial cell monolayers in 6-well plates were incubated with 100 μM L-[3H]arginine (4 μCi ml−1, 30 min, 37°C) in the absence or presence of 10 μM histamine (last 5 min of a 30 min incubation period) and normal or Ca2+-free Krebs solution. Aliquots of 100 g of the cation ion-exchange resin Dowex50W (50X8-200) in its protonated form were converted into the sodium ion form by incubation with 200 ml of 1 n NaOH. After calibration of the Dowex column, 200 μl of endothelial cells digested in 95 % formic acid (∼5 × 105 cells) were passed through the column and eluates of H2O and NaOH were collected (Contreras, Fuentes, Mann & Sobrevia, 1997). The amount of L-[3H]citrulline produced after 30 min incubation with L-[3H]arginine was determined in the H2O eluate and expressed as d.p.m. (106 cells)−1 (30 min)−1.
Paired samples were analysed by thin layer chromatography to determine radioactive L-arginine and L-citrulline levels in endothelial cells. As described previously (Contreras et al. 1997), aliquots of formic acid-digested endothelial cells (200 μl) were desiccated by evaporation (37°C) and resuspended in 100 μl water. Aliquots of 10 μl samples and standards (L-arginine, L-citrulline, L-ornithine and ATP) were chromatographed on silica-gel-coated plates (Whatman, 150A), using the following running solvent: chloroform, methanol, ammonium hydroxide and water in the proportions 5 : 45 : 20 : 10, respectively. Amino acids were localized using ninhydrin (0.2 % ninhydrin in 1:1 methanol : acetone) : 12.5 M pyridine (200 : 1, v/v). After drying, the plates were scanned using a Berthold TLC scanner LB2760, and relative band speed (Rf) values for L-[3H]arginine, L-[3H]ornithine and L-[3H]citrulline were 0.31, 0.60 and 0.79, respectively.
High-performance liquid chromatography
Intracellular contents of L-citrulline and L-arginine in endothelial cell extracts were analysed by high-performance liquid chromatography (HPLC) (see Contreras et al. 1997). Diabetic endothelial cells in 24-well plates were extracted with 0.5 ml methanol (96 %) for 30 min under constant shaking. Samples were exposed to three cycles of freeze-thawing, centrifuged (1500 r.p.m., 2 min), the supernatant evaporated to dryness under a stream of nitrogen gas and resuspended in 100 μl methanol. Aliquots of 20 μl samples or standards were injected onto a Hypersil Ultratechsphere ODS-5 μ reversed-phase HPLC column (Jones Chromatography, Mid-Glamorgan, UK) in a Kontron 400 Series gradient HPLC system (Kontron Instruments Ltd, Watford, UK). Amino acid concentrations were calculated from the peak areas by reference to the area of the internal standard homoserine.
Materials
Newborn and fetal calf serum and all other reagents were purchased from Sigma. Collagenase Type II from Clostridium histolyticum was from Boehringer Mannheim and Bradford protein reagent was from BioRad Laboratories. L-[2,3,-3H]Arginine (36.1 Ci mmol−1), L-[4,5-3H(N)]lysine (87.5 Ci mmol−1), L-[4,5-3H(N)]leucine (70 Ci mmol−1), L-[14C]cystine (322.2 mCi mmol−1), [methyl-3H]thymidine (5 Ci mmol−1), L-[carbamoyl-14C]citrulline (54.3 mCi mmol−1), D-[1-14C or 3H]mannitol (49.3 mCi mmol−1 or 56 Ci mmol−1) and tetraphenylphosphonium bromide[phenyl-3H] (37 Ci mmol−1) were obtained from NEN and 2-deoxy-D- [2,6-3H]glucose (42 Ci mmol−1) and L-[3-3H]-serine (32 Ci mmol−1) were from Amersham International. 3′,5′-cyclic GMP-TME [tyrosine-125I] were from ICN (Thame, Oxon, UK) and [125I]6-keto-PGF1α from Matachem Diagnostics (Northampton, UK).
Statistics
Statistical analyses were carried out on raw data using the Peritz F multiple means comparison test. Student's t test was applied for unpaired raw data and P < 0.05 was considered statistically significant. Values are expressed as means ±s.e.m., where n indicates the number of different endothelial cell cultures with 3-5 replicate measurements per experiment.
RESULTS
Effects of elevated D-glucose on characteristics of gestational diabetic endothelial cells
We have previously reported that incorporation of L-[3H]leucine and [3H]thymidine is markedly inhibited in actively replicating endothelial cells isolated from gestational diabetic pregnancies (Sobrevia et al. 1995a). In the present study, exposure of diabetic endothelial cells to elevated D-glucose (25 mm, 24 h) had no effect on the depressed rates of L-leucine or thymidine incorporation induced by gestational diabetes (Table 1). Human insulin (1 nM, 8 h) restored the decrease in L-[3H]leucine incorporation in diabetic endothelial cells cultured in 5 mm D-glucose to rates measured in non-diabetic cells (Table 1). When diabetic cells were cultured in 25 mm D-glucose (24 h), insulin failed to stimulate L-[3H]leucine incorporation. The diabetes-induced inhibition of [3H]thymidine was insensitive to insulin, and total protein and DNA content, cell number and cell volume in confluent diabetic endothelial cell monolayers were unaffected by elevated D-glucose or insulin (data not shown).
Table 1.
Effects of insulin and elevated D-glucose on incorporation of leucine and thymidine in human umbilical vein endothelial cells isolated from gestational diabetic pregnancies
| Condition | l-[3H]Leucine incorporation (d.p.m. (104 cells)−1) | [3H]Thymidine incorporation (d.p.m. (104 cells)−1) |
|---|---|---|
| Non-diabetic cells | ||
| 5 mm D-glucose † | 17645 ± 1978 * | 10900 ± 1390 * |
| Diabetic cells | ||
| 5 mm D-glucose | 5478 ± 601 | 2599 ± 644 |
| 5 mm D-glucose + insulin | 14793 ± 1670 * | 3610 ± 1266 |
| 25 mm D-glucose | 6782 ± 1700 | 1802 ± 278 |
| 25 mm D-glucose + insulin | 6865 ± 2057 | 2188 ± 1022 |
Human umbilical vein endothelial cells isolated from gestational diabetic pregnancies were cultured initially in M199 containing 20 % serum and either 5 or 25 mm D-glucose in the absence or presence of 1 nM human insulin (added during the last 8 h of a 24 h incubation period). Pretreated confluent third passage endothelial cell monolayers were then washed and incubated in Krebs solution, and contents of protein and DNA, cell number and cell volume measured. Incorporation of L-[3H]leucine and [3H]thymidine was measured over 24 h in actively replicating cells. Values denote the means ± s.e.m. of 3-4 different cell cultures.
P < 0·05 relative to values in diabetic endothelial cells in 5 or 25 mm D-glucose or 25 mm D-glucose + insulin. Values for [3H]thymidine incorporation in diabetic cells are all significantly (P < 0·02) lower than non-diabetic cells cultured in 5 mm D-glucose.
Paired data in endothelial cells obtained from non-diabetic pregnancies and cultured in 5 mm D-glucose.
Effects of elevated D-glucose on L-arginine transport in diabetic endothelial cells
Figure 1 illustrates initial rates of L-arginine transport in diabetic endothelial cells pre-exposed for 0-48 h to M199 containing either 5 or 25 mm D-glucose or 5 mm D-glucose + 20 mm D-mannitol (data not shown) and then in M199 containing 5 mm D-glucose (48-96 h). When L-arginine transport (100 μM) was subsequently measured in cells incubated in Krebs solution, influx was ∼1.8-fold higher in diabetic endothelial cells than values in non-diabetic cells exposed to 5 mm D-glucose (Table 2). Moreover, pre-exposure of diabetic cells to 25 mm D-glucose for 0-48 h or re-exposure of these cells to 5 mm D-glucose had no effect on the elevated rates of L-arginine transport induced by gestational diabetes.
Figure 1. Effects of elevated D-glucose on L-arginine transport in human umbilical vein endothelial cells isolated from gestational diabetic pregnancies.
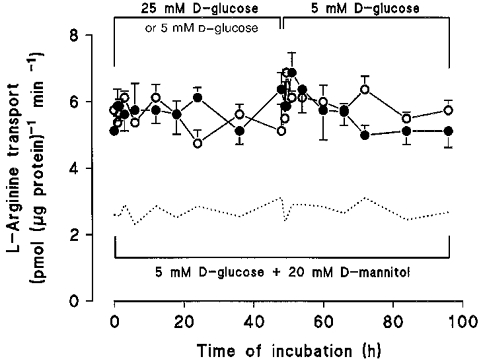
L-Arginine transport (100 μM, 1 min, Krebs buffer) was measured in third passage diabetic endothelial cells after incubation of cells for 0-48 h in M199 containing 20 % serum and either 5 mm (^), 25 mm D-glucose (•) or 5 mm D-glucose + 20 mm D-mannitol (data not shown). After 48 h all culture media were replaced with M199 containing 20 % serum and 5 mm D-glucose (48-96 h) and L-arginine transport was measured in Krebs solution containing 5 mm D-glucose. The dotted line represents L-arginine transport in non-diabetic endothelial cells cultured in 5 mm D-glucose + 20 mm D-mannitol (replotted from Fig. 1 in Sobrevia et al. 1996). Values denote the means ±s.e.m. of experiments in 7 different cell cultures.
Table 2.
Effects of insulin and elevated D-glucose on L-arginine transport, membrane potential and intracellular Ca2+ in human umbilical vein endothelial cells isolated from gestational diabetic pregnancies
| Condition | L-Arginine transport (pmol (μg protein)−1 min−1) | TPP+ influx (pmol (106 cells)−1 min−1) | Membrane potential (mV) | [Ca2+]i (nM) |
|---|---|---|---|---|
| Non-diabetic cells | ||||
| 5 mm D-glucose † | 3·3 ± 0·5 * | 0·49 ± 0·07 * | -67·2 ± 0·4 * | 46 ± 12 * |
| Diabetic cells | ||||
| 5 mm D-glucose | 5·1 ± 0·2 | 0·80 ± 0·1 | -79·4 ± 0·9 | 165 ± 25 |
| 5 mm D-glucose + insulin | 3·7 ± 0·4 * | 0·42 ± 0·04 * | n.d. | n.d. |
| 25 mm D-glucose | 6·9 ± 0·6 | 0·78 ± 0·07 | -78·5 ± 0·5 | 146 ± 22 |
| 25 mm D-glucose + insulin | 5·9 ± 0·3 | 0·69 ± 0·02 | n.d. | n.d. |
Human umbilical vein endothelial cells isolated from gestational diabetic pregnancies were cultured initially in M199 containing 20 % serum and either 5 or 25 mm D-glucose in the absence or presence of 1 nM human insulin (added during the last 8 h of a 24 h incubation period). Pretreated confluent third passage endothelial cell monolayers were then washed and incubated in Krebs solution, and contents of protein and DNA, cell number and cell volume measured. Incorporation of L-[3H]leucine and [3H]thymidine was measured over 24 h in actively replicating cells. Values denote the means ± s.e.m. of 3-4 different cell cultures.
P < 0·05 relative to values in diabetic endothelial cells in 5 or 25 mm D-glucose or 25 mm D-glucose + insulin. Values for [3H]thymidine incorporation in diabetic cells are all significantly (P < 0·02) lower than non-diabetic cells cultured in 5 mm D-glucose.
Paired data in endothelial cells obtained from non-diabetic pregnancies and cultured in 5 mm D-glucose. n.d., not determined.
In kinetic experiments, elevated D-glucose (25 mm) had no effect on Km or KD values for L-arginine transport in diabetic endothelial cells. The Vmax values for saturable L-arginine transport were similar in diabetic endothelial cells cultured in either 5 or 25 mm D-glucose, with transport rates ∼2-fold higher in diabetic cells compared with non-diabetic cells cultured in 5 mm D-glucose (Table 3). Similarly, the stimulation of L-lysine transport induced in cells from gestational diabetic pregnancies was unaffected by elevated D-glucose (Table 4). Transport rates for L-citrulline, L-leucine, L-serine, L-cystine and 2-deoxy-D-glucose were insensitive to gestational diabetes or elevated D-glucose (Table 4).
Table 3.
Effects of insulin and elevated D-glucose on kinetics of L-arginine transport in umbilical vein endothelial cells isolated from gestational diabetic pregnancies
| Condition | Km (μM) | Vmax (pmol (μg protein)−1 min−1) | KD (pmol (μg protein)−1 min−1) | Vmax/Km |
|---|---|---|---|---|
| 5 mm D-glucose | 82 ± 34 | 9·0 ± 1·1 | 0·026 ± 0·003 | 0·11 ± 0·03 |
| 5 mm D-glucose + insulin | 74 ± 48 | 4·0 ± 0·7 * | 0·019 ± 0·002 | 0·05 ± 0·02 |
| 25 mm D-glucose | 86 ± 17 | 9·5 ± 0·5 | 0·026 ± 0·002 | 0·11 ± 0·01 |
| 25 mm D-glucose + insulin | 61 ± 41 | 8·3 ± 1·1 | 0·035 ± 0·005 | 0·14 ± 0·06 |
Diabetic endothelial cells were cultured in M199 containing 20 % serum and either 5 or 25 mm D-glucose in the absence or presence of 1 nM insulin added during the last 8 h of a 24 h incubation period. Cells were then incubated in Krebs solution and the kinetics of saturable L-arginine transport (1 min, 0·015-1 mm) determined in the presence of either 5 or 25 mm D-glucose and absence or presence of 1 nM insulin. Values denote the means ± s.e.m. of n = 4 different cell cultures
P < 0·02 relative to values in 5 or 25 mm D-glucose or 25 mm D-glucose + 1 nM insulin. All other Vmax values for L-arginine transport were significantly (P < 0·03) elevated compared with non-diabetic cells cultured in 5 mm D-glucose (Vmax = 4·9 ± 0·7, Km = 83 ± 11, KD= 0·018 ± 0·002, n = 4 paired experiments).
Table 4.
Effects of insulin and elevated D-glucose on amino acid and 2-deoxy-D-glucose transport in human umbilical vein endothelial cells isolated from gestational diabetic pregnancies
| Transport (pmol (μg protein)−1 min−1) | ||||||
|---|---|---|---|---|---|---|
| Condition | l-Lysine | l-Citrulline | l-Leucine | l-Serine | l-Cystine | 2DG |
| 5 mm D-glucose | 4·3 ± 0·1 | 1·9 ± 0·5 | 5·3 ± 0·7 | 1·8 ± 0·7 | 1·8 ± 0·7 | 3·5 ± 0·9 |
| 5 mm D-glucose + insulin | 2·1 ± 0·1 * | 1·9 ± 0·3 | 4·8 ± 0·6 | 2·1 ± 0·4 | 2·2 ± 0·5 | 3·8 ± 0·8 |
| 25 mm D-glucose | 4·6 ± 0·2 | 1·9 ± 0·9 | 4·6 ± 1·2 | 1·8 ± 0·2 | 2·3 ± 0·3 | 4·1 ± 0·9 |
| 25 mm D-glucose + insulin | 4·5 ± 0·1 | 2·1 ± 0·1 | 4·9 ± 0·3 | 1·8 ± 0·5 | 2·2 ± 0·6 | 3·8 ± 0·7 |
Diabetic endothelial cells were cultured in M199 containing 20 % serum and either 5 or 25 mm D-glucose in the absence or presence of 1 nM insulin added during the last 8 h of a 24 h incubation period. Cells were then incubated in Krebs solution and initial rates of transport measured over 30-120 s. Values denote the means ± s.e.m. of n = 3 different cell cultures
P < 0·04 relative to values in 5 or 25 mm D-glucose or 25 mm D-glucose + insulin. All other values for L-lysine transport were significantly (P < 0·04) elevated compared with non-diabetic cells cultured in 5 mm D-glucose (L-lysine: 2·9 ± 0·5 pmol (μg protein)−1 min−1, n = 4 paired experiments). 2DG, 2-deoxy-D-glucose.
To exclude the possibility that elevated D-glucose altered the specifity of L-arginine transport in diabetic endothelial cells, we screened the inhibitory effects of putative competitor amino acids (1 mm). In diabetic endothelial cells pre-exposed to 25 mm D-glucose for 24 h, L-arginine transport remained stereospecific and inhibited markedly by the cationic amino acids L-lysine (Ki = 116 ± 5 μM, n = 3), L-ornithine (Ki = 150 ± 12 μM) and L-homoarginine (Ki= 133 ± 20 μM) and the potent cationic inhibitor of NO synthase, L-NMMA (Ki = 193 ± 6 μM). Apparent Ki values were similar to those determined in diabetic or non-diabetic endothelial cells cultured in 5 mm D-glucose and were within the range of the corresponding Km for L-arginine (Table 3). In contrast, the neutral amino acids L-serine and MeAIB, and the neutral NO synthase inhibitors L-NNA and its methyl ester L-NAME were poor inhibitors (data not shown).
Effects of elevated D-glucose on TPP+ influx, membrane potential and [Ca2+]i in diabetic endothelial cells
Diabetic endothelial cells cultured in either 5 or 25 mm D-glucose exhibited a small but significant membrane hyperpolarization (P < 0.01) compared with non-diabetic cells (Table 2). In parallel experiments, initial rates of TPP+ influx (an indicator of membrane potential) were increased significantly (P < 0.01) in diabetic endothelial cells cultured in either 5 or 25 mm D-glucose compared with non-diabetic cells cultured in 5 mm D-glucose. To determine whether elevated D-glucose per se altered the sensitivity of TPP+ and L-arginine influx in diabetic endothelial cells to changes in extracellular K+, we screened the effects of elevated K+ on initial rates of influx in diabetic cells cultured in 5 or 25 mm D-glucose. TPP+ and L-arginine influx were inhibited (P < 0.04, n = 4) by elevated K+ (data not shown), suggesting that elevated D-glucose had no direct effect on K+-induced changes in membrane potential.
Previous studies have reported that elevated D-glucose augments agonist-stimulated increases in intracellular Ca2+ in serum-deprived endothelial cells (Graier, Simecek, Kukovetz & Kostner, 1996). When the effects of elevated D-glucose on [Ca2+]i levels were examined in diabetic endothelial cells, basal intracellular Ca2+ levels were increased in cells cultured in the presence of 20 % serum and either 5 or 25 mm D-glucose compared with levels measured previously in non-diabetic cells cultured in 5 mm D-glucose (Table 2).
Effects of elevated D-glucose on basal and histamine-stimulated cGMP accumulation
Figure 2A shows that basal cGMP levels were increased in diabetic endothelial cells exposed to either 5 or 25 mm D-glucose compared with non-diabetic cells cultured in 5 mm D-glucose (0.8 ± 0.3 pmol (106 cells)−1 (5 min)−1, P < 0.02). Unlike our previous findings in non-diabetic endothelial cells (see Sobrevia et al. 1996), elevated D-glucose partially inhibited histamine-stimulated (10 μM, 5 min) cGMP production in diabetic cells. This decrease in histamine-induced cGMP production was not a consequence of impaired Ca2+ signalling, since agonist-induced increases in [Ca2+]i were similar in diabetic endothelial cells cultured in either 5 mm (1.78 ± 0.5 μM, n = 31 cells) or 25 mm (1.67 ± 0.3 μM, n = 31 cells) D-glucose and similar to values measured in non-diabetic cells cultured in 5 mm D-glucose (Sobrevia et al. 1996). Treatment of cells with 100 μM L-NAME abolished both basal and histamine-induced increases in cGMP accumulation in diabetic cells (Fig. 2A). In contrast to NO synthesis, basal and histamine-stimulated PGI2 release was markedly inhibited in diabetic endothelial cells cultured in 5 or 25 mm D-glucose (data not shown).
Figure 2. Effects of elevated D-glucose on basal and histamine-stimulated cGMP accumulation and L-citrulline production in umbilical vein endothelial cells isolated from gestational diabetic pregnancies.
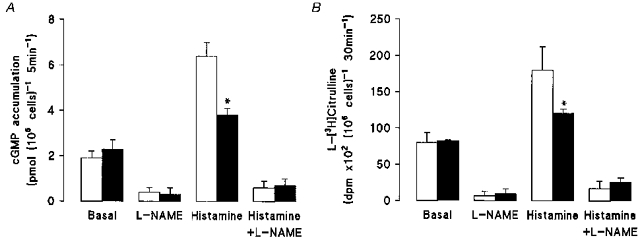
Diabetic cells were cultured for 24 h in M199 containing 5 mm D-glucose (□) or 25 mm D-glucose (▪) and then preincubated for 15 min with 0.5 mm 3-isobutyl-methylxanthine (IBMX, 15 min, 37 °C) and 100 μM L-arginine. A, cGMP accumulation was measured in the presence of 100 μM L-arginine and absence or presence of histamine (10 μM, 5 min, plus 0.5 mm IBMX) and/or 100 μM L-NAME. *P < 0.03 relative to values in diabetic cells cultured in 5 mm D-glucose in the presence or absence of histamine. Basal cGMP accumulation in non-diabetic cells cultursed in 5 mm D-glucose was 0.8 ± 0.3 pmol (106 cells)−1 (5 min−1). B, cells were incubated with 4 μCi ml−1 L-[3H]arginine (100 μM) for 30 min (37 °C) in the absence or presence of 10 μM histamine (last 5 min of a 30 min incubation period) and/or 100 μM L-NAME. Aliquots (300 μl) of formic acid-digested endothelial cells were passed through a cation ion-exchange resin Dowex50W (50X8-200) and radioactivity for L-[3H]citrulline was determined in 200 μl aliquots of H2O eluates. Basal L-citrulline production in non-diabetic cells cultured in 5 mm D-glucose was 3872 ± 339 d.p.m. (106 cells)−1 (30 min)−1, n = 3. Values denote the means ±s.e.m. of 3 different cell cultures.
Formation of L-citrulline from L-arginine
In the presence of 5 mm D-glucose, formation of L-[3H]citrulline from L-[3H]arginine was ∼1.8-fold higher in diabetic endothelial cells (7512 ± 482 d.p.m. (106 cells)−1 (30 min)−1, n = 3) compared with non-diabetic endothelial cells (4171 ± 375 d.p.m. (106 cells)−1 (30 min)−1, n = 3, P < 0.05). Elevated D-glucose had no effect on elevated basal rates of L-citrulline formation in diabetic endothelial cells (Fig. 2B), but, as with the measurements of cGMP accumulation (Fig. 2A), reduced histamine-stimulated L-[3H]citrulline formation (25 mm D-glucose: 12100 ± 589 d.p.m. (106 cells)−1 (30 min)−1vs. 5 mm D-glucose: 18022 ± 322 d.p.m. (106 cells)−1 (30 min)−1. Formation of L-[3H]citrulline was inhibited by ∼80 % when endothelial cells were loaded with L-[3H]arginine in a Ca2+-free Krebs solution (data not shown). Basal intracellular concentrations of L-citrulline, determined by HPLC, were also higher in diabetic endothelial cells (5 mm D-glucose: 0.42 ± 0.03 mm, n = 3 vs. 25 mm D-glucose: 0.47 ± 0.02 mm, n = 3, P < 0.05) compared with values in non-diabetic cells cultured in 5 mm D-glucose (0.18 ± 0.04 mm, n = 3). Elevated D-glucose levels had no effect on intracellular L-arginine levels in diabetic endothelial cells cultured in 5 mm (1.7 ± 0.2 mm) or 25 mm (1.4 ± 0.4 mm, n = 3) D-glucose.
Effects of insulin and elevated D-glucose on L-arginine transport in diabetic endothelial cells
Insulin dose dependently reduced the elevated rates of L-arginine transport in diabetic endothelial cells cultured in 5 mm D-glucose, yet failed to modulate transport in diabetic cells exposed to elevated 25 mm D-glucose (Fig. 3A) or the protein synthesis inhibitor cycloheximide (Fig. 3B). As summarized in Table 3, the reduction in Vmax for L-arginine transport induced by insulin in diabetic endothelial cells cultured in 5 mm D-glucose was abolished upon exposure of these cells to elevated D-glucose for 24 h.
Figure 3. Effects of insulin and elevated D-glucose on L-arginine transport in umbilical vein endothelial cells isolated from gestational diabetic pregnancies.
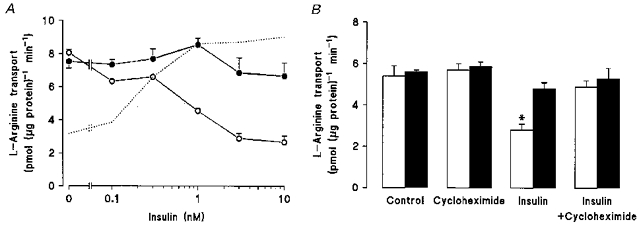
Diabetic cells were cultured in M199 containing either 5 or 25 mm D-glucose in the presence or absence of human insulin (0.1-10 nM, 8 h). In A, L-arginine transport (100 μM, 1 min) was then measured in cells incubated in Krebs solution containing either 5 mm (^) or 25 mm (•) D-glucose and the specified concentrations of human insulin. The dotted line represents the concentration-dependent stimulation of L-arginine transport in non-diabetic umbilical vein endothelial cells cultured in 5 mm D-glucose (see Fig. 1A in Sobrevia et al. 1996). B, effects of cycloheximide (17 μM, 24 h) on L-arginine transport (100 μM, 1 min) in diabetic endothelial cells pretreated with 5 mm (□) or 25 mm (▪) D-glucose in the absence or presence of 1 nM insulin (8 h). Values denote the means ±s.e.m. of 3 different cell cultures. *P < 0.05 relative to all other values.
The inhibition of elevated rates of L-arginine transport by insulin in diabetic endothelial cells cultured in 5 mm D-glucose was paralleled by a reduction in the influx of the membrane potential-sensitive probe TPP+ (Table 2), suggesting that insulin may have depolarized the endothelial cell membrane. In the presence of 25 mm D-glucose, insulin had no such inhibitory effect on either TPP+ or L-arginine influx (Table 2). These actions of insulin appear to be specific for the cationic substrates TPP+, L-arginine and L-lysine, since transport of neutral amino acids and 2-deoxy-D-glucose was unaffected by insulin in endothelial cells cultured in either 5 or 25 mm D-glucose (Table 4).
Modulation of cGMP and PGI2 synthesis by insulin and elevated D-glucose
Elevated rates of cGMP production in diabetic endothelial cells cultured in 5 mm D-glucose were also inhibited by insulin (1 nM, 8 h), and exposure to elevated D-glucose abolished the inhibitory actions of insulin (Fig. 4A). Basal and insulin-stimulated endothelial cGMP accumulation was completely inhibited by the neutral NO synthase inhibitor, L-NAME. PGI2 release was reduced significantly in diabetic endothelial cells cultured in either 5 or 25 mm D-glucose, and the depressed release of PGI2 was unaffected by insulin (Fig. 4B).
Figure 4. Effects of insulin and elevated D-glucose on cGMP accumulation and PGI2 production in human umbilical vein endothelial cells isolated from gestational diabetic pregnancies.
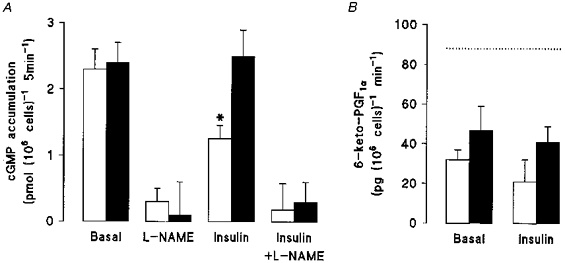
Diabetic cells were cultured in M199 containing 20 % serum and either 5 mm (□) or 25 mm (▪) D-glucose in the absence or presence of human insulin (1 nM, 8 h). Cells were then washed and incubated in Krebs solution containing 0.5 mm IBMX, 100 μM L-arginine and either 5 or 25 mm D-glucose in the absence or presence of 1 nM insulin and/or 100 μM L-NAME. Accumulation of cGMP (A) and release of 6-keto-PGF1α, a stable metabolite of PGI2 (B), were then determined by radioimmunoassay (see Methods). The dotted line represents the basal rate of PGI2 production in non-diabetic endothelial cells cultured in 5 mm D-glucose (see in Sobrevia et al. 1995a). Values denote the means ±s.e.m. of experiments in 4 different cell cultures. *P < 0.05.
DISCUSSION
The present study provides the first evidence that human insulin inhibits the increased activity of system y+ and NO synthesis in human umbilical vein endothelial cells induced by gestational diabetes. Exposure of endothelial cells from gestational diabetic pregnancies to elevated D-glucose attenuated histamine-stimulated NO and L-citrulline production from L-arginine and prevented stimulation by insulin of L-leucine incorporation and inhibition of the L-arginine-NO signalling pathway. Thus, our findings suggest that hyperglycaemia induces insulin insensitivity in fetal vascular endothelial cells derived from gestational diabetic pregnancies.
Effects of gestational diabetes and hyperglycaemia on human endothelial cells
In this study we have confirmed our previous observation that protein and DNA turnover are reduced in endothelial cells from gestational diabetic pregnancies (see Sobrevia et al. 1995a) and established that L-arginine transport, TPP+ influx (index of membrane potential) and basal cytosolic Ca2+ levels are elevated in cells cultured in either 5 or 25 mm D-glucose compared with non-diabetic cells cultured in 5 mm D-glucose. The small membrane hyperpolarization sustained in diabetic cells during prolonged cell culture may in part explain the increased influx of the cation L-arginine (see Sobrevia et al. 1995a; Bogle et al. 1996). Although hyperglycaemia elevated basal cytosolic Ca2+ and NO production threefold (prevented by NO synthase inhibitors) in non-diabetic cells (Sobrevia et al. 1996), high D-glucose had no effect on the increased rates of L-arginine transport and NO production or the membrane hyperpolarization and increased basal [Ca2+]i levels in cells from gestational diabetic pregnancies (see Table 2 and Fig. 3A). The lack of an effect of elevated D-glucose in diabetic endothelial cells may reflect the fact that these processes were already altered during the course of gestational diabetes and insensitive to further modulation by elevated D-glucose.
Unlike our findings of stimulated rates of L-arginine transport and NO synthesis in human endothelial cells from gestational diabetic pregnancies (Sobrevia et al. 1995a), Wu & Meininger (1995) failed to detect changes in L-arginine transport in coronary artery endothelial cells from streptozotocin-diabetic rats but noted that accumulation of nitrite and nitrate (metabolites of NO) was reduced. It is worth pointing out that Wu & Meininger (1995) did not measure initial rates of L-arginine transport nor test the effects of NO synthase inhibitors. In our study, diabetes-induced increases in basal cGMP accumulation were paralleled by increased rates of L-arginine metabolism to L-citrulline (see Fig. 2). Although elevated D-glucose had no effect on cGMP accumulation or L-citrulline production under basal conditions, histamine-stimulated cGMP levels and metabolism of L-arginine to L-citrulline were attenuated in endothelial cells from gestational diabetic pregnancies (Fig. 2). Hyperglycaemia may have altered the interaction of histamine with its receptor and/or modified NO synthase itself. Both basal and histamine-stimulated PGI2 release were markedly depressed in gestational diabetic endothelial cells, confirming earlier studies in human umbilical vein endothelial cells (Karbowski, Bauch & Schneider, 1989).
Modulation of the endothelial L-arginine-NO signalling pathway by insulin
Our recent findings provided the first evidence, at a cellular level, that long-term exposure (8 h) to insulin stimulates NO synthase and system y+/hCAT-1 activity in human umbilical vein endothelial cells isolated from non-diabetic pregnancies (Sobrevia et al. 1996), confirming the vasodilator actions of insulin in man in vivo (Steinberg et al. 1994; Scherrer et al. 1994). Insulin also enhances system y+ activity in gastric glands (Contreras et al. 1997), pancreas (Muñoz, Sweiry & Mann, 1995) and liver cells (Wu, Robinson, Kung & Hatzoglou, 1994). Other studies in umbilical vein endothelial cells have shown that both IGF-1 and insulin stimulate NO production via a Ca2+-independent mechanism (Tsukahara, Gordienko, Tonshoff, Gelato & Goligorsky, 1994). More recent studies in human umbilical vein endothelial cells have shown that insulin results in a rapid stimulation of NO production, which was blocked by either NO synthase or tyrosine kinase inhibitors (Zeng & Quon, 1996). In the same study, wortmannin, an inhibitor of phosphatidylinositol 3-kinase - a necessary effector of insulin signalling related to D-glucose transport (Quon et al. 1995; O'Brien & Granner, 1996) - inhibited insulin-stimulated NO production by only ∼50 %, suggesting that additional signalling events such as insulin-mediated increases in cytosolic Ca2+ (Han, Ouchi, Karaki & Orimo, 1995) may account for the activation of NO synthase.
In the present study, insulin could only restore L-leucine incorporation in diabetic endothelial cells cultured in 5 mm D-glucose, but had no effect on diabetic cells cultured in elevated D-glucose (see Table 1), suggesting that hyperglycaemia impairs the action of insulin in diabetic cells. Studies of endothelial cell function and insulin sensitivity in patients with insulin-dependent diabetes mellitus (IDDM) have established that chronic hyperglycaemia impairs endothelium-dependent forearm vasodilatation and insulin-stimulated D-glucose extraction (Makimatilla et al. 1996). These authors concluded that a defect in D-glucose extraction rather than blood flow characterizes the insulin resistance in uncomplicated IDDM. These findings are consistent with our observation that fetal endothelial cells from gestational diabetic patients are not responsive to insulin in the presence of elevated D-glucose. This insensitivity to insulin may not only be a consequence of endothelial dysfunction but may also be due to a diminished action of insulin in diabetic endothelium exposed to elevated D-glucose.
Umbilical vein endothelial cells isolated from women with IDDM exhibit changes in cellular activity such as increased plasma membrane fluidity, enhanced fluid phase endocytosis, and an increase in mitochondrial area, Weibel-Palade bodies and rough reticulum with wide cisternae (Cester et al. 1996). Moreover, umbilical vein endothelial cells from women with IDDM are also less resistant to shear stress, take up D-glucose more slowly and exhibit an increased expression of major basal membrane components (Sank et al. 1994). These observations support our previous finding that ‘diabetic-like’ characteristics are maintained during prolonged culture of these cells in euglycaemic or hyperglycaemic media (Sobrevia, Jarvis & Yudilevich, 1994; Sobrevia et al. 1995a). Thus, endothelial cells may be the site of a primary defect in IDDM and contribute to the development of vascular disease (Tooke, 1995; Sobrevia & Mann, 1997).
Not all studies concur that insulin influences basal blood flow, since insulin has been reported to either increase basal forearm blood flow (Scherrer et al. 1994) or only to potentiate acetylcholine-induced vasodilatation (Taddei et al. 1995). A recent study has shown that inhibition of Na+-K+-ATPase abolishes the vasodilator action of insulin in the human forearm (Tack, Lutterman, Vervoort, Thien & Smits, 1996), arguing against a direct effect of insulin on endothelial NO synthase in the human forearm vasculature. The mechanisms mediating insulin action in animal models are also not fully resolved (see reviews by Poston & Taylor, 1995; Sobrevia & Mann, 1997).
Insulin insensitivity
The lack of an effect of insulin in gestational diabetic endothelial cells exposed to hyperglycaemia may reflect an inhibitory action of D-glucose on insulin receptor tyrosine kinase activity (Haring, Kellerer & Mosthaf, 1994; Ide, Maegawa, Kashiwagi, Kikkawa & Shigeta, 1995), resulting in an inactivation of the insulin signalling cascade. Acute hyperglycaemia per se can inhibit the tyrosine kinase activity of the insulin receptor within minutes, and this inhibition seems to be mediated by the translocation and activation of isoforms of protein kinase C (PKC; Kellerer & Harding, 1995). Recent studies revealed that hyperglycaemia inhibited insulin receptor phosphorylation and decreased phosphatidylinositol 3-kinase activity (Pillay, Xiao & Olefsky, 1996). These authors further demonstrated that co-incubation of fibroblasts with a PKC inhibitor, but not an inhibitor of the mitogen-activated protein kinase cascade, reversed the impaired insulin receptor phosphorylation induced by hyperglycaemia. Diacylglycerol levels and PKC activity are increased in vascular cells exposed to prolonged hyperglycaemia (Inoguchi et al. 1994; see also review by King, Kunisaki, Nishio, Inoguchi, Shiba & Xia, 1996), and activation of PKC appears to mediate cytokine-induced increases in L-arginine transport in umbilical vein endothelial cells (Pan et al. 1995). Activation of PKC cannot fully explain our previous findings that stimulatory actions of insulin on L-arginine transport and NO synthesis were attenuated in non-diabetic endothelial cells adapted to elevated D-glucose (Sobrevia et al. 1996), since stimulation of PKC does not regulate basal NO release in porcine endothelial cells (Smith & Lang, 1990) and actually stimulates basal PGI2 synthesis in umbilical vein endothelial cells (Carter, Hallam & Pearson, 1989).
Although it is difficult to explain the inhibitory action of insulin on L-arginine transport and NO synthesis in fetal endothelial cells isolated from gestational diabetic pregancies, insulin has been shown to downregulate diabetes-induced stimulation of amino acid transport in hepatocytes (Handlogten & Kilberg, 1984), isolated pancreas (Mann & Norman, 1984) and isolated gastric glands (Contreras et al. 1997). Since cycloheximide abolished the inhibitory action of insulin in gestational diabetic endothelial cells, it is plausible that insulin stimulated synthesis of a factor(s) responsible for the downregulation of the system y+/hCAT-1 transporter and NO synthase.
There is consensus that endothelium-dependent relaxation is impaired in established diabetes mellitus (see Cohen, 1993; Poston & Taylor, 1995; Sobrevia & Mann, 1997), yet in the early and uncomplicated stages of diabetes, peripheral resistance is reduced and blood flow increased in various organs such as the retina, skin and kidney (Mogensen, 1971; Kohner et al. 1975; Houben, Schaper, Slaaf, Tangelder & Niewenhuijzen-Kruseman, 1992). This generalized vasodilatation is influenced by the degree of hyperglycaemia and improves upon lowering of blood glucose levels (Mathiesen et al. 1985), suggesting that D-glucose may directly modulate vascular tone. Our studies in human umbilical vein endothelial cells provide a useful in vitro model for examining the interactions of hyperglycaemia and insulin in the regulation of the L-arginine-NO signalling pathway in fetal endothelial cells derived from non-diabetic and gestational diabetic pregnancies. In preliminary experiments, we have confirmed that mRNA levels of endothelial NO synthase (eNOS) and system y+/hCAT-1 in umbilical vein endothelial cells are not altered by elevated D-glucose (Mann, Siow, Closs & Sobrevia, 1998). Further studies at a molecular level are necessary to elucidate whether hyperglycaemia and/or insulin modulate protein and mRNA levels of the cationic amino acid transporter isoforms (hCAT-1, hCAT-2A, hCAT-2B) and endothelial NO synthase in diabetes mellitus.
Acknowledgments
This work was supported by The Wellcome Trust (040727/Z/94), the British Council (UK), Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT 19711321-1997) and Dirección de Investigación, Universidad de Concepción (DIUC 9733871D, Chile). We thank our colleagues Dr Richard Siow, Dr Angel Nadal and Mr Paolo Cesare for their measurements of intracellular amino acid concentrations, [Ca2+]i and membrane potential. We are indebted to Professor Richard Beard and the midwives of St Mary's Hospital labour ward for their supply of umbilical cords from normal and gestational diabetic pregnancies.
References
- Baron AD. The coupling of glucose metabolism and perfusion in human skeletal muscle: the potential role of endothelium-derived nitric oxide. Diabetes. 1996;45:S105–109. doi: 10.2337/diab.45.1.s105. [DOI] [PubMed] [Google Scholar]
- Bogle RG, Baydoun AR, Pearson JD, Mann GE. Regulation of L-arginine transport and nitric oxide synthesis in superfused porcine aortic endothelial cells. Journal of Physiology. 1996;490:229–241. doi: 10.1113/jphysiol.1996.sp021138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter TD, Hallam TJ, Pearson JD. Protein kinase C activation alters the sensitivity of agonist-stimulated endothelial cell prostacyclin production to intracellular Ca2+ Biochemical Journal. 1989;262:431–437. doi: 10.1042/bj2620431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cester N, Rabini RA, Salvolini E, Staffolani R, Curatola A, Pugnaloni A, Brunelli MA, Biagini G, Mazzanti L. Activation of endothelial cells during insulin-dependent diabetes mellitus: a biochemical and morphological study. European Journal of Clinical Investigation. 1996;26:569–573. doi: 10.1046/j.1365-2362.1996.1750526.x. [DOI] [PubMed] [Google Scholar]
- Cohen RA. Dysfunction of vascular endothelium in diabetes mellitus. Circulation. 1993;87:V67–76. [Google Scholar]
- Contreras R, Fuentes O, Mann GE, Sobrevia L. Diabetes and insulin-induced stimulation of L-arginine transport and nitric oxide synthesis in rabbit isolated gastric glands. Journal of Physiology. 1996;498:787–796. doi: 10.1113/jphysiol.1997.sp021902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornhorst A, Beard RW. Gestational diabetes: a challenge for the future. Diabetic Medicine. 1993;10:897–905. doi: 10.1111/j.1464-5491.1993.tb00004.x. [DOI] [PubMed] [Google Scholar]
- Graier WF, Simecek S, Kukovetz WR, Kostner GM. High D-glucose induced changes in endothelial Ca2+/EDRF signaling are due to generation of superoxide anions. Diabetes. 1996;45:1386–1395. doi: 10.2337/diab.45.10.1386. [DOI] [PubMed] [Google Scholar]
- Han S-Z, Ouchi Y, Karaki H, Orimo H. Inhibitory effects of cytosolic Ca2+ level and contraction in the rat aorta: endothelium-dependent and -independent mechanisms. Circulation Research. 1995;77:673–678. doi: 10.1161/01.res.77.4.673. [DOI] [PubMed] [Google Scholar]
- Handlogten ME, Kilberg MS. Induction and decay of amino acid transport in the liver. Journal of Biological Chemistry. 1984;257:14960–14967. [PubMed] [Google Scholar]
- Haring HU, Kellerer M, Mosthaf I. Modulation of insulin receptor signalling: signficance of altered receptor isoform patterns and mechanism of hyperglycaemia-induced receptor modulation. Diabetologia. 1994;37:S149–154. doi: 10.1007/BF00400838. [DOI] [PubMed] [Google Scholar]
- Houben AJHM, Schaper NC, Slaaf DW, Tangelder GH, Nieuwenhuijzen-Kruseman AC. Skin blood cell flux in insulin-dependent diabetic subjects in relation to retinopathy or incipient nephropathy. European Journal of Clinical Investigation. 1992;22:67–72. doi: 10.1111/j.1365-2362.1992.tb01938.x. [DOI] [PubMed] [Google Scholar]
- Ide R, Maegawa H, Kashiwagi A, Kikkawa R, Shigeta Y. High glucose condition desensitizes insulin action at the level of receptor kinase. Endocrine Journal. 1995;42:1–8. doi: 10.1507/endocrj.42.1. [DOI] [PubMed] [Google Scholar]
- Inoguchi T, Xia P, Kunisaki M, Higashi S, Feener EP, King GL. Insulin's effect on protein kinase C and diacylglycerol induced by diabetes and glucose in vascular tissues. American Journal of Physiology. 1994;267:E369–379. doi: 10.1152/ajpendo.1994.267.3.E369. [DOI] [PubMed] [Google Scholar]
- Karbowski B, Bauch HJ, Schneider HPG. Funktionelle differenzierung des vaskularen endothels bei hochrisikoschwangerschaften. Zeitschrift der Geburtshilfe Perinatologie. 1989;193:8–12. [PubMed] [Google Scholar]
- Kellerer M, Haring HU. Pathogenesis of insulin resistance: modulation of the insulin signal at receptor level. Diabetes Research and Clinical Practice. 1995;28:S173–177. doi: 10.1016/0168-8227(95)01070-t. 10.1016/0168-8227(95)01070-T. [DOI] [PubMed] [Google Scholar]
- King GL, Kunisaki M, Nishio Y, Inoguchi T, Shiba T, Xia P. Biochemical and molecular mechanisms in the development of diabetic vascular complications. Diabetes. 1996;45:S105–108. doi: 10.2337/diab.45.3.s105. [DOI] [PubMed] [Google Scholar]
- Kohner EM, Hamilton AM, Saunders SJ, Sutcliffe BA, Bulpitz CJ. The retinal blood flow in diabetes. Diabetologia. 1975;11:27–33. doi: 10.1007/BF00422814. [DOI] [PubMed] [Google Scholar]
- Makimattila S, Virkamaki A, Groop PH, Cockcroft J, Utrianinen T, Fagerudd J, Ykijarvinen H. Chronic hyperglycemia impairs endothelial function and insulin sensitivity via different mechanisms in insulin-dependent diabetes mellitus. Circulation. 1996;94:1276–1282. doi: 10.1161/01.cir.94.6.1276. [DOI] [PubMed] [Google Scholar]
- Mann GE, Norman PSR. Regulatory effects of insulin and experimental diabetes on neutral amino acid transport in the perfused rat exocrine pancreas: Kinetics of unidirectional L-serine influx and efflux at the basolateral plasma membrane. Biochimica et Biophysica Acta. 1984;778:618–622. doi: 10.1016/0005-2736(84)90415-2. [DOI] [PubMed] [Google Scholar]
- Mann GE, Siow RCM, Closs EI, Sobrevia L. Expression of hCAT-1 (system y+) and NO synthase in human fetal endothelial cells: modulation by elevated D-glucose. In: Moncada S, Toda N, Maeda H, Higg EA, editors. The Biology of Nitric Oxide. Vol. 6. 1998. in the Press. [Google Scholar]
- Mathieson ER, Hilsted J, Feldt-Rasmussen B, Bonde-Petersen F, Christensen NJ, Parving HH. The effects of metabolic control on hemodynamics in short-term insulin-dependent diabetic patients. Diabetes. 1985;34:1301–1305. doi: 10.2337/diab.34.12.1301. [DOI] [PubMed] [Google Scholar]
- Mogensen CE. Glomerular filtration rate and renal plasma flow in short-term and long-term juvenile diabetes mellitus. Scandinavian Journal of Clinical Laboratory Investigation. 1971;28:91–100. doi: 10.3109/00365517109090667. [DOI] [PubMed] [Google Scholar]
- Muñoz M, Sweiry JH, Mann GE. Insulin stimulates cationic amino acid transport activity in the isolated perfused rat pancreas. Experimental Physiology. 1995;80:745–753. doi: 10.1113/expphysiol.1995.sp003883. [DOI] [PubMed] [Google Scholar]
- O'Brien RM, Granner DK. Regulation of gene expression by insulin. Physiological Reviews. 1996;76:1109–1161. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- Pan M, Wasa M, Lind DS, Gertler I, Abbott W, Souba WW, Garrison RN, Wilson MA, Moody PG. TNF-stimulated arginine transport by human vascular endothelium requires activation of protein kinase C. Annals of Surgery. 1995;221:590–601. doi: 10.1097/00000658-199505000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay TS, Xiao S, Olefsky JM. Glucose-induced phosphorylation of the insulin receptor: functional effects and characterization of phosphorylation sites. Journal of Clinical Investigation. 1996;97:613–620. doi: 10.1172/JCI118457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston L, Taylor PD. Endothelium-dependent vascular functions in insulin-dependent diabetes mellitus. Clinical Science. 1995;88:245–255. doi: 10.1042/cs0880245. [DOI] [PubMed] [Google Scholar]
- Quon MJ, Chen H, Ing BL, Liu ML, Zarnowski MJ, Yonezawa K, Kasuga M, Cushman SW, Taylor SI. Roles of 1-phosphatidylinositol 3-kinase and ras in regulating translocation of GLUT4 in transfected rat adipose cells. Molecular and Cellular Biology. 1995;15:5403–5411. doi: 10.1128/mcb.15.10.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sank A, Wei D, Reid J, Ertl D, Nimni M, Weaver F, Yellin A, Tuan TL. Human endothelial cells are defective in diabetic vascular disease. Journal of Surgical Research. 1994;57:647–653. doi: 10.1006/jsre.1994.1195. 10.1006/jsre.1994.1195. [DOI] [PubMed] [Google Scholar]
- Scherrer U, Randin D, Vollenweider P, Vollenweider L, Nicod P. Nitric oxide release accounts for insulin's vascular effects in humans. Journal of Clinical Investigation. 1994;94:2511–2515. doi: 10.1172/JCI117621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Lang D. Release of endothelium-derived relaxing factor from pig cultured aortic endothelial cells, as assessed by changes in endothelial cell cyclic GMP content, is inhibited by a phorbol ester. British Journal of Pharmacology. 1990;99:565–571. doi: 10.1111/j.1476-5381.1990.tb12969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrevia L, Cesare P, Yudilevich DL, Mann GE. Diabetes-induced activation of system y+ and nitric oxide synthase in human endothelial cells: association with membrane hyperpolarization. Journal of Physiology. 1995a;489:183–192. doi: 10.1113/jphysiol.1995.sp021040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrevia L, Jarvis SM, Yudilevich DL. Adenosine transport in cultured human umbilical vein endothelial cells is reduced in diabetes. American Journal of Physiology. 1994;267:C39–47. doi: 10.1152/ajpcell.1994.267.1.C39. [DOI] [PubMed] [Google Scholar]
- Sobrevia L, Mann GE. Dysfunction of the endothelial nitric oxide signalling pathway in diabetes and hyperglycaemia. Experimental Physiology. 1997;82:423–452. doi: 10.1113/expphysiol.1997.sp004038. [DOI] [PubMed] [Google Scholar]
- Sobrevia L, Nadal A, Yudilevich DL, Mann GE. Activation of L-arginine transport (system y+) and nitric oxide synthase by elevated glucose and insulin in human endothelial cells. Journal of Physiology. 1996;490:775–781. doi: 10.1113/jphysiol.1996.sp021185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrevia L, Yudilevich DL, Mann GE. Insulin modulates diabetes-induced stimulation of L-arginine transport in human umbilical vein endothelial cells. Journal of Physiology. 1995b;483:136P. [Google Scholar]
- Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. Journal of Clinical Investigation. 1994;94:1172–1179. doi: 10.1172/JCI117433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack CJJ, Lutterman JA, Vervoort G, Thien T, Smits P. Activation of the sodium-potassium pump contributes to insulin-induced vasodilatation. Hypertension. 1996;28:426–432. doi: 10.1161/01.hyp.28.3.426. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Mattei P, Natali A, Ferrannini E, Salvetti A. Effect of insulin on acetylcholine-induced vasodilation in normotensive subjects and patients with essential hypertension. Circulation. 1995;92:2911–2918. doi: 10.1161/01.cir.92.10.2911. [DOI] [PubMed] [Google Scholar]
- Tooke JE. Microvascular function in human diabetes: a physiological perspective. Diabetes. 1995;44:721–726. doi: 10.2337/diab.44.7.721. [DOI] [PubMed] [Google Scholar]
- Tsukahara H, Gordienko DV, Tonshoff B, Gelato MC, Goligorsky MS. Direct demonstration of insulin-like growth factor-I-induced nitric oxide production by endothelial cells. Kidney International. 1994;45:598–604. doi: 10.1038/ki.1994.78. [DOI] [PubMed] [Google Scholar]
- Van Veen S, Chang PC. Prostaglandins and nitric oxide mediate insulin-induced vasodilation in human forerarm. Cardiovascular Research. 1997;34:223–229. doi: 10.1016/s0008-6363(97)00031-x. 10.1016/S0008-6363(97)00031-X. [DOI] [PubMed] [Google Scholar]
- Wu G, Meininger CJ. Impaired arginine metabolism and NO synthesis in coronary endothelial cells of the spontaneously diabetic BB rat. American Journal of Physiology. 1995;269:H1312–1318. doi: 10.1152/ajpheart.1995.269.4.H1312. [DOI] [PubMed] [Google Scholar]
- Wu JY, Robinson D, Kung H-J, Hatzoglou M. Hormonal regulation of the gene for the type C ecotropic retrovirus receptor in rat liver cells. Journal of Virology. 1994;68:1615–1623. doi: 10.1128/jvi.68.3.1615-1623.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. Journal of Clinical Investigation. 1996;98:894–898. doi: 10.1172/JCI118871. [DOI] [PMC free article] [PubMed] [Google Scholar]