

Abstract
The cardiac muscarinic receptor–K+ channel system was reconstructed in Chinese hamster ovary (CHO) cells by transfecting the cells with the various components of the system. The activity of the muscarinic K+ channel was measured with the cell-attached configuration of the patch clamp technique.
In CHO cells transfected with the channel (Kir3.1/Kir3.4), receptor (hm2) and receptor kinase (GRK2), on exposure to agonist, there was a decline in channel activity as a result of desensitization, similar to that in atrial cells.
Whereas the desensitization was almost abolished by not transfecting with the receptor kinase or by transfecting with a mutant receptor lacking phosphorylation sites, it was only reduced (by ≈39 %) by transfecting with a mutant receptor kinase with little kinase activity.
These results suggest that the receptor kinase is responsible for desensitization of the muscarinic K+ channel and that this involves phosphorylation-dependent and -independent mechanisms.
G-protein-regulated inward rectifier K+ channels (Kir3.0 family) are widely distributed in the brain and heart (Duprat et al. 1995; Doupnik, Davidson & Lester, 1995). The channels are opened by an agonist binding to a G-protein-coupled receptor and the consequent activation of the G-protein (Doupnik et al. 1995). Diverse G-protein-coupled receptors (muscarinic, μ- and δ-opioid, α2-adrenergic, somatostatin, probably GABAB, purinergic, putative sphingosine-1-phosphate) couple to inward rectifier K+ channels (Kurachi, Nakajima & Sugimoto, 1987; Duprat et al. 1995; Bünemann, Brandts, zu Heringdorf, van Koppen, Jakobs & Pott, 1995). When bound to an agonist, G-protein-coupled receptors are known to be phosphorylated by one of a family of G-protein-coupled receptor kinases (GRKs) and this has been shown to be responsible for receptor desensitization by (i) uncoupling the receptor from the G-protein, (ii) causing receptor internalization, and (iii) causing receptor downregulation (Hausdorff, Caron & Lefkowitz, 1990; Tsuga, Kameyama, Haga, Kurose & Nagao, 1994; Lohse, 1995; Freedman & Lefkowitz, 1996). This is expected to affect receptor-linked channels. There are potential candidates: both in the brain and heart there are inward rectifying K+ channels linked to opioid, muscarinic, purinergic and putative sphingosine-1-phosphate receptors, which undergo desensitization in the presence of the agonist (Kurachi et al. 1987; Miyake, Christie & North, 1989; Harris & Williams, 1991; Bünemann et al. 1995; Osborne & Williams, 1995). A well-studied example is the muscarinic K+ channel (a heteromultimer of Kir3.1 and Kir3.4) in the heart. ACh activates the muscarinic K+ channel via the m2 muscarinic receptor and in the continued presence of ACh the channel desensitizes, i.e. channel activity declines (Carmeliet & Mubagwa, 1986; Kurachi et al. 1987; Kim, 1991, 1993; Zang, Yu, Honjo, Kirby & Boyett, 1993; Wang & Lipsius, 1995). This has functional consequences: during tonic vagal nerve activity, the effects of the ACh released from the vagal nerves on heart rate and contractility attenuate (Martin, Levy & Matsuda, 1982), principally as a result of the desensitization of the muscarinic K+ channel (Honjo, Kodama, Zang & Boyett, 1992; Yang, Boyett, Janvier, McMorn, Shui & Karim, 1996). There are various phases of desensitization of the muscarinic K+ channel and these can be divided into short and long term phases. There are at least two phases of short term desensitization: a fast phase that develops over ∼20 s and one or more intermediate phases that develop over several minutes (e.g. Kim, 1991; Zang et al. 1993; Bünemann, Brandts & Pott, 1996). Long term desensitization is a very slow phase that develops over 24–48 h (Bünemann et al. 1996). The fast phase of desensitization is a channel phenomenon and has been suggested to be the result of a dephosphorylation of the channel (Kim, 1991, 1993; Zang et al. 1993; Hong, Pleumsamran & Kim, 1996; Shui, Boyett & Zang, 1997a). Long term desensitization has been suggested to be the result of receptor downregulation (Bünemann et al. 1996; Shui et al. 1997c). Zang et al. (1993) and Shui, Boyett, Zang, Haga & Kameyama (1995) have shown that the intermediate phase of desensitization is a receptor phenomenon and suggested it to be the result of receptor kinase-dependent uncoupling of the receptor from the G-protein. The aim of the present study was to test the hypothesis that receptor kinase is responsible for the intermediate phase of desensitization of the muscarinic K+ channel by reconstructing the cardiac muscarinic receptor- K+ channel system in a cell line.
Abstracts of this work have been presented to The Physiological and Biophysical Societies (Khan, Shui, Tsuga, Haga & Boyett, 1997; Shui, Khan, Tsuga, Haga & Boyett, 1997b).
METHODS
Preparation of cells
Chinese hamster ovary (CHO-K1) cells were cultured in Ham's F12 nutrient mixture (Life Technologies Ltd) supplemented with 10 % fetal bovine serum (Life Technologies Ltd), 50 units ml−1 penicillin G and 50 μg ml−1 streptomycin sulphate (Life Technologies Ltd) at 37°C in 95 % air and 5 % CO2.
The calcium phosphate method of transient transfection (Sambrook, Fritsch & Maniatis, 1989) was used to transfect wild-type cells or cell lines already stably transfected (modified calcium phosphate method; Chen & Okayama, 1987). The following stable cell lines were constructed: (1) cells expressing c-myc-tagged human m2 receptors (plasmid expression vector pEF-myc-hm2) and a selection marker for antibiotic resistant growth in G418 sulphate (pEF-neo); (2) cells expressing hm2 (pEF-myc-hm2), selection marker (pEF-neo) and the G-protein-coupled receptor kinase GRK2 (pEF-GRK2); (3) cells expressing the deletion mutant of the m2 receptor, m2LD/hm2-Δ233–380 (pEF-m2LD) and the selection marker (pEF-neo). These cell lines were transiently transfected with the plasmid expression vectors for Kir3.1/GIRK1 (pEF-GIRK1) and Kir3.4/CIR (pEF-CIR), which together form the functional channel heteromultimer, with or without GRK2 (pEF-GRK2) or its mutant DN-GRK2/GRK2-K220W (pEF-GRK2-K22OW). Kir3.1 and Kir3.4 were gifts from Professor L. Y. Jan (University of California School of Medicine, San Francisco, USA) and Professor R. A. North (Glaxo Institute for Molecular Biology, Geneva, Switzerland), respectively. Some experiments were carried out using m2LD transiently transfected into wild-type cells, rather than the stably transfected cell line carrying that plasmid. All transient transfections included the S65T point mutation of green fluorescent protein (p-GFP-S65T; Clontech) as a marker for successfully transfected cells. The final concentrations of each of the DNA plasmids added during transient transfections were as follows (ng ml−1): m2LD, 400; Kir3.1, 400; Kir3.4, 400; GRK2, 400; DN-GRK2, 400; green fluorescent protein, 200. Ten millilitres of the transfecting solution was added to approximately 1 × 106-2 × 106 cells in a 100 mm plastic tissue culture dish.
Expression levels of stable transfected hm2 receptor were estimated using [3H] QNB (quinuclidinyl benzilate (DuPont NEN) binding, and the expression level of stable transfected GRK2 was estimated by Western blotting and immunostaining with anti-GRK2 antibodies (Tsuga et al. 1994). In the cell line stably transfected with just hm2, the [3H] QNB binding sites in these cells were estimated to be 165 fmol (mg protein)−1 in the total homogenate. Cells stably transfected with hm2 and GRK2 were estimated to have 330 fmol (mg protein)−1 of hm2 in the total homogenate, and 300–600 fmol (mg protein)−1 of GRK2 in the supernatant.
CHO cells were concentrated and prepared for voltage clamp by using 0.02 % EDTA to remove the adherent cell layer from the dish, centrifuging for 3 min at 100 g and resuspending in fresh medium. No enzymatic cleaning of cells was necessary.
Rats were killed by stunning and cervical dislocation and rat atrial cells were prepared as described previously (Harrison, McCall & Boyett, 1992).
Electrophysiology
Cells were placed in a chamber on a Nikon Diaphot microscope. When choosing a CHO cell for study, the cells were illuminated with 470–490 nm light to excite the green fluorescent protein (GFP) in successfully transfected cells. The green fluorescent light from successfully transfected cells was passed through a 515 nm filter before viewing. Successfully transfected CHO cells with a middle level of green fluorescent light were chosen for study.
Experiments were carried out using the cell-attached and inside-out configurations of the patch clamp technique at a holding potential of -60 mV and at room temperature (22–25°C). Sylgard-coated pipettes with a resistance of 5 MΩ were used. In cell-attached experiments, the chamber was filled with extracellular solution containing (mm): KCl, 140; MgCl2, 1.8; EGTA, 5; Hepes, 5; pH 7.4. In both cell-attached and inside-out experiments, the pipette contained extracellular solution plus 10 μm ACh. In inside-out experiments, the chamber was perfused with either control or test intracellular solution. Control intracellular solution contained (mm): potassium aspartate, 120; KCl, 20; KH2PO4, 1; MgCl2, 2.8, (free Mg2+, 1.8); EGTA, 5; Hepes, 5; pH 7.4. Test intracellular solution was made by adding 0.1 mm Na3GTP and 3 mm Na2ATP to the control intracellular solution. Single channel currents were recorded with an Axopatch-1D amplifier and filtered at 5 kHz with an 8-pole Bessel filter. The currents were then digitized at a sampling rate of 0.2 ms with pCLAMP software (Axon Instruments). The channel open probability (NPo) was calculated for consecutive 200 ms episodes as the mean current during an episode divided by the unitary current. Decline in channel activity was fitted with a single exponential function with a least-squares method using SigmaPlot (Jandel Corporation, CA, USA). Statistical tests were carried out using SigmaStat (Jandel Corporation). Results are given as means ± s.e.m. Differences were considered significant if P < 0.05.
RESULTS
We reconstituted the cardiac muscarinic receptor–K+ channel system in a CHO cell line. Cells were stably transfected with DNA for the human cardiac m2 muscarinic receptor (hm2) and the G-protein-coupled receptor kinase, GRK2 (also known as βARK1). GRK2 is known to phosphorylate the m2 muscarinic receptor (Haga, Haga & Kameyama, 1994). The cells were then transiently transfected with DNA for Kir3.1 and Kir3.4 (the two proteins making up the muscarinic K+ channel) and GFP. After transfection and adequate time (> 24 h) for expression of the proteins, recordings were made from cells expressing GFP (i.e. those cells showing green fluorescence when illuminated with ultraviolet light; see Methods for details). The use of GFP was essential, because only a small percentage of cells (∼10 %) were successfully transfected. Muscarinic K+ channel activity was observed in all cells showing green fluorescence.
Muscarinic K+ channel activity was recorded in cell-attached or inside-out patches with ACh in the pipette. Figure 1 shows some of the properties of the transfected muscarinic K+ channel. Figure 1A shows that with receptor, channel and receptor kinase (all wild-type) and ACh in the pipette, channel activity was observed in cell-attached patches (column 4), but if either ACh (column 1) or the receptor (column 2) was excluded little or no channel activity was recorded. No channel activity was also recorded in non-transfected CHO cells (n = 6). This shows that the channel activity was dependent on the presence of ACh, the receptor and the channel as expected. Panels B-D in Fig. 1 were obtained from inside-out patches. Figure 1B shows that the transfected channel exhibited inward rectification (single channel currents during a ramp clamp are shown), Fig. 1C shows that the mean open time was ∼1 ms (open time histogram shown) and Fig. 1D shows that with ACh in the pipette the channel was activated by the application of GTP to the intracellular face of the patch. All of these are characteristic features of the native channel in heart cells (Kurachi, Ito & Sugimoto, 1990; Kaibara, Nakajima, Irisawa & Giles, 1991) and it was concluded that the properties of the transfected channel correspond to those of the native channel in the heart.
Figure 1. Properties of the muscarinic K+ channel in CHO cells.
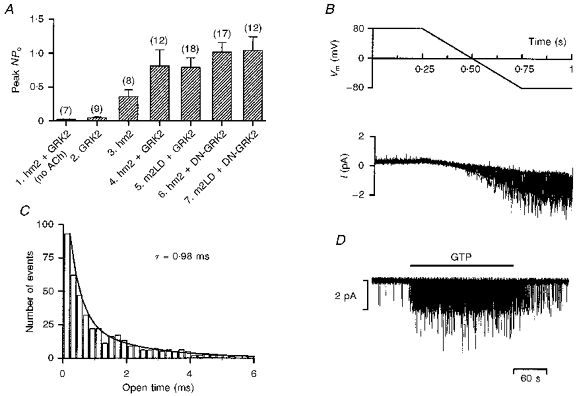
A, mean ( ± s.e.m.) NPo from 7 to 18 cell-attached patches on various groups of cells. The peak value of NPo when the pipette was first attached onto a cell was measured. 1, cells transfected with the wild-type receptor and wild-type receptor kinase. ACh was absent from the pipette. 2, cells transfected with the wild-type receptor kinase, but not the receptor (ACh present). 3, cells transfected with the wild-type receptor, but not the receptor kinase (ACh present). 4, cells transfected with the wild-type receptor and wild-type receptor kinase (ACh present). 5, cells transfected with mutant receptor and wild-type receptor kinase (ACh present). 6, cells transfected with the wild-type receptor and mutant receptor kinase (ACh present). 7, cells transfected with mutant receptor and mutant receptor kinase (ACh present). All groups of cells were also transfected with the muscarinic K+ channel (Kir3.1/Kir3.4). B, 10 superimposed traces of single channel currents (bottom) during repeated voltage ramps (top; Vm, membrane potential). At positive potentials no channel activity was recorded, whereas there was intense channel activity at negative potentials. Inside-out configuration (at least 2 active channels in patch). C, open time histogram. The number of channel openings of a particular open duration is plotted against the open time; 0.2 ms bin width. The data are fitted with an exponential function with a time constant, τ, of 0.98 ms. The data were collected over a 1 s period about 3 min after the attachment of the pipette. Inside-out configuration (at least 2 active channels in patch). D, single channel currents recorded in an inside-out patch (at least 2 active channels in patch). ACh was present in the extracellular solution in the pipette. When 0.1 mm GTP was applied to the intracellular face of the patch during the period indicated by the horizontal bar, the muscarinic K+ channel was activated and channel activity was observed. Transfection: A1 and A4, stable transfection with DNA for hm2 and GRK2 and transient transfection with DNA for Kir3.1, Kir3.4 and GFP; A2, transient transfection with DNA for GRK2, Kir3.1, Kir3.4 and GFP; A3, stable transfection with DNA for hm2 and transient transfection with DNA for Kir3.1, Kir3.4 and GFP;A5, transient transfection with DNA for m2LD, GRK2, Kir3.1, Kir3.4 and GFP or stable transfection with DNA for m2LD and transient transfection with DNA for GRK2, Kir3.1, Kir3.4 and GFP; A6, stable transfection with DNA for hm2 and transient transfection with DNA for DN-GRK2, Kir3.1, Kir3.4 and GFP; A7, stable transfection with DNA for m2LD and transient transfection with DNA for DN-GRK2, Kir3.1, Kir3.4 and GFP; B-D, stable transfection with DNA for hm2 and GRK2 and transient transfection with DNA for Kir3.1, Kir3.4 and GFP (see Methods for details).
Figure 2 shows the activity of the muscarinic K+ channel during the first 3 min after the attachment of an ACh-containing pipette onto a cell (cell-attached configuration used). Data from CHO cells transfected with the receptor, channel and receptor kinase (all wild-type) are shown in Fig. 2A-C and, for comparison, data from rat atrial cells are shown in Fig. 2D-F. Slow time base records of single channel currents from a typical patch are shown in panels A and D (at least five channels were active in the patch in both cases), and single channel currents on a fast time base at various times during the 3 min exposure to ACh are shown in panels B and E. Panels C and F show the mean open probability of the channel in cell-attached patches from at least seven cells. In rat atrial cells (Fig. 2D-F), channel activity was high when the ACh-containing pipette was first attached onto the cell, but during the next 3 min the channel activity declined. In seven patches from different cells, channel activity declined by 52 ± 7 % with a time constant of 166 s during the first 3 min after exposure of the patch to ACh (Fig. 2F). The decline in channel activity in the presence of ACh has been observed before in cardiac cells using various configurations of the patch clamp technique and is referred to as desensitization (Carmeliet & Mubagwa, 1986; Kurachi et al. 1987; Kim, 1991, 1993; Zang et al. 1993; Wang & Lipsius, 1995). A similar decline in channel activity as a result of desensitization was observed in CHO cells transfected with the wild-type receptor and receptor kinase as well as the channel (Fig. 2A-C). In CHO cells, channel activity declined by 75 ± 5 % (n = 11) with a time constant of 113 s during the first 3 min after exposure of the patch to ACh (Fig. 2C).
Figure 2. Desensitization of the muscarinic K+ channel in CHO cells and rat atrial cells.
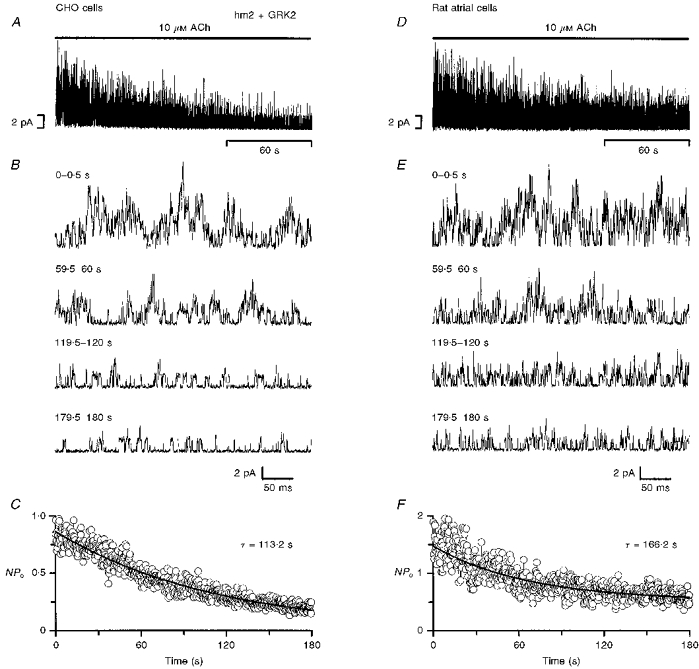
Data were recorded from either CHO cells transfected with wild-type receptor and wild-type receptor kinase (A-C) or rat atrial cells (D-F). Cell-attached configuration. A and D, slow time base recording of single channel currents during the first 3 min after the attachment of an ACh-containing pipette onto a cell. At least 5 active channels were present in the patches. B and E, fast time base records of the single channel currents from the times shown during the first 3 min after the attachment of the pipette. C and F, mean NPo from 11 (C) and 7 (F) patches during the first 3 min after the attachment of the pipette. The data are fitted with single exponential functions with the time constants shown. In A, B, D and E, inward currents are shown in the upwards direction to facilitate comparison with the plots of NPo in C and F. Transfection: the CHO cells were stably transfected with DNA for hm2 and GRK2 and transiently transfected with DNA for Kir3.1, Kir3.4 and GFP (see Methods for details).
To test whether the decline in channel activity in CHO cells was the result of the activity of the receptor kinase, four alternative transfection strategies were used (Fig. 3). In all four cases the cells were transfected with the channel and channel activity was recorded in the cell-attached configuration. Figure 3 shows typical recordings of single channel currents (left) and the mean open probability from eight to eighteen patches (right) during the first 3 min after the attachment of ACh-containing pipettes onto cells. In the first case (Fig. 3A), the cells were transfected with the wild-type receptor (hm2), but not the receptor kinase (GRK2). In the absence of the receptor kinase (GRK2), the channel was still active, but peak channel activity was reduced (although not significantly; compare columns 3 and 4 in Fig. 1A). During the first 3 min after exposure of the patch to ACh, there was little decline in channel activity compared with the control in Fig. 2A-C. In the control cells (with wild-type receptor and receptor kinase - hm2 and GRK2) channel activity declined by 75 ± 5 % during the first 3 min, whereas in cells without the receptor kinase (but with wild-type receptor, hm2) channel activity declined by 12 ± 12 % (Fig. 4, columns 1 and 2). The difference is significant (P < 0.05) and suggests that the receptor kinase, GRK2, is responsible for all or most of the decline in channel activity. In the absence of the receptor kinase some decline in channel activity remained (although not significantly different from zero; P = 0.305) and this could be the result of endogenous G-protein-coupled receptor kinases present in CHO cells (Shih & Malbon, 1994).
Figure 3. Dependence of muscarinic K+ channel desensitization on receptor and receptor kinase.
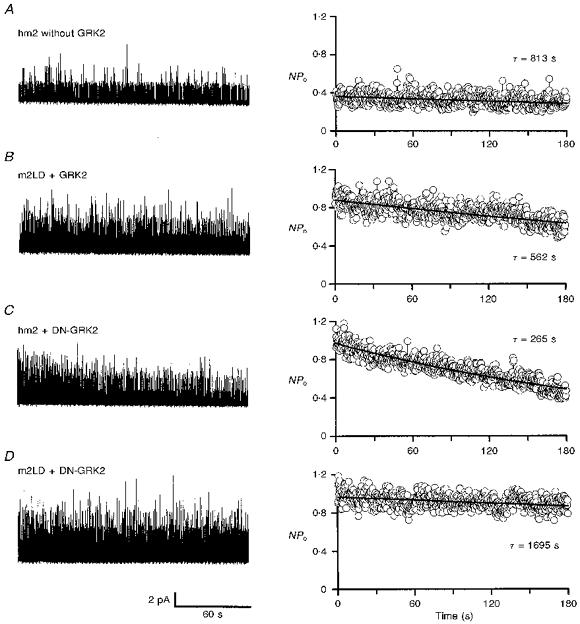
Left, examples of single channel currents during the first 3 min after the attachment of an ACh-containing pipette onto a cell. Right, mean NPo from 8 to 18 patches during the first 3 min after the attachment of the pipette. All data from CHO cells. A, data from cells transfected with the wild-type receptor, but not the receptor kinase. B, data from cells transfected with mutant receptor and wild-type receptor kinase. C, data from cells transfected with the wild-type receptor and mutant receptor kinase. D, data from cells transfected with mutant receptor and mutant receptor kinase. Inward currents are shown in the upwards direction on the left to facilitate comparison with the plots of NPo on the right. Transfection: A, stable transfection with DNA for hm2 and transient transfection with DNA for Kir3.1, Kir3.4 and GFP; B, transient transfection with DNA for m2LD, GRK2, Kir3.1, Kir3.4 and GFP or stable transfection with DNA for m2LD and transient transfection with DNA for GRK2, Kir3.1, Kir3.4 and GFP; C, stable transfection with DNA for hm2 and transient transfection with DNA for DN-GRK2, Kir3.1, Kir3.4 and GFP; D, stable transfection with DNA for m2LD and transient transfection with DNA for DN-GRK2, Kir3.1, Kir3.4 and GFP (see Methods for details).
Figure 4. Muscarinic K+ channel desensitization: summary.
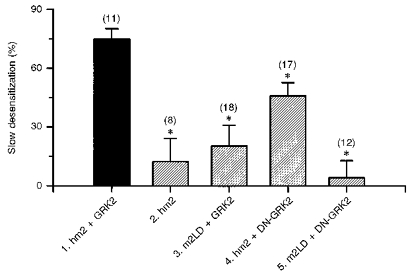
The graph shows the extent of desensitization in different groups of cells. The decline in NPo during the first 3 min after the attachment of an ACh-containing pipette onto a cell is expressed as a percentage of the peak NPo when the pipette was first attached. Values are means ± s.e.m. (n = 8–18). 1, cells transfected with the wild-type receptor and wild-type receptor kinase. 2, cells transfected with the wild-type receptor, but not the receptor kinase. 3, cells transfected with mutant receptor and wild-type receptor kinase. 4, cells transfected with the wild-type receptor and mutant receptor kinase. 5, cells transfected with mutant receptor and mutant receptor kinase. All data from CHO cells. * Significantly different (P < 0.05) from the decline in NPo as a result of desensitization in cells transfected with the wild-type receptor and wild-type receptor kinase (shown in column 1; one-way analysis of variance). Transfection: 1, stable transfection with DNA for hm2 and GRK2 and transient transfection with DNA for Kir3.1, Kir3.4 and GFP; 2, stable transfection with DNA for hm2 and transient transfection with DNA for Kir3.1, Kir3.4 and GFP; 3, transient transfection with DNA for m2LD, GRK2, Kir3.1, Kir3.4 and GFP or stable transfection with DNA for m2LD and transient transfection with DNA for GRK2, Kir3.1, Kir3.4 and GFP; 4, stable transfection with DNA for hm2 and transient transfection with DNA for DN-GRK2, Kir3.1, Kir3.4 and GFP; 5, stable transfection with DNA for m2LD and transient transfection with DNA for DN-GRK2, Kir3.1, Kir3.4 and GFP (see Methods for details).
In the second case (Fig. 3B), the cells were transfected with the wild-type receptor kinase (GRK2). They were also transfected with a large-deletion mutant of the receptor, m2LD (or hm2-Δ233–380). The known phosphorylation sites on the m2 muscarinic receptor are on the third intracellular loop and, in the case of m2LD, residues P233 to S380 encompassing all but one of these sites were deleted (Moro, Lameh & Sadée, 1993). It has been shown previously that the large deletion from the receptor does not prevent the receptor from binding to an agonist (m2LD has the same ligand-binding capacity and selectivity as the wild-type receptor) and activating G-protein in the normal manner (Kameyama, Haga, Haga, Moro & Sadée, 1994). However, the deletion does abolish the phosphorylation of the agonist-bound receptor by the receptor kinase, GRK2 (Kameyama et al. 1994). In cells transfected with m2LD, channel activity was as high as in the control experiments with cells transfected with wild-type receptor (Fig. 1A, columns 4 and 5). This is consistent with m2LD being able to activate the G-protein in the normal way. However, in cells transfected with m2LD the decline in channel activity as a result of desensitization was markedly reduced. Whereas in the control cells (with wild-type receptor, hm2) channel activity declined by 75 ± 5 %, in the cells transfected with m2LD, channel activity declined by just 20 ± 11 % during the first 3 min (Fig. 4, columns 1 and 3). The difference is significant (P < 0.05). This suggests that the third intracellular loop of the receptor is involved in desensitization. The fact there was a small decline in channel activity with m2LD (although not significantly different from zero, P = 0.062; Fig. 4, column 3) perhaps suggests that other sites on the receptor apart from the third intracellular loop (absent in m2LD) are involved in desensitization.
In the third case (Fig. 3C), the cells were transfected with the wild-type receptor (hm2) and a dominant negative mutant of the receptor kinase (GRK2-K220W or DN-GRK2). In the case of DN-GRK2, an invariant lysine residue at position 220 in the protein kinase catalytic domain was substituted by a tryptophan. We have previously shown that this mutation eliminates the ability of the kinase to phosphorylate the m2 receptor (Tsuga et al. 1994). A similar mutant, GRK2-K22OR, also lacks kinase ability (Kong, Penn & Benovic, 1994). If desensitization is exclusively the result of phosphorylation of the receptor by the receptor kinase, transfection with DN-GRK2, like that with m2LD, should largely abolish desensitization. In cells transfected with DN-GRK2, peak channel activity was as high as in cells transfected with the wild-type receptor kinase (GRK2; compare columns 4 and 6 in Fig. 1A). In cells transfected with DN-GRK2, contrary to the prediction above, there was a decline in channel activity during the first 3 min after exposure of the patch to ACh (Fig. 3C). However, the decline in activity was reduced compared with that in the control experiment (Fig. 2A-C). In the cells transfected with DN-GRK2, channel activity declined by 46 ± 7 % during the first 3 min, which is significantly less (P < 0.05) than that (75 ± 5 %) in the control cells (with wild-type receptor kinase, GRK2; Fig. 4, columns 1 and 4). Therefore, use of DN-GRK2 reduced the amount of desensitization by about 39 %. This suggests that about 39 % of the desensitization is the result of the phosphorylation of the receptor by the kinase (as DN-GRK2 lacks the ability to phosphorylate the receptor). However, because a substantial amount of desensitization (about 61 % of normal) still occurred with DN-GRK2, the results suggest that the kinase can cause desensitization via a phosphorylation-independent pathway. This is considered further in the Discussion.
In the final case (Fig. 3D), the cells were transfected with both m2LD and DN-GRK2. Again channel activity was as high as in the control experiments (Fig. 1A, columns 4 and 7). The decline in channel activity during the first 3 min of ACh exposure as a result of desensitization was almost completely abolished. Channel activity declined by 4 ± 9 % only. The decline is significantly different (P < 0.05) from the decline of 75 ± 5 % in the control experiments (Fig. 4, columns 1 and 5). Furthermore, the decline in channel activity in cells transfected with m2LD and DN-GRK2 is significantly less (P < 0.05) than that in cells transfected with the wild-type receptor, hm2, and DN-GRK2 (4 ± 9 vs. 46 ± 7 %; Fig. 4, columns 5 and 4). This indicates that the receptor kinase-dependent, phosphorylation-independent desensitization (present in the cells transfected with hm2 and DN-GRK2) is abolished by the deletion of the third intracellular loop of the receptor.
DISCUSSION
Our results show that the cardiac muscarinic receptor–K+ channel-receptor kinase system can be reconstructed in CHO cells. In cell-attached recordings, the channel activity was dependent on the presence of ACh, the receptor and the channel as expected; the channel properties (single channel conductance, rectification and open time) were also similar to those of the native channel in the heart (Fig. 1). Recordings were also made using the whole-cell configuration (data not shown); again as expected the muscarinic K+ current was activated on application of ACh to the cell and deactivated on wash-off of ACh. In whole-cell recordings, the current also declined during the exposure to ACh as a result of desensitization (in CHO cells transfected with the wild-type receptor, channel and wild-type receptor kinase).
In the heart, there are various phases of desensitization of the muscarinic K+ channel as described in the Introduction: fast, intermediate and slow phases. The fast phase of desensitization develops during the first ∼20 s of an exposure to ACh (e.g. Kim, 1991; Zang et al. 1993). This was not evident in the present study in the cell-attached recordings from either CHO cells or rat atrial cells. However, this is not unexpected, because we have previously shown that in rat atrial cells the fast phase of desensitization is not observed in cell-attached patches as a result of a limitation of the recording technique (after the attachment of an ACh-containing pipette onto a cell, channel activity cannot be recorded immediately, because time is required to optimize recording conditions, and yet fast desensitization will be developing; Shui et al. 1995). The desensitization of the muscarinic K+ channel observed during the first 3 min after the attachment of an ACh-containing pipette onto a rat atrial cell (Fig. 2) is the intermediate phase (Shui et al. 1995). The desensitization of the muscarinic K+ channel in CHO cells during the first 3 min after the attachment of an ACh-containing pipette onto a cell is similar in magnitude and time course to the intermediate phase of desensitization in rat atrial cells (Fig. 2). If atrial cells are exposed to a muscarinic agonist for 24–48 h, long term desensitization develops (Bünemann et al. 1996). Long term desensitization also develops if CHO cells (transfected with the wild-type receptor, channel and wild-type receptor kinase) are exposed to 10 μm carbachol (a muscarinic agonist) for 24 h (Shui et al. 1997c).
In guinea-pig and rat atrial cells, the intermediate phase of desensitization has been attributed to a change in the receptor, because it is abolished if the receptor is bypassed and the muscarinic K+ channel is activated directly by GTPγS (Zang et al. 1993; Shui et al. 1997a). In rat atrial cells, there is also evidence that the intermediate phase of desensitization is the result of receptor kinase; the intermediate phase of desensitization is absent in patch clamp configurations in which the cytoplasm is lost (receptor kinase is a soluble component of the cytoplasm) and is restored if receptor kinase (GRK2) is added back (Shui et al. 1995). This work is supported by the findings of the present study.
In CHO cells, because the desensitization of the muscarinic K+ channel was largely abolished by the omission of the receptor kinase and by the use of a mutant receptor lacking the third intracellular loop of the receptor, we conclude that desensitization of the muscarinic K+ channel is largely the result of the interaction of the receptor kinase with the third intracellular loop of the receptor. However, because the use of the mutant receptor kinase, DN-GRK2, which lacks the ability to phosphorylate the receptor but not the ability to bind to the receptor, only partially reduced desensitization, we conclude that the desensitization is only in part the result of phosphorylation of the third intracellular loop of the receptor by the kinase. The other component of desensitization (still dependent on the receptor kinase and third intracellular loop of the receptor) appears to be phosphorylation independent. It is known that DN-GRK2, like the wild-type receptor kinase, GRK2, binds to the receptor. Furthermore, it is well established that muscarinic receptor kinase and the G-protein αβγ ternary complex compete with each other for interaction with the agonist-bound muscarinic receptor (Haga et al. 1994). Therefore, the binding of DN-GRK2 to agonist-bound receptor could cause a reduction in the activation of G-protein (this would be dependent on the receptor kinase and the third intracellular loop of the receptor, but phosphorylation independent).
The muscarinic K+ channel is activated by G-protein βγ-subunits. Because GRK2 is known to bind G-protein βγ-subunits (it is the binding of G-protein βγ-subunits to GRK2 that leads to the activation of GRK2), GRK2 has been used to disrupt the activation of the muscarinic K+ channel by the muscarinic receptor (presumably GRK2 binds the G-protein βγ-subunits and reduces their concentration below that required to activate the muscarinic K+ channel) (Reuveny et al. 1994). It is possible that the desensitization of the muscarinic K+ channel dependent on the receptor kinase (perhaps all the desensitization; compare columns 1 and 2 in Fig. 4) is the result of binding of G-protein βγ-subunits by GRK2. However, this is unlikely, because in this case the desensitization should not be affected by the deletion of the third intracellular loop of the receptor, whereas desensitization was largely abolished by the deletion (compare columns 1 and 3 in Fig. 4).
In the case of desensitization of β-adrenergic receptors, as well as the receptor kinase, a cofactor, arrestin, is also required for desensitization to take place (Lohse, Benovic, Codina, Caron & Lefkowitz, 1990) and it will be interesting to test whether arrestin is also involved in desensitization of the muscarinic K+ channel. The desensitization of the muscarinic K+ channel represents a novel receptor kinase-induced ‘inactivation’ of the channel. There are other cases of inward rectifier K+ channels in the heart and brain coupled to various receptors (opioid, purinergic, putative sphingosine-1-phosphate) via G-proteins undergoing desensitization and these too could be the result of this novel inactivation mechanism. In failing hearts there is increased expression of the receptor kinase GRK2 (Lohse, 1995; Freedman & Lefkowitz, 1996) and this could in part be responsible for the depression in the activity of the muscarinic K+ channel in the failing heart (Koumi, Arentzen, Backer & Wasserstrom, 1994).
References
- Bünemann M, Brandts B, Pott L. Downregulation of muscarinic M2 receptors linked to K+ current in cultured guinea-pig atrial myocytes. The Journal of Physiology. 1996;494:351–362. doi: 10.1113/jphysiol.1996.sp021497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bünemann M, Brandts B, zu Heringdorf D M, van Koppen CJ, Jakobs KH, Pott L. Activation of muscarinic K+ current in guinea-pig atrial myocytes by sphingosine-1-phosphate. The Journal of Physiology. 1995;489:701–777. doi: 10.1113/jphysiol.1995.sp021084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet E, Mubagwa K. Desensitization of the acetylcholine-induced increase of potassium conductance in rabbit cardiac Purkinje fibres. The Journal of Physiology. 1986;371:239–255. doi: 10.1113/jphysiol.1986.sp015971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Molecular and Cellular Biology. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA. The inward rectifier potassium channel family. Current Opinion in Neurobiology. 1995;5:268–277. doi: 10.1016/0959-4388(95)80038-7. 10.1016/0959-4388(95)80038-7. [DOI] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Guillemare E, Fink M, Hugnot J-P, Bigay J, Lazdunski M, Romey G, Barhanin J. Heterologous multimeric assembly is essential for K+ channel activity of neuronal and cardiac G-protein-activated inward rectifiers. Biochemical and Biophysical Research Communications. 1995;212:657–663. doi: 10.1006/bbrc.1995.2019. 10.1006/bbrc.1995.2019. [DOI] [PubMed] [Google Scholar]
- Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Progress in Hormone Research. 1996;51:319–351. [PubMed] [Google Scholar]
- Haga T, Haga K, Kameyama K. G protein-coupled receptor kinases. Journal of Neurochemistry. 1994;63:400–412. doi: 10.1046/j.1471-4159.1994.63020400.x. [DOI] [PubMed] [Google Scholar]
- Harris GC, Williams JT. Transient homologous mu-opioid receptor desensitization in rat locus coeruleus neurons. Journal of Neuroscience. 1991;11:2574–2581. doi: 10.1523/JNEUROSCI.11-08-02574.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SM, McCall E, Boyett MR. The relationship between contraction and intracellular sodium in rat and guinea-pig ventricular myocytes. The Journal of Physiology. 1992;449:517–550. doi: 10.1113/jphysiol.1992.sp019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausdorff WP, Caron MG, Lefkowitz RJ. Turning off the signal: desensitization of β-adrenergic receptor function. FASEB Journal. 1990;4:2881–2889. [PubMed] [Google Scholar]
- Hong S-G, Pleumsamran A, Kim D. Regulation of atrial muscarinic K+ channel activity by a cytosolic protein via G protein-independent pathway. American Journal of Physiology. 1996;270:H526–537. doi: 10.1152/ajpheart.1996.270.2.H526. [DOI] [PubMed] [Google Scholar]
- Honjo H, Kodama I, Zang W-J, Boyett MR. Desensitization to acetylcholine in single sino-atrial node cells isolated from the rabbit heart. American Journal of Physiology. 1992;263:H1779–1789. doi: 10.1152/ajpheart.1992.263.6.H1779. [DOI] [PubMed] [Google Scholar]
- Kaibara M, Nakajima T, Irisawa H, Giles W. Regulation of spontaneous opening of muscarinic K+ channels in rabbit atrium. The Journal of Physiology. 1991;433:589–613. doi: 10.1113/jphysiol.1991.sp018445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Haga K, Haga T, Moro O, Sadée W. Activation of a GTP-binding protein and a GTP-binding-protein-coupled receptor kinase (β-adrenergic-receptor kinase-1) by a muscarinic receptor m2 mutant lacking phosphorylation sites. European Journal of Biochemistry. 1994;226:267–276. doi: 10.1111/j.1432-1033.1994.tb20050.x. [DOI] [PubMed] [Google Scholar]
- Khan IA, Shui Z, Tsuga H, Haga T, Boyett MR. Reconstitution of the muscarinic receptor, receptor kinase and K+ channel in CHO cells - receptor kinase-dependent desensitization. Biophysical Journal. 1997;72:A51. [Google Scholar]
- Kim D. Modulation of acetylcholine-activated K+ channel function in rat atrial cells by phosphorylation. The Journal of Physiology. 1991;437:133–155. doi: 10.1113/jphysiol.1991.sp018588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. Mechanism of rapid desensitization of muscarinic K+ current in adult rat and guinea-pig atrial cells. Circulation Research. 1993;73:89–97. doi: 10.1161/01.res.73.1.89. [DOI] [PubMed] [Google Scholar]
- Kong G, Penn R, Benovic JL. A β-adrenergic receptor kinase dominant negative mutant attenuates desensitization of the β2-adrenergic receptor. Journal of Biological Chemistry. 1994;269:13084–13087. [PubMed] [Google Scholar]
- Koumi S-I, Arentzen CE, Backer CL, Wasserstrom JA. Alterations in muscarinic K+ channel response to acetylcholine and to G protein-mediated activation in atrial myocytes isolated from failing hearts. Circulation Research. 1994;90:2213–2224. doi: 10.1161/01.cir.90.5.2213. [DOI] [PubMed] [Google Scholar]
- Kurachi Y, Ito H, Sugimoto T. Positive cooperativity in activation of the cardiac muscarinic K+ channel by intracellular GTP. Pflügers Archiv. 1990;416:216–218. doi: 10.1007/BF00370247. [DOI] [PubMed] [Google Scholar]
- Kurachi Y, Nakajima T, Sugimoto T. Short-term desensitization of muscarinic K+ channel current in isolated atrial myocytes and possible role of GTP-binding proteins. Pflügers Archiv. 1987;410:227–233. doi: 10.1007/BF00580270. [DOI] [PubMed] [Google Scholar]
- Lohse MJ. G-protein-coupled receptor kinases and the heart. Trends in Cardiovascular Medicine. 1995;5:63–68. doi: 10.1016/1050-1738(94)00034-4. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. β-arrestin: a protein that regulates β-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Martin P, Levy MN, Matsuda Y. Fade of cardiac responses during tonic vagal stimulation. American Journal of Physiology. 1982;243:H219–225. doi: 10.1152/ajpheart.1982.243.2.H219. [DOI] [PubMed] [Google Scholar]
- Miyake M, Christie MJ, North RA. Single potassium channels opened by opioids in rat locus ceruleus neurons. Proceedings of the National Academy of Sciences of the USA. 1989;86:3419–3422. doi: 10.1073/pnas.86.9.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro O, Lameh J, Sadée W. Serine- and threonine-rich domain regulates internalization of muscarinic cholinergic receptors. Journal of Biological Chemistry. 1993;268:6862–6865. [PubMed] [Google Scholar]
- Osborne PB, Williams JT. Characterization of acute homologous desensitization of μ-opioid receptor-induced currents in locus coeruleus neurones. British Journal of Pharmacology. 1995;115:925–932. doi: 10.1111/j.1476-5381.1995.tb15899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature. 1994;370:143–146. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shih M, Malbon CC. Oligodeoxynucleotides antisense to mRNA encoding protein kinase A, protein kinase C, and β-adrenergic receptor kinase reveal distinctive cell-type-specific roles in agonist-induced desensitization. Proceedings of the National Academy of Sciences of the USA. 1994;91:12193–12197. doi: 10.1073/pnas.91.25.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui Z, Boyett MR, Zang W-J. ATP-dependent desensitization of the muscarinic K+ channel in rat atrial cells. The Journal of Physiology. 1997a;505:77–93. doi: 10.1111/j.1469-7793.1997.077bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui Z, Boyett MR, Zang W-J, Haga T, Kameyama K. Receptor kinase-dependent desensitization of the muscarinic K+ current in rat atrial cells. The Journal of Physiology. 1995;487:359–366. doi: 10.1113/jphysiol.1995.sp020885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui Z, Khan IA, Tsuga H, Haga T, Boyett MR. Expression of the cardiac muscarinic receptor, muscarinic K+ channel and a receptor kinase in CHO cells: desensitization on application of ACh. The Journal of Physiology. 1997b;499.P:131P. [Google Scholar]
- Shui Z, Khan IA, Tsuga H, Haga T, Dobrzynski H, Henderson Z, Boyett MR. The role of G-protein-coupled receptor kinase in long-term desensitization of muscarinic K+ current. The Journal of Physiology. 1997c;504.P:75P. [Google Scholar]
- Tsuga H, Kameyama K, Haga T, Kurose H, Nagao T. Sequestration of muscarinic acetylcholine receptor m2 subtypes. Facilitation by G protein-coupled receptor kinase (GRK2) and attenuation by a dominant-negative mutant of GRK2. Journal of Biological Chemistry. 1994;269:32522–32527. [PubMed] [Google Scholar]
- Wang YG, Lipsius SL. Acetylcholine potentiates acetylcholine-induced increases in K+ current in cat atrial myocytes. American Journal of Physiology. 1995;268:H1313–1321. doi: 10.1152/ajpheart.1995.268.3.H1313. [DOI] [PubMed] [Google Scholar]
- Yang Z-K, Boyett MR, Janvier NC, McMorn SO, Shui Z, Karim F. Regional differences in the negative inotropic effect of acetylcholine within the canine ventricle. The Journal of Physiology. 1996;492:789–806. doi: 10.1113/jphysiol.1996.sp021346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang W-J, Yu X-J, Honjo H, Kirby MS, Boyett MR. On the role of G protein activation and phosphorylation in desensitization to acetylcholine in guinea-pig atrial cells. The Journal of Physiology. 1993;464:649–679. doi: 10.1113/jphysiol.1993.sp019656. [DOI] [PMC free article] [PubMed] [Google Scholar]