

Abstract
The role of protein tyrosine phosphorylation and of G proteins in the activation of a swelling-activated Cl− current (ICl,swell) in calf pulmonary artery endothelial (CPAE) cells was studied using the whole-cell patch clamp technique. ICl,swell was activated by reducing the extracellular osmolality by either 12.5% (mild hypotonicity) or 25% (strong hypotonicity).
The protein tyrosine kinase (PTK) inhibitors tyrphostin B46, tyrphostin A25 and genistein inhibited ICl,swell with IC50 values of, respectively, 9.2 ± 0.2, 61.4 ± 1.7 and 62.9 ± 1.3μM. Tyrphostin A1, a tyrphostin analogue with little effect on PTK activity, and daidzein, an inactive genistein analogue, were without effect on ICl,swell.
The protein tyrosine phosphatase (PTP) inhibitors Na3VO4 (200 μM) and dephostatin (20 μM) potentiated ICl,swell activated by mild hypotonicity by 47 ± 9 and 69 ± 15%, respectively.
Intracellular perfusion with GTPγS (100 μM) transiently activated a Cl− current with an identical biophysical and pharmacological profile to ICl,swell. This current was inhibited by the tested PTK inhibitors and potentiated by the PTP inhibitors. Hypertonicity-induced cell shrinkage completely inhibited the GTPγS-activated Cl− current.
Intracellular perfusion with GDPβS (1 mM) caused a time-dependent inhibition of ICl,swell, which was more pronounced when the current was activated by mild hypotonicity.
Our results demonstrate that the activity of endothelial swelling-activated Cl− channels is dependent on tyrosine phosphorylation and suggest that G proteins regulate the sensitivity to cell swelling.
Outwardly rectifying Cl− currents activated by cell swelling (ICl,swell) have been described in many mammalian and non-mammalian cell types (for a review, see Strange, Emma & Jackson, 1996; Nilius, Eggermont, Voets, Buyse, Manolopoulos & Droogmans, 1997a). They are involved in several important physiological processes such as cell volume regulation, transport of osmolytes, regulation of the membrane potential and are possibly required for the progression through the cell cycle (see Nilius et al. 1997a). In contrast to the large biophysical and pharmacological knowledge of ICl,swell, not much is known about the signal transduction pathways that are involved in the regulation of the underlying Cl− channels.
Several observations point to a link between protein tyrosine phosphorylation and activation of ICl,swell. It has been reported that protein tyrosine kinase (PTK) inhibitors such as genistein can prevent the activation of ICl,swell in cardiac cells (Sorota, 1995) and the hypotonicity-induced efflux of I− in human Intestine 407 cells (Tilly, van den Berghe, Tertoolen, Edixhoven & de Jonge, 1993). Moreover, the protein tyrosine phosphatase (PTP) inhibitor Na3VO4 potentiated the hypotonicity-induced efflux of I− in the latter cell type. However, it has to be noted that others could not detect an effect of PTK inhibitors on ICl,swell (Gosling, Smith & Poyner, 1995; Szücs et al. 1996b).
A role for G proteins in the activation of ICl,swell has also been proposed. One indication comes from the observation that the intracellular application of GTPγS in chromaffin cells (Doroshenko, Penner & Neher, 1991), pigmented ciliary epithelial cells (Mitchell, Zhang, Wang & Jacob, 1997) and endothelial cells (Nilius, Oike, Zahradnik & Droogmans, 1994a; Nilius, Prenen, Voets, Van Den Bremt, Eggermont & Droogmans, 1997d) activates a Cl− current with strong similarities to ICl,swell. In addition, the inhibition of the small G protein p21rho by the Clostridium botulinum C3 exoenzyme reduced the hypotonicity-induced I− efflux in human Intestine 407 cells (Tilly et al. 1996).
In the present study we demonstrate that cell swelling and intracellular GTPγS activate an identical Cl− current in macrovascular endothelial cells. It was found that the activation of this Cl− current by either stimulus requires the phosphorylation of (a) tyrosine residue(s), as the current was efficiently blocked by several PTK inhibitors and potentiated by PTP inhibitors. In addition, we show that intracellular GDPβS, a compound known to inhibit G proteins, reduces the amplitude of ICl,swell. Finally, we demonstrate that the activation of the Cl− current by GTPγS can be prevented by cell shrinkage. Our results suggest that the activation of a G protein causes an increase in the sensitivity to cell swelling, rather than a direct activation of the signalling cascade that results in the activation of ICl,swell.
METHODS
Cells
We used single endothelial cells from an established cell line from calf pulmonary artery (cell line CPAE; American Type Culture Collection, Rockville, MD, USA). Cells were grown in Dulbecco's modified Eagle's medium (Life Technologies, Gibco) containing 20% fetal calf serum, 2 mM L-glutamine, 100 μg ml−1 streptomycin and 100 units ml−1 penicillin. They were then detached by exposure to 0.05% trypsin in a solution containing (mM): 140 NaCl, 6 KCl, 0.5 EDTA, 20 Tris (pH adjusted to 7.4 with HCl), reseeded on gelatine-coated coverslips, and kept in culture for 2–4 days before use. Only non-confluent cells were used.
Solutions
The standard extracellular solution was a Krebs solution, containing (mm): 150 NaCl, 6 KCl, 1 MgCl2, 1.5 CaCl2, 10 glucose, 10 Hepes, pH 7.4 with NaOH. The osmolality of this solution, as measured with a vapour pressure osmometer (Wescor 5500, Schlag, Gladbach, Germany), was 320 ± 5 mosmol kg−1. At the beginning of the patch clamp recordings, the Krebs solution was replaced by an isotonic Cs+ solution, containing (mm): 105 NaCl, 6 CsCl, 1 MgCl2, 1.5 CaCl2, 10 glucose, 90 mannitol, 10 Hepes, pH 7.4 with NaOH (320 ± 5 mosmol kg−1). The 12.5% (280 ± 5 mosmol kg−1) and 25% (240 ± 5 mosmol kg−1) hypotonic solutions were obtained by omitting, respectively, 45 and 90 mm mannitol from this solution. Hypertonic solutions were obtained by addition of 100 mm mannitol, resulting in an osmolality of 410 ± 5 mosmol kg−1. In anion substitution experiments, NaCl was fully replaced by NaI, NaBr or sodium gluconate. In most experiments, a standard pipette solution was used containing (mm): 40 CsCl, 100 caesium aspartate, 1 MgCl2, 1.93 CaCl2, 5 EGTA, 4 Na2ATP, 10 Hepes, pH 7.2 with CsOH (290 mosmol kg−1). The concentration of free Ca2+ in this solution was buffered at 100 nM, which is below the threshold for activation of Ca2+-activated Cl− currents (Nilius et al. 1997c) but sufficient to fully activate volume-activated Cl− currents (Szücs, Heinke, Droogmans & Nilius, 1996c) in CPAE cells. Similar results were, however, obtained when intracellular Ca2+ was less strongly buffered (0.1 mm EGTA, no CaCl2). When indicated, GTPγS and/or GDPβS were included in the standard pipette solution at concentrations of 100 μm and 1 mm, respectively. The Src-kinase activating peptides EPQ(pY)EEIPIA (kindly provided by Dr M. W. Salter, Division of Neuroscience, University of Toronto) and EEEPQ(pY)EEIPIYLELLP (kindly provided by Drs J. Goris and P. Agostinis, Department of Biochemistry, Katholieke Universiteit Leuven) and the inactive forms EPQYEEIPIA and EEEPQYEEIPIYLELLP were added to the pipette solution at a concentration of 1 mm.
Drugs
GTPγS (tetralithium salt), GDPβS (trilithium salt), Na3VO4, genistein, daidzein, quinidine hydrochloride monohydrate and tamoxifen were purchased from Sigma. The tyrphostins A1, A25 and B46 and dephostatin were purchased from Calbiochem. Mibefradil was kindly provided by Dr J.-P. Clozel, Hoffmann-La Roche, Basel, Switzerland.
Genistein, daidzein, the tyrphostins and dephostatin were added to the appropriate extracellular solution from 50 mm stocks in DMSO just before each experiment. It was found that the effects of these compounds were already strongly diminished after 1 h at room temperature (22–25°C) (see also Shuba, Asai, Pelzer & McDonald, 1996), which may explain some of the previous negative results (Szücs et al. 1996b). Quinidine was added from a 500 mm stock in DMSO, tamoxifen from a 100 mm stock in methanol and mibefradil from a 30 mm stock in H2O. Na3VO4 was added from a 20 mm aqueous stock, prepared as described by Gordon (1991). No concentration of DMSO (≤ 0.4%), methanol (≤ 0.01%) or Li+ (≤ 3 mm) had any effect on membrane currents.
Electrophysiological recordings
Currents were monitored with an EPC-7 (List Electronic, Lambrecht/Pfalz, Germany) patch clamp amplifier. Patch electrodes had DC resistances between 2 and 6 MΩ. A Ag-AgCl wire was used as reference electrode. In Cl− substitution experiments, a 3 M KCl-agar bridge was used and membrane voltages were corrected for liquid junction potentials as described by Neher (1992). Whole-cell membrane currents were measured using ruptured patches. Currents were sampled at 1 or 2 ms intervals and filtered at 500 or 200 Hz, respectively. Only patches with series resistances < 10 MΩ were retained to ensure a good diffusion of the pipette solution into the cytosol. Between 50 and 70% of the series resistance was electronically compensated to minimize voltage errors. The following voltage protocol was applied every 15 s from a holding potential of −20 mV: a step to −80 mV for 0.6 s, followed by a step to −100 mV for 0.2 s and a 2.6 s linear voltage ramp to +100 mV. Time courses of the whole-cell current were obtained by averaging the current at −80 mV during the first voltage step. In all experiments, time zero corresponds to the rupture of the membrane. Current-voltage relations were obtained from the currents measured during the linear voltage ramp. To study the voltage-dependent inactivation of the currents a step protocol was used consisting of 2 s voltage steps from a holding potential of −80 mV to potentials ranging from −100 to +100 mV in 20 mV increments.
Taurine efflux measurements
CPAE cells were seeded (20 000 cells per well) on gelatine-coated 12-well culture plates. Pre-confluent monolayers were incubated with 2 μCi [3H]taurine (Amersham, UK) for 3–6 h at 37°C. The cells were washed 6–8 times with normal Krebs solution at room temperature. On removal of the final wash, a further 1 ml of normal Krebs solution was added. After 2 min the solution was removed and transferred directly to a vial containing scintillation liquid. This procedure was repeated throughout the duration of the experiment, with the normal Krebs solution being replaced with isotonic or 25% hypotonic solutions containing the drug under study. Upon termination of an experiment, cells were lysed with 0.5 n NaOH, and radioactivity was counted in a liquid scintillation counter. The counts present in all collected fractions and those remaining in the cells at the end of the experiment were added together to obtain the total count value used for calculations. Efflux at each time point was calculated as fractional release, i.e. the radioactivity measured in each collected sample as a percentage of the total radioactivity found in the cells. Control wells (2 or 3) in each plate were kept in normal Krebs solution throughout the experiment, and the efflux in these wells was set to 1 (isosmotic efflux). Swelling-activated efflux is expressed as fold increase over the isosmotic efflux at each time point (relative efflux).
Analysis
Experiments were performed at room temperature (22–25°C).
Dose-response curves were fitted by a Hill equation of the form:
| (1) |
where Idrug and Icontrol are the current amplitudes in the presence and absence of the drug, respectively, C is the drug concentration, IC50 is the drug concentration for 50% inhibition and nH is the Hill coefficient.
Relative ion permeabilities (PX/PCl) were calculated from the shifts in reversal potential induced by the anion substitution using a modified Goldman-Hodgkin-Katz equation:
| (2) |
where ΔErev is the shift of the reversal potential, [Cl−]n and [Cl−]s are the extracellular Cl− concentrations in the normal and anion substituted hyposmotic solutions, respectively, and [X−]s is the concentration of the substituting anion X−. F is the Faraday constant, R the gas constant and T absolute temperature.
Pooled data are given as means ±s.e.m. from n cells. Significance was tested using Student's paired or unpaired t tests. Differences were considered significant at the level of P < 0.05.
RESULTS
Inhibition of ICl,swell by tyrosine kinase inhibitors
ICl,swell was activated by replacing the external isotonic Cs+ solution by a hypotonic Cs+ solution. This Cl− current has been previously described in endothelial cells (Nilius et al. 1994a; Nilius, Sehrer & Droogmans, 1994b; Szücs, Buyse, Eggermont, Droogmans & Nilius, 1996a). Contamination by other Cl− currents is unlikely, as Ca2+-activated Cl− currents were prevented by the buffering of intracellular Ca2+ at 100 nM, and cAMP- and voltage-activated Cl− currents cannot be detected in CPAE cells (Nilius, Szücs, Heinke, Voets & Droogmans, 1997e). ICl,swell had a mean amplitude of 40.2 ± 6.2 pA pF−1 at −80 mV and 140.8 ± 12.3 pA pF−1 at +100 mV for the 25% hypotonic solution (n = 65) and 12.4 ± 3.2 pA pF−1 at −80 mV and 40.8 ± 4.2 pA pF−1 at +100 mV for the 12.5% hypotonic solution (n = 48).
Figure 1A shows an experiment in which tyrphostin B46 (10 μm), a potent inhibitor of PTK activity (Gazit et al. 1991), was applied to the bath 5 min before superfusion of the cell with the 25% hypotonic solution. Tyrphostin B46 partially inhibited ICl,swell, as the current amplitude, measured at −80 mV, strongly increased after washout of the drug. Tyrphostin B46 also inhibited ICl,swell when applied after full activation of the current (Fig. 1B). The time course in Fig. 1B indicates that both inhibition and recovery from inhibition were rather slow when compared with the effect of presumably direct channel blockers such as 5-nitro-2-(3-phenylpropylamino)-benzoate (Nilius et al. 1994b), quinidine (Voets, Droogmans & Nilius, 1996) or mibefradil (Nilius, Prenen, Kamouchi, Viana, Voets & Droogmans, 1997b). Current-voltage relations obtained at the time points indicated in Fig. 1B show that ICl,swell was inhibited to the same extent at positive and negative potentials (Fig. 1C).
Figure 1. Effects of tyrphostin B46 on ICl,swell.
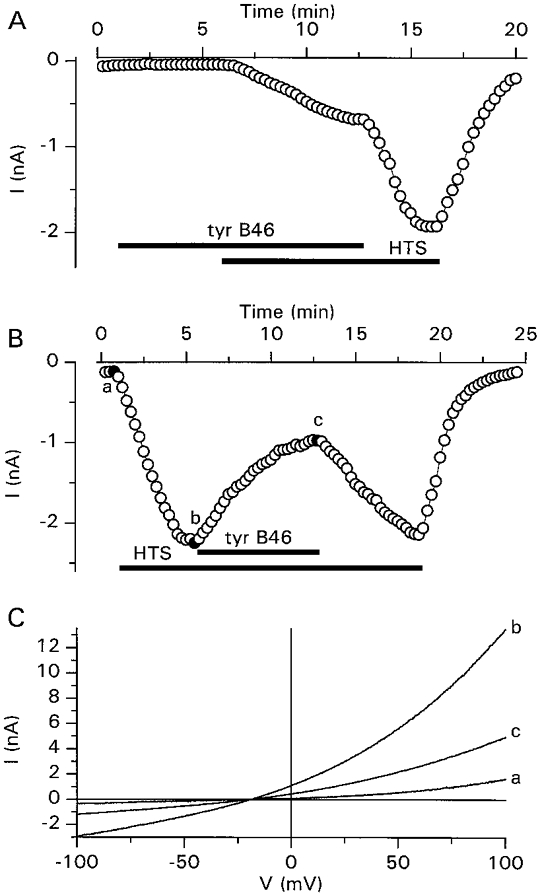
A and B, time course of activation of ICl,swell by extracellular superfusion with the 25% hypotonic solution (HTS) and inhibition of the current by tyrphostin B46 (tyr B46; 10 μm), added at the indicated times. C, current-voltage relations obtained at the time points indicated in B.
As shown in Fig. 2A, ICl,swell was also inhibited by the PTK inhibitor tyrphostin A25 (100 μm), but not by tyrphostin A1, a tyrphostin analogue with minimal effects on PTK activity (Gazit, Yaish, Gilon & Levitzki, 1989). Similarly (Fig. 2B), ICl,swell was inhibited by the PTK inhibitor genistein (100 μm) but not by daidzein, a genistein analogue with minimal effects on PTK activity (Akiyama & Ogawara, 1991). The concentration dependence of the inhibition of ICl,swell by the different PTK inhibitors is represented in Fig. 2C. Values for IC50 and nH, respectively, were 9.2 ± 0.2μm and 2.9 ± 0.2 for tyrphostin B46, 61.4 ± 1.7μm and 2.0 ± 0.1 for tyrphostin A25 and 62.9 ± 1.3μm and 1.6 ± 0.1 for genistein. These PTK inhibitors inhibited ICl,swell with the same potency when the current was activated by the 12.5% hypotonic solution (data not shown).
Figure 2. Inhibition of ICl,swell by tyrosine kinase inhibitors.
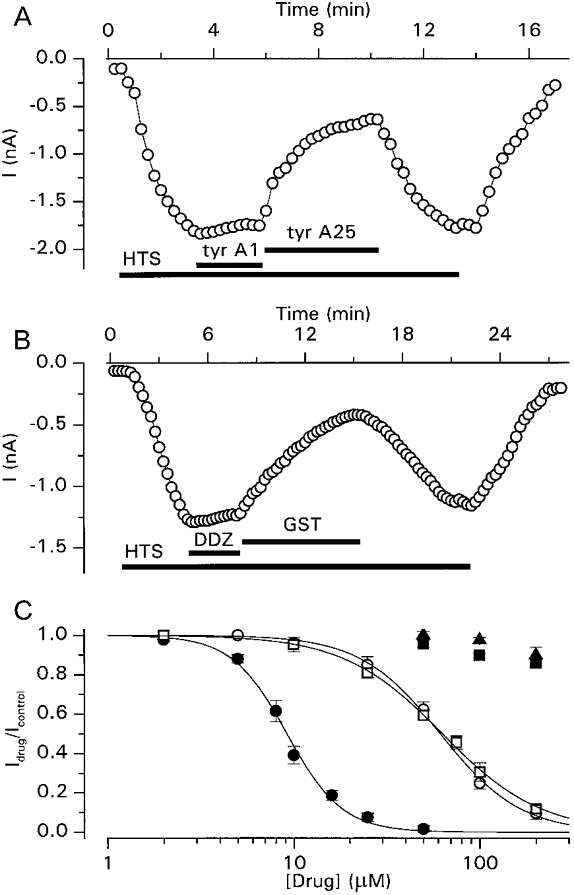
A, time course of activation of ICl,swell by extracellular superfusion with the 25% hypotonic solution, its inhibition by tyrphostin A25 (tyr A25; 100 μm) and the lack of effect of tyrphostin A1 (tyr A1; 100 μm). B, time course of the activation of ICl,swell by extracellular superfusion with the 25% hypotonic solution, its inhibition by genistein (GST; 100 μm) and the lack of effect of daidzein (DDZ; 100 μm). C, dose-response curves for the inhibition of ICl,swell by tyrphostin B46 (•), tyrphostin A25 (○), tyrphostin A1 (▪), genistein (□) and daidzein (▴). The continuous lines represent best fits to eqn (1). Each data point represents observations from at least 4 cells.
We have previously shown that cell swelling induces an efflux of amino acids such as taurine from intact CPAE cells, probably through swelling-activated Cl− channels (Manolopoulos, Voets, Declerq, Droogmans & Nilius, 1997). We therefore examined the effect of PTK inhibitors on the swelling-activated efflux of taurine from CPAE cells. Tyrphostin B46 (10 μm) and genistein (50 μm) inhibited the swelling-activated efflux of taurine by 52.5 ± 3.6% (n = 7) and 50.6 ± 4.2% (n = 6), respectively.
To examine whether direct activation of a tyrosine kinase could induce activation of ICl,swell, we performed experiments in which the cells were intracellularly perfused with a pipette solution containing 1 mm of one of the Src-activating peptides EPQ(pY)EEIPIA and EEE PQ(pY)EEIPIYLELLP (Liu et al. 1993). These peptides stimulate Src tyrosine kinase activity by binding to the SH2-domain and have been used by others to show regulation of NMDA channels by Src (Yu, Askalan, Keil & Salter, 1997). No measurable currents were activated within 30 min after achieving the whole-cell configuration, and subsequent stimulation with 12.5 or 25% hypotonic solutions activated ICl,swell with amplitudes that were not significantly different from those of control cells or from cells perfused with the inactive non-phosphorylated peptides EPQYEEIPIA and eeepqyeeipiy lellp (data not shown).
Potentiation of ICl,swell by tyrosine phosphatase inhibitors
The inhibitory effects on ICl,swell of the PTK inhibitors indicated that PTK activity is necessary to activate and sustain ICl,swell. As the activity of PTK is antagonized by PTP, we were interested in the effects of the PTP inhibitors Na3VO4 (Gordon, 1991) and dephostatin (Imoto et al. 1993) on ICl,swell. Na3VO4 (200 μm) did not significantly affect the ionic currents when applied under isotonic conditions, although in 31% of the cells (4 out of 13) a small and reversible increase of the outward current could be observed (data not shown). However, when applied after activation of ICl,swell by a 12.5% hypotonic solution, Na3VO4 potentiated the current in every tested cell (Fig. 3A). This potentiation, which developed within 2–3 min, amounted to 47 ± 9% (n = 12) and was completely reversible. The current-voltage relations presented in Fig. 3B show that Na3VO4 had in fact two distinct effects on ICl,swell. The first effect was an immediate and voltage-dependent block of the current at positive potentials (compare trace a with b), the second slower effect was the potentiation of ICl,swell. When Na3VO4 was used at a concentration of 500 μm, the immediate blocking effect was so pronounced that the potentiation of ICl,swell was completely masked. The rapid and voltage-dependent nature of the blocking effect suggests that it represents a direct interaction of Na3VO4 with the channel itself rather than a PTP-mediated effect, whereas the slower potentiating effect probably represents the inhibitory effect of Na3VO4 on PTP activity. This is further supported by the finding that the PTP-inhibitor dephostatin (20 μm) potentiated ICl,swell by 69 ± 15% (n = 7), without an initial blocking effect (Fig. 3C). The effect of dephostatin was, however, only partially reversible. In addition, the current level in cells that had been treated with dephostatin mostly did not completely return to basal levels when the extracellular hypotonic solution was replaced by the isotonic solution. These results seem to indicate that dephostatin is only poorly washed out of the cells. Normalized current levels of ICl,swell in the absence and presence of the PTP inhibitors are summarized in Fig. 3D.
Figure 3. Potentiation of ICl,swell by tyrosine phosphatase inhibitors.
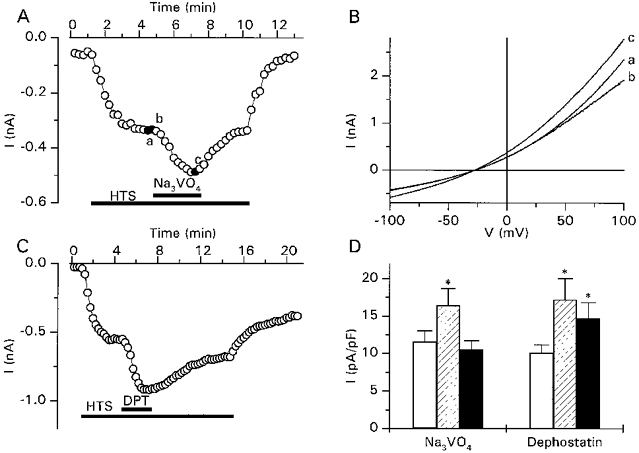
A, time course of activation of ICl,swell by a 12.5% hypotonic solution and subsequent potentiation of the current by Na3VO4 (200 μm). B, current-voltage relations obtained at the time points indicated in A. Note the initial block of ICl,swell at positive potentials. C, time course of activation of ICl,swell by 12.5% hypotonic solution and subsequent potentiation of the current by dephostatin (DPT; 20 μm). D, normalized currents activated by the 12.5% hypotonic solution before (□) and during ( ) addition of the indicated PTP inhibitors and after a washing period of 3 min (▪). * Significantly different from control.
) addition of the indicated PTP inhibitors and after a washing period of 3 min (▪). * Significantly different from control.
Activation of a Cl− current by intracellular perfusion with GTPγS
It has been proposed that activation of ICl,swell occurs via a G protein (Tilly et al. 1996). In CPAE cells, intracellular perfusion with GTPγS (100 μm), a GTP analogue known to activate G proteins (Barritt & Gregory, 1997), induced the transient activation of an inward current at −80 mV (Fig. 4A). The current reached a maximum amplitude (36.1 ± 4.2 pA pF−1 at −80 mV and 133.6 ± 10.2 pA pF−1 at +100 mV) after 198 ± 19 s (n = 23). This activation was followed by a decay phase, as 5 min after reaching this maximum the current decreased to 18 ± 5% of the peak value. Current-voltage relations show that the GTPγS-activated current was outwardly rectifying and reversed close to the reversal potential for Cl− ions (Fig. 4B). During 2 s steps to potentials above +60 mV, the current slightly inactivated (Fig. 4C). Replacing extracellular Cl− by other monovalent anions induced shifts in the reversal potential consistent with a I− > Br− > Cl− ≫ gluconate permeability sequence (Fig. 4D). These biophysical properties are identical to those described for ICl,swell in endothelial cells (Nilius et al. 1994a, b). To further confirm the similarity of both currents, we examined the effect of various inhibitors of ICl,swell on the GTPγS-activated current (Fig. 4E-G). Tamoxifen (10 μm), quinidine (100 μm) and mibefradil (20 μm) inhibited this current by, respectively, 81 ± 5, 82 ± 2 and 65 ± 5%, well in agreement with their effects on ICl,swell (Voets, Szücs, Droogmans & Nilius, 1995; Voets et al. 1996; Nilius et al. 1997b). Taken together, the biophysical and pharmacological data strongly suggest that the GTPγS-activated Cl− current and ICl,swell are identical. It should, however, be noted that the GTPγS-activated current had a transient nature, whereas ICl,swell could be sustained for at least 15 min (Fig. 5). As can be appreciated from this figure, intracellular GTPγS did not exclude a subsequent activation of ICl,swell by hypotonic cell swelling.
Figure 4. Activation of a Cl− current by intracellular GTPγS.
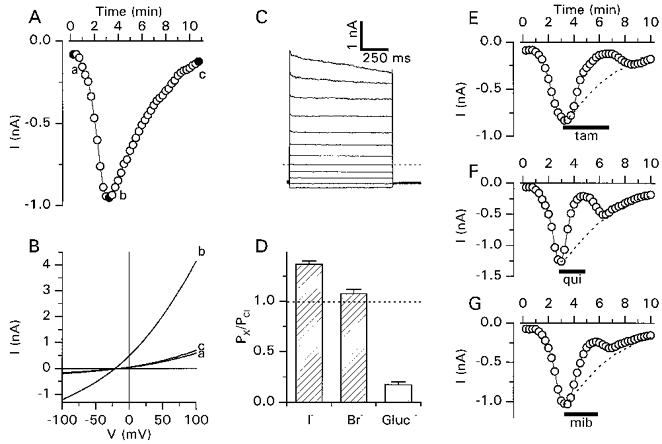
A, time course of the transient activation of a whole-cell current after breaking into the cell with a pipette solution containing 100 μm GTPγS. Time zero corresponds to the rupture of the cell membrane. B, current-voltage relations obtained at the time points indicated in A. C, GTPγS-activated currents measured using the step voltage protocol. The dotted line indicates the zero current level. D, relative permeabilities (PX/PCl) for the indicated anions were calculated from the shifts in the reversal potential using eqn (2). Gluc, gluconate. Data were obtained from 5 different cells. E-G, inhibition of the GTPγS-activated current by tamoxifen (tam; 10 μm), quinidine (qui; 100 μm) and mibefradil (mib; 20 μm). For comparison, the time course of the GTPγS-activated Cl− current in the absence of blockers is shown (dashed lines, rescaled from A).
Figure 5. GTPγS-activated and swelling-activated Cl− currents in the same cell.
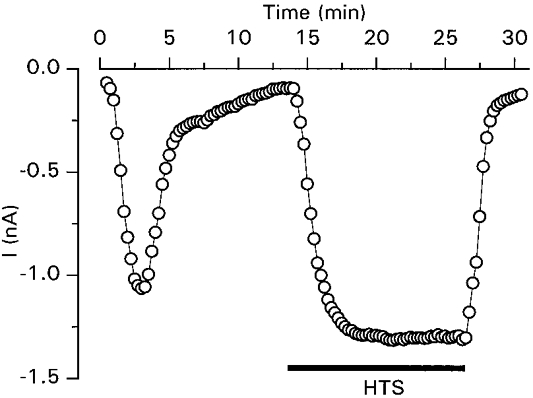
Transient activation of a Cl− current by intracellular GTPγS and subsequent activation of ICl,swell by extracellular superfusion with the 25% hypotonic solution.
The GTPγS-activated current is inhibited by PTK inhibitors and potentiated by PTP inhibitors
To examine whether tyrosine phosphorylation is necessary for the activation of the Cl− current by GTPγS, we tested the effects of the above-described PTK and PTP inhibitors on this current. Pre-incubation of the cells with 100 μm genistein almost completely prevented the activation of the Cl− current (Fig. 6A). Addition of 25 μm tyrphostin B46 after maximal development of the current caused a complete and partially reversible inhibition (Fig. 6B). A similar inhibitory effect was obtained with tyrphostin A25 but not with daidzein or tyrphostin A1 (data not shown).
Figure 6. Modulation of the GTPγS-activated Cl− current by tyrosine kinase/phosphatase inhibitors.
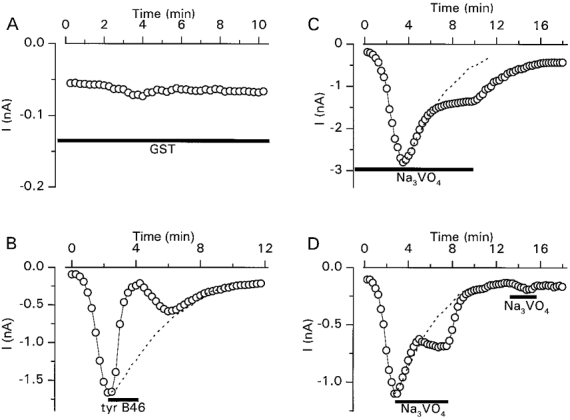
A, inhibition of the GTPγS-activated current by pre-incubation (1 min) with 100 μm genistein. B, inhibition of the GTPγS-activated current by 25 μm tyrphostin B46 applied when the current reached its peak. C, potentiation of the GTPγS-activated current by pre-incubation (1 min) with 200 μm Na3VO4. D, potentiation of the GTPγS-activated current by 200 μm Na3VO4 applied when the current reached its peak. For comparison, the time course of the GTPγS-activated Cl− current in the absence of blockers is shown (dashed lines, rescaled from Fig. 4A).
Pre-incubation with Na3VO4 (200 μm) significantly potentiated the GTPγS-activated current as the peak amplitude at −80 mV was 76.9 ± 8.9 pA pF−1 (n = 6) versus36.1 ± 4.2 pA pF−1 in the absence of Na3VO4. Moreover, 5 min after reaching this maximum the current decreased to 48 ± 9% of the peak value which was significantly higher than control. Washout of Na3VO4 caused a further decrease of the current to basal levels (Fig. 6C). Similarly, Na3VO4 potentiated the GTPγS-activated current when applied during the decay phase, but had no significant effect when applied after the current had returned to basal levels (Fig. 6D). Pre-incubation with dephostatin (20 μm) also caused a significant potentiation of the GTPγS-activated current as the peak current was 65.2 ± 8.1 pA pF−1 (n = 5).
The GTPγS-activated current is volume sensitive
Although we did not observe volume changes during the intracellular perfusion with GTPγS, the concomitant activation of the Cl− current was clearly volume sensitive. Pre-incubation of the cells in hypertonic solution almost completely prevented the activation of the current (Fig. 7A). Once activated, the GTPγS-activated current was also rapidly and completely abolished by increasing the extracellular osmolality (Fig. 7B).
Figure 7. Inhibition of the GTPγS-activated Cl− current by hypertonic cell shrinkage.
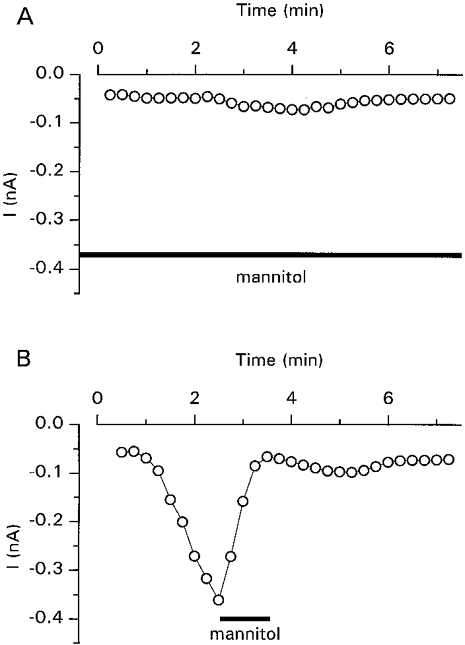
A, inhibition of the GTPγS-activated current by pre-incubation (1 min) with the hypertonic extracellular solution (410 ± 5 mosmol kg−1 by addition of 100 mm mannitol). B, inhibition of the GTPγS-activated current by hypertonic extracellular solution added after partial activation of the current.
Effect of GDPβS on Cl− current activation by GTPγS and cell swelling
We examined the effect of intracellular GDPβS, a GDP analogue known to inhibit G proteins (Barritt & Gregory, 1997), on the Cl− current activated by intracellular GTPγS or cell swelling. A comparison of the effect of intracellular perfusion with a pipette solution containing either GTPγS (100 μm) alone or in combination with GDPβS (1 mm) is shown in Fig. 8. GDPβS caused a small but significant reduction of the peak current from 33.3 ± 3.1 pA pF−1 (n = 10) to 23.4 ± 2.9 pA pF−1 (n = 10). In addition, in the presence of intracellular GDPβS the GTPγS-activated current decreased at a much slower rate than in its absence.
Figure 8. Effect of GDPβS on the GTPγS-activated Cl− current.
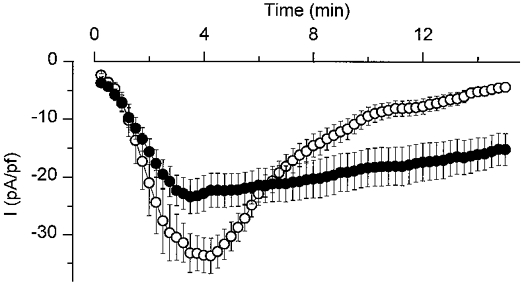
Average time course for the activation of a whole-cell current after breaking into the cell with a pipette solution containing 100 μm GTPγS (○; n = 10) or 100 μm GTPγS + 1 mm GDPβS (•; n = 10, cells from the same coverslips).
Intracellular GDPβS also caused a time-dependent inhibition of ICl,swell. Figure 9A shows the effect of repetitive stimulation with the 25% hypotonic solution on a cell which was intracellularly perfused with GDPβS (1 mm). The amplitude of ICl,swell clearly decreased during this experiment, reaching an apparent steady state of about 40% of the initial amplitude. In the absence of intracellular GDPβS, ICl,swell could be repetitively activated for at least 1 h without significant reduction of the amplitude (data not shown; see also Oike, Droogmans & Nilius, 1994; Nilius et al. 1996). The effect of intracellular GDPβS was even more pronounced when the 12.5% hypotonic solution was used (Fig. 9B). It can be seen that in this experiment the second application of the 12.5% hypotonic solution did not evoke any response, whereas a subsequent stimulation with the 25% hypotonic solution still did. The effects of intracellular GDPβS on ICl,swell are summarized in Fig. 9C.
Figure 9. Effect of GDPβS on ICl,swell.
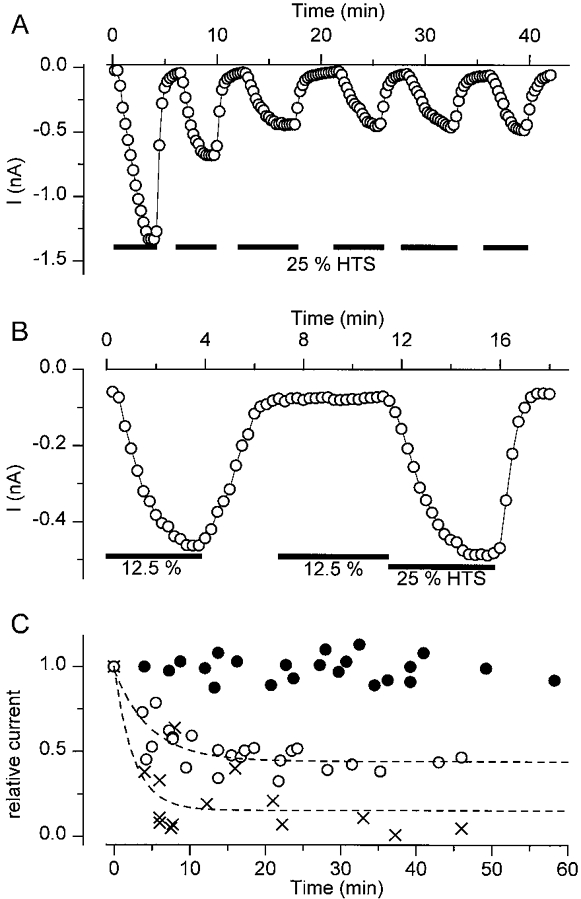
A, time course of the whole-cell current during repetitive superfusion with the 25% hypotonic solution. The pipette solution contained 1 mm GDPβS. B, same as A but using the 12.5% hypotonic solution. Note that the second application is no longer able to activate ICl,swell, whereas the 25% hypotonic solution still does. C, summary of several experiments as in A. Peak current amplitudes during the different hypotonic challenges were normalized to the amplitude during the first challenge. •, normal pipette solution, 25% hypotonic solution; ○, normal pipette solution + 1 mm GDPβS, 25% hypotonic solution; X, normal pipette solution + 1 mm GDPβS, 12.5% hypotonic solution. The dashed lines represent monoexponential fits through the data points. Time constants were 282 ± 63 s (25%) and 205 ± 75 s (12.5%).
DISCUSSION
The effects of PTK and PTP inhibitors on swelling-activated Cl− currents have until now not been studied in detail. It has been shown previously that the activation of ICl,swell in cardiac cells (Sorota, 1995) and of swelling-induced efflux of I− from human Intestine 407 cells (Tilly et al. 1993) could be prevented by pre-incubation of the cells with the PTK inhibitors herbimycin A (> 24 h) or genistein (> 10 min). In the latter cells, pre-incubation with Na3VO4 caused a potentiation of the swelling-induced efflux of I−.
We demonstrate here that the PTK inhibitors tyrphostin B46, tyrphostin A25 and genistein inhibit ICl,swell in CPAE cells in a time- and concentration-dependent manner. PTK inhibitors precluded activation of ICl,swell if they were added prior to the hypotonic stimulus. In addition, application of PTK inhibitors after complete activation of ICl,swell reduced the amplitude of the current. Our results are in some aspects at variance with previous data on the effects of PTK inhibitors on ICl,swell; Gosling et al. (1995) reported that tyrphostin A25 (200 μm) was without effect on ICl,swell in rat osteoblast-like (ROS 17/2.8) cells and Szücs et al. (1996b) could not detect an effect of genistein (100 μm) on ICl,swell in the same endothelial cell line as used in the present study. The low solubility and stability of these compounds (see Methods) may be a possible explanation for these conflicting results.
The PTP inhibitors Na3VO4 and dephostatin potentiated ICl,swell when applied after activation of the current by a mild hypotonic shock. However, under isotonic conditions the PTP inhibitors were not able to activate ICl,swell in a reproducible manner. These effects of PTK and PTP inhibitors point to a reversible tyrosine phosphorylation step as a critical step in the activation of ICl,swell. The identity of the phosphorylated substrate remains unknown since our data cannot distinguish whether the channel itself or a critical regulator thereof is the target for tyrosine phosphorylation.
It is also not known which PTK is required for the activation of ICl,swell. To investigate whether a PTK from the Src family is involved, we made use of two high-affinity peptides that are known to activate this family of kinases (Liu et al. 1993). However, neither of them activated ICl,swell under isotonic conditions nor modulated the amplitude of ICl,swell activated by hypotonic cell swelling. It therefore seems unlikely that a Src-like PTK is involved in the activation of ICl,swell.
It has been shown by others (Doroshenko et al. 1991; Nilius et al. 1994a; Mitchell et al. 1997; Nilius et al. 1997d) that intracellular perfusion with GTPγS, the non-hydrolyzable GTP analogue which irreversibly activates G proteins, induces a Cl− current. These authors proposed that this current is identical to ICl,swell, based on biophysical and pharmacological similarities, although, for example, the anion selectivity of both currents has never been compared. We show here that in CPAE cells the GTPγS-activated Cl− current and ICl,swell have identical biophysical parameters (outward rectification, slight inactivation at positive potentials, I− > Br− > Cl− ≫ gluconate permeability sequence) and pharmacological properties (sensitivity to tamoxifen, quinidine and mibefradil). An additional similarity between both currents is their inhibition by PTK inhibitors and potentiation by PTP inhibitors, indicating that the activation of Cl− channels by either cell swelling or G proteins requires the phosphorylation of tyrosine residues.
An intriguing aspect of the Cl− current activated by GTPγS is its transient nature. The decay of the current cannot be attributed to a depletion of GTPγS, as the pipette solution contained a virtually unlimited amount of the GTP analogue. A possible explanation could be that intracellular GTPγS activates two signalling cascades, one leading to the opening of Cl− channels and the other resulting in a subsequent closing of the same channels. The observed permanent activation of the Cl− current in the presence of both GTPγS and GDPβS might indicate that GDPβS inhibits the second pathway more efficiently than the first pathway. It has, for example, been described that GDPβS may bind more tightly to trimeric G proteins than to small monomeric G proteins (Barritt & Gregory, 1997).
An important question is whether cell volume acts via a G protein to activate ICl,swell. It was recently proposed by Tilly et al. (1996) that cell swelling activates the small G protein p21rho leading to increased PTK activity and subsequent opening of swelling-activated Cl− channels. This model was based on the finding that the hypotonicity-induced I− efflux in human Intestine 407 cells was strongly reduced by the Clostridium botulinum C3 exoenzyme, an inhibitor of p21rho, and by the PTK inhibitors genistein and herbimycin A (Tilly et al. 1993, 1996). In such a model, direct activation of G proteins by intracellular GTPγS should activate ICl,swell independent of cell volume. However, we show here that hypertonic cell shrinkage completely inhibits the activation of the Cl− current by GTPγS. In addition, we show that inhibition of G proteins by intracellular GDPβS causes an almost complete inhibition of ICl,swell activated by a mild hypotonic shock but only a ∼50% reduction of ICl,swell activated by a strong hypotonic shock. We therefore propose an alternative model whereby G proteins modulate the sensitivity to cell swelling, rather than directly activating the signalling cascade that results in the activation of ICl,swell. In this model, activation of the G protein by intracellular GTPγS increases the sensitivity such that ICl,swell becomes activated under isovolumetric conditions.
Acknowledgments
We thank Drs M. Salter, J. Goris and P. Agostinis for providing the Src-activating peptides. J. E. is a Research Associate of the Flemish Fund for Scientific Research (F.W.O.-Vlaanderen). V. M. received a fellowship from the European Commission (contract ERB-CHBG-CT94-0754). This research was supported by the European Grant BMH4-CT96-0602 (co-ordinator B. N.).
References
- Akiyama T, Ogawara H. Use and specificity of genistein as inhibitor of protein-tyrosine kinases. Methods in Enzymology. 1991;201:362–370. doi: 10.1016/0076-6879(91)01032-w. [DOI] [PubMed] [Google Scholar]
- Barritt GJ, Gregory RB. An evaluation of strategies available for the identification of GTP-binding proteins required in intracellular signalling pathways. Cellular Signalling. 1997;9:207–218. doi: 10.1016/s0898-6568(96)00131-3. [DOI] [PubMed] [Google Scholar]
- Doroshenko P, Penner R, Neher E. A novel chloride conductance in the membrane of bovine chromaffin cells activated by intracellular GTPγS. Journal of Physiology. 1991;436:711–724. doi: 10.1113/jphysiol.1991.sp018575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit A, Osherov N, Posner I, Yaish P, Poradosu E, Gilon C, Levitzki A. Tyrphostins. 2. Heterocyclic and alpha-substituted benzylidenemalononitrile tyrphostins as potent inhibitors of EGF receptor and ErbB2/neu tyrosine kinases. Journal of Medicinal Chemistry. 1991;34:1896–1907. doi: 10.1021/jm00110a022. [DOI] [PubMed] [Google Scholar]
- Gazit A, Yaish P, Gilon C, Levitzki A. Tyrphostins I: synthesis and biological activity of protein tyrosine kinase inhibitors. Journal of Medicinal Chemistry. 1989;32:2344–2352. doi: 10.1021/jm00130a020. [DOI] [PubMed] [Google Scholar]
- Gordon JA. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods in Enzymology. 1991;201:477–482. doi: 10.1016/0076-6879(91)01043-2. [DOI] [PubMed] [Google Scholar]
- Gosling M, Smith JW, Poyner DR. Characterization of a volume-sensitive chloride current in rat osteoblast-like (ROS 17/2.8) cells. Journal of Physiology. 1995;485:671–682. doi: 10.1113/jphysiol.1995.sp020761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto M, Kakeya H, Sawa T, Hayashi C, Hamada M, Takeuchi T, Umezawa K. Dephostatin, a novel protein tyrosine phosphatase inhibitor produced by Streptomyces. I. Taxonomy, isolation, and characterization. Journal of Antibiotics. 1993;46:1342–1346. doi: 10.7164/antibiotics.46.1342. [DOI] [PubMed] [Google Scholar]
- Liu X, Brodeur SR, Gish G, Songyang Z, Cantley LC, Laudano AP, Pawson T. Regulation of c-Src tyrosine kinase activity by the Src SH2 domain. Oncogene. 1993;8:1119–1126. [PubMed] [Google Scholar]
- Manolopoulos V, Voets T, Declerq P, Droogmans G, Nilius B. Swelling-activated efflux of taurine and other organic osmolytes in endothelial cells. American Journal of Physiology. 1997;273:C214–222. doi: 10.1152/ajpcell.1997.273.1.C214. [DOI] [PubMed] [Google Scholar]
- Mitchell CH, Zhang JJ, Wang L, Jacob TJC. Volume-sensitive chloride current in pigmented ciliary epithelial cells: role of phospholipases. American Journal of Physiology. 1997;272:C212–222. doi: 10.1152/ajpcell.1997.272.1.C212. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods in Enzymology. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, Droogmans G. Properties of volume-regulated anion channels in mammalian cells. Progress in Biophysics and Molecular Biology. 1997a doi: 10.1016/s0079-6107(97)00021-7. in the Press. [DOI] [PubMed] [Google Scholar]
- Nilius B, Gerke V, Prenen J, Szücs G, Heinke S, Weber K, Droogmans G. Annexin II modulates volume-activated chloride currents in vascular endothelial cells. Journal of Biological Chemistry. 1996;271:30631–30636. doi: 10.1074/jbc.271.48.30631. 10.1074/jbc.271.48.30631. [DOI] [PubMed] [Google Scholar]
- Nilius B, Oike M, Zahradnik I, Droogmans G. Activation of a Cl− current by hypotonic volume increase in human endothelial cells. Journal of General Physiology. 1994a;103:787–805. doi: 10.1085/jgp.103.5.787. 10.1085/jgp.103.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Kamouchi M, Viana F, Voets T, Droogmans G. Mibefradil, a novel calcium channel antagonist, inhibits Ca2+- and volume-activated Cl− channels in macrovascular endothelial cells. British Journal of Pharmacology. 1997b;121:547–555. doi: 10.1038/sj.bjp.0701140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Szücs G, Wei L, Tanzi F, Voets T, Droogmans G. Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. Journal of Physiology. 1997c;497:95–107. doi: 10.1113/jphysiol.1997.sp021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Voets T, Van Den Bremt K, Eggermont J, Droogmans G. Kinetic and pharmacological properties of the calcium-activated chloride current in macrovascular endothelial cells. Cell Calcium. 1997d;22:53–63. doi: 10.1016/s0143-4160(97)90089-0. 10.1016/S0143-4160(97)90089-0. [DOI] [PubMed] [Google Scholar]
- Nilius B, Sehrer J, Droogmans G. Permeation properties and modulation of volume-activated Cl− currents in human endothelial cells. British Journal of Pharmacology. 1994b;112:1049–1056. doi: 10.1111/j.1476-5381.1994.tb13189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Szücs G, Heinke S, Voets T, Droogmans G. Multiple types of chloride channels in bovine pulmonary artery endothelial cells. Journal of Vascular Research. 1997e;34:220–228. doi: 10.1159/000159226. [DOI] [PubMed] [Google Scholar]
- Oike M, Droogmans G, Nilius B. The volume-activated chloride current in human endothelial cells depends on intracellular ATP. Pflügers Archiv. 1994;427:184–186. doi: 10.1007/BF00585960. [DOI] [PubMed] [Google Scholar]
- Shuba LM, Asai T, Pelzer S, McDonald TF. Activation of cardiac chloride conductance by the tyrosine kinase inhibitor, genistein. British Journal of Pharmacology. 1996;119:335–345. doi: 10.1111/j.1476-5381.1996.tb15991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorota S. Tyrosine protein kinase inhibitors prevent activation of cardiac swelling-induced chloride current. Pflügers Archiv. 1995;431:178–185. doi: 10.1007/BF00410189. [DOI] [PubMed] [Google Scholar]
- Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive anion channels. American Journal of Physiology. 1996;270:C711–730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Szücs G, Buyse G, Eggermont J, Droogmans G, Nilius B. Characterisation of volume-activated chloride currents in endothelial cells from bovine pulmonary artery. Journal of Membrane Biology. 1996a;149:189–197. doi: 10.1007/s002329900019. [DOI] [PubMed] [Google Scholar]
- Szücs G, Heinke S, De Greef C, Raeymaekers L, Eggermont J, Droogmans G, Nilius B. The volume-activated chloride current in endothelial cells from bovine pulmonary artery is not modulated by phosphorylation. Pflügers Archiv. 1996b;431:540–548. doi: 10.1007/BF02191901. [DOI] [PubMed] [Google Scholar]
- Szücs G, Heinke S, Droogmans G, Nilius B. Activation of the volume-sensitive chloride current in vascular endothelial cells requires a permissive intracellular Ca2+ concentration. Pflügers Archiv. 1996c;431:467–469. doi: 10.1007/BF02207289. [DOI] [PubMed] [Google Scholar]
- Tilly BC, Edixhoven MJ, Tertoolen LGJ, Morii N, Saitoh Y, Narumiya S, de Jonge HR. Activation of the osmo-sensitive chloride conductance involves p21rho and is accompanied by a transient reorganization of the F-actin cytoskeleton. Molecular Biology of the Cell. 1996;7:1419–1427. doi: 10.1091/mbc.7.9.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly BC, van den Berghe N, Tertoolen LG, Edixhoven MJ, de Jonge HR. Protein tyrosine phosphorylation is involved in osmoregulation of ionic conductances. Journal of Biological Chemistry. 1993;268:19919–19922. [PubMed] [Google Scholar]
- Voets T, Droogmans G, Nilius B. Potent block of volume-activated chloride currents in endothelial cells by the uncharged form of quinine and quinidine. British Journal of Pharmacology. 1996;118:1869–1871. doi: 10.1111/j.1476-5381.1996.tb15616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Szücs G, Droogmans G, Nilius B. Blockers of volume-activated Cl− currents inhibit endothelial cell proliferation. Pflügers Archiv. 1995;431:132–134. doi: 10.1007/BF00374387. [DOI] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil GJ, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]