

Abstract
The whole-cell patch-clamp technique was used to examine the participation of nitric oxide synthase (NOS) and soluble guanylyl cyclase in the muscarinic regulation of the L-type Ca2+ current (ICa) in freshly isolated human atrial myocytes.
Acetylcholine (ACh, 1 μM) decreased basal ICa by 39.1 ± 5.5 % (n= 8) under control conditions, and by 38.0 ± 6.1 % (n= 6) in the presence of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one (ODQ, 10 μM), a potent guanylyl cyclase inhibitor, and NG-monomethyl-L-arginine (L-NMMA, 1 mM), a competitive NOS inhibitor. L-NMMA alone had no effect on ICa, whilst ODQ increased ICa in 50 % of the cells.
The accentuated antagonism of ACh on ICa, i.e. its ability to antagonize the stimulatory effect of β-adrenergic agonists and, by extension, of other cAMP-elevating agents, was examined after the current was stimulated by either the β-adrenergic agonist isoprenaline (Iso) or serotonin (5-HT). ACh (100 nM or 1 μM) completely blocked the stimulatory effects of 10 nM Iso or 10 nM 5-HT on ICa.
Extracellular application of Methylene Blue (MBlue, 10 μM), a guanylyl cyclase inhibitor, antagonized the inhibitory effect of 1 μM ACh on Iso- or 5-HT-stimulated ICa. However, this effect was overcome by a 100-fold higher ACh concentration and was not mimicked by an intracellular application of MBlue.
Inhibition of NOS and soluble guanylyl cyclase activities by addition of ODQ (10 μM) and L-NMMA (1 mM) to both extracellular and intracellular solutions, or by a 2 h pre-incubation of the cells with these inhibitors, modified neither the Iso (10 nM) response nor the inhibitory effect of ACh (100 nM or 1 μM) on Iso-stimulated ICa.
Extracellular application of the NO donor SNAP (S-nitroso-N-acetyl-d,l-penicillamine) at 100 nM produced a stimulatory effect on ICa in control conditions. This stimulatory effect was abolished by intracellular MBlue (20 μM) or by intracellular and extracellular application of ODQ (10 μM) in combination with L-NMMA (1 mM).
We conclude that the NO-cGMP pathway does not contribute significantly to the muscarinic regulation of ICa in human atrial myocytes.
The cardiac L-type calcium current (ICa) plays a determinant role in the dual regulation of the inotropic state of the heart by the autonomic nervous system (McDonald, Pelzer, Trautwein & Pelzer, 1994). Whereas considerable evidence supports the view that the cAMP cascade is the unique route for the β-adrenoceptor-mediated sympathetic stimulation of ICa (reviewed in Hove-Madsen, Méry, Jurevicius, Skeberdis & Fischmeister, 1996), the intracellular signalling pathways that underlie the acetylcholine (ACh)-mediated parasympathetic antagonism of the β-adrenergic response, i.e. the indirect negative inotropic effect of vagal stimulation, are less clear (reviewed in Méry, Abi-Gerges, Vandecasteele, Jurevicius, Eschenhagen & Fischmeister, 1997).
The cardiac negative inotropic effect of ACh is generally accompanied by a reduction in the concentration of cAMP (Fleming, Strawbridge & Watanabe, 1987). This effect mainly results from an inhibition of adenylyl cyclase via a pertussis toxin-sensitive G protein (Breitwieser & Szabo, 1985; Hescheler, Kameyama & Trautwein, 1986; Fleming et al. 1987; Jurevicius & Fischmeister, 1996). However, other mechanisms such as a muscarinic activation of a cyclic nucleotide phosphodiesterase or activation of a phosphatase have also been documented in heart muscle (for a brief review, see Méry et al. 1997). Moreover, ACh and other muscarinic receptor agonists have been shown to elevate cGMP levels in the heart muscle (George, Polson, O'Toole & Goldberg, 1970) as well as in isolated cardiac myocytes (Stein, Drögemüller, Mülsch, Schmitz & Scholz, 1993; MacDonnel, Tibbits & Diamond, 1995). The recent discovery of the existence of a nitric oxide synthase (NOS) in a variety of cardiac cells including cardiomyocytes (Balligand et al. 1995; Kelly, Balligand & Smith, 1996) led to the hypothesis that ACh-mediated increases in cGMP level might result from a stimulation of NOS activity (Kelly et al. 1996).
Since cGMP and nitric oxide (NO) donors were shown to produce negative inotropic effects and an inhibition of ICa in several cardiac preparations (for review see Fischmeister & Méry, 1996), it was tempting to speculate that the NO-cGMP pathway might participate in the cardiac effects of ACh. This hypothesis was recently confirmed in cardiac preparations from different animal species using inhibitors of the NO-cGMP pathway which were found to attenuate or even abolish the inhibitory response to muscarinic agonists (Balligand, Kelly, Mardsen, Smith & Michel, 1993; Han, Shimoni & Giles, 1994, 1995; Levi, Alloatti, Penna & Gallo, 1994; Balligand et al. 1995; Wang & Lipsius, 1995; Han, Kobzik, Balligand, Kelly & Smith, 1996). However, this hypothesis was contradicted in several other cardiac preparations in which the muscarinic response was found to be insensitive to these inhibitors (Stein et al. 1993; Kennedy, Hicks, Brian & Seiffen, 1994; Nawrath, Baumner, Rupp & Oelert, 1995; Habuchi, Nishio, Tanaka, Yamamoto, Lu & Yoshimura, 1996; Méry, Hove-Madsen, Chesnais, Hartzell & Fischmeister, 1996; Abi-Gerges, Méry & Fischmeister, 1997c). For example, in human papillary muscle strips, the anti-adrenergic effect of carbachol remained unaffected by a pre-treatment of the preparation with the competitive NOS inhibitor NG-monomethyl-L-arginine (L-NMMA), or the guanylyl cyclase inhibitor Methylene Blue (MBlue: Kilter et al. 1995). Moreover, in human atria, where transcripts of the constitutive endothelial NOS (NOS 3) were detected by in situ hybridization (see Kelly et al. 1996), the participation of this enzyme in the muscarinic response was challenged by the observation that ACh inhibits contraction in this preparation (Böhm, Gierschik, Schwinger, Uhlmann & Erdmann, 1994) but NO donors have no effect (Nawrath et al. 1995) or even stimulate ICa in isolated human atrial myocytes (Kirstein, Rivet-Bastide, Hatem, Bénardeau, Mercadier & Fischmeister, 1995).
To get further insights into the mechanism of action of ACh in the human heart, we have re-examined in the present study the effect of ACh on ICa recorded in freshly isolated human atrial myocytes using the whole-cell patch-clamp technique. The effects of ACh on basal and stimulated ICa were investigated in the absence or presence of extracellular and/or intracellular L-NMMA (a NOS inhibitor) and of two guanylyl cyclase inhibitors, MBlue and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one (ODQ). This allowed us to examine whether NO and/or cGMP participate in the muscarinic regulation of basal and phosphorylated ICa.
Preliminary results have appeared in abstract form (Vandecasteele, Eschenhagen & Fischmeister, 1997).
METHODS
Surgery
All protocols for obtaining human cardiac tissue were approved by the ethics committee of our institution (GREBB, Hôpital de Bicêtre, Université de Paris-Sud). Specimens of right atrial appendages were obtained from twenty-two patients (aged 8-78 years) undergoing heart surgery for congenital defects, coronary artery diseases or valve replacement at the Hôpital Marie-Lannelongue, Le Plessis-Robinson, France. Most patients received a pharmacological pretreatment (Ca2+ channel blockers, digitalis/glycosides, β-adrenergic antagonists, diuretics, angiotensin converting enzyme (ACE) inhibitors, NO donors and/or anti-arrhythmic drugs). In addition, all patients received sedatives, anaesthesia, and antibiotics. Four patients did not receive any cardiovascular pharmacotherapy. We found no obvious correlation between the calcium current density or the effects on ICa of the drugs tested here and the therapy received (if any) by the patient. Dissociation of the cells was realized immediately after surgery.
Human atrial cell dissociation
Myocytes were isolated as described previously (Kirstein et al. 1995) with some modifications. Briefly, after excision of the atrial tissue, the tissue was cut up and washed in a calcium-free Tyrode solution supplemented with 30 mM 2,3-butanedionemonoxime for 10 min and then incubated in the same solution containing 40 i.u. ml −1 collagenase, 15 i.u. ml−1 protease and 5 mg ml−1 BSA for 30 min. The solution was then replaced by fresh enzymatic solution containing only collagenase for 15-60 min until a satisfactory cell yield was obtained. All steps were carried out at 37°C, with continuous gassing with 95 % O2 and 5 % CO2. The digestion mix was then poured on a nylon filter (pore diameter, 250 μm) in order to separate the dissociated myocytes from the non-digested part of the tissue. The resulting cell suspension was then centrifuged, and the pellet resuspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10 % fetal calf serum, non-essential amino acids, insulin (1 nM) and antibiotics (penicillin, 100 i.u. ml−1 and streptomycin, 0.1 μg ml−1). For patch-clamp experiments, 100-200 μl of this cell suspension were put in a Petri dish containing control external solution.
Electrophysiological experiments
The whole-cell configuration of the patch-clamp technique (Hamill, Marty, Neher, Sakmann & Sigworth, 1981) was used to record the high-threshold calcium current (ICa) on Ca2+-tolerant human atrial myocytes. In the routine protocols the cells were depolarized every 8 s from a holding potential of -50 mV to 0 mV for 400 ms. This holding potential was chosen to completely inactivate the fast Na+ current. K+ currents were blocked by replacing all K+ ions with intracellular and extracellular Cs+. Occasionally (as in Fig. 2), the cell was held at -80 mV and depolarized to -50 mV for 50 ms, prior to the test pulse to 0 mV. Voltage-clamp protocols were generated by a challenger/09-VM programmable function generator (Kinetic Software, Atlanta, GA, USA). The cells were voltage clamped using a patch-clamp amplifier (model RK-400; Biologic, Claix, France). Currents were filtered at 3 kHz and sampled at a frequency of 10 kHz using a 12-bit analog-to-digital converter (DT2827, Data Translation, Marlboro, MA, USA) connected to a PC-compatible computer (386/33 System-pro, Compaq, Houston, TX, USA). While all points were used for data analysis (see below), only one out of four points was used for the drawings of current traces (Figs 1 and 2). All experiments were done at room temperature (19-25°C).
Figure 2. Accentuated antagonism of ACh on ICa in human atrial myocytes.
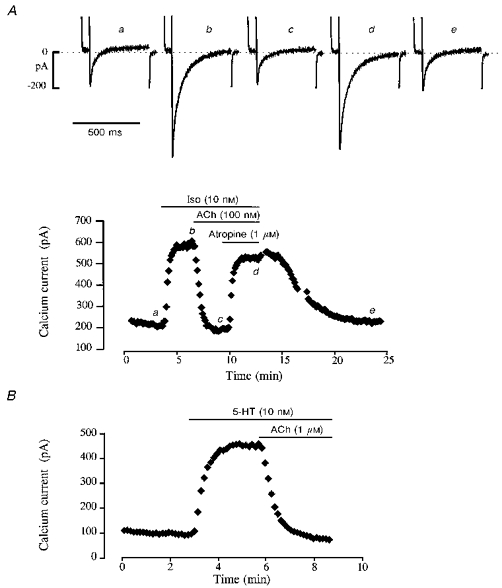
The cells were first superfused with control solution and then exposed to the drugs during the periods indicated in the graphs by the bars. A, superfusion of the cell with 10 nM Iso produced a nearly maximal stimulation of ICa, which was fully abolished by 100 nM ACh. Addition of the muscarinic antagonist atropine (1 μM) restored ICa to an amplitude which was close to the Iso-stimulated level. Upon washout of the drugs and return to control conditions, ICa slowly recovered its control amplitude. The individual current traces shown in the upper part of the figure were obtained at the times indicated by the corresponding letters in the graph. The dotted line indicates the zero current level. B, application of 10 nM 5-HT to another myocyte also produced a strong stimulation of ICa. Application of 1 μM ACh in the continuous presence of 5-HT induced a complete inhibition of the serotoninergic response.
Figure 1. Effect of ACh on basal ICa in a human atrial myocyte.
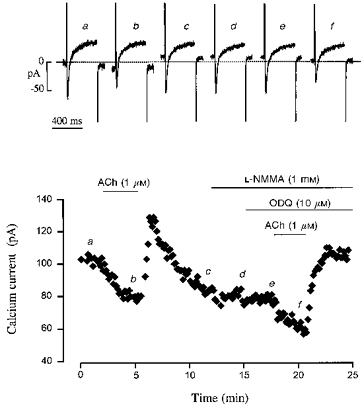
Each symbol on the graph corresponds to a measure of ICa at 0 mV, obtained every 8 s. The cell was first superfused with control solution and then exposed to the drugs during the periods indicated by the bars. 1 μM ACh alone, or together with ODQ (10 μM) + L-NMMA (1 mM) produced the same inhibition of ICa, which was followed in both cases by a rebound stimulation of the current. The individual current traces shown in the upper part of the figure were obtained at the times indicated by the corresponding letters in the bottom graph. The dotted line indicates the zero current level.
Solutions
Control external solution contained (mM): 107.1 NaCl, 10 Hepes, 40 CsCl, 4 NaHCO3, 0.8 NaH2PO4, 1.8 CaCl2, 1.8 MgCl2, 5 D-glucose, 5 sodium pyruvate, pH adjusted to 7.4 with NaOH. Control or drug-containing solutions were applied to the exterior of the cell, at a flow rate of ∼10 μl min−1, by placing the cell at the opening of a 250 μm (inner diameter) capillary tube. Patch electrodes (0.8-1.5 MΩ) were filled with control internal solution that contained (mM): 119.8 CsCl, 5 EGTA (acid form), 4 MgCl2, 5 creatine phosphate disodium salt, 3.1 Na2-ATP, 0.42 Na2-GTP, 10 Hepes, 62 μM CaCl2 (pCa, 8.5), pH adjusted to 7.3 with CsOH. In some experiments, MBlue (10 μM) or L-NMMA (1 mM) + ODQ (10 μM) were added to the intracellular solution.
Materials
DMEM was obtained from Gibco-BRL. L-NMMA, ODQ and SNAP were from Tocris Cookson (Bristol, UK). All other drugs, including collagenase Type V and protease Type XXIV were purchased from Sigma. Stock solution of ODQ (100 mM) was made in DMSO. All other drugs were dissolved in ionic aqueous solutions. All stock solutions were made fresh daily and kept at 4°C until use.
Data analysis
The maximum amplitude of ICa was measured as the difference between the peak inward current and the end-pulse current (I400), i.e. the current amplitude at the end of the 400 ms duration pulse (Kirstein et al. 1995). Currents were not compensated for capacitive and leak currents. Cell membrane capacitance and series resistance were measured by exponential analysis of current responses to 1 mV step changes in membrane potential. Membrane capacitance was 64.6 ± 3.1 pF (mean ±s.e.m.) and series resistance was 4.3 ± 0.3 MΩ (n= 47). The on-line analysis was made possible by programming a PC-compatible computer in PASCAL language to determine, for each depolarization, peak and steady-state current values.
The results are expressed as means ±s.e.m. In each experimental condition, the effects of the drugs tested on ICa are expressed as percentage change with respect to the values of the current under basal conditions, i.e. in the absence of any hormonal stimulation. The variations in ICa induced by the different drugs were tested for statistical significance by Student's t test.
RESULTS
ICa was recorded in human atrial myocytes using the whole-cell configuration of the patch-clamp technique (Hamill et al. 1981). Basal ICa amplitude was measured 3-15 min after patch break to allow for equilibration between intracellular and pipette solutions. When the cells were superfused with control external solution and the patch pipette was filled with control internal solution, basal ICa amplitude at 0 mV membrane potential was on average -257 ± 32 pA, and ICa density, which represents the ratio of ICa amplitude to membrane capacitance, was -4.1 ± 0.5 pA pF−1 (n= 28). As in our previous studies (Kirstein et al. 1995; Rivet-Bastide, Vandecasteele, Hatem, Bénardeau, Mercadier & Fischmeister, 1997), ICa densities showed a large scatter between different patients and between individual cells from the same patient, with no obvious correlation with the diagnosis, sex, age or pretreatment of the patients.
Effect of L-NMMA and ODQ on basal ICa
To examine whether NOS and/or guanylyl cyclase activities modulate basal ICa, the cells were exposed to L-NMMA and ODQ. Superfusion of human atrial myocytes with 1 mM L-NMMA did not significantly modify the basal amplitude of ICa (-4.6 ± 3.5 % variation, n= 4; data not shown). The effect of 10 μM ODQ was examined in eight atrial myocytes and showed a high variability. In four cells, ODQ produced an increase of basal ICa ranging from 12 to 104 % above the control amplitude, whereas ODQ had no effect in the four other cells. Consequently, the mean effect of ODQ on the eight cells (+22.8 ± 13.7 % increase over basal amplitude) was not statistically significant (P= 0.66). Thus, in our experimental conditions, NOS does not participate in the regulation of basal ICa, whereas soluble guanylyl cyclase may play a role, at least in some cells.
ACh inhibition of basal ICa in the presence of L-NMMA and ODQ
Figure 1 shows the effect of ACh (1 μM) on basal ICa in a human atrial myocyte. The effect of ACh was first examined under control conditions. Superfusion of the cell with a 1 μM concentration of the muscarinic agonist decreased the amplitude of ICa from -100 to -80 pA within 2-3 min. Upon washout of ACh, ICa increased rapidly to a value which was ∼20 % above control, and this rebound was followed by a progressive reduction towards a new steady-state amplitude, which was reached after ∼5 min. The cell was subsequently exposed to 1 mM L-NMMA for 3 min and then to a combination of L-NMMA plus 10 μM ODQ for the rest of the experiment. As shown in Fig. 1, exposure of the cell to L-NMMA or to L-NMMA + ODQ had no apparent effect on ICa. In addition, it did not prevent either the inhibitory effect of ACh, or the rebound stimulation upon ACh washout elicited by a second application and washout of the agonist in the continuing presence of L-NMMA and ODQ. In a total of eight experiments, ACh (1 μM) alone decreased basal ICa by 39 ± 7 %. In six of them, the effect of 1 μM ACh was tested in the presence of extracellular L-NMMA (1 mM) + ODQ (10 μM) and similarly decreased ICa (38 ± 6 %). In these eight cells, a rebound stimulation of ICa upon ACh washout was observed in only five cells, and this phenomenon was also not modified by the presence of the NOS and guanylyl cyclase inhibitors. Thus, inhibition of NOS and soluble guanylyl cyclase activities does not modify the muscarinic regulation of basal ICa in human atrial myocytes.
Accentuated antagonism of ACh on stimulated ICa
In the ventricular muscle, activation of muscarinic receptors has relatively little inhibitory effect on the force of contraction and Ca2+ current amplitude in the absence of β-adrenergic stimulation. However, activation of muscarinic receptors after a stimulation of the β-adrenergic receptors leads to a profound inhibition of the β-response. As shown in Fig. 2, this phenomenon, referred to as accentuated antagonism of ACh, is also present in human atrial myocytes. In the experiment shown in Fig. 2A, 10 nM of the non-selective β-adrenergic agonist isoprenaline (Iso) produced an ∼3-fold increase in ICa. This stimulation was completely abolished by the addition of 100 nM ACh to the Iso-containing extracellular solution (inhibition of the β-response was 106 %). The inhibitory effect of ACh was due to activation of muscarinic receptors, since application of 1 μM atropine, a muscarinic receptor antagonist, antagonized most of the response to ACh (88 ± 6 %, n= 3). Activation of β-adrenergic receptors was not a prerequisite for the accentuated antagonism of ACh to take place. Indeed, as shown in Fig. 2B, application of ACh (1 μM) also fully antagonized (102 % inhibition) the 5-HT4 receptor-mediated stimulatory effect induced by serotonin (5-HT, 10 nM) on ICa (Ouadid, Seguin, Dumuis, Bockaert & Nargeot, 1991). On average, 5-HT (10 nM) and Iso (10 nM) increased ICa by +376 ± 77 % (n= 3) and +170 ± 22 % (n= 14) over basal ICa amplitude, respectively, and both responses were fully antagonized by 1 μM ACh (to, respectively, -19 ± 27 % (n= 3) and +4 ± 6 % (n= 3) of basal amplitude).
Accentuated antagonism of ACh in the presence of Methylene Blue
To determine whether a soluble guanylyl cyclase activity was implicated in the accentuated antagonism of ACh on ICa, we examined the effect of ACh in the presence of Methylene Blue (MBlue), a classical inhibitor of this enzyme (for review, see Tremblay, Gerzer & Hamet, 1988). In the experiment shown in Fig. 3A, ICa was stimulated by 10 nM Iso. After ICa reached its maximal amplitude, 10 μM MBlue was added to the Iso-containing solution, which had little effect on the current. However, the presence of MBlue prevented the inhibitory effect of 1 μM ACh added subsequently to the solution. MBlue inhibited the effect of ACh in a competitive manner, since addition of ACh at a concentration of 100 μM overcame the inhibitory effect of 10 μM MBlue, and ICa was decreased to basal level. Figure 3B summarizes the results of three similar experiments. On average, Iso (10 nM) enhanced the current by 116 ± 26 and 123 ± 16 % in the absence and presence of 10 μM MBlue, respectively, demonstrating that the guanylyl cyclase inhibitor did not interfere with the β-adrenergic response. The subsequent addition of ACh at 1 μM in the presence of MBlue produced a slight decrease in ICa (to 100 ± 34 % above control level) which was not statistically significant. However, a 100-fold higher concentration of the muscarinic agonist clearly inhibited ICa to -15 ± 6 % below basal amplitude.
Figure 3. Accentuated antagonism of ACh on ICa in the presence of extracellular Methylene Blue.
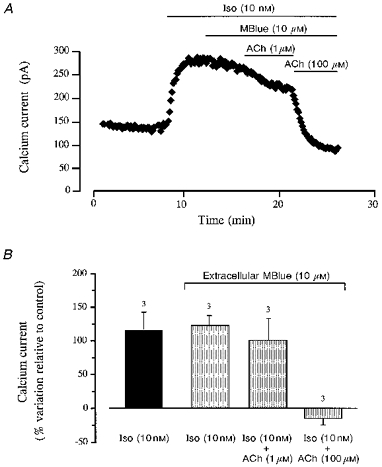
A, the cell was superfused for several minutes with a control solution and then challenged with different drugs during the periods indicated by the bars. Application of 10 nM Iso induced an ≈2-fold increase in ICa, which was unchanged when the cell was superfused with the same solution containing 10 μM MBlue. In the presence of MBlue, 1 μM ACh had only a weak inhibitory effect on ICa, whilst 100 μM ACh completely blocked the Iso response. B, summary of the results of three experiments similar to that shown in A (means ±s.e.m.; number of experiments indicated above the vertical columns).
MBlue was shown recently to behave as a muscarinic receptor antagonist in frog and rat cardiac myocytes (Abi-Gerges, Eschenhagen, Hove-Madsen, Fischmeister & Méry, 1997a; Abi-Gerges, Hove-Madsen, Fischmeister & Méry, 1997b). Thus, the possibility exists that the antagonistic effect of MBlue on the muscarinic inhibition of ICa in human atrial myocytes seen here was not due to guanylyl cyclase inhibition but rather to the binding of MBlue to the muscarinic receptor. In order to test this hypothesis, we performed experiments in which MBlue was applied intracellularly to exclude any direct interaction of MBlue with the agonist-binding site on the muscarinic receptors. To do this, the patch-pipette was filled with an intracellular solution which contained 20 μM MBlue. As shown in Fig. 4A, the presence of MBlue in the intracellular solution did not interfere with the 5-HT stimulatory effect on ICa. On average (Fig. 4B), 10 nM 5-HT increased ICa by 251 ± 9 % (n= 5) in the presence of intracellular MBlue. In contrast to the previous experiments, the presence of intracellular MBlue did not prevent the inhibitory effect of 1 μM ACh. In the experiment shown in Fig. 4A, 1 μM ACh completely and reversibly inhibited the 5-HT response of ICa. This result was confirmed in a total of five identical experiments (Fig. 4B) where the 5-HT stimulation of ICa was fully antagonized by 1 μM ACh in the presence of intracellular MBlue. In these experiments, 5-HT (10 nM)-stimulated ICa was decreased to a value which was -18 ± 18 % of basal amplitude after addition of 1 μM ACh, an effect which was not different from the effect of ACh in the absence of MBlue (Fig. 4B). The inhibitory effect of ACh (1 μM) was also complete in three cells perfused with intracellular MBlue where Iso (10 nM) was used instead of 5-HT to stimulate ICa (data not shown).
Figure 4. Accentuated antagonism of ACh on ICa in the presence of intracellular Methylene Blue.
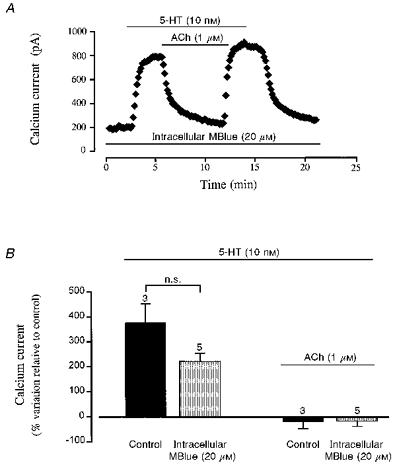
A, the cell was internally perfused with a pipette solution containing 20 μM MBlue throughout the entire experiment, as indicated by the bar. After superfusion of the cell with control solution, the cell was exposed to 5-HT (10 nM) and subsequently to the same solution containing 1 μM ACh during the periods indicated by the upper bars. B, comparison of the effects of 5-HT and ACh in the absence (Control) and presence of 20 μM intracellular MBlue (means ±s.e.m.; number of experiments indicated above the vertical columns). n.s., not significant.
Accentuated antagonism of ACh in the presence of ODQ and L-NMMA
The lack of effect of intracellular MBlue on the inhibition of ICa by ACh suggests that this effect is independent of its action on soluble guanylyl cyclase. However, MBlue may not be the most efficient guanylyl cyclase inhibitor. Unlike MBlue, ODQ is not a superoxide anion generator, and inhibits, selectively, the NO-sensitive isoform of guanylyl cyclase (Garthwaite, Southam, Boulton, Nielsen, Schmidt & Mayer, 1995; Abi-Gerges et al. 1997a). Thus, in the following experiments, we re-examined the effect of ACh in human atrial cells in the presence of ODQ. Moreover, to eliminate a possible NO-dependent, guanylyl cyclase-independent effect of ACh, the cells were also exposed to L-NMMA, a competitive NOS inhibitor. Figure 5A shows a typical experiment in which ODQ (10 μM) and L-NMMA (1 mM) were introduced both in the intracellular and extracellular solutions. As shown, application of 10 nM Iso produced a large increase in ICa in the presence of the two inhibitors. The mean stimulation of ICa by 10 nM Iso was not different in control experiments (+170 ± 22 % above basal ICa amplitude, n= 14) and in the presence of ODQ and L-NMMA (+205 ± 38 %, n= 11; Fig. 5B). Figure 5A also shows that the presence of ODQ and L-NMMA did not interfere with the muscarinic regulation of ICa. Indeed, addition of 1 μM ACh fully antagonized the Iso-response in the presence of the two inhibitors. The summary data shown in Fig. 5B indicate that for two different concentrations of ACh (100 nM and 1 μM), the presence of intracellular and extracellular ODQ (10 μM) and L-NMMA (1 mM) did not significantly modify the inhibitory effect of the muscarinic agonist.
Figure 5. Accentuated antagonism of ACh on ICa in the presence of ODQ and L-NMMA.
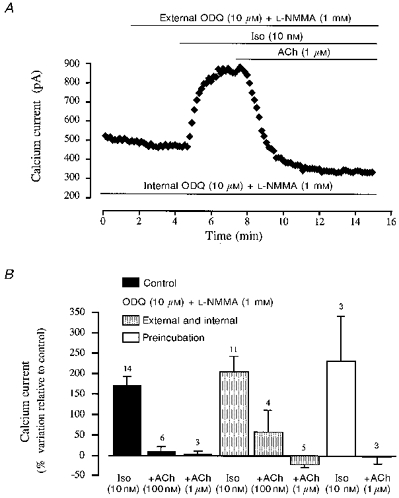
A, ODQ (10 μM) and L-NMMA (1 mM) were added to the pipette solution (lowermost bar) throughout the entire experiment, and applied extracellularly during the period indicated by the uppermost bar. The cell was then superfused with Iso (10 nM) and Iso + ACh (1 μM). B, comparison of the effect of Iso (10 nM) and ACh (100 nM and 1 μM) in the absence (Control, ▪) or presence of intracellular and extracellular ODQ (10 μM) and L-NMMA (1 mM) ( ), and after > 2 h preincubation of the cells with these compounds and introduction of the compounds in both extracellular and intracellular solutions (□). (Means ±s.e.m.; number of experiments indicated above the vertical columns.)
), and after > 2 h preincubation of the cells with these compounds and introduction of the compounds in both extracellular and intracellular solutions (□). (Means ±s.e.m.; number of experiments indicated above the vertical columns.)
Although L-NMMA is supposed to act rapidly (Han et al. 1995), the possibility remained that, in the relative short time of a patch-clamp experiment (usually around 20 min), ODQ and/or L-NMMA were unable to completely block guanylyl cyclase and/or NOS activity. Thus, a residual activity of these enzymes might remain in the presence of the two inhibitors, which could be sufficient to support the action of ACh on ICa. To test this hypothesis, we pre-incubated the cells with ODQ (10 μM) and L-NMMA (1 mM) for at least 2 h before the beginning of the patch-clamp experiment. Furthermore, the two inhibitors remained present in both intracellular and extracellular solutions throughout the experiments. However, this protocol did not prevent the accentuated antagonism of ACh (1 μM) on Iso (10 nM)-enhanced ICa. Indeed, as illustrated in Fig. 5B, in three cells taken from three different patients, the mean stimulatory effect of Iso (10 nM) was a 231 ± 111 % increase above basal amplitude, and ACh (1 μM) reduced the current to a value which was -2 ± 18 % below basal amplitude.
Positive control for the efficiency of MBlue and ODQ
Although unlikely, the above results do not rule out the possibility that MBlue and/or ODQ are ineffective in blocking NO-mediated pathways in human atrial myocytes under our experimental conditions. To address this question specifically, we have examined the effect of MBlue and ODQ on the regulation of ICa by NO. We have shown previously that the NO donor SIN-1 (3-morpholino-sydnonimine) stimulates basal ICa in human atrial myocytes under control conditions (Kirstein et al. 1995). This effect is probably due to an increase in intracellular cGMP concentration, since it is mimicked by intracellular dialysis of cGMP (Rivet-Bastide et al. 1997). Here, we tested the effect of another NO donor, SNAP (S-nitroso-N-acetyl-d,l-penicillamine) on ICa in human atrial myocytes. Extracellular application of 100 nm SNAP increased ICa by 52 ± 13 % above basal level (n= 16, P < 0.01; data not shown) under control conditions. However, when 20 μM MBlue was dialysed inside the cells, the same concentration of SNAP had no significant effect on ICa (12.9 ± 9.3 % increase above basal ICa, P= 0.14, n= 7). Similarly, the stimulatory effect of SNAP was abolished in the presence of intracellular and extracellular ODQ (10 μM) and L-NMMA (1 mM) (1.4 ± 16.8 % increase above basal ICa, n= 4).
DISCUSSION
In the present study, we used several inhibitors of the NO-cGMP pathway to examine its role in the effect of ACh on the L-type calcium current in human atrial myocytes. The main conclusion of our study is that neither NOS nor guanylyl cyclase activity appeared to be involved in the muscarinic regulation of ICa in this preparation.
The constitutive form of endothelial NOS (NOS 3 or eNOS) was found recently to be expressed in intact myocytes from rat ventricular and human atrial tissues (Balligand et al. 1995; Kelly et al. 1996). Since NO donors modulate cardiac ICa in human heart (Kirstein et al. 1995), as in a number of other animal species (Fischmeister & Méry, 1996), it was legitimate to question the potential role of NOS activity in the regulation of ICa. For this reason, we investigated the effect of L-NMMA on ICa in human atrial myocytes. L-NMMA is an L-arginine analogue that was shown to fully inhibit both the constitutive and inducible forms of NOS at submillimolar concentrations (Marletta, 1994). We used L-NMMA at a high concentration (1 mM) to ensure a complete inhibition of NOS activity (Marletta, 1994). However, we did not observe any effect on basal ICa in human atrial myocytes. This suggests that, either NOS activity is absent in our experimental conditions, or it is not functionally coupled to L-type Ca2+ channels in the absence of any exogenous modulator.
One main effector of NO in cardiac myocytes is a soluble form of guanylyl cyclase. Since cGMP was shown to regulate cardiac ICa in a number of animal species including human (Rivet-Bastide et al. 1997), it was of interest to examine whether an endogenous production of cGMP might regulate basal ICa. Recently, we found that inhibition of cGMP-stimulated phosphodiesterase (PDE2), one of the main targets of intracellular cGMP in heart (Fischmeister & Méry, 1996), with erythro-9-[2-hydroxy-3-nonyl]-adenine (EHNA) stimulates basal ICa in human atrial myocytes (Rivet-Bastide et al. 1997). Since the stimulatory effect of EHNA on ICa required the presence of intracellular GTP (the substrate of guanylyl cyclase), it was concluded that human atrial myocytes possess a basal guanylyl cyclase activity that provides a sufficient amount of cGMP to stimulate PDE2 (Rivet-Bastide et al. 1997). Inhibition of guanylyl cyclase should lead to an effect similar to PDE2 inhibition, i.e. to a stimulation of ICa. However, cardiac myocytes possess other cGMP-effectors besides PDE2, including a cGMP-inhibited phosphodiesterase (PDE3) and a cGMP-dependent protein kinase (PKG), which regulate ICa in different directions (Fischmeister & Méry, 1996; Méry et al. 1997). In the present study, we found that inhibition of soluble guanylyl cyclase with ODQ induced a variable stimulatory effect on basal ICa in 50 % of human atrial cells and had no effect in the other cells. We used a 10 μM concentration, which was ∼500 times higher than the IC50 reported by Garthwaite et al. (1995) for the inhibition of soluble guanylyl cyclase in slices of cerebellum. At the same concentration, ODQ had no effect on basal, Iso-, forskolin- or cAMP-stimulated ICa in frog ventricular myocytes but only reversed the inhibitory effect of NO donors on Iso-stimulated ICa (Abi-Gerges et al. 1997b). These results in frog and the stimulatory effect of ODQ observed in half of the cells in human suggests that ODQ was also acting as a guanylyl cyclase inhibitor in human atrial myocytes, but that a cell-to-cell variability may exist with respect to the relative efficacy of the different cGMP effectors; namely PDE2, PDE3 and PKG, and/or their coupling to Ca2+ channels (Rivet-Bastide et al. 1997).
Our finding that ACh inhibits basal ICa in human atrial myocytes may indicate that the intracellular concentration of cAMP, in the absence of any agonist, is sufficient to ensure a substantial phosphorylation of the L-type Ca2+ channels through cAMP-dependent protein kinase. Abrupt washout of ACh elicited a rebound stimulation of ICa, a phenomenon already observed by Wang & Lipsius (1995) in cat atrial myocytes, and by Tareen, Ono, Noma & Ehara (1991) on the Iso-stimulated Cl− current in guinea-pig ventricular myocytes. However, in our study, the rebound was not systematically observed in human atrial myocytes. Moreover, in contrast with cat atrial myocytes (Wang & Lipsius, 1995), superfusion of human atrial cells with ODQ and L-NMMA did not modify the inhibitory effect of ACh or the rebound stimulation (when present). Thus, NOS and guanylyl cyclase activities do not seem to play an important role in the regulation of basal ICa by muscarinic receptors in human atrial myocytes.
The finding that, in ventricular myocytes from frog (Hartzell & Fischmeister, 1986) and rat (Méry et al. 1991), cGMP antagonizes the effect of a β-adrenergic stimulation on ICa suggested a potential role for cGMP in the anti-adrenergic effect of ACh. However, studies specifically addressing this question led to conflicting results (see Méry et al. 1997). Since some of the discrepancies may be due to species differences, it was important to test this hypothesis directly in human tissue. In human atrial myocytes, it was reported that both 5-HT and Iso produce a similar maximal increase in ICa, and that 5-HT4 receptors, like β-adrenergic receptors, mediate their effect via cAMP and cAMP-dependent protein kinase (Ouadid et al. 1991). Our results showing that ACh antagonized in a similar manner both 5-HT- and Iso-stimulation of ICa confirm these findings. Extracellular application of MBlue, a soluble guanylyl cyclase inhibitor, had no effect on the β-adrenergic stimulation of ICa. However, at the same concentration (10 μM), MBlue prevented the accentuated antagonism of ACh on ICa in human atrial myocytes. This is similar to results obtained in guinea-pig (Levi et al. 1994) and rat (Balligand et al. 1993) ventricular myocytes as well as in rabbit nodal myocytes (Han et al. 1995, 1996). However, this antagonistic effect of MBlue is unlikely to be due to guanylyl cyclase inhibition since it was not mimicked by an intracellular perfusion with MBlue. Moreover, MBlue was shown recently to bind to muscarinic M2 receptors in rat cardiac myocytes (Abi-Gerges et al. 1997a). Thus, the anti- muscarinic effect of extracellular MBlue seen here is probably due to this detrimental side effect of the drug. In a second series of experiments, we used ODQ, a more selective guanylyl cyclase inhibitor (Garthwaite et al. 1995; Abi-Gerges et al. 1997b), in combination with L-NMMA to inhibit the two key enzymes of the NO-cGMP pathway. However, no difference in the β-adrenergic stimulation or in the accentuated antagonism of ACh was found in comparison with control experiments. In several studies in which the muscarinic regulation of ICa was found to depend upon NO (Han et al. 1994, 1995, 1996), pre-incubation of the myocytes with NOS antagonists was used. However, incubation of human atrial myocytes with ODQ and L-NMMA for as long as 2 h before the experiment did not attenuate the efficacy of ACh. Finally, the lack of effect of MBlue and ODQ on the muscarinic response of ICa was not due to an inefficiency of these compounds in blocking NO-mediated pathways, since both drugs antagonized the stimulatory effect of the NO donor SNAP on ICa.
In conclusion, our present results demonstrate that NOS and guanylyl cyclase activities do not play a determinant role in the muscarinic regulation of basal and stimulated ICa in human atrial myocytes. This conclusion may not only apply to atrial tissue since Kilter et al. (1995) reported recently that pre-treatment of human papillary muscle strips with L-NMMA or MBlue modified neither the basal force of contraction, nor the positive inotropic effect of Iso, nor the anti-adrenergic effect of carbachol. Moreover, a similar conclusion was reached recently in a study performed in frog ventricular myocytes (Méry et al. 1996). The discrepancy with other results may partly be due to species differences in the respective contributions of the NO-cGMP pathway and the inhibition of adenylyl cyclase (Hescheler et al. 1986; Jurevicius & Fischmeister, 1996) to the signalling cascades mediated by activation of cardiac muscarinic receptors. Nevertheless, species- and tissue-independent discrepancies remain (Balligand et al. 1993; Habuchi et al. 1996; Han et al. 1996; Abi-Gerges et al. 1997c) that argue against a determinant role of the NO-cGMP pathway for the muscarinic control of the heart.
Acknowledgments
We wish to thank Mr Patrick Lechêne, Mrs Florence Lefebvre and Mrs Catherine Rücker-Martin for skilful technical assistance, Dr Pierre-François Méry for helpful discussions, Drs Thierry Folliguet, Patrice Dervanian, Jean-Yves Neveux, and Loïc Macé (Service de Chirurgie Cardiaque, Hôpital Marie Lannelongue, Le Plessis-Robinson, France) for their assistance in obtaining the tissues used in these experiments, and Drs Jean-Jacques Mercadier, Stéphane Hatem and Agnès Bénardeau (CNRS URA 1159, Hôpital Marie Lannelongue, Le Plessis-Robinson, France) for their assistance in providing the isolated cells and for continual support. This work was supported by the Association Française contre les Myopathies, the Fondation pour la Recherche Médicale, the Ministère de la Recherche et de l'Enseignement Supérieur (ACC-SV9), and the Association Recherche et Partage. Thomas Eschenhagen was supported by a Heisenberg grant of the Deutsche Forschungsgemeinschaft.
References
- Abi-Gerges N, Eschenhagen T, Hove-Madsen L, Fischmeister R, Méry P-F. Methylene blue is a muscarinic antagonist in cardiac myocytes. Molecular Pharmacology. 1997 a;52:482–490. doi: 10.1124/mol.52.3.482. [DOI] [PubMed] [Google Scholar]
- Abi-Gerges N, Hove-Madsen L, Fischmeister R, Méry P-F. A comparative study of the effects of three guanylyl cyclase inhibitors on the L-type Ca2+ and muscarinic K+ currents in frog cardiac myocytes. British Journal of Pharmacology. 1997 b;121:1369–1377. doi: 10.1038/sj.bjp.0701249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi-Gerges N, Méry P-F, Fischmeister R. The NO-sensitive guanylyl cyclase does not participate in the muscarinic regulation of rat Ca2+ current. Biophysical Journal. 1997c;72:A34. [Google Scholar]
- Balligand J-L, Kelly RA, Mardsen PA, Smith TW, Michel T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proceedings of the National Academy of Sciences of the USA. 1993;90:347–351. doi: 10.1073/pnas.90.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balligand J-L, Kobzik L, Han XQ, Kaye DM, Belhassen L, Ohara DS, Kelly RA, Smith TW. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. Journal of Biological Chemistry. 1995;270:14582–14586. doi: 10.1074/jbc.270.24.14582. 10.1074/jbc.270.24.14582. [DOI] [PubMed] [Google Scholar]
- Böhm M, Gierschik P, Schwinger RHG, Uhlmann R, Erdmann E. Coupling of M-cholinoceptors and A1 adenosine receptors in human myocardium. American Journal of Physiology. 1994;266:H1951–1958. doi: 10.1152/ajpheart.1994.266.5.H1951. [DOI] [PubMed] [Google Scholar]
- Breitwieser GE, Szabo G. Uncoupling of cardiac muscarinic and β-adrenergic receptors from ion channels by a guanine nucleotide analogue. Nature. 1985;317:538–540. doi: 10.1038/317538a0. [DOI] [PubMed] [Google Scholar]
- Fischmeister R, Méry P-F. Regulation of cardiac calcium channels by cGMP and NO. In: Morad M, Ebashi S, Trautwein W, Kurachi Y, editors. Molecular Physiology and Pharmacology of Cardiac Ion Channels and Transporters. Dordrecht, Boston, London: Kluwer Academic Publishers; 1996. pp. 93–105. [Google Scholar]
- Fleming JW, Strawbridge RA, Watanabe AM. Muscarinic receptor regulation of cardiac adenylate cyclase activity. Journal of Molecular and Cellular Cardiology. 1987;19:47–61. doi: 10.1016/s0022-2828(87)80544-8. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one. Molecular Pharmacology. 1995;48:184–188. [PubMed] [Google Scholar]
- George WJ, Polson JB, O'Toole AG, Goldberg N. Elevation of 3′,5′-cyclic phosphate in rat heart after perfusion with acetylcholine. Proceedings of the National Academy of Sciences of the USA. 1970;66:398–483. doi: 10.1073/pnas.66.2.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habuchi Y, Nishio H, Tanaka H, Yamamoto T, Lu LL, Yoshimura M. Regulation by acetylcholine of Ca2+ current in rabbit atrioventricular node cells. American Journal of Physiology. 1996;271:H2274–2282. doi: 10.1152/ajpheart.1996.271.6.H2274. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recordings from cell and cell-free patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Han X, Kobzik L, Balligand J-L, Kelly RA, Smith TW. Nitric oxide synthase (NOS3)-mediated cholinergic modulation of Ca2+ current in adult rabbit atrioventricular nodal cells. Circulation Research. 1996;78:998–1008. doi: 10.1161/01.res.78.6.998. [DOI] [PubMed] [Google Scholar]
- Han X, Shimoni Y, Giles WR. An obligatory role for nitric oxide in autonomic control of mammalian heart rate. The Journal of Physiology. 1994;476:309–314. doi: 10.1113/jphysiol.1994.sp020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Shimoni Y, Giles WR. A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. Journal of General Physiology. 1995;106:45–65. doi: 10.1085/jgp.106.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC, Fischmeister R. Opposite effect of cyclic GMP and cyclic AMP on Ca2+ current in single heart cells. Nature. 1986;323:273–275. doi: 10.1038/323273a0. [DOI] [PubMed] [Google Scholar]
- Hescheler J, Kameyama M, Trautwein W. On the mechanism of muscarinic inhibition of the cardiac Ca current. Pflügers Archiv. 1986;407:182–189. doi: 10.1007/BF00580674. [DOI] [PubMed] [Google Scholar]
- Hove-Madsen L, Méry P-F, Jurevicius J, Skeberdis A, Fischmeister R. Regulation of myocardial calcium channels by cyclic AMP metabolism. Basic Research in Cardiology. 1996;91(suppl. 2):1–8. doi: 10.1007/BF00795355. [DOI] [PubMed] [Google Scholar]
- Jurevicius J, Fischmeister R. Acetylcholine inhibits Ca2+ current by acting exclusively at a site proximal to adenylyl cyclase in frog cardiac myocytes. The Journal of Physiology. 1996;491:669–675. doi: 10.1113/jphysiol.1996.sp021248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RA, Balligand JL, Smith TW. Nitric oxide and cardiac function. Circulation Research. 1996;79:363–380. doi: 10.1161/01.res.79.3.363. [DOI] [PubMed] [Google Scholar]
- Kennedy RH, Hicks KK, Brian JE, Jr, Seiffen E. Nitric oxide has no chronotropic effect in rat atria isolated from rat heart. European Journal of Pharmacology. 1994;255:149–156. doi: 10.1016/0014-2999(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Kilter H, Lenz O, La Rosée K, Flesch M, Schwinger RHG, Mädge M, Kuhn-Regnier F, Böhm M. Evidence against a role of nitric oxide in the indirect negative inotropic-effect of M-cholinoceptor stimulation in human ventricular myocardium. Naunyn-Schmiedebergs's Archives of Pharmacology. 1995;352:308–312. doi: 10.1007/BF00168562. [DOI] [PubMed] [Google Scholar]
- Kirstein M, Rivet-Bastide M, Hatem S, Bénardeau A, Mercadier J-J, Fischmeister R. Nitric oxide regulates the calcium current in isolated human atrial myocytes. Journal of Clinical Investigation. 1995;95:794–802. doi: 10.1172/JCI117729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi RC, Alloatti G, Penna C, Gallo MP. Guanylate-cyclase-mediated inhibition of cardiac ICa by carbachol and sodium nitroprusside. Pflügers Archiv. 1994;426:419–426. doi: 10.1007/BF00388305. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- MacDonnel KL, Tibbits GF, Diamond J. cGMP elevation does not mediate muscarinic-induced negative inotropy in rat ventricular cardiomyocytes. American Journal of Physiology. 1995;269:H1905–1912. doi: 10.1152/ajpheart.1995.269.6.H1905. [DOI] [PubMed] [Google Scholar]
- Marletta MA. Approaches towards selective inhibition of nitric oxide synthase. Journal of Medical Chemistry. 1994;37:1899–1907. doi: 10.1021/jm00039a001. [DOI] [PubMed] [Google Scholar]
- Méry P-F, Abi-Gerges N, Vandecasteele G, Jurevicius J, Eschenhagen T, Fischmeister R. Muscarinic regulation of the L-type calcium current in isolated cardiac myocytes. Life Sciences. 1997;60:1113–1120. doi: 10.1016/s0024-3205(97)00055-6. [DOI] [PubMed] [Google Scholar]
- Méry P-F, Hove-Madsen L, Chesnais JM, Hartzell HC, Fischmeister R. Nitric oxide synthase does not participate in the negative inotropic effect of acetylcholine in frog heart. American Journal of Physiology. 1996;39:H1178–1188. doi: 10.1152/ajpheart.1996.270.4.H1178. [DOI] [PubMed] [Google Scholar]
- Nawrath H, Baumner D, Rupp J, Oelert H. The ineffectiveness of the NO-cyclic GMP signaling pathway in the atrial myocardium. British Journal of Pharmacology. 1995;116:3061–3067. doi: 10.1111/j.1476-5381.1995.tb15964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouadid H, Seguin J, Dumuis A, Bockaert J, Nargeot J. Serotonin increases calcium current in human atrial myocytes via the newly described 5-hydroxytryptamine4 receptors. Molecular Pharmacology. 1991;41:346–351. [PubMed] [Google Scholar]
- Rivet-Bastide M, Vandecasteele G, Hatem S, Bénardeau A, Mercadier JJ, Fischmeister R. cGMP-stimulated phosphodiesterase regulates the basal calcium current in human atrial myocytes. Journal of Clinical Investigation. 1997;99:111–119. doi: 10.1172/JCI119460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein B, Drögemüller A, Mülsch A, Schmitz W, Scholz H. Ca2+-dependent constitutive nitric oxide synthase is not involved in the cyclic GMP-increasing effects of carbachol in ventricular cardiomyocytes. Journal of Pharmacological and Experimental Therapeutics. 1993;266:919–925. [PubMed] [Google Scholar]
- Tareen FM, Ono K, Noma A, Ehara T. β-Adrenergic and muscarinic regulation of the chloride current in guinea-pig ventricular myocytes. The Journal of Physiology. 1991;440:225–241. doi: 10.1113/jphysiol.1991.sp018705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay J, Gerzer R, Hamet P. Cyclic GMP in cell function. Advances in Second Messenger and Protein Phosphorylation Research. 1988;22:320–383. [PubMed] [Google Scholar]
- Vandecasteele G, Eschenhagen T, Fischmeister R. Muscarinic regulation of the L-type calcium current in human atrial myocytes. Journal of Molecular and Cellular Cardiology. 1997;29:A94. [Google Scholar]
- Wang YG, Lipsius SL. Acetylcholine elicits a rebound stimulation of the Ca2+ current mediated by pertussis toxin-sensitive G protein and cAMP-dependent protein kinase A in atrial myocytes. Circulation Research. 1995;76:634–644. doi: 10.1161/01.res.76.4.634. [DOI] [PubMed] [Google Scholar]