

Abstract
Using whole-cell and perforated-patch recordings, we have examined the part played by endogenous G-protein βγ subunits in neurotransmitter-mediated inhibition of N-type Ca2+ channel current ICa) in dissociated rat superior cervical sympathetic neurones.
Expression of the C-terminus domain of β-adrenergic receptor kinase 1 (βARK1), which contains the consensus motif (QXXER) for binding Gβγ, reduced the fast (pertussis toxin (PTX)-sensitive) and voltage-dependent inhibition of ICa by noradrenaline and somatostatin, but not the slow (PTX-insensitive) and voltage-independent inhibition induced by angiotensin II. βARK1 peptide reduced GTP-γ-S-induced voltage-dependent and PTX-sensitive inhibition of ICa but not GTP-γ-S-mediated voltage-independent inhibition.
Overexpression of Gβ1γ2, which mimicked the voltage-dependent inhibition by reducing ICa density and enhancing basal facilitation, occluded the voltage-dependent noradrenaline- and somatostatin-mediated inhibitions but not the inhibition mediated by angiotensin II.
Co-expression of the C-terminus of βARK1 with β1 and γ2 subunits prevented the effects of Gβγ dimers on basal Ca2+ channel behaviour in a manner consistent with the sequestering of Gβγ.
The expression of the C-terminus of βARK1 slowed down reinhibition kinetics of ICa following conditioning depolarizations and induced long-lasting facilitation by cumulatively sequestering βγ subunits.
Our findings identify endogenous Gβγ as the mediator of the voltage-dependent, PTX-sensitive inhibition of ICa induced by both noradrenaline and somatostatin but not the voltage-independent, PTX-insensitive inhibition by angiotensin II. They also support the view that voltage-dependent inhibition results from a direct Gβγ-Ca2+ channel interaction.
Modulation of Ca2+ influx through voltage-gated N-type Ca2+ channels (ICa) is a powerful mechanism for controlling neurotransmitter release (Lipscombe, Kongsamut & Tsien, 1989). Sympathetic superior cervical ganglion (SCG) neurones, which express primarily N-type Ca2+ channels (Plummer, Logothetis & Hess, 1989; Regan, Sah & Bean, 1991), have proved to be an excellent preparation with which to investigate G-protein-mediated ICa regulation. In these cells, ICa is a convergent target for inhibition by multiple G-protein-coupled neurotransmitter receptors (Hille, 1994). These include α-adrenoceptors (Schofield, 1990, 1991) and somatostatin receptors (Ikeda & Schofield, 1989; Shapiro & Hille, 1993). Inhibition by these receptors is dependent on voltage (i.e. involves a gating-shift), is ‘membrane-delimited’ (i.e. does not involve a cytoplasmic messenger) and involves Bordetella pertussis toxin (PTX)-sensitive G-proteins (see Hille, 1994).
In the case of noradrenaline (α-adrenoceptor)-induced inhibition, the responsible G-protein is probably Go (Caulfield et al. 1994). Further, recent experiments involving the overexpression of exogenous G-protein βγ subunits in SCG neurones, which replicated and occluded the effect of noradrenaline, suggest that G-protein βγ subunits, rather than the α subunit, might constitute the inhibitory subunit (Ikeda, 1996; Herlitze, Garcia, Mackie, Hille, Scheuer & Catterall, 1996). However, it has not yet been fully established that this is true for endogenous Gα-associated βγ subunits, nor has the role of βγ subunits been assessed in other forms of Ca2+ current inhibition, such as that produced by angiotensin II, which is not voltage sensitive, uses a different (PTX-insensitive) G-protein and involves a diffusible messenger (Shapiro, Wollmuth & Hille, 1994).
Accordingly, in the present experiments, we have used a complementary approach to that employed by Ikeda (1996) and Herlitze et al. (1996) involving forced expression of the C-terminal domain of β-adrenergic receptor kinase (βARK1) (GRK2), a kinase which binds Gβγ (Koch, Inglese, Stone & Lefkowitz, 1993). The βARK1 peptide has previously been shown to attenuate Gβγ-mediated activation of type II adenylate cyclase (Koch, Hawes, Inglese, Luttrell & Lefkowitz, 1994) and inhibition of the G-protein-activated inward rectifier channel GIRK1 (Reuveny et al. 1994). We find that the C-terminus domain of βARK1 antagonizes the voltage-dependent inhibitory effects of noradrenaline and somatostatin, but not the voltage-independent inhibitory effects of angiotensin II.
METHODS
Cell culture
Sympathetic neurones were isolated from superior cervical ganglia of 15- to 19-day-old Sprague-Dawley rats and cultured using standard procedures (Marrion, Smart & Brown, 1987). Briefly, rats were killed by CO2 inhalation and immediately decapitated. Following removal, the ganglia were desheathed, incubated initially in collagenase and then in trypsin. Triturated, centrifuged and resuspended cells were plated onto laminin-coated glass coverslips and incubated at 37°C and 5% CO2 in culture medium (L-15 plus 10% fetal bovine serum, 2 mm glutamine, 24 mm NaHCO3, 38 mm glucose, 50 U ml−1 penicillin, 50 U ml−1 streptomycin, 25 ng ml−1 7S nerve growth factor). Cells were refed the day following culture. All culture reagents were from Gibco except laminin, collagenase, trypsin, nerve growth factor (Sigma) and fetal bovine serum (Hyclone).
DNA plasmids
The human cDNA sequence of βARK1 coding for Gly495 to Leu689 was cloned into the vector pCIN1 using a strategy previously described (Koch et al. 1994). The bovine β1 and γ2 cDNAs were subcloned into pCDNA3. Plasmids were propagated in XL-1 Blue (Stratagene, Cambridge, UK), purified using Qiagen maxiprep columns (Hilden, Germany) and the presence of the inserts was confirmed by restriction analysis. The plasmids were then diluted (100–400 μg ml−1) in Ca2+-free KCl-based solution (composition (mm): KCl, 140; MgCl2, 1; Hepes, 10; 290 mosmol l−1, pH 7.3) containing 0.5% fluoroscein isothiocyanate-conjugated dextran (70 kDa, Molecular Probes) and microinjected into the nucleus of SCG neurones 2 days in culture as described elsewhere (Abogadie, Vallis, Buckley & Caulfield, 1997). Cells were maintained in culture for a further 24–48 h and identified for recording by fluorescence microscopy.
Electrophysiology
Ca2+ currents were measured from SCG neurones 3–4 days in culture using patch-clamp techniques. Patch electrodes (2–4 MΩ) for whole-cell recording were filled with the following solution (mm): CsCl, 130; MgCl2, 1; BAPTA, 10; CaCl2, 0.1; Na2ATP, 2; Na3GTP, 0.12; Hepes, 10; pH 7.2–7.3 with CsOH. The external solution consisted of (mm): NaCl, 130; KCl, 3; MgCl2, 1; Hepes, 10; tetrodotoxin (TTX), 0.0005; CaCl2, 2; glucose, 11 (pH 7.3 with NaOH). The osmolarity of all solutions was ∼300 mosmol l−1. Recordings were obtained with an Axopatch 200A amplifier (Axon Instruments) and filtered at 2–5 kHz. After seal rupture, the cell membrane capacitance and series resistance (4–8 MΩ) were compensated (80–90%) and periodically monitored. Only small (< 40 pF) SCG neurones were recorded to improve clamp. Ca2+ currents were typically elicited by the use of a double-pulse voltage protocol which consisted of a 5–10 ms test pulse applied before and after a 10–20 ms conditioning depolarizing step to +90 mV (see inset in Fig. 1) and were corrected for leak and capacitive currents. The amplitude of ICa was measured isochronally 4 ms after the onset of a test pulse from the current remaining in the presence of 500 μm Cd2+ (see inset in Fig. 3).
Figure 1. The C-terminus of βARK1 reduces ICa inhibition by noradrenaline and somatostatin but not by angiotensin II.
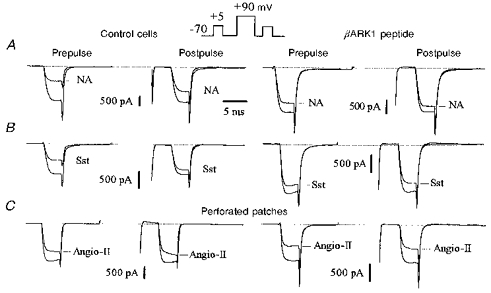
Superimposed Ca2+ current traces recorded in the absence or presence of noradrenaline (NA; 10 μm, A), somatostatin (Sst; 500 nm, B) and angiotensin II (Angio-II; 500 nm, C) in uninjected neurones (left panels, Control cells) and in neurones preinjected with 200 μg ml−1 of the βARK1 construct (right panels). Ca2+ currents were elicited by the double-pulse protocol as illustrated in the inset. The outward currents elicited by the conditioning voltage pulse to +90 mV (at break) are omitted. In this and subsequent figures we have referred to the test depolarizations before and after the conditioning voltage pulse as ‘prepulse’ and ‘postpulse’, respectively. Responses to angiotensin II in C were recorded using the perforated-patch method where access resistances were 10.5 MΩ (left traces) and 8 MΩ (right traces). ICa inhibition was measured at steady state, 4 s after application of noradrenaline or somatostatin and 15–18 s after application of angiotensin II. Cells were recorded 48 h after injection. In this and subsequent figures, the horizontal dotted lines at the top of the traces indicate the zero current level.
Figure 3. βARK1 peptide expression prevents voltage-dependent inhibition of ICa.
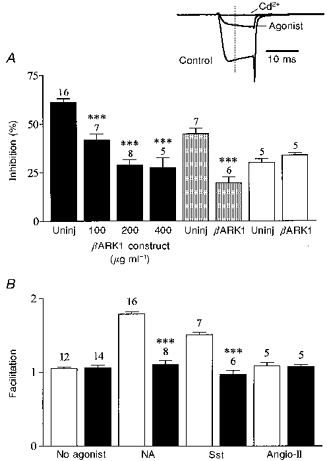
A, bar graph summarizing the effects of βARK1 peptide expression on ICa inhibition induced by noradrenaline (10 μm, filled bars), somatostatin (500 nm, shaded bars) and angiotensin II (500 nm, open bars). Uninj, uninjected control cells. Bars represent means ±s.e.m. for the number of cells indicated. Somatostatin and angiotensin II inhibition was recorded in cells preinjected with 200 μg ml−1βARK1 construct. Cells were recorded 48 h after injection. *** P < 0.0001, compared with the respective controls. Inset, ICa amplitude was measured isochronally 4 ms after the onset of the test pulse (dashed line) from the zero current level obtained in the presence of cadmium (Cd2+, 500 μm). ICa was elicited from −70 to +5 mV. B, bar graph summarizing the effects of the βARK1 peptide on the facilitation ratio (postpulse : prepulse) in the absence or presence of neurotransmitters as indicated. □, control; ▪, βARK1 peptide. *** P < 0.0001.
Some whole-cell Ca2+ currents were recorded using the amphotericin B perforated-patch method (Rae, Cooper, Gates & Watsky, 1991). Patch pipettes (2–3 MΩ) were filled by dipping the tip into a filtered standard Cs+-based solution (composition (mm): CsCl, 130; MgCl2, 1; Hepes, 10) for 40 s, after which the pipette was backfilled with the above solution containing 0.07–0.1 mg ml−1 amphotericin B. Access resistances after permeabilization ranged between 8 and 11 MΩ.
Drug solutions were made just before the experiments and applied for 10–30 s using a gravity-fed perfusion system at 5–10 ml min−1. All chemical compounds were from Sigma. Data were expressed as means ±s.e.m. Student's unpaired t test was applied to determine the statistical significance and differences were considered significant if P < 0.05. All experiments were performed at 33–34°C.
Immunocytochemistry
Following recordings, SCG neurones were acetone-fixed and stained using selective antibodies, alkaline phosphatase-conjugated secondary antibodies and the substrate BCIP/NBT (Dako) as previously described (Abogadie et al. 1997). The affinity-purified rabbit polyclonal anti-carboxy terminus of βARK1 (sc-562), anti-GαoA/B (sc-387) and anti-Gβ1-4 (sc-378) antibodies were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
RESULTS
Expression of βARK1 C-terminus reduced α-adrenoceptor and somatostatin receptor-mediated inhibition of ICa but not that induced by angiotensin II
N-type Ca2+ currents (ω-conotoxin GVIA sensitive) in rat SCG neurones are inhibited by noradrenaline (Schofield, 1990, 1991), somatostatin (Ikeda & Schofield, 1989; Shapiro & Hille, 1993) as well as by angiotensin II (Shapiro et al. 1994). Using whole-cell recording, noradrenaline (10 μm) and somatostatin (500 nm) rapidly (time to steady-state inhibition, ∼2-3 s) and reversibly inhibited ICa by 61.2 ± 1.9% (n = 16) and 44.8 ± 2.8% (n = 7), respectively. As previously reported, inhibition by noradrenaline and somatostatin was voltage dependent in that it was partly reversed (Elmslie, Zhou & Jones, 1990; Ikeda, 1991) following conditioning depolarization to +90 mV (see Fig. 1). Thus, in the presence of noradrenaline and somatostatin, the facilitation ratio (postpulse : prepulse) was 1.79 ± 0.5 and 1.51 ± 0.03, respectively. No facilitation (1.05 ± 0.04, n = 12) was observed in the absence of agonists. The ICa inhibition induced by angiotensin II (500 nm) was recorded using a perforated-patch electrode in order to limit perturbation of the intracellular milieu. By contrast with the PTX-sensitive inhibition, the angiotensin II-mediated inhibition (29.9 ± 1.9%, n = 5) of ICa was slow (time to steady-state inhibition, 15 ± 2 s) and completely voltage insensitive (facilitation ratio, 1.08 ± 0.05) (see Fig. 1). Pretreatment of cells with PTX (500 ng ml−1 for 27 h) reduced inhibition by somatostatin, noradrenaline and angiotensin II by 98 ± 2% (n = 5), 89 ± 4% (n = 4) and 16 ± 3% (n = 4), respectively.
To investigate whether neurotransmitter-induced inhibition involved Gβγ, we microinjected SCG neurones intranuclearly with plasmids (100–400 μg ml−1) containing cDNA inserts coding for the C-terminus domain of βARK1, which includes the Gβγ-binding domain (Koch et al. 1994; see Introduction). Cellular expression of the βARK1 peptide was immunochemically assessed 24–48 h later by using an antibody specific for the C-terminus of βARK1. All injected neurones showed strong immunoreactivity (Fig. 2), which increased as the concentration of the βARK1 construct was increased (data not shown). The expression of the βARK1 peptide had no appreciable effect on the synthesis of Gαo or Gβ subunits as immunoreactivity for GαoA/B or Gβ1-4 antibody was unchanged.
Figure 2. SCG neurone preinjected with the βARK1 construct (100 μg ml−1) displays strong immunoreactivity for the C-terminus of βARK1.

Upper panel, fluorescence image of an intranuclearly injected neurone. Lower panel, immunostaining for the C-terminus domain of βARK1 48 h after injection. The star indicates the injected cell shown in the corresponding fluorescence image. Scale bar, 20 μm.
In whole-cell recordings, expression of the C-terminus of βARK1 had no significant effect on ICa density (54.2 ± 7 pA pF−1, n = 7; 57.3 ± 5 pA pF−1, n = 8; and 53.4 ± 8 pA pF−1, n = 5; in cells injected with 100, 200 and 400 μg ml−1βARK1 construct, respectively; 59.1 ± 2.2 pA pF−1 in uninjected cells, n = 10), current kinetics (τonset=1.3 ± 0.2 ms, n = 6; 1.2 ± 0.1 ms in uninjected cells), basal facilitation (1.06 ± 0.05, n = 6) or current- voltage relationships. However, the βARK1 peptide reduced the voltage-dependent noradrenaline-mediated inhibition of ICa in a dose-dependent fashion: by 31% (41.8 ± 3%, n = 7), 52% (29 ± 2.5%, n = 8) and 56% (27.5 ± 5%, n = 5) in cells injected with 100, 200 and 400 μg ml−1βARK1 construct, respectively (Figs 1 and 3A). At 200 μg ml−1, the βARK1 peptide construct also attenuated the inhibition induced by 500 nm somatostatin by 56%, to 19.6 ± 3% (n = 6; Figs 1 and 3A). In the presence of βARK1, the residual inhibition induced by noradrenaline and somatostatin was no longer reversed by large depolarization and the facilitation ratios were reduced to 1.1 ± 0.06 and 0.97 ± 0.05, respectively (Figs 1 and 3B). By contrast, expression of the βARK1 peptide had no significant effect (P = 0.12) on the inhibition of ICa in perforated-patch recordings by 500 nm angiotensin II (controls, 29 ± 1.9%; βARK1, 33.7 ± 1.3%; n = 5; Fig. 3). In cells preinjected with 200 μg ml−1βARK1 construct, a second application of agonist ∼5 min after wash out of the first application produced a similar amount of inhibition to that produced by the first application (angiotensin II, 30.2 ± 4%, n = 3; noradrenaline, 31 ± 5%, n = 4).
βARK1 peptide selectively prevents GTP-γ-S-induced voltage-dependent inhibition of ICa
Because of the ability of the βARK1 C-terminus to bind βγ dimers (Koch et al. 1993, 1994), the above results suggest that the βARK1 peptide acts by sequestering βγ subunits freed from their associated GTP-bound α subunits. Alternatively, the effects of the βARK1 peptide could result from a reduction in the amount of Gαβγ heterotrimer available for the receptors to activate (though the unaffected angiotensin II-mediated responses argue against this). This possibility was addressed by examining the effects of intracellular dialysis of GTP-γ-S on ICa in both uninjected cells and in cells overexpressing βARK1 peptide (400 μg ml−1 construct). We included GTP-γ-S (500 μm, GTP omitted) in the patch pipette and ICa was recorded as soon as possible after achieving whole-cell access. GTP-γ-S dialysis in control neurones produced a strong (83 ± 3%, n = 5) inhibition of ICa which had both fast (17 ± 3 s) and slow (95 ± 8 s) time constant phases (Fig. 4). The GTP-γ-S-mediated fast component of inhibition was accompanied by a change in the voltage dependence of ICa as judged by the parallel increase in the facilitation ratio, and was nearly abolished by PTX treatment (1 μg ml−1 for 24 h; n = 3). The latter finding was not expected since PTX is regarded as preventing receptor-G-protein interaction but not GTP-γ-S-induced G-protein dissociation (Huff & Neer, 1986). We interpret the data as indicating that uncoupling of receptor and G-protein induced by PTX treatment may have eliminated the ‘basal’ GTPase activation of Go/i-type G-proteins by unoccupied receptors, as previously suggested (Dolphin, 1991; Ito et al. 1991).
Figure 4. Modulation of ICa by GTP-γ-S in neurones overexpressing βARK1 peptide.
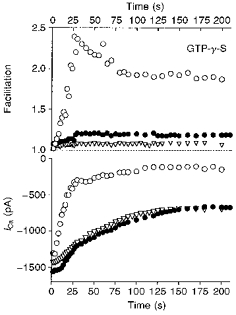
Changes in facilitation (upper panel) and peak Ca2+ currents (lower panel) during dialysis of GTP-γ-S (500 μm) in an uninjected neurone (○), in an uninjected neurone incubated with PTX (1 μg ml−1 for 24 h; ▿) and in a neurone preinjected with 400 μg ml−1βARK1 construct (•). Time 0 refers to the time at which an adequate access resistance was obtained. ICa was elicited by voltage steps from −70 to +5 mV as in Fig. 3.
The slow inhibitory component was mostly PTX resistant, voltage independent and reminiscent of inhibition by M1 muscarinic receptors and angiotensin II receptors (see Hille, 1994). In cells preinjected with the βARK1 construct, GTP-γ-S-induced inhibition of ICa exhibited only a slow time constant phase (82 ± 7 s; n = 4), showing that the fast PTX-sensitive component of inhibition was selectively prevented by the βARK1 peptide expression. These results indicate that βARK1 peptide expression mainly reduces PTX-sensitive and voltage-dependent pathways. In addition, they favour the view that the βARK1 peptide owes its effects solely to its ability to bind freed βγ subunits released from GTP-bound α subunits and is unlikely to disrupt G-protein-receptor interaction in a non-specific fashion as a PTX-insensitive G-protein activated by GTP-γ-S still inhibited ICa in the presence of βARK1 peptide (see also Koch et al. 1994).
βγ overexpression occludes noradrenaline and somatostatin inhibition but not angiotensin II inhibition
The above data suggest that endogenous βγ dimers mediate PTX- and voltage-sensitive inhibition of ICa but not PTX-insensitive and voltage-independent inhibition. If so, excess free Gβγ would be expected to occlude only the PTX-sensitive pathways (Ikeda, 1996). To test this, we co-injected plasmids containing cDNA inserts (400 μg ml−1) coding for β1 and γ2 subunits and ICa was recorded 24 h later using the perforated-patch method. As previously reported (Ikeda, 1996), expression of Gβ1γ2 induced a voltage-dependent inhibition of basal ICa (Fig. 5B). In these cells, the mean basal ICa density was significantly (P < 0.005) reduced from 49 ± 2 pA pF−1 (n = 6) to 30 ± 5 pA pF−1 (n = 4) and basal ICa was strongly facilitated (facilitation ratio, 1.66 ± 0.08) following conditioning depolarizations (Fig. 5B, lower traces). No change in basal ICa density was observed in cells overexpressing only β1 (n = 3; data not shown) or γ2 subunits (n = 3) (Fig. 5A). In agreement with the hypothesis that PTX-sensitive inhibition involved Gβγ, we observed that the responses to noradrenaline (see Ikeda, 1996; Herlitze et al. 1996) and somatostatin, but not to angiotensin II, were strongly prevented in cells overexpressing β1γ2 dimers (Fig. 5B) whereas no significant change was seen in neurones injected with Gβ1 (n = 3) or Gγ2(n = 3) cDNA alone (Fig. 5A). On average, in Gβ1γ2-overexpressing cells, inhibition of ICa was reduced by 60% (from 51 ± 2 to 19 ± 3%) and by 70% (from 43 ± 2 to 12.5 ± 4%) in the case of noradrenaline (300 nm; n = 4) and somatostatin (50 nm; n = 3), respectively, whereas it remained unchanged (from 31 ± 2 to 29 ± 3%) in the case of angiotensin II (500 nm; n = 3).
Figure 5. β1γ2 overexpression specifically occludes PTX-sensitive inhibition.
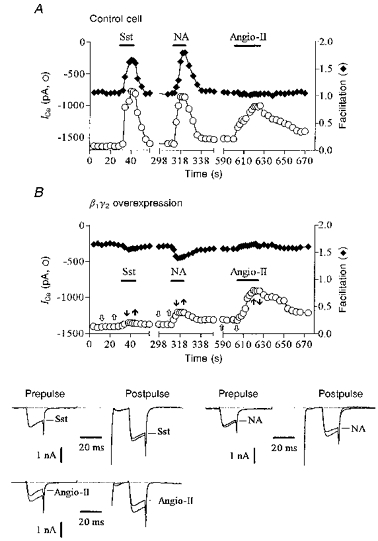
A and B, prepulse ICa amplitude (○) and facilitation (♦) plotted as a function of time in a control neurone (A; preinjected with 800 μg ml−1γ2 construct, 31 pF) and in a neurone co-expressing β1 and γ2 subunits (B; 400 μg ml−1 each of β1 and γ2 constructs, denoted β1γ2, 40 pF). Horizontal bars indicate the time and duration of application of somatostatin (50 nm), noradrenaline (300 nm) and angiotensin II (500 nm). The lower panel in B shows superimposed ICa traces selected before (open arrows) and during (filled arrows) application of agonists as indicated in the upper panel in B. ICa was recorded using the perforated-patch method and evoked using the double voltage pulse as illustrated in Fig. 1. Cells were recorded 24 h after injection.
βARK1 peptide antagonizes the effects of exogenous βγ subunits
We then tested whether the βARK1 peptide expression could antagonize overexpressed Gβγ. The plasmid encoding the C-terminus domain of βARK1 (400 μg ml−1) was therefore co-injected with β1 and γ2 cDNA plasmids (400 μg ml−1). Co-expression of the βARK1 peptide together with β1γ2 subunits effectively prevented the effects of Gβγ (Fig. 6). In these cells, the basal ICa density (45 ± 4 pA pF−1, n = 3) was not significantly different from that in uninjected neurones (49 ± 2 pA pF−1, n = 4) or neurones preinjected with Gβ1 (47 ± 4 pA pF−1, n = 3) or Gγ2 (51 ± 2 pA pF−1, n = 3) cDNAs and the Gβγ-dependent facilitation of basal ICa was absent (facilitation ratio, 1.09 ± 0.05). Decreasing the concentration of βARK1 construct (to 100–150 μg ml−1) co-injected with β1 and γ2 constructs was not able to prevent the βγ-induced basal ICa inhibition (34 ± 4 pA pF−1, n = 3).
Figure 6. Co-expression of the C-terminus domain of βARK1 prevents the effects of Gβ1γ2 dimers.
A, perforated-patch clamp recordings of Ca2+ currents evoked using the double-pulse voltage protocol (as in Fig. 1) in a neurone preinjected with β1- and γ2-encoding plasmids (400 μg ml−1, each; upper traces) and in a neurone in which the βARK1-generating plasmid (400 μg ml−1; lower traces) was co-injected with the β1 and γ2 constructs. The dashed lines delineate the level of facilitated ICa following conditioning depolarization. B, bar graph summarizing the effects of β1γ2 and β1γ2+βARK1 overexpression on basal ICa density (□) and facilitation (▪). Bars represent means ±s.e.m. for the number of cells indicated. ** P < 0.001. Cells were recorded using the perforated-patch method, 24 h after injection.
βARK1 peptide slows down reinhibition kinetics and causes long-lasting facilitation of ICa
It has been suggested that the voltage dependence of inhibition results from the voltage-dependent binding of a G-protein subunit (presumably the βγ subunit) to the Ca2+ channel (Lopez & Brown, 1991; Boland & Bean, 1993; Golard & Siegelbaum, 1993; Elmslie & Jones, 1994). If so, the rate of reinhibition following voltage-dependent facilitation should be slowed when the concentration of βγ subunits is reduced. We tested this by comparing the time course of reinhibition of ICa (decay of facilitation) during modulation by 10 μm noradrenaline in control cells and in cells expressing the βARK1 peptide (150 μg ml−1 construct, which gave 37.4 ± 4% inhibition of ICa, n = 4). The decay of facilitation was measured at the holding potential of −70 mV by applying test postpulses separated from the conditioning steps to +90 mV by a variable interval (Elmslie et al. 1990). The reinhibition developed exponentially with a 51 ± 6 ms time constant in the presence of βARK1 peptide (n = 4); this was significantly (P < 0.05) slower than that (37 ± 4 ms, n = 5) in control cells (Fig. 7A). A comparable slowing in the kinetics of reinhibition (τ= 48 ± 5 ms, n = 4) was observed in uninjected cells by lowering the noradrenaline concentration to 660 nm. These results indicate that the Ca2+ channels sense the concentration of free βγ subunits.
Figure 7. The βARK1 peptide modifies decay of facilitation.
A, representative examples of the decay of facilitation in an uninjected neurone (○) and in a neurone preinjected with 150 μg ml−1βARK1 construct (•) in the presence of 10 μm noradrenaline. Peak currents were normalized to the current measured with a 2.5 ms interval between the conditioning step to +90 mV and the test step to +5 mV. Smooth curves are exponential fits to the data. The inset shows the protocol used to measure the time course of inhibition and some superimposed current trace recordings of the βARK1-expressing cell. B, ICa amplitude plotted as a function of time in an uninjected neurone (left panel) and in a neurone expressing the βARK1 peptide (100 μg ml−1, right panel). The horizontal filled bars indicate the application of 10 μm noradrenaline and the dashed lines the time and duration for which iterative (4 Hz) depolarizations to +90 mV were applied. ICa was activated every 0.5–1 s, except immediately after the +90 mV conditioning depolarization (dashed line) where it was activated 10 ms, 80 ms, 200 ms and 1 s after the offset of the depolarization. Ca2+ currents were recorded in the whole-cell mode and elicited by voltage steps from −70 to +5 mV. C, superimposed current traces selected at the times indicated in B.
Another series of experiments was aimed at investigating whether Ca2+ channel modulation by Gβγ involves a ‘direct’ interaction of βγ with the channel. If this were the case, the βARK1 peptide should be able to cumulatively sequester βγ dimers freed from their interaction with Ca2+ channels during conditioning depolarization. Such an experiment is displayed in Fig. 7B and C. Upon noradrenaline application (25–30 s), ICa amplitude evoked at +5 mV was measured before and after repetitive (4 Hz for 5 s), long-duration (100 ms) membrane depolarizations to +90 mV. In control cells (n = 5), iterative depolarizations transiently relieved ICa inhibition (as did the double-pulse voltage protocol), which recovered in less than ∼100 ms (Fig. 7B and C, left panels). By contrast, in five out of six cells preinjected with 100 μg ml−1βARK1 construct (which only partly reduced ICa modulation, see above), the conditioning protocol caused a long-lasting facilitation of ICa throughout the noradrenaline application, which slowly (≫ 5 s) decayed (Fig. 7B and C, right panels). In two cells where noradrenaline was applied long enough for the inhibition to redevelop, recovery of inhibition (∼90%) required 18 and 23 s. These findings show that, in the presence of βARK1 peptide, some Ca2+ channels become unmodulated (facilitated) for some time following long depolarizations. This probably resulted from the cumulative sequestration of βγ dimers by βARK1 peptide as depolarization uncouples βγ subunits from Ca2+ channels, thereby identifying the βγ subunits as the modulators that interact, presumably directly, in a voltage-dependent fashion with the Ca2+ channel.
DISCUSSION
The principal new information from these experiments concerns the effects produced by forced expression of the carboxyl terminus domain (495–689) of βARK1. We found that the βARK1 polypeptide selectively antagonizes the voltage-dependent inhibition of the N-type Ca2+ current produced by both exogenous G-protein β1γ2 combination and by stimulation of the endogenous PTX-sensitive G-proteins with either noradrenaline or somatostatin. This provides strong evidence to support the view (derived from previous observations on the effects of overexpressing exogenous Gβγ subunits; Ikeda, 1996; Herlitze et al. 1996) that Ca2+ current inhibition produced by noradrenaline (see also Ikeda, 1996) and somatostatin results from the involvement of endogenous βγ subunits. This conclusion is reinforced by the finding that overexpression of βγ dimers selectively occludes noradrenaline and somatostatin inhibition. Thus, ICa inhibition by noradrenaline probably results from the inhibitory effect of endogenous Go-associated βγ subunits released from the αoβγ trimer following adrenoceptor-mediated activation of αo (see Caulfield et al. 1994). It also implies an equivalent effect of somatostatin, though the specific G-protein activated by somatostatin in sympathetic neurones has not yet been positively identified.
It is interesting to note that the residual inhibition induced by noradrenaline and somatostatin in the presence of βARK1 peptide was mostly voltage insensitive. This would suggest that the residual inhibition results from a distinct transducing signal which does not involve βγ dimers, and therefore might be mediated by GTP-bound α subunits. Alternatively, if one assumes that βγ dimers are responsible for this voltage-independent inhibition, this suggests that a higher ratio of βγ dimers to Ca2+ channels is required to produce voltage-dependent inhibition.
The C-terminal domain of βARK1 contains the signature sequence QXXER for Gβγ binding and has been shown to interact physically with Gβγ (Koch et al. 1993). This signature sequence is also present in the I-II cytoplasmic linker connecting the first and second transmembrane domains of the Ca2+ channel α1 subunit, thought to be the site of G-protein interaction (De Waard, Liu, Walker, Scott, Gurnett & Campbell, 1997; Zamponi, Bourinet, Nelson, Nargeot & Snutch, 1997; Page, Stephens, Berrow & Dolphin, 1997; see, however, Zhang, Ellinor, Aldrich & Tsien, 1996). Since we have shown that overexpression of this domain in SCG neurones antagonizes the effects of exogenous Gβγ, it is most likely that the βARK1 peptide exerts its effects by sequestering free Gβγ dimers, thereby competing with the Ca2+ channel protein for available Gβγ.
An alternative possibility is that the combination of βγ subunits with the βARK1 peptide might reduce the amount of αβγ heterotrimer available for receptor activation. However, this seems unlikely because the effects of the βARK1 peptide were specific for the voltage-dependent, presumed βγ-mediated, pathways (noradrenaline and somatostatin) and it did not affect the voltage-insensitive G-protein-dependent inhibition of ICa by GTP-γ-S, angiotensin II receptors and M1 muscarinic receptors (P. Delmas, unpublished data). This is in agreement with other studies using overexpressed βγ-sequestering agents (Koch et al. 1994; Macrez-Leprêtre, Kalkbrenner, Schultz & Mironneau, 1997), which have shown comparable specific effects of βARK1 C-terminus. Indeed, it seems likely that the affinity of βγ for the βARK1 peptide is quite low relative to its affinity for the GDP-bound α subunit. Thus, the slow recovery of inhibition we observed following depolarization-induced facilitation in cells expressing low concentrations of βARK1 peptide (Fig. 7B and C) seems to indicate that βγ dimers scavenged by βARK1 peptide are slowly released, and the fact that we did not observe any obvious use-dependence of the effects of the βARK1 peptide reinforces the idea that βARK1-βγ interaction is reversible.
Strong depolarization quickly relieves Ca2+ channels from G-protein-dependent inhibition (Elmslie et al. 1990; Ikeda, 1991). Reinhibition following depolarization has been attributed to the rebinding of the active G-protein subunit to the Ca2+ channels (Elmslie et al. 1990; Lopez & Brown, 1991; Boland & Bean, 1993; Golard & Siegelbaum, 1993; Elmslie & Jones, 1994). Using the double-pulse voltage protocol, we found that the βARK1 peptide slows the kinetics of reinhibition following voltage-dependent facilitation in a manner comparable to the slowing of reinhibition caused by decreasing the concentration of noradrenaline. The simplest explanation may be that the peptide reduced the rate of association of Gβγ with Ca2+ channels by transiently sequestering βγ subunits, thereby mimicking the concentration dependence of reinhibition (Golard & Siegelbaum, 1993; Elmslie & Jones, 1994; Ehrlich & Elmslie, 1995). Then, from the bimolecular reaction scheme: βγ+ C = Cβγ (where C is channel), the time constant for reinhibition (τ) would be governed by the concentration of free βγ according to the expression: τ−1=k1[βγ]+k2, where k1 and k2 are the forward and backward rate constants, respectively, and steady-state inhibition would be: I/Imax=k2/(k2+k1[βγ]). Thus, the change in steady-state inhibition produced by 10 μm noradrenaline following preinjection of 150 μg ml−1βARK1 construct (from 61 to 37%) implies a 62% reduction of [βγ], in which case, the reinhibition time constant would be expected to lengthen from 37 to 59 ms, which is not greatly different from the observed slowing (to 51 ms). This provides good evidence that the decay of facilitation reflects concentration-dependent reinhibition by βγ subunits, and is compatible with a direct interaction of βγ dimers with the Ca2+ channel (see De Waard et al. 1997; Zamponi et al. 1997).
However, during the continued presence of agonist and following repeated depolarization, there was a prolonged period of facilitation (in cells expressing a low concentration of βARK1 peptide; Fig. 7B), implying an additional, very slow phase of reinhibition lasting many seconds. One explanation for this might be that reinhibition normally involves the rebinding of the same βγ subunits released from the channels by the depolarizing prepulse. If some of these are sequestered by the βARK1 peptide, the rate of reinhibition will depend either on the binding of new receptor-generated βγ subunits (and hence will follow the same kinetics for the formation and diffusion of βγ following the original activation of the receptors, which is much slower (∼1 s); Jones, 1991; Beech, Bernheim & Hille, 1992; ∼2 s under our conditions) or on the rate of release of βγ from the βARK1 peptide.
In contrast to inhibition produced by noradrenaline and somatostatin, the PTX-insensitive and voltage-independent inhibition of ICa induced by angiotensin II was unaffected by βARK1 C-terminus expression. This suggests that βγ subunits are not involved in this regulation and raises the possibility that (related) voltage- and PTX-insensitive pathways (Hille, 1994) use α subunits to carry the signal. However, we cannot exclude the possibility that the βγ dimer of the G-protein coupled to angiotensin II receptors in sympathetic neurones might have a low affinity for the βARK1 Gβγ-binding domain, since various βγ combinations have different abilities to translocate the β-adrenergic receptor kinase (Muller, Hekman & Lohse, 1993; Daaka, Pitcher, Richardson, Stoffel, Robishaw & Lefkowitz, 1997). Nevertheless, our proposal that βγ subunits are not involved in the angiotensin II-mediated inhibition is reinforced by the finding that overexpression of Gβγ did not occlude angiotensin II responses. This implies that the effect of Gβγ and the ultimate effect of the (unknown) diffusible messenger system activated by angiotensin II are independent and directed at physically distinct sites on the Ca2+ channel protein (Page et al. 1997).
Thus, the effects of βARK1 peptide support the view (derived from previous experiments with exogenous βγ subunits) that voltage-dependent Ca2+ current inhibition following receptor activation of PTX-sensitive α subunit(s) results from the action of released endogenous βγ subunits, and would also be compatible with a direct interaction of the βγ subunit with the Ca2+ channel. On the other hand, the more ‘remote’ inhibition mediated by PTX-insensitive G-protein(s) probably involves the α subunit, rather than the βγ subunits.
Acknowledgments
We are grateful to Professor Graeme Milligan (University of Glasgow) who provided the β1 and γ2 constructs and to Dr Carol Harris and Dr Carol Scorer (Receptor Systems, Glaxo-Wellcome) for the βARK1 construct. We thank Dr Jane E. Haley for helpful discussions. This work was supported by The Wellcome Trust and the UK Medical Research Council.
References
- Abogadie FC, Vallis Y, Buckley NJ, Caulfield MP. Use of antisense-generating plasmids to probe the function of signal transduction proteins in primary neurons. In: Challiss RAJ, editor. Receptor Signal Transduction Protocols. Totowa, NJ, USA: Humana Press; 1997. pp. 217–225. 10.1385/0-89603-495-X:217. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bernheim L, Hille B. Pertussis toxin and voltage dependence distinguish multiple pathways modulating calcium channels of rat sympathetic neurones. Neuron. 1992;8:97–106. doi: 10.1016/0896-6273(92)90111-p. [DOI] [PubMed] [Google Scholar]
- Boland LM, Bean BP. Modulation of N-type calcium channels in bullfrog sympathetic neurons by luteinizing hormone releasing hormone: kinetics and voltage dependence. Journal of Neuroscience. 1993;13:516–533. doi: 10.1523/JNEUROSCI.13-02-00516.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Jones S, Vallis Y, Buckley NJ, Kim G-D, Milligan G, Brown DA. Muscarinic M-current inhibition via Gαq/11 and α-adrenoceptor inhibition of Ca2+ current via Gαo in rat sympathetic neurones. Journal of Physiology. 1994;477:415–422. doi: 10.1113/jphysiol.1994.sp020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Pitcher JA, Richardson M, Stoffel RH, Robishaw JD, Lefkowitz RJ. Receptor and Gβγ isoform-specific interactions with G protein-coupled receptor kinases. Proceedings of the National Academy of Sciences of the USA. 1997;94:2180–2185. doi: 10.1073/pnas.94.6.2180. 10.1073/pnas.94.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard M, Liu H, Walker D, Scott VES, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Regulation of calcium channel activity by GTP binding proteins and second messengers. Biochimica et Biophysica Acta. 1991;1091:68–80. doi: 10.1016/0167-4889(91)90224-l. 10.1016/0167-4889(91)90224-L. [DOI] [PubMed] [Google Scholar]
- Ehrlich E, Elmslie KS. Neurotransmitters acting via different G proteins inhibit N-type calcium current by an identical mechanism in rat sympathetic neurons. Journal of Neurophysiology. 1995;74:2251–2257. doi: 10.1152/jn.1995.74.6.2251. [DOI] [PubMed] [Google Scholar]
- Elmslie KS, Jones SW. Concentration dependence of neurotransmitter effects on calcium current kinetics in frog sympathetic neurones. Journal of Physiology. 1994;481:35–46. doi: 10.1113/jphysiol.1994.sp020417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmslie KS, Zhou W, Jones SW. LHRH and GTP-γ-S modify calcium current activation in bullfrog sympathetic neurons. Neuron. 1990;5:75–80. doi: 10.1016/0896-6273(90)90035-e. 10.1016/0896-6273(90)90035-E. [DOI] [PubMed] [Google Scholar]
- Golard A, Siegelbaum SA. Kinetic basis for the voltage-dependent inhibition of N-type calcium current by somatostatin and norepinephrine in chick sympathetic neurons. Journal of Neuroscience. 1993;13:3884–3894. doi: 10.1523/JNEUROSCI.13-09-03884.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G protein-coupled receptors. Trends in Neurosciences. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Huff RM, Neer EJ. Subunit interactions of native and ADP-ribosylated α39 and α41, two guanine nucleotide-binding proteins from bovine cerebral cortex. Journal of Biological Chemistry. 1986;261:1105–1110. [PubMed] [Google Scholar]
- Ikeda SR. Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. Journal of Physiology. 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, Schofield GG. Somatostatin blocks a calcium current in rat sympathetic ganglion neurones. Journal of Physiology. 1989;409:221–240. doi: 10.1113/jphysiol.1989.sp017494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Sugimoto T, Kobayashi I, Takahashi K, Katada T, Ui M, Kurachi Y. On the mechanism of basal and agonist-induced activation of the G protein-gated muscarinic K+ channel in atrial myocytes of guinea pig heart. Journal of General Physiology. 1991;98:517–533. doi: 10.1085/jgp.98.3.517. 10.1085/jgp.98.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SW. Time course of receptor coupling in frog sympathetic neurons. Biophysical Journal. 1991;60:502–507. doi: 10.1016/S0006-3495(91)82077-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gβγ-mediated signaling. Journal of Biological Chemistry. 1994;269:6193–6197. [PubMed] [Google Scholar]
- Koch WJ, Inglese J, Stone WC, Lefkowitz RJ. The binding site for the βγ subunits of heterotrimeric G proteins on the β-adrenergic receptor kinase. Journal of Biological Chemistry. 1993;268:8256–8260. [PubMed] [Google Scholar]
- Lipscombe D, Kongsamut S, Tsien RW. α-Adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N-type calcium-channel gating. Nature. 1989;340:639–642. doi: 10.1038/340639a0. 10.1038/340639a0. [DOI] [PubMed] [Google Scholar]
- Lopez HS, Brown AM. Correlation between G protein activation and reblocking kinetics of Ca2+ channel currents in rat sensory neurons. Neuron. 1991;7:1061–1068. doi: 10.1016/0896-6273(91)90350-9. 10.1016/0896-6273(91)90350-9. [DOI] [PubMed] [Google Scholar]
- Macrez-Leprêtre N, Kalkbrenner F, Schultz G, Mironneau J. Distinct functions of Gq and G11 proteins in coupling α1-adrenoreceptors to Ca2+ release and Ca2+ entry in rat portal vein myocytes. Journal of Biological Chemistry. 1997;272:5261–5268. doi: 10.1074/jbc.272.8.5261. 10.1074/jbc.272.8.5261. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Smart TG, Brown DA. Membrane currents in adult rat superior cervical ganglia in dissociated tissue culture. Neuroscience Letters. 1987;77:55–60. doi: 10.1016/0304-3940(87)90606-9. 10.1016/0304-3940(87)90606-9. [DOI] [PubMed] [Google Scholar]
- Muller S, Hekman M, Lohse MJ. Specific enhancement of β-adrenergic receptor kinase activity by defined G-protein β and γ subunits. Proceedings of the National Academy of Sciences of the USA. 1993;90:10439–10443. doi: 10.1073/pnas.90.22.10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page KM, Stephens GJ, Berrow NS, Dolphin AC. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. Journal of Neuroscience. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer MR, Logothetis DE, Hess P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron. 1989;2:1453–1463. doi: 10.1016/0896-6273(89)90191-8. 10.1016/0896-6273(89)90191-8. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of Neuroscience Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. 10.1016/0165-0270(91)90017-T. [DOI] [PubMed] [Google Scholar]
- Regan LJ, Sah DWY, Bean BP. Ca2+ channels in rat central and peripheral neurons: high-threshold current resistant to dihydropyridine blockers and ω-conotoxin. Neuron. 1991;6:269–280. doi: 10.1016/0896-6273(91)90362-4. 10.1016/0896-6273(91)90362-4. [DOI] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iñiguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature. 1994;370:143–146. doi: 10.1038/370143a0. 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- Schofield GG. Norepinephrine blocks a calcium current of adult rat sympathetic neurons via an α2-adrenoceptor. European Journal of Pharmacology. 1990;180:37–47. doi: 10.1016/0014-2999(90)90590-3. 10.1016/0014-2999(90)90590-3. [DOI] [PubMed] [Google Scholar]
- Schofield GG. Norepinephrine inhibits a Ca2+ current in rat sympathetic neurons via a G-protein. European Journal of Pharmacology. 1991;207:195–207. doi: 10.1016/0922-4106(91)90031-c. 10.1016/0922-4106(91)90031-C. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Hille B. Substance P and somatostatin inhibit calcium channels in rat sympathetic neurons via different G protein pathways. Neuron. 1993;10:11–20. doi: 10.1016/0896-6273(93)90237-l. 10.1016/0896-6273(93)90237-L. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Wollmuth LP, Hille B. Angiotensin II inhibits calcium and M current channels in rat sympathetic neurons via G proteins. Neuron. 1994;12:1319–1329. doi: 10.1016/0896-6273(94)90447-2. 10.1016/0896-6273(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 1996;17:991–1003. doi: 10.1016/s0896-6273(00)80229-9. 10.1016/S0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]