

Abstract
We have investigated the interactions between intracellular pH (pHi) and the intracellular free calcium concentration ([Ca2+]i) in isolated rat pancreatic acinar cells. The fluorescent dyes fura-2 and BCECF were used to measure [Ca2+]i and pHi, respectively.
Sodium acetate and ammonium chloride (NH4Cl) were used to acidify and alkalinize pHi, respectively. Cytosolic acidification had no effect on [Ca2+]i in resting pancreatic acinar cells, whereas cytosolic alkalinization released Ca2+ from intracellular stores.
Cytosolic acidification using either acetate or a CO2-HCO3−-buffered medium enhanced Ca2+ signals evoked by acetylcholine (ACh) and cholecystokinin (CCK). In contrast, both NH4Cl and trimethylamine (TMA) inhibited Ca2+ signals during stimulation with either ACh or CCK. This inhibitory effect was also observed in the absence of extracellular Ca2+, and was therefore not due to changes in Ca2+ entry.
Calcium oscillations evoked by physiological concentrations of CCK were enhanced by cytosolic acidification and inhibited by cytosolic alkalinization.
In order to determine the effects of pHi upon Ca2+ handling by intracellular Ca2+ stores, intraorganellar [Ca2+] was monitored using the low affinity Ca2+ indicator mag-fura-2 in permeabilized cells. Addition of NH4Cl, which is expected to alkalinize intraorganellar pH, did not alter intraorganellar [Ca2+] in permeabilized cells, suggesting that changing intraorganellar pH does not release Ca2+ from intracellular stores. Addition of NH4Cl or acetate also did not affect the rate of Ca2+ release induced by inositol 1,4,5-trisphosphate (InsP3).
Modification of extraorganellar (‘cytosolic’) pH did not affect the rate of ATP-dependent Ca2+ uptake into stores, but did modify the rate of Ca2+ release evoked by submaximal concentrations of InsP3. The rate of Ca2+ release was increased at more alkaline extraorganellar pHs. These results would suggest that manipulation of intraorganellar pH does not affect Ca2+ handling by the intracellular stores. In contrast, extraorganellar (‘cytosolic’) pH does affect InsP3-induced Ca2+ release from the stores.
In conclusion, changes in intracellular pH in pancreatic acinar cells can profoundly alter cytosolic [Ca2+]. This may shed light on earlier observations whereby cell-permeant weak acids and bases can modulate fluid secretion in epithelia.
Alteration of pHi using a wide range of weak acids, including HCO3−, acetate, pyruvate and butyrate, has been shown to stimulate fluid and electrolyte transport in a number of epithelia (Ullrich, Radtke & Rumrich, 1971; Petersen, Wood, Schulze & Heintze, 1981; Case, Conigrave, Favaloro, Novak, Thompson & Young, 1982). In exocrine acinar cells, weak bases have been found to activate Ca2+-dependent K+ and Cl− channels (Hayashi et al. 1992), and to modify the rate of Cl− fluxes (Seagrave, Barker, Curry & Martinez, 1992). The mechanism(s) underlying these effects are unknown. However, one attractive possibility is the involvement of changes in cytosolic Ca2+ ([Ca2+]i). Changes in [Ca2+]i in acinar cells following receptor stimulation result in activation of luminal and basolateral membrane ion channels, and subsequent fluid secretion (Petersen, 1992). A large number of studies on a variety of cell types, including acinar cells, have illustrated interactions between changes in pHi and Ca2+ signals. For example, cytosolic acidification increases [Ca2+]i in collecting duct cells (Slotki, Schwartz & Alexander, 1993) and pancreatic acinar cells (Tsunoda, 1990). Furthermore, cytosolic alkalinization was also found to increase [Ca2+]i in lymphocytes (Grinstein & Goetz, 1985), cultured smooth muscle cells (Siskind, McCoy, Chobanian & Schwartz, 1989), lacrimal acinar cells (Yodozawa, Speake & Elliott, 1997) and endothelial cells (Danthuluri, Kim & Brock, 1990).
In the present study we have investigated the relationship between pHi and Ca2+ signalling in detail in isolated pancreatic acinar cells. We report that in resting cells, acetate has no effect on [Ca2+]i, whereas NH4Cl releases Ca2+ from intracellular stores. In contrast, cytosolic acidification during sustained or oscillatory Ca2+ signalling increases [Ca2+]i, whereas cytosolic alkalinization inhibits Ca2+ signals. These effects appear to occur at the level of the intracellular Ca2+ store.
METHODS
Pancreatic acinar cell isolation
Small clusters of acinar cells were prepared from the pancreas of Sprague-Dawley rats, which were killed by fluothane inhalation. Isolated pancreata were digested with collagenase (680 U (g tissue)−1) in physiological medium (see below for composition) supplemented with 0.12 mg ml−1 soybean trypsin inhibitor (Sigma), 1% (w/v) bovine serum albumin (BSA), and an amino acid mixture (containing minimum essential amino acids; 2% v/v). Following 30 min incubation at 37°C, the tissue suspension was washed several times with enzyme-free medium and dissociated into a fine suspension of acini by repeatedly passing the suspension through a 5 ml plastic pipette tip. The resulting cell suspension was layered onto medium supplemented with 4% BSA, and centrifuged at 1000 g for 2 min. After discarding the supernatant and repeating the centrifugation step once more, cells were resuspended in 0.1% BSA medium. Cells were either used immediately or stored on ice until use.
Loading of fluorescent dyes
Portions (1 ml) of the cell suspension were incubated with one of the following dyes: 2 μM of the acetoxymethyl ester form of fura-2 (fura-2 AM) for 30 min at room temperature, 1 μM of the acetoxymethyl ester form of 2′,7′-bis(carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF AM) for 30 min at room temperature, or 5 μM mag-fura-2 AM for 45 min at 37°C. Loaded cells were then washed twice with 0.1% BSA containing-medium, and resuspended in physiological medium.
Fluorescence measurements
Dye fluorescence was measured using a system based on a Nikon Diaphot inverted microscope and was described previously (Berrie & Elliott, 1994). Dye-loaded acinar cells were allowed to adhere to a coverslip forming the base of a perfusion chamber and observed with a × 40 oil immersion lens (numerical aperture 1.3). Individual cells, or small clusters of cells were isolated from the remaining cells in view using an adjustable diaphragm. Cells were excited at wavelengths of 350 and 380 nm (emission wavelength, 500 ± 20 nm) for fura-2 and mag-fura-2, and 488 and 436 nm (emission wavelength, 526 ± 12.5 nm) for BCECF, using a filter wheel spinning at 40 Hz (Cairn Research Ltd, Faversham, Kent, UK).
Permeabilization of acinar cells
Mag-fura-2-loaded acinar cells were allowed to settle onto the glass coverslip and were then superfused with Ca2+-free ‘cytosolic’ medium containing (mm): 135 KCl, 1.2 KH2PO4, 1.39 MgCl2, 0.5 EGTA, 0.5 HEDTA, 0.5 nitriloacetic acid (NTA) and 20 Hepes (pH 7.1 with KOH), supplemented with Streptolysin-O (SLO; 0.4 u ml−1) to permeabilize the cells, as described in detail in van de Put & Elliott (1996). In those experiments where the effects of modifying extraorganellar pH on inositol 1,4,5-trisphosphate (InsP3)-induced Ca2+ release were examined, the EGTA, HEDTA, and NTA Ca2+ buffers were replaced by 1.5 mm BAPTA, since the ability of BAPTA to bind Ca2+ is only very weakly dependent on pH (Tsien, 1980). The permeabilization process was followed on-line by monitoring the 350 nm fluorescence signal (near the isosbestic point for mag-fura-2, 360 nm). The 350 nm signal dropped significantly as the dye was lost from the cytosol (van de Put & Elliott, 1996). Acinar cells were subsequently perfused with Ca2+-uptake ‘cytosolic’ medium with the same composition as the Ca2+-free ‘cytosolic’ medium but with the addition of 0.175 mm CaCl2. This gave free [Mg2+] and [Ca2+] of 0.9 mm and 0.2 μm, respectively (calculated according to Schoenmakers, Visser, Flik & Theuvenet, 1992). Calcium uptake into intracellular stores was initiated by superfusing the permeabilized cells with Ca2+-uptake ‘cytosolic’ medium containing 1 mm ATP. After a Ca2+ store loading period of 10 min permeabilized cells were stimulated with InsP3.
Calibration of ratio values
In experiments measuring [Ca2+]i, the uncalibrated 350:380 nm ratio signal is shown as an index of [Ca2+]i, since absolute estimates of [Ca2+]i were not routinely derived from the values of the 350:380 nm fluorescence ratio. However, a two-point calibration of the fura-2 signal was carried out on a number of pancreatic acinar cells (Grynkiewicz, Poenie & Tsien, 1985). Individual cells were initially treated with 1 μm thapsigargin and 1 μm ionomycin in Ca2+-free medium (containing 2 mm EGTA) to obtain fluorescence parameters for fura-2 under Ca2+-free conditions (Rmin). The cells were subsequently superfused with thapsigargin and ionomycin in Ca2+-supplemented medium to obtain fluorescence parameters for Ca2+-saturated fura-2 (Rmax). The values obtained were: Rmax, 5.74 ± 0.58; Rmin, 0.92 ± 0.03; and the ratio SF,380/SB,380, 6.34 ± 0.41 (all values n = 4); where SF,380/SB,380 refers to the fluorescence signals measured at 380 nm for Ca2+-free and Ca2+-saturated dye. The 350:380 nm ratio signal varied between 1.0 and 1.5 in resting cells, corresponding to an estimated [Ca2+]i of between 15 and 120 nM (the Kd of fura-2 at 22°C was taken as 135 nM; Grynkiewicz et al. 1985). Submaximal concentrations of ACh (1–2 μm) typically raised the 350:380 fluorescence ratio to around 3.0, which corresponds to an estimated [Ca2+]i of around 0.65 μm, while supramaximal ACh concentrations (10 μm) raised the 350:380 fluorescence ratio to around 4.5, corresponding to a [Ca2+]i of around 2.5 μm.
Intracellular pH was calibrated from the 488:436 nm ratio signal using the high extracellular K+/nigericin technique (Thomas, Buchsbaum, Zimniak & Racker, 1979), as previously described (Yodozawa et al. 1997).
Effects of changes in pHi on fura-2 and mag-fura-2
Since many of the experiments in the present paper involve treatments which simultaneously change both pHi and [Ca2+]i, it is necessary to consider whether changes in pHi alter the behaviour of the Ca2+-sensing dyes. For mag-fura-2, a detailed in vitro study by Lattanzio & Bartschat (1991) found that the dye's Kd for Ca2+ was essentially unaltered by changes in pH over the range 5.5–7.5. For fura-2, the effects of pH on the dye's Kd have been extensively characterized both in solution and inside intact cells in a series of papers by Martínez-Zaguilán and co-workers (Martínez-Zaguilán, Martínez, Lattanzio & Gillies, 1991; Martínez-Zaguilán, Gurulé & Lynch, 1996a; Martínez-Zaguilán, Parnami & Lynch, 1996b). These authors showed that the Kd of fura-2 rises as pH becomes more acidic, with the effect being particularly marked at pH values below around 6.8 (see also Lattanzio, 1990). The values of pHi recorded in the present study ranged from 8.0 to 6.4. However, in most experiments pHi was between 7.8 and 7.0 (mean pHi in unstimulated cells was around 7.3, while acetate acidified cells to a mean pHi close to 7.0 and NH4Cl alkalinized cells to a mean pHi around 7.8; see Results). The data of Martínez-Zaguilán et al. (1995) show that the fura-2 Kd will vary only slightly over this pH range. Calculation from their data gives a change in the fura-2 Kd from 124 nM at pH 7.8 to 139 nM at pH 7.3 and 161 nM at pH 7.0.
Changes in the fura-2 Kd will lead to ‘apparent’ changes in [Ca2+]i of equal magnitude. However, under most conditions in the present study these effects are small compared with the actual changes in 350:380 ratio/[Ca2+]i recorded, and thus the actions of pHi on the Kd of fura-2 do not materially alter any of the conclusions reached. To give a quantitative example, application of NH4Cl caused a mean increase in the fura-2 ratio of about 0.4 ratio units in resting cells (see Results). Using the calibration parameters given above, and neglecting any effects of pHi this corresponds to an increase in calculated [Ca2+]i of at least 100%. The change in pHi that accompanied this increase in [Ca2+]i, an alkalosis of around 0.5 pH units, will cause the Kd of fura-2 to decrease by 10%, which will in turn cause an ‘apparent’ increase in [Ca2+]i of around 10%. The contaminating effects of pHi on the fura-2 Kd would thus account for 10% or less of the observed change in [Ca2+]i in this situation. Overall, when cells are alkalinized with NH4Cl, any increase in [Ca2+]i measured using fura-2 will be overestimated by a maximum of around 10%, while any decrease in [Ca2+]i will be underestimated by a similar amount. Conversely, when acetate is used to acidify the cell, any increase in [Ca2+]i will be underestimated by a maximum of around 15%.
The experimental situation producing the largest change in pHi in this study is wash-out of NH4Cl, which typically reduced pHi to values near 6.5–6.6. At a pHi of 6.5 the fura-2 Kd is expected to be around 250 nM, almost doubled from 139 nM at pHi 7.3 (Martínez-Zaguilán et al. 1996a). Therefore values of [Ca2+]i recorded during the NH4Cl wash-out phase (see Figs 2B, 7, 8 and 10) will almost certainly be substantially underestimated due to the accompanying reduction in fura-2 Ca2+ sensitivity. This effect can be expected to be most marked for wash-out following longer (6–7 min) exposures to NH4Cl (Figs 2B and 10), where acid loading will be greater than following short (2–4 min) exposures (Figs 7 and 8).
Figure 2. The effect of acetate and NH4Cl on [Ca2+]i in resting pancreatic acinar cells.

Traces show records of the 350:380 nm fura-2 ratio signal as an index of [Ca2+]i. A, the effect of 20 mm acetate; B, the effect of 20 mm NH4Cl. A and B from two different cells.
Figure 7. Effect of NH4Cl-induced cytosolic alkalinization on [Ca2+]i during agonist stimulation.
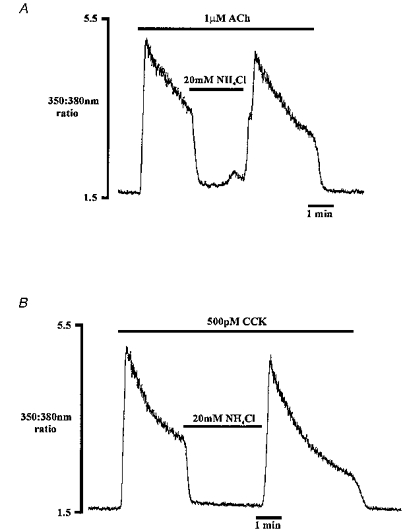
These traces show the effect of NH4Cl on ACh-evoked (A) and CCK-evoked (B) Ca2+ signals. The traces are from two different cells (both traces, n = 5).
Figure 8. Effect of NH4Cl on Ca2+ entry.
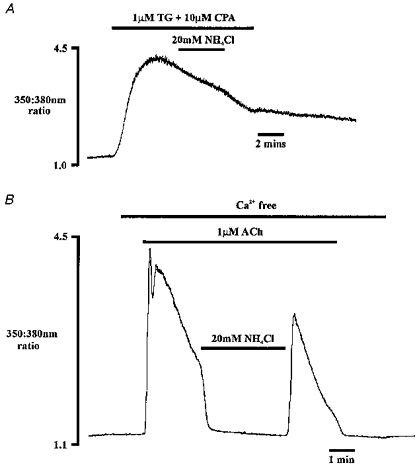
A, the effect of NH4Cl on [Ca2+]i during Ca2+ entry induced by microsomal Ca2+-ATPase inhibition using thapsigargin (TG, 1 μm) and cyclopiazonic acid (CPA, 10 μm). B, NH4Cl is applied during ACh stimulation in the absence of extracellular Ca2+. Traces shown are from two different cells.
Figure 10. Effect of cytosolic alkalinization on Ca2+ oscillations.

NH4Cl was applied during a series of Ca2+ oscillations induced by 10 pm CCK.
Materials
The standard experimental medium contained (mm): 104 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 2 CaCl2, 15 glucose and 25 Hepes. For Ca2+-free medium, Ca2+ was omitted and the medium also contained 0.5 mm EGTA. Media containing acetate, NH4Cl or trimethylamine were made by equimolar replacement of 20 mm NaCl with sodium acetate, NH4Cl, or trimethylamine. Experimental media were equilibrated with 100% O2 prior to use, and pH was set to 7.4 with NaOH. The CO2-HCO3−-buffered medium was made by replacement of 25 mm NaHepes with 25 mm NaHCO3, and was gassed continuously with 95% O2-5% CO2 to give a pH of 7.4. All experiments were carried out at room temperature (22–24°C).
Chemicals were obtained from the following suppliers: ACh, InsP3, thapsigargin, cyclopiazonic acid and nigericin from Sigma; CCK-8 from Bachem UK Ltd; fluorescent dyes from Molecular Probes, Europe; Streptolysin-O from Difco. All other chemicals were of analytical grade.
Statistics
Results are given as means ±s.e.m. Tests for differences between two means were made using Student's t test.
RESULTS
Effects of acetate/ammonium chloride on intracellular pH
In order to establish the size of the pHi changes evoked in experiments using acetate and NH4Cl, pancreatic acinar cells were loaded with the H+-sensitive dye BCECF. The resting pHi of pancreatic acinar cells in Hepes-buffered medium was found to be 7.33 ± 0.06 (n = 22), in good agreement with a previous study with BCECF-loaded pancreatic acinar cells (7.28; Muallem & Loessberg, 1990a). Addition of the weak acid acetate (20 mm) to the extracellular medium (extracellular pH constant at 7.4) decreased pHi by 0.30 ± 0.02 pH units (n = 11) as shown in Fig. 1A. Intracellular pH recovery during acetate exposure was extremely slow, with a rate of only 0.002 ± 0.001 pH units min−1 (n = 11). Application of a CO2-HCO3−-buffered medium acidified pHi to a similar extent as 20 mm acetate (not shown; decrease in pHi, 0.33 ± 0.04 pH units, n = 9).
Figure 1. The effect of acetate and NH4Cl on intracellular pH.

Representative records of BCECF fluorescence calibrated as pHi from two different pancreatic acinar cells. A, the effect of 20 mm acetate (extracellular pH, 7.4); B, the effect of 20 mm NH4Cl.
Figure 1B shows that application of 20 mm NH4Cl to pancreatic acinar cells resulted in a large alkaline shift in pHi of 0.47 ± 0.04 pH units (n = 11). During exposure to NH4Cl, there was some recovery towards resting pHi (rate of reacidification, 0.03 ± 0.003 pH units min−1, n = 11), probably resulting from the entry of NH4+ (Roos & Boron, 1981). On removal of NH4Cl, pHi rapidly acidified as NH4+ dissociated, releasing protons into the cytoplasm. Intracellular pH then slowly recovered towards resting pHi at a rate of 0.016 ± 0.007 pH units min−1 (n = 11). The slow pHi recovery would suggest that the Na+-H+ exchanger is relatively inactive in unstimulated pancreatic acinar cells under these conditions. A different weak base, trimethylamine (20 mm), also alkalinized pHi, although the pHi change was less than with NH4Cl (0.27 ± 0.02 pH units, n = 9; not shown).
Effects of intracellular pH change on [Ca2+]i in resting pancreatic acinar cells
Figure 2A illustrates that acidifying the cytosol with acetate had no effect on cytosolic [Ca2+] in resting pancreatic acinar cells (n = 5). Cytosolic acidification with a CO2-HCO3−-buffered medium also had no effect on [Ca2+]i (n = 4; not shown). In contrast, alkalinizing the cytoplasm with NH4Cl transiently increased [Ca2+]i (Fig. 2B). The mean change in ratio with 20 mm NH4Cl was 0.41 ± 0.05 ratio units (n = 11). Similar changes in [Ca2+]i were also noted with 20 mm trimethylamine (not shown; mean increase in ratio, 0.41 ± 0.07 pH units, n = 8). These observations are in contrast to an earlier study using pancreatic acinar cells, where addition of the weak acid propionate (20 mm) resulted in a small increase in [Ca2+]i, while NH4Cl failed to increase [Ca2+]i (Tsunoda, 1990). Washout of NH4Cl caused a slight but noticeable undershoot of the 350:380 nm ratio in approximately 50% of the cells studied (see e.g. Fig. 2B). However, this may represent an effect of the acidic pHi on the fura-2 Kd rather than a real undershoot in [Ca2+]i (see Methods for a discussion of this issue).
The ability of NH4Cl to increase [Ca2+]i in resting pancreatic acinar cells was examined in greater detail. In lacrimal acinar cells, NH4Cl was suggested to release Ca2+ from the same internal store as agonists (Yodozawa et al. 1997). This possibility was explored in pancreatic acinar cells using a combination of NH4Cl and ACh. Figure 3A illustrates that, following supramaximal stimulation with 10 μm ACh in a Ca2+-free medium, application of 20 mm NH4Cl failed to release any additional intracellular Ca2+. In contrast, application of NH4Cl prior to ACh stimulation resulted in clear intracellular Ca2+ release (Fig. 3B). No significant difference was observed between the magnitude of the ACh-induced increase in [Ca2+]i in the presence and absence of NH4Cl (change in ratio with ACh in the presence of NH4Cl: 1.81 ± 0.11 ratio units, n = 6, compared with 1.79 ± 0.1 ratio units, n = 7, in the absence of NH4Cl). These results suggest that ACh and NH4Cl release Ca2+ from the same intracellular stores. The protocol adopted in Fig. 3B also demonstrates the ability of NH4Cl to increase cytosolic Ca2+ in the absence of any extracellular Ca2+, implying that cytosolic alkalinization increases [Ca2+]i via Ca2+ release from internal stores.
Figure 3. The effect of NH4Cl on [Ca2+]i under Ca2+-free conditions.

The effect of NH4Cl following (A) and prior to (B) store depletion with 10 μm ACh. Traces shown are from two different cells and are each representative of five experiments.
Calcium release evoked by acetate during agonist stimulation
In pancreatic acinar cells, the secretagogues ACh and CCK induce both Ca2+ release from intracellular stores, via the generation of InsP3, and subsequent Ca2+ entry from the external medium (Williams & Yule, 1993). In Fig. 4A and B, application of these agonists, at submaximal concentrations (ACh, 0.5 μm; CCK, 100–500 pm), resulted in a rapid increase in cytosolic [Ca2+]. When acetate was applied during the falling phase of the Ca2+ signal, there was a second transient increase in [Ca2+]i, as previously observed in lacrimal acinar cells stimulated with ACh (Yodozawa, Grant & Elliott, 1994). Cytosolic acidification therefore enhanced agonist-evoked Ca2+ signals during stimulation with both ACh and CCK. The increase in [Ca2+]i evoked by acetate varied considerably between different cells (compare Fig. 4A and B).
Figure 4. The effect of ACh/CCK and acetate on [Ca2+]i.
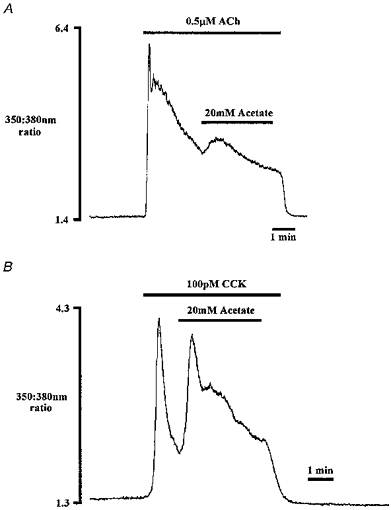
These traces show the effect of acetate-induced cytosolic acidification during the falling phase of theACh-evoked (A) and CCK-evoked (B) Ca2+ signal. Traces shown are from two different cells and are each representative of five experiments.
Acetate could produce this increase in [Ca2+]i by promoting Ca2+ entry from the external medium during agonist stimulation. This possibility was tested by repeating the protocol in Fig. 4A in the absence of extracellular Ca2+. In Fig. 5A, acetate application again resulted in a transient increase in [Ca2+]i during ACh stimulation, arguing against any involvement of Ca2+ entry (n = 8). The transient increase in [Ca2+]i evoked by acetate often resembled that of the initial phase of agonist stimulation (see Figs 5A and 4B), suggesting that the source of Ca2+ may be the intracellular store(s). The concentration effects of acetate were therefore investigated with respect to the degree of fullness of these stores. In Fig. 5B, the stores were maximally depleted using supramaximal ACh concentrations (10 μm), after which no second increase in [Ca2+]i was observed upon acetate application (n = 11). The increase in [Ca2+]i evoked by acetate during submaximal (2 μm) ACh stimulation (0.739 ± 0.15, n = 8; ratio change with ACh was 2.13 ± 0.31) was significantly larger (P < 0.0001) than during supramaximal (10 μm) ACh stimulation (0.05 ± 0.02, n = 11; ratio change with ACh was 3.39 ± 0.26). This would suggest that in agonist-stimulated pancreatic acinar cells the concentration effects of acetate are dependent upon the ‘fullness’ of the intracellular stores. The variable size of the acetate-evoked increase in [Ca2+]i (compare Figs 4A, 4B, and 5A) thus probably reflects varying degrees of prior Ca2+ store emptying by agonists in different individual cells.
Figure 5. Effect of acetate on [Ca2+]i during ACh stimulation.
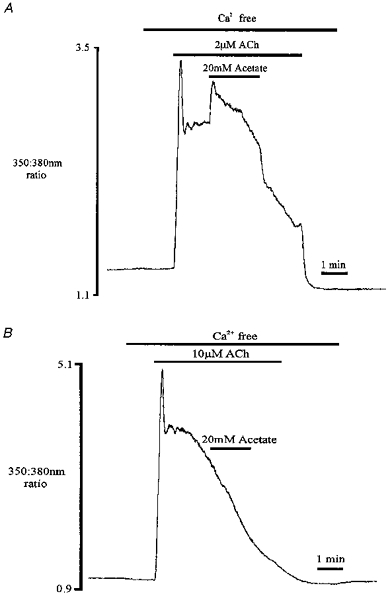
These traces show the effect of acetate addition during submaximal (2 μm; A) and supramaximal (10 μm; B) ACh stimulation under Ca2+-free conditions. Traces shown are from two different cells.
It was important to ascertain if the acetate-evoked increase in [Ca2+]i in agonist-stimulated cells resulted from a specific effect of acetate rather than an effect of intracellular acidification. This question was addressed using a CO2-HCO3−-buffered medium to acidify the cytosol (Roos & Boron, 1981). The Hepes-buffered medium was exchanged for a CO2-HCO3−-buffered medium during the plateau phase of agonist stimulation with 0.5 μm ACh. Figure 6 shows a representative experiment, illustrating that acidifying pHi with CO2 also increases cytosolic Ca2+ during agonist stimulation (n = 5). This therefore argues against any specific effects of acetate during the process of enhanced agonist-induced Ca2+ release.
Figure 6. Effect of cytosolic acidification on [Ca2+]i during ACh stimulation.
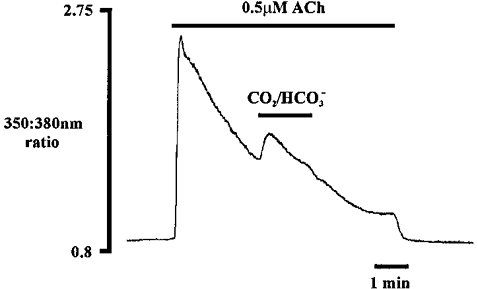
Cytosolic acidification was achieved with a CO2-HCO3−-buffered medium (n = 5).
Calcium signals are inhibited by NH4Cl during agonist stimulation
When NH4Cl was applied to cells stimulated with submaximal concentrations of ACh or CCK (1 μm and 500 pm respectively), there was a strikingly rapid decrease in [Ca2+]i back to the prestimulation level (Fig. 7A and B). Removal of NH4Cl, increased [Ca2+]i back to, or close to, the peak level. Indeed, this [Ca2+]i‘overshoot’ may be substantially underestimated due to the increase in the Kd of fura-2 under these conditions. A comparable degree of inhibition of Ca2+ signals was observed upon application of 20 mm trimethylamine during ACh stimulation (not shown; n = 6).
The second, sustained phase of Ca2+ signalling during agonist stimulation of non-excitable cells is generally attributed to Ca2+ entry from the external medium (Putney, 1986). The inhibitory effect exerted by NH4Cl during agonist stimulation in Fig. 7A and B might therefore reflect inhibition of Ca2+ entry from the external medium. This possibility was examined under conditions where Ca2+ entry could be studied in isolation, achieved using the microsomal Ca2+-ATPase pump inhibitors thapsigargin and cyclopiazonic acid (Mason, Garcia-Rodriguez & Grinstein, 1991). These inhibitors release Ca2+ from intracellular stores in the absence of any changes in inositol polyphosphate levels (Mason et al. 1991). As the stores become progressively depleted, Ca2+ entry is stimulated, i.e. capacitative or depletion-activated Ca2+ entry occurs (Putney, 1986). Surprisingly, 1 μm thapsigargin alone was unable to release Ca2+ from intracellular stores in our hands. Intracellular stores were effectively depleted only when using a combination of 1 μm thapsigargin and 10 μm cyclopiazonic acid, resulting in the observed increase in [Ca2+]i in Fig. 8A. During the period of sustained raised [Ca2+]i, the cytosol was alkalinized with NH4Cl. An inhibitory effect of NH4Cl alkalinization on Ca2+ entry would be expected to decrease [Ca2+]i. However, this manoeuvre had no effect on [Ca2+]i (n = 5). Therefore the decrease in [Ca2+]i observed during agonist stimulation in Fig. 7A and B is most likely not due to impaired Ca2+ entry.
The experiment illustrated in Fig. 7A was also repeated in the absence of extracellular Ca2+. Figure 8B shows that cytosolic alkalinization with NH4Cl still rapidly decreased [Ca2+]i during stimulation with 1 μm ACh (n = 6). This observation reinforces the suggestion that inhibition of Ca2+ signals by NH4Cl is not due to impaired Ca2+ entry.
A smaller return of ACh-induced response was observed upon removal of NH4Cl in Ca2+-free medium (Fig. 7A) than in Ca2+-containing medium (Fig. 8B) even though the changes in pHi should be very similar. This may reflect a lack of Ca2+ entry into the cell in the absence of extracellular Ca2+. Alternatively, this observation may be explained by greater store depletion under these conditions, since the cytosolic Ca2+ stores cannot be replenished in the absence of extracellular Ca2+.
Calcium oscillations and intracellular pH modification
In a number of cells, submaximal concentrations of CCK (10–150 pm) were used to induce Ca2+ oscillations. The exact CCK concentration required to elicit oscillations varied, probably reflecting both cell-to-cell variability and slight day-to-day variation in cell responsiveness. Figure 9A shows a typical experiment, where addition of 20 pm CCK elicited a series of small transient oscillations, or Ca2+ spikes, arising from baseline [Ca2+]i. When acetate was applied during stimulation with CCK, there was a large increase in the amplitude of the Ca2+ spikes (n = 5). This enhancement of amplitude persisted for the period of cytosolic acidification associated with acetate application. Upon removal of acetate, but in the continued presence of CCK, Ca2+ signals were inhibited for approximately 8 min in this particular cell.
Figure 9. Effect of cytosolic acidification on Ca2+ oscillations.
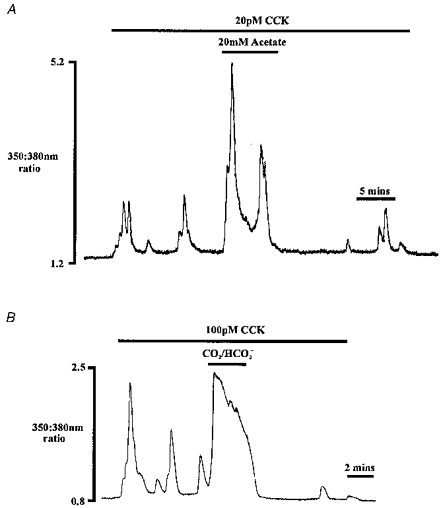
The effect of 20 mm acetate (A) and CO2-HCO3−-buffered medium (B) on oscillations evoked by 20 pm and 100 pm CCK, respectively. Traces shown are from two different cells.
Similar observations were made using a CO2-HCO3−-buffered medium to acidify cytosolic pH during a series of Ca2+ oscillations. Figure 9B shows an experiment where application of CO2 increased cytosolic Ca2+ above the amplitude of the spikes induced by 100 pm CCK alone. The elevated [Ca2+]i persisted for the duration of CO2 addition (n = 5). In three out of five cells, Ca2+ oscillations were inhibited for several minutes following removal of CO2 from the extracellular medium. In the remaining two cells, spike amplitude was reduced upon return to a Hepes-buffered medium.
Experiments were also performed using NH4Cl to alkalinize pHi. Figure 10 shows an experiment in which regular large transient oscillations in [Ca2+]i were produced using 10 pm CCK. Addition of NH4Cl to the extracellular medium abolished the oscillations, and [Ca2+]i transiently increased above the basal level (n = 5). Upon removal of NH4Cl, the oscillations in [Ca2+]i resumed. The first spike following removal of NH4Cl took noticeably longer to return to baseline levels of [Ca2+]i. The degree to which the basal [Ca2+] increased during NH4Cl exposure, and the enhancement of Ca2+ spikes upon NH4Cl removal, varied between the five individual cells tested, but the general pattern seen in Fig. 10 was observed in all five cells.
Calcium handling by the intracellular stores
The above data suggest that the application of acetate, and the subsequent cytosolic acidification, increases [Ca2+]i in agonist-stimulated pancreatic acinar cells by an action at the level of the intracellular Ca2+ store. The intracellular store(s) involved are presumably those sensitive to InsP3, as the acetate effect was only observed in the presence of an InsP3-generating secretagogue. The situation with NH4Cl is more complex, in that cytosolic alkalinization increased [Ca2+]i in resting cells, whereas during agonist stimulation Ca2+ signals were inhibited. However, the increase in cytosolic [Ca2+] generated by cytosolic alkalinization in resting cells is clearly due to Ca2+ release from intracellular stores, since removal of extracellular Ca2+ did not inhibit the [Ca2+]i response to NH4Cl. Modification of pHi could alter Ca2+ handling by the intracellular stores during agonist stimulation by acting on either the InsP3-receptor Ca2+ release channel or the microsomal Ca2+-ATPase pump. In order to assess whether pHi alters these processes we used the low-affinity Ca2+ indicator mag-fura-2 to monitor intraorganellar [Ca2+] in permeabilized cells. We examined the affects of altering both intraorganellar pH (using acetate and NH4Cl) and extraorganellar pH (using medium of various pHs) upon ATP-driven Ca2+ uptake and InsP3-induced Ca2+ release.
Intraorganellar pH and InsP3-induced calcium release
The resting mag-fura-2 ratio of the permeabilized cells was 0.94 ± 0.02 (n = 52). Addition of ATP (1 mm) to the superfusate resulted in an increase in the 350:380 nm ratio (Fig. 11), indicating an increase in organellar [Ca2+] as a result of ATP-dependent Ca2+ uptake. The actual change in ratio with the addition of ATP was an increase of 0.74 ± 0.04 ratio units (n = 26). A steady-state level of Ca2+ uptake was attained within 4–5 min. We first attempted to examine the effects of changing intraorganellar pH on Ca2+ handling by the Ca2+ stores. The Ca2+ stores loaded by mag-fura-2 in permeabilized pancreatic acinar cells are located in the endoplasmic reticulum (ER; van de Put & Elliott, 1996, 1997). Although there are no direct measurements of ER lumen pH in intact cells, the pH within all vesicular subcompartments is thought to be acidic with respect to the cytosol (see e.g. Anderson & Pathak, 1985, for references), and recent direct measurements of pH inside the Golgi apparatus, the organelle most closely related to the ER, show a pH of 6.2–6.5 (Seksek, Biwersi & Verkman, 1995; Kim, Lingwood, Williams, Furuya, Manolson & Grinstein, 1996). The pH gradients between acidic organellar subcompartments and the cytosol can be disrupted by addition of permeant weak bases (e.g. methylamine, NH4Cl, chloroquine), which accumulate in the acidic compartment and dissipate the pH gradient (see e.g. Niederau, Van Dyke, Scharschmidt & Grendell, 1986; De Lisle & Williams, 1987; Kim et al. 1996). However, when permeabilized cells were perfused with NH4Cl (Fig. 11) or acetate (both 20 mm, medium pH constant at 7.1), there was no change in the ratio signal and hence in intraorganellar Ca2+ content (acetate, n = 6; NH4Cl, n = 6). Figure 11 also shows that addition of 0.5 μm InsP3 caused a large drop in the mag-fura-2 fluorescence ratio, demonstrating that the Ca2+ stores in the permeabilized cells were capable of releasing Ca2+ in response to stimuli.
Figure 11. Effect of ATP, NH4Cl and IP3 on the Ca2+ content of intracellular Ca2+ stores.

The trace shows the 350:380 nm mag-fura-2 fluorescence ratio in a cluster of Streptolysin-O permeabilized pancreatic acinar cells loaded with mag-fura-2.
We went on to examine the rate of InsP3-induced Ca2+ release in combination with changing intraorganellar pH using acetate and NH4Cl. A final concentration of 0.5 μm InsP3 was added together with either 20 mm acetate or 20 mm NH4Cl to alter intraorganellar pH. The initial rates of InsP3-induced Ca2+ release were compared in the presence and absence of acetate/NH4Cl. The rate of Ca2+ release obtained with 0.5 μm InsP3 alone (0.46 ± 0.09 ratio units min−1, n = 11) was not significantly different from the rate obtained in the presence of acetate (0.33 ± 0.08 ratio units min−1, n = 10; P = 0.33 by an un-paired t test) or NH4Cl (0.34 ratio units min−1± 0.04, n = 11; P = 0.27 by unpaired t test). These results suggest that modifying intraorganellar pH does not affect either the steady-state intraorganellar [Ca2+] or the rate of InsP3-induced Ca2+ release.
Extraorganellar pH and InsP3-induced calcium release
Three different medium pH values were selected to examine the effects of extraorganellar (cytosolic) pH on ATP-dependent Ca2+ uptake and InsP3-induced Ca2+ release - pH 6.7, 7.1 (control) and 7.6. Medium pHs of 6.7 and 7.6 were selected to reflect the changes in pHi observed in the earlier experiments on applying acetate and NH4Cl respectively to pancreatic acinar cells. The permeabilized acinar cells were continuously superfused with experimental medium at a given pH following permeabilization and during application of ATP (1 mm) and InsP3 (0.5 μm).
There was no significant difference in the extent of ATP-dependent Ca2+ uptake by the intracellular stores (as judged by the steady-state change in ratio) at the different pHs. Pooling the data from all three pHs, the actual change in ratio with the addition of ATP was an increase of 0.76 ± 0.04 ratio units (n = 24). As described above (see Fig. 11), a steady-state level of Ca2+ uptake was achieved within 4 min, and subsequent application of InsP3 rapidly released Ca2+ from the intracellular stores.
The rates of Ca2+ uptake and release at the different medium pHs are given in Table 1. The rate of Ca2+ uptake varied markedly between different cell clusters at any given value of pH, and mean Ca2+ uptake rates at each of the medium pHs were not significantly different from one another. In contrast, rates of InsP3-induced Ca2+ release were fairly consistent between different cell clusters. Table 1 gives averaged data indicating that the rate of InsP3-induced Ca2+ release increased with increasing extraorganellar pH. The rate of Ca2+ release at pH 6.7 was significantly slower than at pH 7.1 (P < 0.05, unpaired t test) and pH 7.6 (P < 0.02, unpaired t test). There was no significant difference between the rate of Ca2+ release at pH 7.1 and pH 7.6. In contrast to the above results, when permeabilized cells were treated with a fivefold higher concentration of InsP3 (2.5 μm, a near-maximal concentration; see van de Put & Elliott, 1996), altering the pH had no effect on the rate of Ca2+ release (data not shown). This suggests that changing extraorganellar pH alters the sensitivity of the InsP3 receptor to InsP3 in pancreatic acinar cells.
Table 1.
Results of the study comparing the rate of Ca2+ uptake and release induced by 1 mm ATP and 0.5 μm InsP3, respectively, in single permeabilized pancreatic acinar cells
| Medium pH | Rate of Ca2+ uptake (t1/2) (s) | Initial rate of Ca2+ release (ratio units min−1) |
|---|---|---|
| 6.7 | 246 ± 92 (7) | 0.28 ± 0.08 (8)* |
| 7.1 | 403 ± 167 (6) | 1.55 ± 0.56 (6) |
| 7.6 | 575 ± 127 (9) | 2.93 ± 1.08 (6) |
Assessments of uptake and release rates were carried out at different medium pHs - 6.7, 7.1 (control) and 7.6. The rate of Ca2+ uptake was analysed by fitting a mono-exponential time course by non-linear regression to the data. The rate of Ca2+ release was not well fitted by a mono-exponential time course and is therefore given as the initial rate of change of the 350:380 ratio/Δtime (minutes).
Significantly different from rate at pH 7.1 and 7.6. Values of n given in parentheses.
DISCUSSION
The data described above illustrate that there is a relationship between pHi and [Ca2+]i in pancreatic acinar cells both under conditions of sustained Ca2+ signalling and during Ca2+ oscillations. In the present study, acetate application evoked a cytosolic acidification but had no effect on [Ca2+]i in unstimulated pancreatic acinar cells. In contrast, acidification of pHi during agonist stimulation resulted in a second increase in [Ca2+]i, as previously observed in lacrimal acinar cells (Yodozawa et al. 1994). In unstimulated cells, the weak base NH4Cl transiently increased cytosolic [Ca2+] by mobilizing Ca2+ from internal stores. However, cytosolic alkalinization with NH4Cl/trimethylamine dramatically inhibited Ca2+ signalling in the presence of secretory agonists. In addition, CCK-induced Ca2+ oscillations were enhanced by cytosolic acidification, and inhibited by cytosolic alkalinization. Therefore the effects of changing pHi upon [Ca2+]i in pancreatic acinar cells are essentially opposite depending upon whether the cells are resting or simultaneously stimulated with secretory agonists.
Ca2+ release in unstimulated cells
Application of NH4Cl to resting pancreatic acinar cells induced a transient increase in [Ca2+]i. Similar observations have been made in a number of other cell types (Siskind et al. 1989; Danthuluri et al. 1990), including our recent study in lacrimal acinar cells (Yodozawa et al. 1997). The resulting increase in [Ca2+]i in resting cells was much smaller than that evoked by ACh stimulation and also occurred in the absence of extracellular Ca2+, indicating that intracellular stores act as the source of Ca2+ (Fig. 3B). As in lacrimal acinar cells, NH4Cl appears to release Ca2+ from the same intracellular stores released by ACh, since application of NH4Cl had no effect on [Ca2+]i when the agonist-sensitive intracellular Ca2+ stores had been depleted with maximal concentrations of ACh under Ca2+-free conditions (Fig. 3A).
The Ca2+-releasing effect of alkalinization in resting cells might be explained by an increase in InsP3 concentration above basal levels. In lacrimal acinar cells, NH4Cl increased total inositol phosphate production, suggesting that NH4Cl may have released Ca2+ via an increase in InsP3 concentration (Yodozawa et al. 1997). However, intracellular alkalinization can also release Ca2+ from agonist-sensitive stores without altering basal InsP3 levels, e.g. in bovine aortic endothelial cells (Danthuluri et al. 1990). Our experiments in permeabilized cells clearly show that cytosolic alkalinization would be expected to enhance the rate of Ca2+ release for a given intracellular InsP3 concentration.
Ca2+ entry
Calcium entry has previously been reported to be sensitive to both pHo and pHi (Muallem, Pandol & Beeker, 1989). In pancreatic acinar cells, Tsunoda, Stuenkel & Williams (1990) found that intracellular alkalinization or acidification during Ca2+ influx, respectively, inhibited or enhanced Ca2+ influx. In the present study cytosolic acidification during agonist stimulation induced a secondary transient increase in [Ca2+]i in pancreatic acinar cells. If acidification/H+ were enhancing Ca2+ entry, one might expect the resulting increase in [Ca2+]i to be of a more sustained (rather than transient) nature, given that H+ extrusion during acetate exposure is relatively slow (Fig. 1A and B).
In both pancreatic (this study) and lacrimal acinar cells (Yodozawa et al. 1997), NH4Cl application during prolonged agonist stimulation produced a rapid reduction in [Ca2+]i. Upon removal of NH4Cl, the Ca2+ signals showed an ‘overshoot’ and returned to the initial peak level (Fig. 7A and B), probably as a result of the acid loading associated with NH4Cl removal (Fig. 1B). The ‘overshoot’ in [Ca2+]i may indeed be substantially larger than indicated by the traces shown, since the Kd of fura-2 increases during large acidifications, which has the effect of making the dye less sensitive to [Ca2+]i. The [Ca2+]i response to agonists during the plateau phase of sustained signalling is derived from increased Ca2+ influx (Marty & Tan, 1989; Kwan, Takemura, Obie, Thastrup & Putney, 1990). Since these observations were made in the presence of extracellular Ca2+, they could relate to a reduction in Ca2+ entry during agonist stimulation as a result of NH4Cl-induced cytosolic alkalinization. To test whether cytosolic alkalinization inhibited Ca2+ entry, microsomal Ca2+-ATPase inhibitors were employed to study the Ca2+ entry process in isolation following store depletion. Using a combination of cyclopiazonic acid and thapsigargin to deplete Ca2+ stores and promote Ca2+ entry, alkalinization with NH4Cl had no effect on [Ca2+]i (Fig. 8A). In addition, cytosolic alkalinization with NH4Cl rapidly decreased [Ca2+]i during agonist stimulation in the absence of extracellular Ca2+ (Fig. 8B) also arguing against any inhibitory effect of NH4Cl on Ca2+ entry in pancreatic acinar cells.
Since agonists and antagonists of the muscarinic receptor are quarternary NH4+ ions, it is conceivable that NH4+ might interfere with muscarinic receptor function, thereby explaining the rapid decrease in [Ca2+]i during ACh stimulation in pancreatic acinar cells. However, this would not explain why NH4Cl also inhibited the [Ca2+]i response to CCK (Fig. 7B).
Acetate enhances calcium signalling
The transient nature of the acetate-evoked increase in [Ca2+]i during agonist stimulation, together with the observation that this increase in [Ca2+]i does not persist following store depletion, suggests that acetate acts to release Ca2+ from intracellular stores. In addition, the intracellular store(s) involved are presumably those sensitive to InsP3, since the acetate effect is only observed in the presence of ACh or CCK.
There are several possible mechanisms by which acetate might enhance Ca2+ release from the intracellular stores and thus account for the observed rise in [Ca2+]i. Acetate/H+ might reduce Ca2+ re-uptake via inhibition of the microsomal Ca2+-ATPase. Inhibition of this type may produce a net increase in Ca2+ release from the intracellular stores. The data in Table 1 do not show any significant differences in the rate of Ca2+ uptake into stores in permeabilized pancreatic acinar cells at the various medium pHs. However, cell-to-cell variation in the rate of Ca2+ uptake was quite large, and this may have obscured the effect of pH.
Inositol 1,4,5-trisphosphate-mediated Ca2+ release from intracellular stores has been reported to be sensitive to H+ in a number of studies. Firstly, the binding of InsP3 to the InsP3 receptor increases at more alkaline pH in both brain and smooth muscle (Worley, Baraban, Supattapone, Wilson & Snyder, 1987; Joseph, Rice & Williamson, 1989; Varney, Rivera, Lopez Bernal & Watson, 1990). In addition, permeabilized human platelets released 30, 50 and 75% of stored Ca2+ when suspended in medium at pH 6.9, 7.1 and 7.4, respectively, showing that the affinity of the InsP3 receptor for InsP3 increases at more alkaline pHs (Brass & Joseph, 1985). In permeabilized smooth muscle cells, the sensitivity of the Ca2+ release mechanism to InsP3 and the rate of InsP3-induced Ca2+ release were both increased at more alkaline pHs in the presence of Ca2+, but pH had little effect in the absence of Ca2+ (Tsukioka, Iino & Endo, 1994). The fact that effects of pHi were only observed in the presence of Ca2+ in the latter study suggests some degree of cross-talk between InsP3, Ca2+ and H+ binding sites.
The available literature thus consistently shows an enhancement of the effects of InsP3 at more alkaline pHi. However, from the results on intact cells presented in this study, one might expect the converse to be true, i.e. the sensitivity and/or rate of InsP3-induced Ca2+ release to be reduced at more alkaline pHs. This is suggested by the fact that the weak base NH4Cl inhibits Ca2+ signalling during agonist stimulation, while the weak acid acetate enhances agonist-evoked Ca2+ release. This theory was examined directly by measuring intraorganellar [Ca2+] using mag-fura-2 in permeabilized pancreatic acinar cells. Addition of acetate/NH4Cl at a constant medium pH did not affect the rate of InsP3-evoked Ca2+ release. Since NH4Cl is known to alkalinize vesicular compartments, this suggests that intraorganellar pH is unlikely to have a dramatic effect on Ca2+ release. Consistent with this conclusion, the collapse of the zymogen granule pH gradient in pancreatic acinar cells induced by a number of lysosomotropic agents, including methylamine, did not affect CCK- or carbachol-stimulated amylase secretion (De Lisle & Williams, 1987).
We went on to examine the effects of extraorganellar ‘cytosolic’ pH on InsP3-evoked Ca2+ release. In single-cell intraorganellar [Ca2+] measurements, a concentration of 0.5 μm InsP3 was used to release Ca2+ from the stores. Increasing extraorganellar pH between 6.7 and 7.6 enhanced the rate of InsP3-induced Ca2+ release, probably by altering the sensitivity of the InsP3 receptor to InsP3. This is consistent with the work of Tsukioka et al. (1994). Calcium is a known co-agonist of InsP3-induced Ca2+ release from intracellular stores (Finch, Turner & Goldin, 1991), and the coupling domain of the InsP3 receptor contains putative binding sites for Ca2+ (Bezprozvanny & Ehrlich, 1995). It is possible that competition between Ca2+ and H+ could occur at the Ca2+ binding site(s). For example, an acidic pHi may result in decreased Ca2+ binding to the InsP3 receptor and hence a reduction in the open probability of the InsP3 receptor-linked channel. This may explain some of the earlier observations by Tsukioka et al. (1994), as well as our own results in permeabilized cells. However, these data clearly cannot explain the enhanced Ca2+ release from intracellular stores observed with acetate during agonist stimulation, or the rapid inhibition of Ca2+ signals by cytosolic alkalinization.
The question remains, then, of how acidification enhances Ca2+ release from the intracellular stores. An increase in InsP3 formation via an action of pHi at some point in the phosphatidylinositol pathway is a plausible option. Unfortunately, we were unable to investigate this possibility because of recurring technical problems with measuring InsP3 levels. In rat IMCD cells, NH4Cl removal and nigericin-induced cytosolic acidification significantly raised basal InsP3 levels under resting conditions (Slotki et al. 1993), while alkalinization raised the basal rate of inositol phosphate turnover in lacrimal acinar cells (Yodozawa et al. 1997).
Fluid secretion
The results of this study may be relevant to observations in many epithelia where a number of weak acids can substitute for HCO3− in fluid and electrolyte transport (Ullrich et al. 1971; Petersen et al. 1981; Case et al. 1982). For instance, in the rabbit mandibular salivary gland, acetate perfusion increased ACh-stimulated fluid secretion by more than 100% (Novak & Young, 1989). An increase in [Ca2+]i, such as that observed with acetate during ACh application, would be expected to lead to an increase in fluid secretion by causing activation of Ca2+-dependent Cl− and K+ channels. Interestingly, the acetate effect observed in Novak & Young's study was abolished in mandibular glands stimulated with supramaximal concentrations of ACh. In the present study, the Ca2+ releasing effect of acetate was also reduced with 10 μm ACh. An observation which argues against a mechanism based on changes in [Ca2+]i is that salivary flow rate in the mandibular gland remained elevated above the control value for several hours during perfusion with acetate (Novak & Young, 1989). However, in pancreatic acinar cells (this study) and lacrimal acinar cells (Yodozawa et al. 1994), the effects of acetate on [Ca2+]i were short lived. It therefore seems unlikely that enhancement of agonist-evoked intracellular Ca2+ release by acetate could fully explain the prolonged elevation in secretion in rabbit mandibular glands.
In perfused rabbit mandibular glands, the weak base NH4Cl had dramatic inhibitory effects upon ACh-evoked fluid secretion (Lau, Donohue & Case, 1996). Even at concentrations as low as 0.5 mm, NH4Cl totally inhibited fluid secretion. This observation is reminiscent of the inhibitory effects of NH4Cl upon Ca2+ signalling in this study and in lacrimal acinar cells (Yodozawa et al. 1997), although the concentrations of NH4Cl employed were quite different. Cytosolic alkalinization has been reported to influence electrolyte transport in a number of epithelia (see Lau, Elliott, Brown & Case, 1990; Yodozawa et al. 1997, for references).
In summary, many earlier observations indicate that cell permeant weak acids and bases, including acetate and NH4Cl, significantly modulate acinar cell function. These effects have not generally been attributed to altered cytosolic Ca2+ handling. However, the present study clearly shows that alterations in pHi induced by weak acids and bases can alter agonist-evoked [Ca2+]i signals. Changes in pHi have been reported to occur during agonist stimulation in many acinar cells, with the typical pattern of a transient intracellular acidosis followed by a sustained alkalosis (Muallem & Loessberg, 1990b; Seo, Larcombe-McDouall, Case & Steward, 1995; Steward, Poronnik & Cook, 1996). The data presented in this study suggest that these cytosolic pH changes occurring in acinar cells during stimulation are likely to modify Ca2+ signalling.
Acknowledgments
This work was supported by Wellcome Trust Programme Grant No. 035280/Z/92. T. S. held a Wellcome Prize Studentship.
References
- Anderson RGW, Pathak RK. Vesicles and cisternae in the trans Golgi apparatus of human fibroblasts are acidic compartments. Cell. 1985;40:635–643. doi: 10.1016/0092-8674(85)90212-0. [DOI] [PubMed] [Google Scholar]
- Berrie CP, Elliott AC. Activation of protein kinase C does not cause desensitization in rat and rabbit mandibular acinar cells. Pflügers Archiv. 1994;428:163–172. doi: 10.1007/BF00374854. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Ehrlich BE. The inositol 1,4,5-trisphosphate (InsP3) receptor. Journal of Membrane Biology. 1995;145:205–216. doi: 10.1007/BF00232713. [DOI] [PubMed] [Google Scholar]
- Brass LF, Joseph SK. A role for inositol triphosphate in intracellular Ca2+ mobilization and granule secretion in platelets. Journal of Biological Chemistry. 1985;260:15172–15179. [PubMed] [Google Scholar]
- Case RM, Conigrave AD, Favaloro EJ, Novak I, Thompson CH, Young JA. The role of buffer anions and protons in secretion by the rabbit mandibular salivary gland. Journal of Physiology. 1982;322:273–286. doi: 10.1113/jphysiol.1982.sp014037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthuluri NR, Kim D, Brock TA. Intracellular alkalinization leads to Ca2+ mobilization from agonist-sensitive pools in bovine aortic endothelial cells. Journal of Biological Chemistry. 1990;265:19071–19076. [PubMed] [Google Scholar]
- De Lisle RC, Williams JA. Zymogen granule acidity is not required for stimulated pancreatic protein secretion. American Journal of Physiology. 1987;253:G711–719. doi: 10.1152/ajpgi.1987.253.6.G711. [DOI] [PubMed] [Google Scholar]
- Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- Grinstein S, Goetz JD. Control of free cytoplasmic calcium by intracellular pH in rat lymphocytes. Biochimica et Biophysica Acta. 1985;819:267–270. doi: 10.1016/0005-2736(85)90183-x. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hayashi T, Shigetomi T, Ueda M, Kaneda T, Matsumoto T, Tokuno H, Tomita T. Effects of ammonium chloride on membrane currents of acinar cells dispersed from the rat parotid gland. Pflügers Archiv. 1992;420:297–301. doi: 10.1007/BF00374462. [DOI] [PubMed] [Google Scholar]
- Joseph SK, Rice HL, Williamson JR. The effect of external calcium and pH on inositol trisphosphate-mediated calcium release from cerebellum microsomal fractions. Biochemical Journal. 1989;258:261–265. doi: 10.1042/bj2580261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lingwood CA, Williams DB, Furuya W, Manolson MF, Grinstein S. Dynamic measurement of the pH of the Golgi complex in living cells using retrograde transport of the verotoxin receptor. Journal of Cell Biology. 1996;134:1387–1399. doi: 10.1083/jcb.134.6.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan CY, Takemura H, Obie J, Thastrup O, Putney JW., Jr Effects of MeCh, thapsigargin, and La3+ on plasmalemmal and intracellular Ca2+ transport in lacrimal acinar cells. American Journal of Physiology. 1990;258:C1006–1015. doi: 10.1152/ajpcell.1990.258.6.C1006. [DOI] [PubMed] [Google Scholar]
- Lau KR, Donohue M, Case RM. An ammonium pulse inhibits fluid secretion. Pflügers Archiv. 1996;431:415–418. doi: 10.1007/BF02207280. [DOI] [PubMed] [Google Scholar]
- Lau KR, Elliott AC, Brown PD, Case RM. Bicarbonate transport by salivary gland acinar cells. In: Young JA, Wong PYD, editors. Epithelial Secretion of Water and Electrolytes. Berlin: Springer; 1990. pp. 171–187. [Google Scholar]
- Lattanzio FA., Jr The effects of pH and temperature on fluorescent calcium indicators as determined with Chelex-100 and EDTA buffer systems. Biochemical and Biophysical Research Communications. 1990;171:102–108. doi: 10.1016/0006-291x(90)91362-v. [DOI] [PubMed] [Google Scholar]
- Lattanzio FA, Jr, Bartschat DK. The effect of pH on rate constants, ion selectivity and thermodynamic properties of fluorescent calcium and magnesium indicators. Biochemical and Biophysical Research Communications. 1991;177:184–191. doi: 10.1016/0006-291x(91)91966-g. [DOI] [PubMed] [Google Scholar]
- Martínez-Zaguilán R, Gurulé MW, Lynch RM. Simultaneous measurement of intracellular pH and Ca2+ in insulin-secreting cells by spectral imaging microscopy. American Journal of Physiology. 1996a;270:C1438–1446. doi: 10.1152/ajpcell.1996.270.5.C1438. [DOI] [PubMed] [Google Scholar]
- Martínez-Zaguilán R, Martínez GM, Lattanzio F, Gillies RJ. Simultaneous measurement of intracellular pH and Ca2+ using the fluorescence of SNARF-1 and fura-2. American Journal of Physiology. 1991;260:C297–307. doi: 10.1152/ajpcell.1991.260.2.C297. [DOI] [PubMed] [Google Scholar]
- Martínez-Zaguilán R, Parnami G, Lynch RM. Selection of fluorescent ion indicators for simultaneous measurements of pH and Ca2+ Cell Calcium. 1996b;19:337–349. doi: 10.1016/s0143-4160(96)90074-3. [DOI] [PubMed] [Google Scholar]
- Marty A, Tan YP. The initiation of calcium release following muscarinic stimulation in rat lacrimal glands. Journal of Physiology. 1989;419:665–687. doi: 10.1113/jphysiol.1989.sp017892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MJ, Garcia-Rodriguez C, Grinstein S. Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Journal of Biological Chemistry. 1991;266:20856–20862. [PubMed] [Google Scholar]
- Muallem S, Pandol SJ, Beeker TJ. Modulation of agonist-activated calcium influx by extracellular pH in rat pancreatic acini. American Journal of Physiology. 1989;257:G917–924. doi: 10.1152/ajpgi.1989.257.6.G917. [DOI] [PubMed] [Google Scholar]
- Muallem S, Loessberg PA. Intracellular pH-regulatory mechanisms in pancreatic acinar cells. I. Characterization of H+ and HCO3− transporters. Journal of Biological Chemistry. 1990a;265:12806–12812. [PubMed] [Google Scholar]
- Muallem S, Loessberg PA. Intracellular pH-regulatory mechanisms in pancreatic acinar cells. II. Regulation of H+ and HCO3− transporters by Ca2+ mobilizing agonists. Journal of Biological Chemistry. 1990b;265:12813–12819. [PubMed] [Google Scholar]
- Niederau C, Van Dyke RW, Scharschmidt BF, Grendell JH. Rat pancreatic zymogen granules. Gastroenterology. 1986;91:1433–1442. doi: 10.1016/0016-5085(86)90197-6. [DOI] [PubMed] [Google Scholar]
- Novak I, Young JA. Acetate stimulates secretion in the rabbit mandibular gland. Pflügers Archiv. 1989;414:68–72. doi: 10.1007/BF00585628. [DOI] [PubMed] [Google Scholar]
- Petersen K-U, Wood JR, Schulze G, Heintze K. Stimulation of gallbladder fluid and electrolyte absorption by butyrate. Journal of Membrane Biology. 1981;62:183–193. doi: 10.1007/BF01998164. [DOI] [PubMed] [Google Scholar]
- Petersen OH. Stimulus-secretion coupling: cytoplasmic calcium signals and the control of ion channels in exocrine acinar cells. Journal of Physiology. 1992;448:1–51. doi: 10.1113/jphysiol.1992.sp019028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiological Reviews. 1981;61:297–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Schoenmakers TJM, Visser GJ, Flik G, Theuvenet APR. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. BioTechniques. 1992;12:870–879. [PubMed] [Google Scholar]
- Seagrave JC, Barker S, Curry M, Martinez JR. Effects of NH4Cl and dimethylamine on Cl− fluxes in resting and stimulated rat submandibular acinar cells. American Journal of Physiology. 1992;263:G558–565. doi: 10.1152/ajpgi.1992.263.4.G558. [DOI] [PubMed] [Google Scholar]
- Seksek O, Biwersi J, Verkman AS. Direct measurement of trans Golgi pH in living cells and regulation by second messengers. Journal of Biological Chemistry. 1995;270:4967–4970. doi: 10.1074/jbc.270.10.4967. [DOI] [PubMed] [Google Scholar]
- Seo JT, Larcombe-McDouall JB, Case RM, Steward MC. Modulation of Na+-H+ exchange by altered cell volume in perfused rat mandibular salivary gland. Journal of Physiology. 1995;487:185–195. doi: 10.1113/jphysiol.1995.sp020870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskind MS, McCoy CE, Chobanian A, Schwartz JH. Regulation of intracellular calcium by cell pH in vascular smooth muscle cells. American Journal of Physiology. 1989;256:C234–240. doi: 10.1152/ajpcell.1989.256.2.C234. [DOI] [PubMed] [Google Scholar]
- Slotki I, Schwartz JH, Alexander EA. Inter-relationship between cell pH and cell calcium in rat inner medullary collecting duct cells. American Journal of Physiology. 1993;34:C432–438. doi: 10.1152/ajpcell.1993.265.2.C432. [DOI] [PubMed] [Google Scholar]
- Steward MC, Poronnik P, Cook DI. Bicarbonate transport in rat parotid secretory cells. Journal of Physiology. 1996;494:819–830. doi: 10.1113/jphysiol.1996.sp021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry. 1979;18:2210–2218. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry. 1980;19:2396–2404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- Tsukioka M, Iino M, Endo M. pH dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in permeabilized smooth muscle cells of the guinea-pig. Journal of Physiology. 1994;475:369–375. doi: 10.1113/jphysiol.1994.sp020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda Y. Cytosolic free calcium spiking affected by intracellular pH change. Experimental Cell Research. 1990;188:294–301. doi: 10.1016/0014-4827(90)90173-8. [DOI] [PubMed] [Google Scholar]
- Tsunoda Y, Stuenkel EL, Williams JA. Characterization of sustained [Ca2+]i increase in pancreatic acinar cells and its relation to amylase secretion. American Journal of Physiology. 1990;259:G792–801. doi: 10.1152/ajpgi.1990.259.5.G792. [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Radtke HW, Rumrich G. The role of bicarbonate and other buffers on isotonic fluid absorption in the proximall convolution of the rat kidney. Pflügers Archiv. 1971;330:149–161. doi: 10.1007/BF00643031. [DOI] [PubMed] [Google Scholar]
- van de Put FHMM, Elliott AC. Imaging of intracellular stores in permeabilized pancreatic acinar cells. Journal of Biological Chemistry. 1996;271:4999–5006. doi: 10.1074/jbc.271.9.4999. [DOI] [PubMed] [Google Scholar]
- van de Put FHMM, Elliott AC. The endoplasmic reticulum can act as a functional Ca2+ store in all subcellular regions of pancreatic acinar cell. Journal of Biological Chemistry. 1997;272:27764–27770. doi: 10.1074/jbc.272.44.27764. [DOI] [PubMed] [Google Scholar]
- Varney MA, Rivera J, Lopez Bernal A, Watson SP. Are there subtypes of the inositol 1,4,5-trisphosphate receptor? Biochemical Journal. 1990;269:211–216. doi: 10.1042/bj2690211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Yule DI. Stimulus-secretion coupling in pancreatic acinar cells. In: Go VLW, Dimagno EP, Gardner JD, Lebenthal E, Reber HA, Scheele GA, editors. The Pancreas: Biology, Pathobiology, and Disease. 2. New York: Raven Press; 1993. pp. 167–189. [Google Scholar]
- Worley PF, Baraban JM, Supattapone S, Wilson VS, Snyder SH. Characterization of inositol trisphosphate receptor binding in brain. Journal of Biological Chemistry. 1987;262:12132–12136. [PubMed] [Google Scholar]
- Yodozawa S, Grant T, Elliott AC. Effects of acetate on intracellular calcium release in isolated rat lacrimal acinar cells. Journal of Physiology. 1994;479.P:10P. [Google Scholar]
- Yodozawa S, Speake T, Elliott AC. Intracellular alkalinization mobilizes calcium from agonist-sensitive pools in rat lacrimal acinar cells. Journal of Physiology. 1997;499:601–611. doi: 10.1113/jphysiol.1997.sp021953. [DOI] [PMC free article] [PubMed] [Google Scholar]