

Abstract
Using whole-cell voltage-clamp recordings of dissociated hippocampal CA1 neurones, we demonstrated that 17β-oestradiol rapidly potentiates kainate-induced currents when applied either to the outside or the inside of the neurone. However, when the steroid was conjugated to bovine serum albumin (E2-BSA), application to either the extracellular plasma membrane (E2-BSAout) or the cytosolic side of the cell (E2-BSAin) had no observable effect on kainate-induced currents. However, when applied stimultaneously to both sides of the plasma membrane, E2-BSA potentiated kainate-induced currents.
Application of E2-BSAout and GTPγSin potentiated kainate-induced currents. The potentiation of kainate-induced currents by 17β-oestradiol was occluded by cholera toxin pretreatment and appeared to be pertussis toxin insensitive.
E2-BSAin prolonged the effect of 8-bromoadenosine 3′,5′ cyclic monophosphate (8-bromo-cAMP) on kainate-induced currents. The recovery from the 8-bromo-cAMP response was found to be a function of the concentration of E2-BSAin. The application of ATPγSin occluded the effect of 17β-oestradiol.
These results suggest that the non-genomic action of 17β-oestradiol in the potentiation of kainate-induced currents is mediated via an action on Gs protein-coupled receptors. This operates in concert with an internal action of 17β-oestradiol on a cAMP-dependent phosphorylation.
Oestrogen (17β-oestradiol; E2), a lipophilic steroid hormone, regulates female reproductive functions through a nuclear receptor that belongs to a superfamily of ligand-activated transcriptional factors. In addition to this classical transcriptional action, oestrogen has been shown to modulate cellular activity in a variety of cells from endocrine to nerve cells. The rapid onset and time course of these effects, seen in seconds to minutes, precludes activation of the transcriptional pathway to produce the change in cellular activity (for reviews see Wong, Thompson & Moss, 1996; Moss, Gu & Wong, 1997). Evidence for the non-transcriptional actions of oestrogen on nerve cells is accumulating from studies on different regions of the brain (Nabekura, Oomura, Minami, Mizuno & Fukuda, 1986; Becker, 1990a, b; Castner, Xiao & Becker, 1993; Thompson & Moss, 1994; Lagrange, Ronnekleiv & Kelly, 1994; Zhou, Watters & Dorsa, 1996), but the sites and mechanisms of action have not been addressed directly. These non-transcriptional actions are thought to be neuromodulatory and critical for cell-cell communications.
In hippocampal CA1 neurones, 17β-oestradiol can rapidly increase the amplitude of glutamate-induced or Schaffer collateral-activated EPSPs possibly via non-NMDA receptors (Wong & Moss, 1991, 1992, 1994). In experiments employing perforated whole-cell recording techniques, 17β-oestradiol was demonstrated to potentiate kainate-induced currents in a dose-dependent manner. 17β-Oestradiol did not affect the kinetics or reversal potential of the kainate-induced response, suggesting that the potentiation did not involve direct interaction of 17β-oestradiol with kainate receptors or activation of ion channels other than kainate receptor- channels. The effect of oestrogen in increasing the conductance of kainate currents was mimicked by 8-bromoadenosine 3′,5′ cyclic monophosphate (8-bromo-cAMP), and could be blocked by the specific protein kinase A (PKA) inhibitor, RP-cyclic 3′,5′-hydrogen phosphorothioate adenosine triethylammonium salt (Rp-cAMPS). These results point to the potentiation of kainate-induced currents by 17β-oestradiol as a cAMP-PKA-mediated event (Gu & Moss, 1996). However, the sites and mechanisms for the short-term actions of 17β-oestradiol have remained elusive.
Recent studies have demonstrated that steroid hormones may act upon cellular second messenger systems via a specific membrane receptor(s). For example, 17β-oestradiol reduces Ba2+ currents in rat neostriatal neurones by coupling a membrane receptor to a G protein (Mermelstein, Becker & Surmeier, 1996), and ffrench-Mullen and co-workers found that some neurosteroids can access their binding site from the extracellular surface, and modulate voltage-gated calcium currents in hippocampal CA1 neurones via a pertussis toxin-sensitive G protein-coupled mechanism (ffrench-Mullen, Danks & Spence, 1994; ffrench-Mullen, 1995). Studies on hypothalamic neurones suggest that 17β-oestradiol induces rapid changes in the pharmacodynamics of the G protein-coupled mechanism of another ligand by decreasing the potency of opioid agonists in opening inwardly rectifying potassium channels (Lagrange et al. 1994). Furthermore, immunological studies have provided evidence for the localization of membrane oestrogen receptors in GH3 pituitary tumour cells using well-characterized antibodies (Pappas, Gametchu & Watson, 1995). These antibodies recognized multiple oestrogen receptor epitopes along the full length of the protein. The authors concluded that membrane oestrogen receptors may share identity with intracellular oestrogen receptors. High affinity membrane oestrogen-binding sites have also been identified in several specific brain areas, namely, the hypothalamus, cerebellum, olfactory bulbs (Ramirez, Zheng & Siddique, 1996; Zheng, Ali & Ramirez, 1996) and hippocampus (Horvat, Nikezic & Martinovic, 1995).
The goal of the present study was to examine the effects of a membrane-impermeant form of 17β-oestradiol on kainate-induced currents in dissociated CA1 hippocampal neurones using whole-cell voltage-clamp techniques. We sought specifically to test the hypothesis that the potentiation of kainate-induced currents by oestrogen is a G protein-coupled and cAMP-dependent process. The data presented here provide evidence for a novel mechanism of steroid action. In order for 17β-oestradiol potentiation of kainate-induced currents to occur, oestrogen must be present on both sides of the plasma membrane. On the extracellular surface, 17β-oestradiol appears to activate a Gs-coupled receptor, while on the cytosolic side, extracellular oestrogen operates in concert with an internal action of 17β-oestradiol on cAMP-dependent phosphorylation.
METHODS
Animals and preparation of acutely dissociated neurones
Hippocampal CA1 pyramidal neurones were acutely dissociated using modified procedures of Kay & Wong (1986). Sprague-Dawley male and female rats (2-4 weeks old) were decapitated with a guillotine, the skull was opened and the hippocampus was quickly removed from the brain. These procedures were approved for use by the Animal Review Committee (Animal Welfare Assurance no. A3472-01). Hippocampi were sectioned (450 μm thick sections) with a vibratome (Oxford) while being bathed in 4°C oxygenated Pipes-saline solution comprising (mM): NaCl, 120; KCl, 5.0; CaCl2, 1.0; MgCl2, 1.0; D-glucose, 25; Pipes, 20; pH 7.4. The slices were placed in a culture dish, and ‘punches’ were taken from the CA1 area with a capillary tube. The punches were incubated at room temperature (20-22°C) in Pipes-saline solution containing 1.5 mg ml−1 protease Type XIV. The incubation medium was stirred slowly, and exposed to 95 % O2-5 % CO2 at its surface. After 30-45 min of enzymatic digestion, punches were rinsed 3 times in oxygenated Pipes-saline and triturated with a fire-polished Pasteur pipette for mechanical dissociation. The cell suspension was then plated into the central concave area of a slide containing the standard extracellular solution comprising (mM): NaCl, 140; KCl, 3.0; CaCl2, 2.0; Hepes, 10; pH 7.3.
Electrophysiological recordings
Whole-cell recordings were performed under voltage-clamp mode according to standard techniques (Hamill, Marty, Neher, Sakmann & Sigworth, 1981). Both conventional and perforated whole-cell recordings were employed in isolated hippocampal CA1 neurones. The neurones were visualized with a Nikon inverted phase-contrast microscope equipped with Nomarski optics. The cell body was approximately 30-40 μm in diameter and pyramidal in shape with relatively intact apical dendrites. Whole-cell recording electrodes were pulled from borosilicate glass with a Sutter Flaming-Brown electrode puller, and fire polished with a CPM-2 coating/polishing microforge. The electrode resistance was typically 2-5 MΩ in bath solution. The standard internal solution for the recording electrode consisted of (mM): CsCl, 140; NaCl, 4.0; EGTA, 10; Hepes, 10; CaCl2, 1; pH adjusted to 7.3 with CsOH. For perforated whole-cell recording, the electrode was first submerged in standard internal solution for 3-5 s. A small amount of solution would reach the tip by capillary action. Then additional standard internal solution containing 100 μg ml−1 nystatin was added by backfilling the electrode.
After formation of a gigaohm (> 1 GΩ) seal, the membrane was ruptured by slight suction in conventional whole-cell recording configuration. In the case of perforated whole-cell recording, series resistance and capacitive transient current evoked by a voltage pulse (-10 mV, 20 ms) were monitored during the development of nystatin perforation. Usually within 25-30 min, the amplitude of the capacitive transient increased and the time constant of the transient decreased. Whole-cell currents induced by 20 mV steps (from -80 to +80 mV) gradually became larger with time, and the series resistance decreased to a stable minimal value (< 20 MΩ), indicating the establishment of perforated whole-cell recording. The holding potential was -60 mV in both configurations. Series resistance was compensated electronically and monitored periodically. Hippocampal neurones with lower seal resistance or high series resistance were not selected for further study.
Application of chemicals
A multiple-barrel pipette with a total tip diameter < 10 μm was used to apply individual substances on the recorded neurone. Ejection of each chemical was made with a Picospritzer unit (General Valve Corporation). The kainate current was induced by ejection of kainate (20 ms, 0.1-1.0 p.s.i.) at the dendrites of the CA1 neurone. The application commenced immediately after the patch was ruptured, and was repeated once every 30 s. Other chemicals were assigned randomly to one of the multiple barrels. Their effects on kainate currents were tested by extracellularly perfusing the cell for 3 min.
Kainate was made as a concentrated stock solution and diluted in standard extracellular bath solution prior to use at a concentration of 100 μM. This concentration represents the EC50 value for kainate (Gu & Moss, 1996). 17β-Oestradiol was initially dissolved in dimethylsulphoxide (DMSO) and diluted in standard extracellular bath solution at a concentration of 100 nM with the final DMSO concentration not exceeding 0.01 %. 17β-Oestradiol conjugated to bovine serum albumin (E2-BSA) on the seventeenth carbon was made up as a concentrated stock (33 mM) in the extracellular bath solution. The ratio of oestradiol to BSA in this preparation was 30 : 1. The conjugate was serially diluted to its final concentration before the experiment. 8-Bromo-cAMP and 8-bromoguanosine 3′,5′ cyclic monophosphate (8-bromo-cGMP) were made up as stock solutions and frozen. Aliquots were diluted in standard extracellular bath solution immediately prior to use to concentrations of 500 μM. GTPγS and ATPγS were stored in aliquots at -70°C, and diluted to 500 μM in standard internal solution immediately before use.
In some experiments, E2, E2-BSA, GTPγS or ATPγS was added to the standard internal solution and allowed to diffuse into the cell. These experiments were conducted under the standard whole-cell recording configuration, and an ATP regeneration system consisting of 4 mM Tris-ATP, 20 mM phosphocreatine and 50 U ml−1 creatine phosphokinase was added to the internal solution to minimize the washout effect on kainate currents. In experiments involving cholera toxin or pertussis toxin treatment, tissue punches were incubated in the extracellular solution containing cholera toxin or pertussis toxin for 1-4 h. Control preparations were treated using the same protocol in a toxin-free extracellular solution. All chemicals were purchased from Sigma except GTPγS which was a kind gift from Drs Susan Mumby and Al Gilman (Department of Pharmacology, University of Texas Southwestern Medical Center).
Data analysis
Whole-cell currents were recorded under voltage-clamp configuration with an Adams/List EPC-9 amplifier, sampled at 2 kHz and filtered at 2.3 kHz. Data were digitized and stored on an Atari Mega 4 computer. Analysis of recordings was performed with an Atari data analysis program. Peak currents were normalized as I/I0, where I represents the amplitude of kainate-induced currents at any test time point and I0 is the initial value at the beginning of the recording. The percentage change in the amplitude of kainate currents was determined according to the formula: (Idrug/I0 - 1)100, where Idrug represents the peak amplitude of the kainate current in the presence of the test drug. Prolonged potentiation of the effect induced by 8-bromo-cAMP by E2-BSA was described as percentage recovery of the response at 15 min after removal of the chemical. Dose-response data were fitted with the logistic equation of the form:
where Rmax is the response to a saturating concentration of agonist, EC50 is the concentration that evokes a half-maximal response, D is the agonist concentration and nH is the estimated Hill coefficient that describes the steepness of the curve. Fitting was performed with SigmaPlot (Jandel Scientific). All quantitative data are expressed as means ±s.e.m.; n indicates the number of cells tested. Statistical analysis was performed using Student's paired or unpaired t test. Results were considered significant only for P < 0.05.
RESULTS
Comparison of the effects of 17β-oestradiol and E2-BSA on kainate-induced currents
Kainate currents were induced by repeated application of kainate (100 μM, 20 ms) onto the cell surface of isolated hippocampal CA1 neurones. The initial amplitude of the response was 620 ± 253 pA and varied less than ± 10 % during the recording period. The amplitude of kainate-induced currents could be increased by 40 ± 7 % in the presence of 17β-oestradiol (100 nM, 3 min). 17β-Oestradiol was effective in the potentiation of kainate-induced currents in nearly 38 % of the cells tested (52 out of 136). No morphological difference was observed between the cells responding to 17β-oestradiol and those not responding. The effect was rapid in onset (within 3 min of application) but readily reversible upon the removal of the steroid. The effect of 17β-oestradiol was dose dependent but sex independent. The aforementioned data corroborate our earlier results (Gu & Moss, 1996). To identify the possible sites of 17β-oestradiol action on kainate-induced currents, 17β-oestradiol conjugated to bovine serum albumin (E2-BSA) was applied to cells that were responsive to 17β-oestradiol. Although extracellular application of 17β-oestradiol (100 nM), which readily crosses the plasma membrane, could potentiate kainate currents in these cells (12 out of 34), application of the membrane-impermeant E2-BSA (3.3 nM) to the extracellular surface (E2-BSAout) caused no obvious change in the amplitude of kainate-induced currents (Fig. 1A). To test the possibility that 17β-oestradiol acts solely on the cytosolic side, a group of cells were intracellularly dialysed with 17β-oestradiol or E2-BSA (E2-BSAin). Kainate-induced currents were gradually increased by 35 ± 7 % when 17β-oestradiol (100 nM) was diffused into the cell (5 out of 18). The enhancement was slow in onset and relatively irreversible compared with the response of cells to extracellular application of 17β-oestradiol. E2-BSA (3.3 nM), however, had no observable effect on kainate-induced currents when applied intracellularly for more than 20 min (n= 25; Fig. 1B). These negative results from the application of E2-BSA implied that the presence of 17β-oestradiol on both sides of the cellular membrane might be a requisite for oestradiol modulation of kainate-induced currents in hippocampal neurones.
Figure 1. Comparison of the effects of 17β-oestradiol and E2-BSA on kainate-induced currents.

A, E2-BSA (3.3 nM, 3 min) was applied extracellularly (E2-BSAout) to the cells responsive to 17β-oestradiol (E2,out, 100 nM, 3 min), and caused no obvious change in the amplitude of kainate-induced currents. B, kainate-induced currents were gradually increased by 35.1 ± 7.2 % when the cell was dialysed with 17β-oestradiol (100 nM). The enhancement was slow in onset and irreversible (•). E2-BSA (3.3 nM) had no observable effect on kainate-induced currents when applied intracellularly for more than 20 min (○). In A and B, actual current traces corresponding to the specific time points in the graphs (arrows) are displayed at the top.
Potentiation of kainate-induced currents by simultaneous application of E2-BSA on the extra- and intracellular side of the membrane
To test further whether E2-BSA is required on both sides of the plasma membrane, experiments were conducted by exposing hippocampal neurones to E2-BSA simultaneously on both the extracellular surface and cytosolic side of the membrane. After E2-BSA (3.3 nM) was allowed to dialyse into the cell interior for at least 7 min, the external application of E2-BSA (3.3 nM, 3 min) to the cell surface induced an increase in the amplitude of kainate-induced currents by 36 ± 7 % in twelve of thirty-one cells tested (Fig. 2). This enhancement was similar to the potentiation of kainate-induced currents by 17β-oestradiol in the rapidity of onset and the magnitude of the current increase.
Figure 2. Potentiation of kainate-induced currents by simultaneous application of E2-BSA on the extracellular surface and cytosolic side of the membrane.
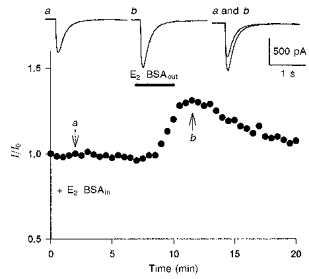
After E2-BSA (3.3 nM) was allowed to dialyse into the cell interior for at least 7 min (E2-BSAin), the extracellular application of E2-BSA (3.3 nM, 3 min) induced an increase in the amplitude of kainate-induced currents. Representative current traces from sample points (arrows) are shown and compared at the top.
Activation of a G protein by 17β-oestradiol in the potentiation of kainate-induced currents
It has been shown that E2-BSAout alone caused no observable action on kainate-induced currents. However, when cells (n= 22) were intracellularly dialysed with GTPγS (500 μM), a non-hydrolysable GTP analogue, the application of E2-BSAout (3.3 nM, 3 min) induced an increase in the amplitude of kainate-induced currents by 34 ± 2 % (n= 8; Fig. 3). This effect on kainate-induced currents persisted even after the removal of external E2-BSA. When applied alone, GTPγSin had no observable effect on kainate-induced currents. This response pattern suggests that E2-BSAout activated a G protein-coupled receptor in the potentiation of kainate-induced currents. This concept was explored further by testing cells that were pretreated with cholera toxin or pertussis toxin. Pretreatment of the cells with cholera toxin (250 ng ml−1) increased the amplitude of kainate-induced currents compared with those of cells incubated in bath solution. Application of 17β-oestradiol (100 nM) to these cells caused no further potentiation. Incubation of cells with pertussis toxin (250 ng ml−1), however, did not elicit any obvious change in kainate-induced currents (n= 8), and application of 17β-oestradiol could enhance the amplitude in a pattern similar to that induced in the control group (Table 1). The results indicate that activation of a G protein, which is sensitive to cholera toxin, could potentiate kainate-induced currents. Since incubation in cholera toxin could occlude the action of 17β-oestradiol on kainate-induced currents, the activation induced by 17β-oestradiol may share a common signalling pathway with that of cholera toxin.
Figure 3. Activation of a G protein by 17β-oestradiol in the potentiation of kainate-induced currents.
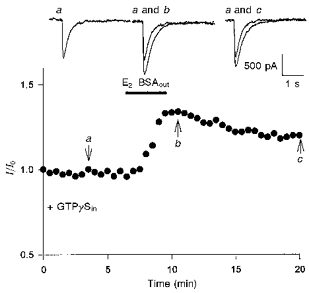
The external presence of E2-BSA (3.3 nM, 3 min) induced an increase in the amplitude of kainate-induced currents when the illustrated cell was intracellularly dialysed with GTPγS (GTPγSin; 500 μM). The effect on kainate-induced currents persisted even after the removal of external E2-BSA. Representative current traces selected at specific time points (arrows) are displayed at the top.
Table 1.
Effects of cholera toxin and pertussis toxin on kainate-induced currents and potentiation by 17β-oestradiol
| Current response ( %) | ||
|---|---|---|
| Bath solution | Before E2 application | After E2 application |
| Control | 100 (n= 18) | 129.0 ± 6.4 (n= 5) a |
| Cholera toxin | 122.6 ± 4.5 (n= 10) b | 119.4 ± 8.2 (n= 10) |
| Pertussis toxin | 98.1 ± 8.6 (n= 8) | 115 ± 5.7 (n= 3)c |
Current response was normalized to that measured in the control solution.
Amplitude of kainate-induced currents was increased significantly (P < 0.01) compared with the control group before E2 application.
P < 0.05, compared with the control group before E2 application.
P < 0.05, compared with the pertussis toxin group before E2 application.
Prolonged potentiation of cAMP action on kainate-induced currents by 17β-oestradiol
In order to evaluate the contribution of PKA-dependent phosphorylation/dephosphorylation to the effect of 17β-oestradiol on kainate-induced currents, a series of experiments was performed using 8-bromo-cAMP, 8-bromo-cGMP or ATPγS. Application of 8-bromo-cAMP (500 μM, 3 min), a membrane-permeant analogue of cAMP, enhanced the amplitude of kainate-induced currents by 35 ± 4 % (n= 6). The enhancement showed latency and magnitude characteristics similar to those observed in the potentiation induced by cell surface application of 17β-oestradiol and the effect was readily reversible when 8-bromo-cAMP was removed (Fig. 4A). When cells were intracellularly dialysed with E2-BSA (3.3 nM), the recovery after removal of 8-bromo-cAMP was significantly reduced (P < 0.01). A portion (30 ± 5 %) of the potentiation was recovered at 15 min after the removal of 8-bromo-cAMP (n= 3; Fig. 4B). By varying the intracellular concentration of E2-BSA in a population of E2-BSA-sensitive neurones, it was found that the decrease in recovery induced by E2-BSA was dose dependent; it was evident at 0.1 nM and saturated at 0.1 μM (Fig. 5). A Hill plot derived from the dose-response data gave an EC50 of 2.51 ± 3.18 nM and an nH of 0.39 ± 0.07. It should be noted that a persistent recovery (about 20 %) from cAMP-induced potentiation existed independently of E2-BSA concentration. The application of 8-bromo-cGMP had no observable effect on kainate-induced currents (n= 10; data not shown).
Figure 4. Prolongation of cAMP action on kainate-induced currents by 17β-oestradiol.

A, extracellular application of 8-bromo-cAMP (500 μM, 3 min) reversibly increased the amplitude of kainate-induced currents. The enhancement showed similar latency and magnitude to the potentiation induced by 17β-oestradiol (n= 6). B, the recovery from enhancement induced by 8-bromo-cAMP (500 μM, 3 min) was significantly retarded when cells were intracellularly dialysed with E2-BSA (3.3 nM; n= 3).
Figure 5. The decrease in recovery induced by intracellular E2-BSA was dose dependent.
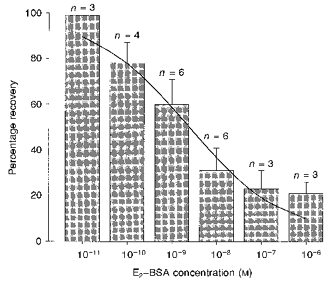
The decrease in recovery was evident at 0.1 nM and was saturated at 0.1 μM. A Hill plot derived from the dose-response data gave an EC50 of 2.51 ± 3.18 nM and an nH of 0.39 ± 0.07. Prolongation of cAMP action is described as the percentage recovery at 15 min after removal of 8-bromo-cAMP. It should be noted that a persistent recovery (about 20 %) existed independent of E2-BSA concentration. n indicates the number of the cells tested.
In another set of experiments, cells were dialysed with the non-hydrolysable ATP analogue, ATPγS (500 μM). By introducing stable phosphate groups onto relevant proteins, ATPγS can affect the phosphorylation state of the proteins. The amplitude of kainate-induced currents was slowly enhanced in eight of the twenty-one neurones tested under this recording configuration. The enhancement usually started ∼10 min after the whole-cell recording formation and dialysis, and gradually reached a steady-state level after 20-30 min (Fig. 6A). During the initial period of dialysis, extracellular application of 17β-oestradiol (100 nM, 3 min) was capable of affecting kainate-induced currents in four of the eight cells that were sensitive to ATPγS. The potentiation induced by 17β-oestradiol exhibited the same pattern of onset and amplitude as that observed previously. However, addition of 17β-oestradiol had no further potentiating effect after the amplitude increased by ATPγS dialysis reached the steady level (Fig. 6B). Thus, the action of ATPγS may have occluded the effect of 17β-oestradiol on kainate-induced currents. It should be noted that, in a subset of cells that was not responsive to ATPγS dialysis (n= 8), no potentiation of kainate-induced currents was observed on application of 17β-oestradiol.
Figure 6. Enhancement induced by ATPγS dialysis occludes 17β-oestradiol potentiation of kainate-induced currents.
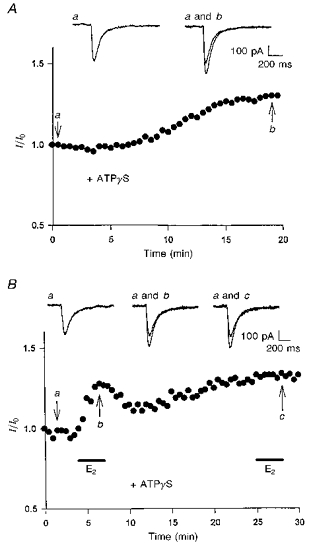
A, intracellular dialysis of ATPγS (500 μM) increased the amplitude of kainate-induced currents. The illustrated example shows that the increase occurred ≈10 min after ATPγS dialysis and gradually reached steady state at ≈20 min. B, the illustrated whole-cell recording was obtained from a CA1 neurone responsive to ATPγS (500 μM) dialysis. Application of 17β-oestradiol (100 nM, 3 min) during the initial period of recording elicited a rapid potentiation in the peak currents. The potentiation was partially reversible upon removal of 17β-oestradiol. After the amplitude of the currents increased by ATPγS dialysis reached a steady level, addition of 17β-oestradiol caused no further potentiation. In A and B, actual current traces corresponding to the specific time points (arrows) are displayed at the top.
DISCUSSION
Our results, based on the impermeant form of oestrogen and several potential modulatory molecules including GTP, ATP, cAMP and cGMP analogues, suggest that 17β-oestradiol is required on both sides of the plasma membrane to potentiate kainate-induced currents. In the present study, E2-BSA applied to either the extracellular surface or the cytosolic side of the plasma membrane failed to induce an observable action on kainate-induced currents. If applied simultaneously to both sides of the membrane, E2-BSA was able to potentiate kainate-induced currents. The finding that oestrogen must be present on both sides of the membrane in order for potentiation to occur is intriguing and puzzling and indeed may represent a completely novel mechanism of steroid hormone action. Thus, the lack of effect of 17β-oestradiol when on the outside does not appear to be due to the concentration of oestrogen. Since 17β-oestradiol is lipophilic and readily crosses the membrane, its presence on both sides of the membrane is not unexpected in vivo.
Several groups have shown that steroids tethered to BSA could still affect neuronal activities with uncompromised potency (Lieberherr, Grosse, Kachkache & Balsan, 1993; Tesarik & Mendoza, 1995). Extracellular E2-BSA could inhibit neostriatal Ba2+ currents, suggesting that the action of 17β-oestradiol occurs at a membrane site accessible from the outside of the cell (Mermelstein et al. 1996). Progesterone immobilized on BSA was even more effective than free progesterone in releasing dopamine from striatal slices (Dluzen & Ramirez, 1989). Although it is difficult to estimate the actual amount of effective oestrogen molecules per E2-BSA available to the cells, the maximum available oestrogen in a solution of 3.3 nM E2-BSA is 100 nM (considering the 30 : 1 ratio of oestrogen to BSA in this preparation). Theoretically, oestrogen packed onto a carrier molecule should present a higher local concentration of oestrogen to either the cell surface or the interior of the cell. In addition, a single BSA molecule binding many steroid molecules could cross-link and activate receptors in close proximity to the plasma membrane. Clustering of receptors has been shown to be capable of activating certain signal transduction systems (Cadena & Gill, 1992). It should be noted that the effect of E2-BSA was irreversible in some cases. One explanation is that the covalent attachment of 17β-oestradiol to a membrane-impermeant protein molecule might prevent the signal from being processed away or downregulated by subsequent degradation of the steroid.
The results from the present study also indicate that a G protein is involved in the transduction pathway by which 17β-oestradiol modulates kainate receptor-channels. In the presence of GTPγSin, application of E2-BSAout could elicit an irreversible, prolonged potentiation of kainate-induced currents, suggesting that 17β-oestradiol does trigger a G protein-coupled signal pathway. Furthermore, results from the experiments utilizing cells pretreated with cholera toxin or pertussis toxin suggest the involvement of Gs in this signalling pathway. Cholera toxin prevents the deactivation of Gs by catalysing the ADP-ribosylation of the Gα subunit near the GTP-binding site and thereby inhibits GTP hydrolysis. The cellular response induced by treatment with cholera toxin or 17β-oestradiol may result from activation of a common pathway, since incubation of cells with cholera toxin occluded the effect of 17β-oestradiol on kainate-induced currents. In any case, a G protein appears to be essential for coupling the activation of an oestrogen membrane-binding site to the functional modulation of kainate receptor-channels. On the other hand, the prolonged potentiation of the kainate currents suggests an additional mechanism. Apparently, some maintenance in the signalling pathway, as provided by GTPγS in this case, is required for the triggering action to promulgate a persistent and extensive effect on its target.
A variety of studies have suggested that non-NMDA glutamate receptors can be functionally regulated by protein phosphorylation (Raymond, Blackstone & Huganir, 1993a, b; Raymond, Tingley, Blackstone, Roche & Huganir, 1994). These observations are consistent with the present findings from the experiments in which cells were dialysed with ATPγS. The increase in the amplitude of kainate-induced currents by ATPγS dialysis suggests that kainate receptor-channels or relevant proteins are constitutively phosphorylated under a balanced condition, and that the shift of the equilibrium towards phosphorylation may enhance the conductance of the channels. The effect of ATPγS occluded the action of 17β-oestradiol, suggesting that both may share a common mechanism in changing the phosphorylation state of kainate receptor-channels. The potentiation induced by 17β-oestradiol was only observed in the cells that were sensitive to ATPγS dialysis, in other words, no potentiation was observed on application of 17β-oestradiol to cells that were not responsive to ATPγS dialysis. Thus, the basal phosphorylation level of the cell might be critical for the responsiveness of kainate receptor-channels to 17β-oestradiol modulation. This may also provide a possible explanation of why only 38 % of kainate-sensitive neurones are responsive to 17β-oestradiol.
Our previous data have shown that application of 8-bromo-cAMP could mimic the action of 17β-oestradiol, and that the PKA inhibitor Rp-cAMPS completely blocked the effect of 17β-oestradiol on kainate-induced currents. These findings suggest that the effect of 17β-oestradiol on kainate-induced currents may involve cAMP-dependent phosphorylation of kainate receptor-channels or relevant cellular components (Gu & Moss, 1996). The potentiation induced by 8-bromo-cAMP was readily reversible upon its removal in the present study. In the presence of E2-BSAin, however, recovery after 8-bromo-cAMP action was substantially retarded. Since 8-bromo-cAMP is rather membrane permeant and phosphodiesterase resistant, free diffusion away from the cell causes a decrease in its intracellular concentration. This reduction in the level of cAMP may affect the cAMP-dependent protein phosphorylation process, which in turn alters the frequency and duration of the kainate receptor-channel opening (Knapp, Schmidt & Dowling, 1990; Greengard, Jen, Nairn & Stevens, 1991). Another possible reason for the recovery from 8-bromo-cAMP action is dephosphorylation by protein phosphatases, such as phosphatase 1 (PP-1). PP-1 can be activated by PKA and play a part in modulating kainate currents by counterbalancing the phosphorylation process (Wang, Salter & MacDonald, 1991). As there is no evidence that E2-BSA can prevent 8-bromo-cAMP diffusion, it is possible that intracellular 17β-oestradiol could influence the balance between phosphorylation and dephosphorylation, especially by inhibiting the dephosphorylation process, thereby prolonging the effect of 8-bromo-cAMP on kainate-induced currents.
Reversible protein phosphorylation might play an important role in excitatory synaptic transmission in hippocampus (Wang et al. 1991; Wang, Orser, Brautigan & MacDonald, 1994). As physiological responses of cells to neurotransmitters are determined by the phosphorylation state of key proteins, co-ordinating the opposing actions of protein kinases and phosphatases becomes important for cells to exhibit appropriate synaptic activities. In many tissues, hormones that elevate intracellular [cAMP] increase the activity of phosphoprotein, inhibitor-1 (Foulkes & Cohen, 1979; Khatra, Chiasson, Shikama, Exton & Soderling, 1980; Foulkes, Cohen, Strada, Everson & Jefferson, 1982; MacDougall, Campbell, Hubbard & Cohen, 1989; Neumann, Gupta, Schmitz, Scholz, Nairn & Watanabe, 1991; Snyder et al. 1992). The protein inhibitor-1 can inhibit protein phosphatase activity when phosphorylated by PKA. This protein provides a potential mechanism for cross-talk between protein kinase and phosphatase that amplifies hormonal signals mediated by the second messenger cAMP (Huang & Glinsmann, 1976). Such a cascade of phosphatases controls synaptic plasticity in hippocampal neurones (Mulkey, Endo, Shenolikar & Malenka, 1994). Future studies are required to investigate the mechanism by which 17β-oestradiol can exert an effect on these events.
Glutamate is one of the most important excitatory neurotransmitters in the mammalian central nervous system. Modulation of non-NMDA receptor function has been considered as a critical mechanism underlying fast excitatory synaptic transmission and use-dependent synaptic plasticity. Oestrogen has been shown to modulate excitatory synaptic transmission, especially in the hippocampus where the glutamate receptors are implicated in memory, epilepsy and some neurodegenerative diseases (Collingridge & Lester, 1989; Meldrum & Garthwaite, 1990). As we investigate the complex actions of 17β-oestradiol on kainate-induced currents, it is becoming increasingly clear that oestrogen may modulate alternative sites of a signalling pathway to alter membrane excitability. An explanation for the dual presence requirements of oestrogen on both sides of the plasma membrane could be to provide a means by which modulation of the membrane excitability by 17β-oestradiol can be maintained for longer periods of time.
Acknowledgments
We are grateful to Carol Dudley for her valuable advice and assistance throughout the project. We thank Greg Goldmakher and Corina Morrison for their comments and technical assistance. We also wish to thank Drs Susan Mumby and Al Gilman for their generous gift of purified GTPγS. This work was supported by National Institutes of Health Grant RO1-MH47418 awarded to R.L.M.
References
- Becker JB. Direct effect of 17β-estradiol on striatum: sex differences in dopamine release. Synapse. 1990a;5:157–164. doi: 10.1002/syn.890050211. [DOI] [PubMed] [Google Scholar]
- Becker JB. Estrogen rapidly potentiates amphetamine-induced striatal dopamine release and rotational behavior during microdialysis. Neuroscience Letters. 1990b;118:169–171. doi: 10.1016/0304-3940(90)90618-j. [DOI] [PubMed] [Google Scholar]
- Cadena DL, Gill GN. Receptor tyrosine kinases. FASEB Journal. 1992;6:2332–2337. doi: 10.1096/fasebj.6.6.1312047. [DOI] [PubMed] [Google Scholar]
- Castner SA, Xiao L, Becker JB. Sex differences in striatal dopamine: in vivo microdialysis and behavioral studies. Brain Research. 1993;610:127–134. doi: 10.1016/0006-8993(93)91225-h. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Lester RA. Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacological Reviews. 1989;41:143–210. [PubMed] [Google Scholar]
- Dluzen DE, Ramirez VD. Progesterone effects upon dopamine release from the corpus striatum of female rats. II. Evidence for a membrane site of action and the role of albumin. Brain Research. 1989;476:338–344. doi: 10.1016/0006-8993(89)91255-9. 10.1016/0006-8993(89)91255-9. [DOI] [PubMed] [Google Scholar]
- ffrench-Mullen JM. Cortisol inhibition of calcium currents in guinea pig hippocampal CA1 neurons via G-protein-coupled activation of protein kinase C. Journal of Neuroscience. 1995;15:903–911. doi: 10.1523/JNEUROSCI.15-01-00903.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ffrench-Mullen JM, Danks P, Spence KT. Neurosteroids modulate calcium currents in hippocampal CA1 neurons via a pertussis toxin-sensitive G-protein-coupled mechanism. Journal of Neuroscience. 1994;14:1963–1977. doi: 10.1523/JNEUROSCI.14-04-01963.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes JG, Cohen P. The hormonal control of glycogen metabolism. Phosphorylation of protein phosphatase inhibitor-1 in vivo in response to adrenaline. European Journal of Biochemistry. 1979;97:251–256. doi: 10.1111/j.1432-1033.1979.tb13109.x. [DOI] [PubMed] [Google Scholar]
- Foulkes JG, Cohen P, Strada SJ, Everson WV, Jefferson LS. Antagonistic effects of insulin and β-adrenergic agonists on the activity of protein phosphatase inhibitor-1 in skeletal muscle of the perfused rat hemicorpus. Journal of Biological Chemistry. 1982;257:12493–12496. [PubMed] [Google Scholar]
- Greengard P, Jen J, Nairn AC, Stevens CF. Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons. Science. 1991;253:1135–1138. doi: 10.1126/science.1716001. [DOI] [PubMed] [Google Scholar]
- Gu Q, Moss RL. 17β-Estradiol potentiates kainate-induced currents via activation of the cAMP cascade. Journal of Neuroscience. 1996;16:3620–3629. doi: 10.1523/JNEUROSCI.16-11-03620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Horvat A, Nikezic G, Martinovic JV. Estradiol binding to synaptosomal plasma membranes of rat brain regions. Experientia. 1995;51:11–15. [PubMed] [Google Scholar]
- Huang FL, Glinsmann WH. Separation and characterization of two phosphorylase phosphatase inhibitors from rabbit skeletal muscle. European Journal of Biochemistry. 1976;70:419–426. doi: 10.1111/j.1432-1033.1976.tb11032.x. [DOI] [PubMed] [Google Scholar]
- Kay AR, Wong RK. Isolation of neurons suitable for patch-clamping from adult mammalian central nervous systems. Journal of Neuroscience Methods. 1986;16:227–238. doi: 10.1016/0165-0270(86)90040-3. [DOI] [PubMed] [Google Scholar]
- Khatra BS, Chiasson JL, Shikama H, Exton JH, Soderling TR. Effect of epinephrine and insulin on the phosphorylation of phosphorylase phosphatase inhibitor 1 in perfused rat skeletal muscle. FEBS Letters. 1980;114:253–256. doi: 10.1016/0014-5793(80)81127-6. [DOI] [PubMed] [Google Scholar]
- Knapp AG, Schmidt KF, Dowling JE. Dopamine modulates the kinetics of ion channels gated by excitatory amino acids in retinal horizontal cells. Proceedings of the National Academy of Sciences of the USA. 1990;87:767–771. doi: 10.1073/pnas.87.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrange AH, Ronnekleiv OK, Kelly MJ. The potency of μ-opioid hyperpolarization of hypothalamic arcuate neurons is rapidly attenuated by 17β-estradiol. Journal of Neuroscience. 1994;14:6196–6204. doi: 10.1523/JNEUROSCI.14-10-06196.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberherr M, Grosse B, Kachkache M, Balsan S. Cell signaling and estrogens in female rat osteoblasts: a possible involvement of unconventional nonnuclear receptors. Journal of Bone and Mineral Research. 1993;8:1365–1376. doi: 10.1002/jbmr.5650081111. [DOI] [PubMed] [Google Scholar]
- MacDougall LK, Campbell DG, Hubbard MJ, Cohen P. Partial structure and hormonal regulation of rabbit liver inhibitor-1; distribution of inhibitor-1 and inhibitor-2 in rabbit and rat tissues. Biochimica et Biophysica Acta. 1989;1010:218–226. doi: 10.1016/0167-4889(89)90164-x. [DOI] [PubMed] [Google Scholar]
- Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends in Pharmacological Sciences. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Becker JB, Surmeier DJ. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. Journal of Neuroscience. 1996;16:595–604. doi: 10.1523/JNEUROSCI.16-02-00595.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss RL, Gu Q, Wong M. Estrogen: non-transcriptional signaling pathway. Recent Progress in Hormone Research. 1997;52:33–69. [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Oomura Y, Minami T, Mizuno Y, Fukuda A. Mechanism of the rapid effect of 17β-estradiol on medial amygdala neurons. Science. 1986;233:226–228. doi: 10.1126/science.3726531. [DOI] [PubMed] [Google Scholar]
- Neumann J, Gupta RC, Schmitz W, Scholz H, Nairn AC, Watanabe AM. Evidence for isoproterenol-induced phosphorylation of phosphatase inhibitor-1 in the intact heart. Circulation Research. 1991;69:1450–1457. doi: 10.1161/01.res.69.6.1450. [DOI] [PubMed] [Google Scholar]
- Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB Journal. 1995;9:404–410. doi: 10.1096/fasebj.9.5.7896011. [DOI] [PubMed] [Google Scholar]
- Ramirez VD, Zheng J, Siddique KM. Membrane receptors for estrogen, progesterone, and testosterone in the rat brain: fantasy or reality. Cellular and Molecular Neurobiology. 1996;16:175–198. doi: 10.1007/BF02088175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond LA, Blackstone CD, Huganir RL. Phosphorylation of amino acid neurotransmitter receptors in synaptic plasticity. Trends in Neurosciences. 1993a;16:147–153. doi: 10.1016/0166-2236(93)90123-4. [DOI] [PubMed] [Google Scholar]
- Raymond LA, Blackstone CD, Huganir RL. Phosphorylation and modulation of recombinant GluR6 glutamate receptors by cAMP-dependent protein kinase. Nature. 1993b;361:637–641. doi: 10.1038/361637a0. [DOI] [PubMed] [Google Scholar]
- Raymond LA, Tingley WG, Blackstone CD, Roche KW, Huganir RL. The Journal of Physiology. Vol. 88. Paris: 1994. Glutamate receptor modulation by protein phosphorylation; pp. 181–192. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Girault JA, Chen JY, Czernik AJ, Kebabian JW, Nathanson JA, Greengard P. Phosphorylation of DARPP-32 and protein phosphatase inhibitor-1 in rat choroid plexus: regulation by factors other than dopamine. Journal of Neuroscience. 1992;12:3071–3083. doi: 10.1523/JNEUROSCI.12-08-03071.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesarik J, Mendoza C. Nongenomic effects of 17β-estradiol on maturing human oocytes: relationship to oocyte developmental potential. Journal of Clinical Endocrinology and Metabolism. 1995;80:1438–1443. doi: 10.1210/jcem.80.4.7714121. [DOI] [PubMed] [Google Scholar]
- Thompson TL, Moss RL. Estrogen regulation of dopamine release in the nucleus accumbens: genomic- and nongenomic-mediated effects. Journal of Neurochemistry. 1994;62:1750–1756. doi: 10.1046/j.1471-4159.1994.62051750.x. [DOI] [PubMed] [Google Scholar]
- Wang LY, Orser BA, Brautigan DL, MacDonald JF. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature. 1994;369:230–232. doi: 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- Wang LY, Salter MW, MacDonald JF. Regulation of kainate receptors by cAMP-dependent protein kinase and phosphatases. Science. 1991;253:1132–1135. doi: 10.1126/science.1653455. [DOI] [PubMed] [Google Scholar]
- Wong M, Moss RL. Electrophysiological evidence for a rapid membrane action of the gonadal steroid, 17β-estradiol, on CA1 pyramidal neurons of the rat hippocampus. Brain Research. 1991;543:148–152. doi: 10.1016/0006-8993(91)91057-8. [DOI] [PubMed] [Google Scholar]
- Wong M, Moss RL. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. Journal of Neuroscience. 1992;12:3217–3225. doi: 10.1523/JNEUROSCI.12-08-03217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Moss RL. Patch-clamp analysis of direct steroidal modulation of glutamate receptor-channels. Journal of Neuroendocrinology. 1994;6:347–355. doi: 10.1111/j.1365-2826.1994.tb00592.x. [DOI] [PubMed] [Google Scholar]
- Wong M, Thompson TL, Moss RL. Non-genomic actions of estrogen in the brain: physiological significance and cellular mechanisms. Critical Reviews in Neurobiology. 1996;10:189–203. doi: 10.1615/critrevneurobiol.v10.i2.30. [DOI] [PubMed] [Google Scholar]
- Zheng J, Ali A, Ramirez VD. Steroids conjugated to bovine serum albumin as tools to demonstrate specific steroid neuronal membrane binding sites. Journal of Psychiatry and Neuroscience. 1996;21:187–197. [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Watters JJ, Dorsa DM. Estrogen rapidly induces the phosphorylation of the cAMP response element binding protein in rat brain. Endocrinology. 1996;137:2163–2166. doi: 10.1210/endo.137.5.8612562. [DOI] [PubMed] [Google Scholar]