

Abstract
The cross-modulation of glycine responses by cyclic-AMP-dependent protein kinase (PKA) and protein kinase C (PKC) was determined in acutely dissociated trigeminal neurons.
Whole-cell glycine-evoked Cl− current (IGly) was recorded using the patch clamp technique. Protein kinases and their inhibitors were intracellularly perfused into the cells.
Both PKA and PKC when applied separately potentiated IGly.
When PKA and PKC were sequentially applied, PKC could not increase the IGly any further after the glycine responses were enhanced by PKA.
In 42% of our cells, IGly increased spontaneously. Endogenous PKA was found to mediate the increase. PKC had no effects on IGly in these cells.
The effect of PKA on IGly was studied in PKC-pretreated cells. PKA failed to potentiate IGly in these cells, suggesting that the PKA action also depends on the activity of PKC inside the cells.
These results suggest that the PKC action on IGly is conditional upon the modulation of the currents by PKA and vice versa. This cross-regulation of ligand-gated channel activity by protein kinases may play a role in neuronal integration and synaptic plasticity.
The glycine receptor (GlyR) is one of the major receptors mediating inhibitory synaptic transmission in nociceptive (pain) systems, including the spinal cord and the trigeminal nuclei. It consists of 48 kDa (α) and 58 kDa (β) subunits, forming a pentameric (3α : 2β) channel structure (Langosch, Thomas & Betz, 1988; Grenningloh et al. 1990), and an additional 93 kDa associated protein, gephyrin, essential for anchoring the receptor in the postsynaptic membrane (Betz et al. 1994). The α1 subunits of GlyRs can be phosphorylated by protein kinase C (PKC) in vitro and in intact cells (Ruiz-Gómez, Vaello, Valdivieso & Mayor, 1991; Vaello, Ruiz-Gómez, Lerma & Mayor, 1994). The phosphorylation site is located in the large intracellular loop, close to the predicted fourth transmembrane segment. Activation of endogenous cAMP-dependent protein kinase (PKA) also induces GlyR phosphorylation in adult rat spinal cord cells (Vaello et al. 1994). The phosphorylation is thought to occur in an alternative spliced form of the α1 subunit, α1ins, of the receptor (Malosio et al. 1991a; Vaello et al. 1994). The α1ins subunit contains eight additional amino acids located in the cytoplasmic loop following residue 325. The first serine residue of the insertion is presumably the phosphorylation site for PKA (Malosio et al. 1991a).
Functional studies of GlyR phosphorylation in various preparations have shown that both PKA and PKC change the amplitude of glycine currents (IGly). The direction of the changes, however, is not always consistent. For instance, cAMP potentiates IGly in spinal trigeminal neurons (Song & Huang, 1990) and in oocytes injected with rat brain poly (A)+ mRNA (Vaello et al. 1994) whereas, the same treatment results in a reduction of IGly in ventromedial hypothalamic neurons (Agopyan, Tokutomi & Akaike, 1993). Similarly, activation of PKC by phorbol esters is shown to reduce IGly in oocyte expression systems (Uchiyama, Hirai, Hishinuma & Akagi, 1994; Vaello et al. 1994; Nishizaki & Ikeuchi, 1995), but enhances IGly in cultured hippocampal neurons (Schönrock & Bormann, 1995). In addition, activation of endogenous PKC by serum potentiates IGly in the oocyte expressing αGlyR subunits (Nishizaki & Ikeuchi, 1995); intracellular application of PKC increases IGly in isolated trigeminal neurons (Gu & Huang, 1994) (Fig. 1) and substantia nigra neurons (Nabekura, Omura, Horimoto, Ogawa & Akaike, 1996).
Figure 1. PKC potentiates IGly in trigeminal neurons.

A, IGly was recorded 3 min before and 25 min after PKC was intracellularly perfused into the cell. The peak amplitude of IGly was increased by 2.5-fold in PKC. The current decay was fitted with a single exponential function (thicker continuous lines). The decay rate increased: the decay time constant τ was 14.5 s in control and 6.0 s in PKC. The τPKC: τcontrol was 0.4 in this cell. The membrane potential was held at −10 mV. The reversal potential of IGly was −46 mV. The lower graph shows changes in peak (•) and late (○) IGly in response to repeated applications of glycine. The duration of PKC application is indicated by a dotted horizontal line. Note the larger differences between peak and steady currents after PKC treatment. B, the bar graph summarizes the effect of PKC on the peak IGly in cells that do not display run-up (cf. Fig. 4). Peak current ratio was 2.00 ± 0.16 (n = 15) in PKC and 0.99 ± 0.04 (n = 3) in PKC plus the PKC inhibitor, PKCI(19–36). I0, Igly in absence of PKC.
Since glycine responses can be modulated by both PKA and PKC, it is important to determine if cross-modulation exists between these two second messenger systems. Vaello et al. (1994) observed that the phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA) produced a similar inhibitory action on IGly in control and in dibutyryl cAMP-treated oocytes and thus concluded that the modulatory effects of PKA and PKC were independent. This conclusion, however, could be obscured by non-specific actions of phorbol esters (Wilkinson & Hallam, 1994; Nishizaki & Ikeuchi, 1995). We therefore studied the effects of intracellularly applied PKA and PKC on IGly in trigeminal neurons and report here that PKA and PKC indeed cross regulate the activity of GlyR channels.
METHODS
Long-Evans rats (aged 10–15 days) were anaesthetized with methoxyflurane and killed by decapitation. Trigeminal neurons were acutely isolated according to previously described methods (Huang, 1989; Gu & Huang, 1991). In brief, the lower medulla was quickly removed and put in an ice-cold, oxygenated dissecting solution, consisting of (mm): NaCl, 120; KCl, 10; CaCl2, 1; MgCl2, 6; glucose, 10; and Pipes, 10; pH, 7.15; osmolarity, 305–315 mosmol l−1. The tissue was cut into 300 μm thick transverse slices with a vibratome slicer and incubated in the dissecting solution for 30 min at 34.5°C. The slices were then incubated with 2.7 unit ml−1 papain (Sigma P-3250) in the dissecting solution for 40–50 min at 34.5°C. After incubation, the tissue was washed with enzyme-free dissecting solution and stored at room temperature (21–23°C). Prior to an experiment, the spinal trigeminal nuclei in the caudal medulla were isolated from the tissue slice with a scalpel. Neurons were dissociated by triturating the tissue with a series of fire-polished Pasteur pipettes.
Whole-cell currents were recorded using the patch clamp technique. The patch electrode had a diameter of ∼2-3 μm and a resistance between 3 and 4 MΩ. The currents were filtered at 3 kHz and sampled at 200 μs per point. The external solution contained (mm): NaCl, 140; KCl, 4; CaCl2, 2; MgCl2, 1; glucose, 10; Hepes, 10; pH, 7.4. Unless stated otherwise, 20 μm glycine was used to evoke IGly. The basic internal solution contained (mm): caesium methanesulphonate, 120; CsCl, 20; glucose, 10; Mg-ATP, 5; Hepes, 10; pH, 7.2. In PKC experiments, 2 mm CaCl2 and 5 mm BAPTA were added to the internal solution, whereas 0.1 mm CaCl2 and 1 mm BAPTA were used in PKCM experiments. All chemicals were of ultrapure grade. Amino acid-containing solutions were delivered to the recorded cell by gravity. A solenoid valve, which was controlled by computer pulses, was used to start and stop the solution flow. By keeping the dead volume small and the flow rate high, the solution change could be accomplished within 10–20 ms (Gu & Huang, 1991). Glycine solution was washed out completely after each application, and glycine pulses were applied less frequently than once every 2 min. Under these conditions, the run-down of the IGly was minimal. Protein kinases and their inhibitors were intracellularly perfused into the recording cell through two thin glass tubes inserted into the patch pipette (Chen & Huang, 1991; Chen & Huang, 1992); the ends of the tubes were placed within 100 μm of the tip of the pipette. The concentration of chemicals used was as follows: 20 μm glycine, 100 unit ml−1 PKA, 10 μm wiptide (Peninsula Lab., Belmont, CA, USA), 10 μm of the PKC inhibitor PKCI(19–36), 6.6 μm chelerythrine. In earlier experiments, PKC (0.252 unit ml−1) was used. Diolein (2 μg ml−1, Sigma) was added to the internal solution to activate PKC. In later experiments, a catalytically active form of PKC, PKCM, was used. PKCM did not require Ca2+ or phospholipid for its activity and was more potent (i.e. it gave a similar potentiation of IGly as PKC at a 5-fold less concentration (0.05 unit ml−1)). PKA, PKC, PKCM, PKCI(19–36) and chelerythrine were from Calbiochem (La Jolla, CA, USA). The χ2 method was used to analyse the data. The data are expressed as means ±s.e.m. The late current is defined as the last sampling point of a current record.
RESULTS
PKC potentiates glycine-activated Cl− currents
Figure 1A shows the effects of intracellularly perfused PKC on IGly. With 20 mm Cl− in the internal solution, glycine induced an outward current at the holding potential of −10 mV. As PKC perfused into the cell, the IGly amplitude increased from 1 to 2.5 nA. The effect usually lasted more than 30 min. The decay of the IGly in both control and with PKC infusion could be fitted with a single exponential. The decay of the IGly in PKC was faster; the ratio of the decay time constants (τPKC: τcontrol) in this case was 0.4. The change in the current kinetics could also be seen in the IGlyvs. time plot (Fig. 1A) where the differences between the peak and late currents grew following the PKC treatment. The increase in the rate of current decay is reminiscent of an increase in the rate of desensitization following phosphorylation of acetylcholine receptors (Hoffman, Ravindran & Huganir, 1994).
The potentiating effect of PKC was observed in 80% of the cells that did not display run-up (discussed later). The amplitude of the IGly increased 2-fold (Fig. 1B). The decay time constants were τcontrol of 7.5 ± 2.3 s and τPKC of 3.6 ± 1.1 s (n = 8). The mean τPKC: τcontrol was 0.47. These effects of PKC were specific because changes in current amplitude and kinetics were not observed in cells treated with PKC plus the PKC inhibitors, PKCI(19–36) (Fig. 1B) or chelerythrine (data not shown).
The effect of PKC on the voltage dependence of IGly was examined next. Figure 2A shows the peak IGly recorded at different holding potentials with and without PKC. The currents in both the control and the PKC-treated cells rectified slightly. PKC potentiated IGly to the same extent at various holding potentials; the reversal potential of the glycine responses was not affected. These results suggest that PKC does not change the voltage dependence of the IGly. We also studied the dose-response curves for glycine in cells treated with PKC (Fig. 2B). PKC enhanced glycine responses similarly at different glycine concentrations and did not change the EC50 of glycine (26.8 ± 3.6μm (n = 8) in control; 25.0 ± 3.6μm (n = 8) in PKC).
Figure 2. Voltage and concentration dependence of IGly in PKC.

A, peak current-voltage curves in control (○) and with PKC (•). Points are the mean values from 5 cells. For each cell, the currents at different potentials were normalized to the IGly measured at −80 mV before the PKC application. s.e.m. values larger than the symbol size are indicated by bars. PKC did not change the voltage dependence of IGly. B, dose-response curves for glycine. Symbols as A. The mean IGly values from 8 cells are presented. All the values were normalized to the IGly recorded with 20 μm glycine in the absence of PKC (i.e. Io). The continuous lines were drawn according to the equation I/Io=Rmax[Gly]nH/(EC50nH+[Gly]nH), where Rmax is the maximal current ratio and [Gly] is the concentration of glycine. The Hill coefficient, nH, was 2 in this case. The mean EC50 from the 8 cells was 26.8 ± 3.6 μm in control solution and 25.0 ± 3.6 μm in PKC. The change in the EC50 was not significant (P > 0.05). A lower concentration (0.168 unit ml−1) of PKC was used in this set of experiments.
PKC fails to potentiate glycine responses in PKA-pretreated cells
As we have shown previously (Song & Huang, 1990), intracellular application of the PKA catalytic subunit enhanced glycine responses (Fig. 3A). The enhancement was blocked by cAMP-dependent protein kinase inhibitor (PKAI) (data not shown). The potentiation by PKA could be observed in 77% of our cells that did not exhibit run-up. The peak currents were increased by about 1.5-fold (n = 10).
Figure 3. PKA occludes the enhancing effect of PKC.

A, PKA was first perfused into the cell. The IGly was potentiated 1.68-fold. After PKA treatment, PKC could not produce any additional enhancing effect on the IGly. The current traces shown were recorded at 8, 33 and 63 min. B, a bar graph summarizes the effect of PKA and PKA plus PKC. With PKA, the peak current increased 1.7-fold (i.e. I/Io=1.66 ± 0.14 (n = 4) where Io is the IGly before PKA application). In the same set of cells, additional PKC produced no further enhancement in IGly (I/Io=1.56 ± 0.12, P > 0.05).
To determine if cross-modulation of GlyRs exists between PKC and PKA, we studied the effect of PKC on cells pretreated with PKA. In those cells where PKA had an effect, we found that treatment with PKC did not produce any enhancement in the IGly (Fig. 3A). The peak current ratio in PKC plus PKA was 1.56 ± 0.12 (n = 4) which was similar to the current ratio (1.66 ± 0.14; n = 4) obtained with PKA alone (Fig. 3B). Since PKC modulated IGly in 80% of the cells, we expected that < 20% of PKA pretreated cells would not respond to PKC if the effect of PKC and of PKA were independent. Instead, none of the cells pretreated with PKA responded to PKC. This result has led us to conclude that PKC cannot potentiate the GlyR-mediated responses when the receptors are previously modulated by PKA.
PKA is not necessary for the PKC action
To test if PKA is required for the action of PKC, we studied the effect of PKC when the PKA activity was abolished by wiptide, a PKA inhibitor. Figure 4 gives an example of such an experiment. Wiptide was first perfused into the cell and the action of PKC was then studied. IGly changed very little with the wipetide treatment. Thus, the level of basal phosphorylation of GlyRs by PKA in those cells that showed no run-up was low. Under such conditions, PKC potentiated IGly to the same extent as in the control, suggesting that PKA phosphorylation is not necessary for the action of PKC.
Figure 4. PKCM potentiates IGly in the presence of PKA inhibitor.

After blocking PKA activity with wipetide, PKCM still enhanced IGly. The same result was observed in 3 cells.
The run-up of glycine currents results from activation of endogenous PKA
In thirty out of seventy-two cells that we studied, IGly increased spontaneously (i.e. run-up) in the initial period of whole-cell recordings. The run-up process took more than 15–20 min to reach a steady state. The increases were often substantial (> 2-fold; Fig. 5). In addition, the rate of current decay speeded up and the differences between peak and late currents increased with time (Fig. 5). These results suggest that GlyRs may be phosphorylated spontaneously by endogenous protein kinases. It has been suggested that endogenous PKC is responsible for the run-up of IGly (Schönrock & Bormann, 1995). To test if this was the case in our cells, we perfused PKCI(19–36) into those cells showing run-up within 10 min of the recording and before a steady state was reached. The inhibitor changed neither the rate nor the extent of the run-up (n = 5; Fig. 6A). On the other hand, when we perfused wiptide into the cells instead, the rate of run-up was sharply reduced (n = 14; Fig. 6B). The current amplitude remained constant afterwards (Fig. 5B) or was even reduced in some cases, depending presumably on the activity of phosphatases inside the cells. These results indicate that the run-up process is mediated by PKA, not by PKC, in trigeminal neurons. In our previous report (Song & Huang, 1990), the run-up of IGly and the change in the decay constant of IGly following PKA treatment were not as prominent. The major difference in the experimental conditions between the previous and the current study is that caesium methanesulphonate, instead of potassium fluoride, is the major component of the internal solution used here.
Figure 5. Spontaneous increases (run-up) in IGly.

A cell showed a spontaneous increase in IGly with repeated applications of glycine. The current reached a new steady value within 15–20 min. The peak IGly increased about 2.75-fold. Note again the faster decay of IGly and the gradual increase in the differences between the peak (•) and late (○) currents with time, suggesting the spontaneous phosphorylation of GlyRs.
Figure 6. PKA mediates the spontaneous increase in IGly.
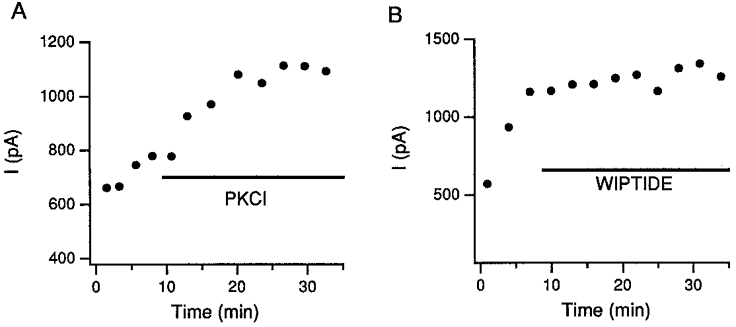
A, the rate and the extent of IGly increase did not change when PKCI(19–36) was injected into the cell during the run-up period. Similar results were observed in 5 cells. B, when a cell was perfused with wiptide during the run-up period (within 10 min of the recording), the increase in the IGly was stopped. Similar results were observed in 14 different cells.
PKC cannot potentiate glycine responses in cells showing run-up
If our conclusion that PKA modulation prevents further enhancement of IGly by PKC is correct, application of PKC should not enhance IGly in those cells that have exhibited run-up. This was indeed the case. If we let the current run up until it reached a new steady state, treatment with the PKC catalytic subunit, PKCM, had no effect on IGly (n = 4; Fig. 7A). To show that PKC could still modulate those GlyRs that had not been affected by endogenous PKA, the run-up process was stopped with wiptide before a new steady state was established. Adding PKCM afterwards in those cells resulted in additional increases in IGly (n = 4; Fig. 7B). Thus, PKC when applied separately readily enhanced glycine currents, but it could not increase the current any further when all the GlyRs had been modulated by PKA.
Figure 7. PKCM does not affect the IGly in cells that exhibit run-up.
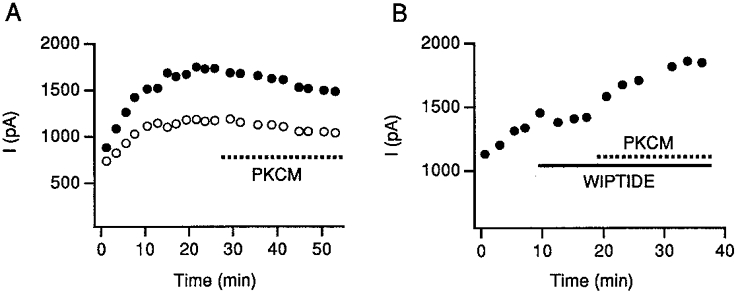
A, PKCM was applied to this run-up cell after allowing IGly to reach a new steady state. The kinase produced no additional effect on IGly. Both peak (•) and late (○) currents are shown. B, when a cell showing run-up was treated with wiptide before the IGly could reach a new steady state, PKCM elicited an additional enhancing effect on the glycine responses.
PKA cannot potentiate glycine currents in PKC-pretreated cells either
To determine if PKC also occludes the action of PKA, we studied the effect of PKA in cells pretreated with PKCM. PKCM was added to the patch pipette solution. As PKCM diffused into the cell, IGly increased gradually (Fig. 8A). Subsequent perfusion of PKA could not produce further increases in IGly (Fig. 8A and B). Thus, PKCM blocks the action of PKA. We therefore conclude, that the PKC action on glycine currents is occluded once the channels are phosphorylated by PKA and vice versa.
Figure 8. PKC pretreatment occludes the action of PKA.

A, PKA cannot potentiate IGly further in cells pretreated with PKCM. As PKCM in the patch pipette diffused into the cell, the amplitude of IGly gradually increased and the rate of current decay was enhanced. After the amplitude of IGly reached a steady state, adding PKA into the cell could not potentiate the IGly any further. B, the bar graph summarizes the results from 8 cells. PKA did not potentiate IGly in cells pretreated with PKC. Io is the IGly recorded in the presence of PKCM.
DISCUSSION
In this study, we show that intracellularly applied PKC and PKCM enhances IGly in isolated trigeminal neurons (Figs 1 and 4). We also confirm our previous finding that the catalytic subunit of PKA increases the glycine responses (Fig. 3). Moreover, using sequential applications of PKA and PKC, we demonstrate that the action of PKC depends on the activity of PKA inside the cells. When exogenous (Fig. 3) or endogenous (Fig. 6) PKA is elevated, PKC can no longer potentiate the glycine responses. Furthermore, we found that PKCM also occludes the action of PKA (Fig. 8). Although our results do not provide evidence for direct phosphorylation of GlyR channels in trigeminal neurons, the characteristic faster decay of IGly (Figs 1, 5 and 8) and the close correlation between phosphorylation of glycine, GABA and ACh receptors and the rate of desensitization of these receptors reported by others (Bouvier, 1990; Swope, Moss, Blackstone & Huganir, 1992; Hoffman et al. 1994; Vaello et al. 1994) have tempted us to suggest that the potentiation of IGly results from phosphorylation of GlyRs by PKA or PKC.
Activation of PKA and of PKC is found to change the amplitude of IGly in various preparations. But the direction of the changes is not always the same. This may result from the varying subunit composition of GlyRs in different cells. In situ hybridization experiments have shown that αGlyR subunit mRNAs are expressed in distinct regional patterns in the brain and in the spinal cord (Malosio, Marquèze-Pouey, Kuhase & Betz, 1991b). The amino acid residues in the transmembrane domain II determine the conductance of GlyR channels (Bormann, Rundström, Betz & Langosch, 1993) and serine/threonine residues in the intracellular loop flanked by the transmembrane domains III and IV are important for the phosphorylation of GlyRs (Ruiz-Gómez et al. 1991; Vaello et al. 1994). It is reasonable to assume that differences in subunit composition or phosphorylation sites could contribute to the different PKA and PKC effects. In addition, the expression of α1 and α2 GlyR subunits undergoes temporal changes: α2 transcripts are abundant in embryonic and neonatal rats, but are largely replaced by α1 transcripts after 2 weeks of postnatal development (Malosio et al. 1991b). A change in subunit composition could alter the phosphorylation properties of GlyRs. For instance, activation of PKA by forskolin results in phosphorylation of the α subunits of GlyRs only in adult spinal cord neurons (Vaello et al. 1994). PKA appears to phosphorylate specifically the α1ins subunit because an alternatively spliced form of the α1 subunit, α1ins, which represents ∼30% of the total α subunit mRNA, is found in the adult but not in the embryonic rat spinal cord and brainstem (Malosio et al. 1991a), and the α1ins possesses a potential phosphorylation site (Vaello et al. 1994). Since the subunit composition of GlyRs in most of the neurons studied is unknown, its determination in the future would help to explain the discrepancies in different preparations.
Another possible cause for the variability is the compounds that were used to activate second messengers. It is well known that the actions of forskolin, cAMP analogues and phorbol esters are complex, many of which are not related to the activation of protein kinases (Wagoner & Pallotta, 1988; Leidenheimer, Browning & Harris, 1991; Wilkinson & Hallam, 1994). Particularly relevant, Nishizaki et al. (1995) found that treatment with TPA reduced IGly in oocytes expressing α1 and α2 GlyRs, but activation of endogenous PKC by serum enhanced IGly in the same preparation. Since a specific PKC inhibitor could reverse only the effect of serum, but not the effect of TPA, PKC activation might not underlie the reduction of IGly by TPA in oocytes (Nishizaki & Ikeuchi, 1995).
The non-PKC related action of TPA could also explain why cross-modulation of GlyRs by PKA and PKC was not observed in Xenopus oocytes expressing GlyRs. In that preparation, Vaello et al. (1994) found that dibutyryl cAMP increased IGly, whereas TPA decreased glycine responses. Since the reduction of IGly by the phorbol ester remained unchanged in the oocytes pretreated with dibutyryl cAMP, they concluded that PKA and PKC modulations were independent (Vaello et al. 1994). If TPA had a direct action on GlyRs unrelated to PKC activation as suggested by Nishizaki et al. (1994), the blocking action of TPA would mask the enhancing effect associated with the PKC activation, thus obscuring the interaction between PKC and PKA. Moreover, phorbol esters are known to stimulate adenylyl cyclase activity (Jacobowitz & Iyengar, 1994). This could further complicate the data interpretation. By perfusing specific PKC or PKCM directly into cells in this study, we avoided the problems that might arise from using phorbol esters as PKC activators. Although the activation of phosphatase by PKA could explain the disappearance of the PKC effect (Hunter, 1995), this was unlikely in our case because we have not observed a decrease in peak glycine responses or a slowing of IGly decays following PKA application, signs that might accompany the dephosphorylation of GlyRs.
There are several possible mechanisms by which the cross-modulation between PKA and PKC could occur. One is that both PKC and PKA phosphorylate an intermediate protein which in turn modulates the glycine responses. Since GlyRs are directly phosphorylated by both kinases in vitro, (Ruiz-Gómez et al. 1991; Vaello et al. 1994), an intermediate protein is not necessary for the modulation. One cannot, however, eliminate the possibility that such a protein may be present in intact cells. Another potential mechanism for the cross-modulation is that PKC and PKA phosphorylate identical sites on a GlyR molecule. This mechanism is not likely because PKC has been shown to phosphorylate serine 391 of the α1 subunit (Vaello et al. 1994), while PKA appears to phosphorylate serine residue within the α1ins subunit (Malosio et al. 1991; Vaello et al. 1994). A third mechanism, which we favour, is that PKA and PKC phosphorylate their respective sites and phosphorylation of only one site is sufficient to result in the maximal change in IGly.
Cross-talk between PKA and PKC has been convincingly demonstrated biochemically. It has been known for some time that activation of PKC by phorbol esters phosphorylates and stimulates the activity of adenylyl cyclases, thus resulting in the activation of PKA (Sugden, Vanacek, Klein, Thomas & Anderson, 1985; Yoshimasa, Sibley, Lefkowitz & Caron, 1987; Bouvier, 1990; Yoshimura & Cooper, 1993). More recently, some reports have suggested that stimulation of cAMP production inhibits phosphoinositide breakdown, presumably leading to a decrease in PKC activity (Wen, Anwer, Singh & Sanborn, 1992; Liu & Simon, 1996). The discovery of an anchoring protein to which PKA, PKC and phosphatase 2B (calcineurin) can bind further suggests that the actions of these signalling enzymes are co-ordinated (Coghlan et al. 1995; Klauck, Faux, Labudda, Langeberg, Jaken & Scott, 1996).
The functional consequences of interactions between PKA and PKC pathways have also begun to emerge. For example, the reduction of peak Na+ currents by PKA requires phosphorylation of voltage-gated Na+ channels by PKC in CHO cells that express brain Na+ channels (Li, West, Numann, Murphy, Scheuer & Catterall, 1993). Elevation of cAMP is necessary for initiating Ca2+-calmodulin-mediated long term potentiation in hippocampal neurons (Blitzer, Wong, Nouranifar, Iyengar & Landau, 1995). Our results here suggest that even if both PKC and PKA are fully activated, their effects on IGly are not additive. Thus, the modulatory effect of a second messenger on channel activity is plastic; it depends on the phosphorylated state of the channel in the signalling pathways. The co-ordination among different second messenger systems allows a neuron to integrate inputs and adapt to changes in the external environment.
Following tissue damage, spinal and trigeminal neurons often develop super-sensitivity to noxious stimuli (Willis, 1992). Concurrently, the levels of PKC and possibly PKA are elevated (Coderre, 1992; Mao, Price, Mayer & Hayes, 1992; Lin, Peng & Willis, 1996). Under these conditions, phosphorylation of GlyRs should lead to a more effective inhibitory control. It is conceivable that cells may use a regulatory mechanism such as the one described here to self-limit channel activation and to prevent possible harmful effects of excessive phosphorylation.
Acknowledgments
The authors thank Drs H. Pant and M. Vasko for discussions and S.-Y. Wong for preparing trigeminal neurons. The work is supported by National Institutes of Health grants NS 30045 and NS 11255.
References
- Agopyan N, Tokutomi N, Akaike N. Protein kinase A mediated phosphorylation reduces only the fast desensitizing glycine current in acutely dissociated ventromedial hypothalamic neurons. Neuroscience. 1993;56:605–615. doi: 10.1016/0306-4522(93)90360-r. 10.1016/0306-4522(93)90360-R. [DOI] [PubMed] [Google Scholar]
- Betz H, Kuhse J, Fischer M, Schmieden V, Laube B, Kuryatov A, Langosch D, Meyer G, Bormann J, Rundstrom N, Matzenbach B, Kirsch J, Ramming M. Structure, diversity and synaptic localization of inhibitory glycine receptors. Journal of Physiology Paris. 1994;88:243–248. doi: 10.1016/0928-4257(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Bormann J, Rundström N, Betz H, Langosch D. Residues within transmembrane segment M2 determine chloride conductance of glycine receptor homo- and hetero-oligomers. EMBO Journal. 1993;12:3729–3737. doi: 10.1002/j.1460-2075.1993.tb06050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier M. Cross-talk between second messengers. Neuropeptides and Immunopeptides: Messengers in a Neuroimmune axis. Annals of the New York Academy of Sciences. 1990;594:120–129. doi: 10.1111/j.1749-6632.1990.tb40473.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Huang L-YM. Sustained potentiation of NMDA receptor mediated glutamate responses through activation of protein kinase C by a μ opioid. Neuron. 1991;7:319–326. doi: 10.1016/0896-6273(91)90270-a. [DOI] [PubMed] [Google Scholar]
- Chen L, Huang L-YM. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- Coderre TJ. Contribution of protein kinase C to central sensitization and persistent pain following tissue injury. Neuroscience Letters. 1992;140:181–184. doi: 10.1016/0304-3940(92)90097-q. [DOI] [PubMed] [Google Scholar]
- Coghlan VM, Perrino BA, Howard ML, Langeberg K, Hicks JB, Gallatin WM, Scott JD. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science. 1995;267:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- Grenningloh G, Pribilla I, Prior P, Multhaup G, Beyreuther K, Taleb O, Betz H. Cloning and expression of the 58 kd β subunit of the inhibitory glycine receptor. Neuron. 1990;4:963–970. doi: 10.1016/0896-6273(90)90149-a. 10.1016/0896-6273(90)90149-A. [DOI] [PubMed] [Google Scholar]
- Gu Y, Huang L-YM. Block of kainate receptor channels by Ca2+ in isolated spinal trigeminal neurons of rat. Neuron. 1991;6:777–784. doi: 10.1016/0896-6273(91)90174-x. 10.1016/0896-6273(91)90174-X. [DOI] [PubMed] [Google Scholar]
- Gu Y, Huang L-YM. Modulation of glycine receptor mediated chloride responses by protein kinase C. Neuroscience Abstracts. 1994;20:933. [Google Scholar]
- Hoffman PW, Ravindran A, Huganir RL. Role of phosphorylation in desensitization of acetylcholine receptors expressed in Xenopus oocytes. Journal of Neuroscience. 1994;14:4185–4195. doi: 10.1523/JNEUROSCI.14-07-04185.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L-YM. Calcium channels in isolated rat dorsal horn neurones including labelled spinothalamic and trigeminothalamic cells. Journal of Physiology. 1989;411:161–177. doi: 10.1113/jphysiol.1989.sp017566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- Jacobowitz O, Iyengar R. Phorbol ester-induced stimulation and phosphorylation of adenylyl cyclase 2. Proceedings of the National Academy of Sciences of the USA. 1994;91:10630–10634. doi: 10.1073/pnas.91.22.10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- Langosch D, Thomas L, Betz H. Conserved quaternary structure of ligand gated ion channels: the postsynaptic glycine receptor is a pentamer. Proceedings of the National Academy of Sciences of the USA. 1988;85:7394–7398. doi: 10.1073/pnas.85.19.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidenheimer NJ, Browning MD, Harris RA. GABAA receptor phosphorylation: multiple sites, actions and artifacts. Trends in Pharmacological Sciences. 1991;12:84–87. doi: 10.1016/0165-6147(91)90509-q. 10.1016/0165-6147(91)90509-Q. [DOI] [PubMed] [Google Scholar]
- Li M, West J, Numann WR, Murphy BJ, Scheuer T, Catterall WA. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- Lin Q, Peng YB, Willis WD. Possible role of protein kinase C in the sensitization of primate spinothalamic tract neurons. Journal of Neuroscience. 1996;16:3026–3034. doi: 10.1523/JNEUROSCI.16-09-03026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Simon MI. Regulation by cAMP-dependent protein kinase of a G-protein mediated phospholipase C. Nature. 1996;382:83–87. doi: 10.1038/382083a0. 10.1038/382083a0. [DOI] [PubMed] [Google Scholar]
- Malosio M-L, Grenningloh G, Kuhase J, Schmieden V, Schmitt B, Prior P, Betz H. Alternative splicing generates two variants of the α1 subunits of the inhibitory glycine receptor. Journal of Biological Chemistry. 1991a;266:2048–2053. [PubMed] [Google Scholar]
- Malosio M-L, Marquèze-Pouey B, Kuhase J, Betz H. Widespread expression of glycine receptor subunit mRNAs in the adult and developing rat brain. EMBO Journal. 1991b;10:2401–2409. doi: 10.1002/j.1460-2075.1991.tb07779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ, Hayes RL. Pain-related increases in spinal cord membrane-bound protein kinase C following peripheral nerve injury. Brain Research. 1992;588:144–149. doi: 10.1016/0006-8993(92)91354-h. 10.1016/0006-8993(92)91354-H. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Omura T, Horimoto N, Ogawa T, Akaike N. Alpha 1-adrenoceptor activation potentiates taurine response mediated by protein kinase C in substantia nigra neurons. Journal of Neurophysiology. 1996;76:2455–2460. doi: 10.1152/jn.1996.76.4.2455. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Ikeuchi Y. Activation of endogenous protein kinase C enhances currents through α1 and α2 glycine receptor channels. Brain Research. 1995;687:214–216. doi: 10.1016/0006-8993(95)00543-y. 10.1016/0006-8993(95)00543-Y. [DOI] [PubMed] [Google Scholar]
- Ruiz-Gómez A, Vaello M-L, Valdivieso F, Mayor F., Jr Phosphorylation of the 48-kDa subunit of the glycine receptor by protein kinase C. Journal of Biological Chemistry. 1991;266:559–566. [PubMed] [Google Scholar]
- Schönrock B, Bormann J. Modulation of hippocampal glycine receptor channels by protein kinase C. NeuroReport. 1995;6:301–304. doi: 10.1097/00001756-199501000-00019. [DOI] [PubMed] [Google Scholar]
- Song Y, Huang L-YM. Modulation of glycine receptor chloride channels by cAMP-dependent protein kinase in spinal trigeminal neurons. Nature. 1990;348:242–245. doi: 10.1038/348242a0. 10.1038/348242a0. [DOI] [PubMed] [Google Scholar]
- Sugden D, Vanacek J, Klein DC, Thomas TP, Anderson WB. Activation of protein kinase C potentiates isoprenaline-induced cyclic AMP accumulation in rat pinealocytes. Nature. 1985;314:359–361. doi: 10.1038/314359a0. [DOI] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Blackstone CD, Huganir RL. Phosphorylation of ligand-gated ion channels: a possible mode of synaptic plasticity. FASEB Journal. 1992;6:2514–2523. [PubMed] [Google Scholar]
- Uchiyama M, Hirai K, Hishinuma F, Akagi H. Downregulation of glycine receptor channels by protein kinase C in Xenopus oocytes injected with synthetic RNA. Molecular Brain Research. 1994;24:295–300. doi: 10.1016/0169-328x(94)90142-2. 10.1016/0169-328X(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Vaello M-L, Ruiz-Gómez A, Lerma J, Mayor F., Jr Modulation of inhibitory glycine receptors by phosphorylation by protein kinase C and cAMP-dependent protein kinase. Journal of Biological Chemistry. 1994;269:2002–2008. [PubMed] [Google Scholar]
- Wagoner PK, Pallotta BS. Modulation of acetylcholine receptor desensitization by forskolin is independent of cAMP. Science. 1988;240:1655–1657. doi: 10.1126/science.2454507. [DOI] [PubMed] [Google Scholar]
- Wen Y, Anwer K, Singh SP, Sanborn BM. Protein kinase-A inhibits phospholipase-C activity and alters protein phosphorylation in rat myometrial plasma membranes. Endocrinology. 1992;131:1377–1382. doi: 10.1210/endo.131.3.1324160. 10.1210/en.131.3.1377. [DOI] [PubMed] [Google Scholar]
- Wilkinson SE, Hallam TJ. Protein kinase C: is its pivotal role in cellular activation over-stated? Trends in Pharmacological Sciences. 1994;15:53–57. doi: 10.1016/0165-6147(94)90110-4. 10.1016/0165-6147(94)90110-4. [DOI] [PubMed] [Google Scholar]
- Willis WD. Hyperalgesia and Allodynia. 1992. pp. 1–400. The Bristol-Myers Squibb Symposium on Pain Research.
- Yoshimasa T, Sibley DR, Lefkowitz RJ, Caron MG. Cross-talk between cellular signalling pathways suggested by phorbol-ester-induced adenylate cyclase phosphorylation. Nature. 1987;327:67–70. doi: 10.1038/327067a0. 10.1038/327067a0. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Cooper DMF. Type-specific stimulation of adenylylcyclase by protein kinase C. Journal of Biological Chemistry. 1993;268:4604–4607. [PubMed] [Google Scholar]