

Abstract
The effects of chloride channel blockers on pressure-induced constriction, K+-induced force, and whole-cell calcium channel currents were tested in rat cerebral arteries using isobaric and isometric myography, and patch clamp.
Under isobaric conditions at 75 mmHg, 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB), a chloride channel blocker, reversibly depressed the myogenic constriction with an IC50 of 32.8 ± 0.52 μm (mean ± s.e.m., n = 5). Blockers of Ca2+-activated chloride channels, flufenamic acid (100 μm) and 9-anthracene chloride (9-AC; 1 mm), and the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel blocker, glibenclamide (100 μm), were without effect in this tissue (n = 3).
Under isobaric conditions at 20 mmHg, 37 °C, raising [K+]o to 45 mm induced a constriction which was unaffected by 100 μm NPPB (n = 4). In contrast, at 75 mmHg and 18–21 °C, 100 μm NPPB completely and reversibly blocked a 45 mm K+-induced constriction (n = 3).
Under isometric conditions, NPPB reversibly depressed a 45 mm K+-induced force with an IC50 of 10.0 ± 0.76 μm (mean ± s.e.m., n = 5). Indanyloxyacetic acid 94 (IAA-94), another chloride channel blocker, depressed the K+-induced force with an IC50 of 17.0 ± 1.2 μm (mean ± s.e.m., n = 4).
Using whole-cell patch clamp, 100 μm NPPB or 200 μm IAA-94 blocked calcium channel currents carried by 10 mm Ba2+ by 79.1 ± 1.7 and 39.8 ± 7.0 %, respectively (mean ± s.e.m., n = 6).
In summary, chloride channel blockers depress calcium channel currents in rat cerebral arteries, which could contribute to a reduction in myogenic contraction.
Resistance arteries respond to increases in transmural pressure by actively contracting, thus maintaining a constant blood flow to an organ despite changes in blood pressure. This mechanism, termed the myogenic response, was first described in the early years of the century (Bayliss, 1902). In pressurized vessels, the myogenic response is strongly temperature dependent, in that it is absent at room temperature (18–21°C). At 37°C, a pressure-dependent membrane depolarization from approximately -65 to -40 mV associated with the myogenic response has been observed in pressurized renal and cerebral arteries (Harder, 1984; Harder, Gilbert & Lombard, 1987). This depolarization is sufficient to increase significantly the open probability (Po) of dihydropyridine (DHP)-sensitive calcium channels, thus leading to a calcium-dependent contraction. However, the ionic mechanism underlying this pressure-dependent depolarization is poorly understood. Evidence that chloride channels are present in smooth muscle (e.g. Pacaud, Loirand, Baron, Mironneau & Mironneau, 1991; Large & Wang, 1996), combined with an estimated reversal potential for chloride of -20 to -30 mV (Aickin, 1990) makes a chloride conductance an obvious candidate for mediating the pressure-induced depolarization. Indeed, chloride channel blockers have been demonstrated to inhibit depolarization and the myogenic response in pressurized cerebral arteries (Nelson, Conway, Knot & Brayden, 1997). In this paper evidence is presented that some chloride channel blockers are not selective in isolated cerebral resistance arteries, and effects of chloride channel blockers on the myogenic response may be, at least in part, due to a block of calcium channels. Thus, chloride channel blockers should be used with caution in evaluating the possible function of a chloride conductance in the myogenic response.
METHODS
Wistar rats (weight, 250–300 g) were killed by an intraperitoneal injection of sodium pentobarbitone (500 mg kg−1). The brain was removed into an ice-cold physiological saline solution (PSS) containing (mm): NaCl, 119; KCl, 4.7; NaHCO3, 25; KH2PO4, 1.18; CaCl2, 1.8; MgSO4, 1.2; glucose, 11; EDTA, 0.027; and gassed with 95 % O2-5 % CO2. Posterior or middle cerebral arteries were dissected from the surface of the brain and used for myograph studies. Basilar cerebral arteries were dissociated for patch clamp studies on single myocytes.
Halpern pressure myography
Leak-free segments of artery of at least 1 mm in length were mounted between two glass cannulae in an arteriograph (Living Systems Instrumentation, Burlington, Vermont, USA; Halpern, Osol & Coy, 1984) at room temperature (18–21°C) and pressurized to 75 mmHg, under conditions of no lumenal flow. A set constant pressure was maintained via a pressure servo control system (PS200, Living Systems Instrumentation). Arteries were viewed through a Nikon TMS inverted microscope and a measurement of the internal diameter was made from a video image using a video dimension analyser (V91, Living Systems Instrumentation). The arteriograph was continually superfused with the standard PSS (see above) at a rate of 25 ml min−1. The superfusing PSS was warmed to 37°C, and if a sustained reduction in artery diameter was seen then a myogenic response was considered to be present and an artery was used for further experiments. The artery was allowed to equilibrate for at least 30 min before experiments were started. Pressure and diameter measurements were recorded to computer via a Digidata 1200 interface using Axoscope software (Axon Instruments).
Mulvany wire myography
Isometric recordings of tension were made from 1–2 mm segments of rat middle cerebral artery mounted on 20 μm tungsten wires in a Mulvany Wire Myograph 410A at 37°C (J. P. Trading, Aarhus, Denmark) and bathed in PSS containing (mm): NaCl, 119; KCl, 4.7; Hepes, 10; MgSO4, 1.2; CaCl2, 1.8; Na2HCO3, 4; KH2PO4, 1.18; and glucose, 11 (pH 7.4 with NaOH), and continuously bubbled with air. High K+ (45 mm) PSS was made by isosmotic replacement of Na+. Length-tension curves were constructed as previously described, and the vessel normalized to 90 % of the passive diameter of the vessel at 100 mmHg (Mulvany & Halpern, 1977). Isometric increases in force in response to high K+ PSS were recorded and measured on a chart recorder.
Cell dissociation and patch clamp recording
Single smooth muscle myocytes were acutely isolated from cerebral arteries by enzymatic digestion. Arteries were placed in a standard PSS containing (mm): NaCl, 134; KCl, 5; Hepes, 10; MgCl2, 1; and CaCl2, 0.1 (pH 7.4 with NaOH). Arteries were digested with papain (Sigma, 14 U ml−1) and dithioerythritol (Sigma, 1 mg ml−1) at 36–37°C for 10 min. This was followed by a 36–37°C digestion in collagenase (Worthington CLS-2, 570 U ml−1) and hyaluronidase (Sigma, 320 U ml−1). Digestion was stopped by placing the tissue into fresh PSS. Tissue was kept at 4°C for up to 8 h before cells were dissociated by teasing the tissue apart with forceps.
For whole-cell patch clamp recording, pipettes were pulled from filamented borosilicate glass tubing (o.d., 1.5 mm; Clarke Electromedical, Pangbourne, UK), fire polished and coated with dental sticky wax (Kemdent, Swindon, UK) to reduce pipette capacitance. Pipettes had resistances of 2–5 MΩ when filled with an intracellular solution containing (mm): CsCl, 135; Hepes, 10; EGTA, 5; MgCl2, 1; glucose, 10; Na-ATP, 5; Li-GTP, 0.5, (pH 7.2 with CsOH). Experiments were carried out in a stage-mounted experimental chamber on an inverted microscope (Nikon Diaphot 200). The chamber was constantly perfused by a Hepes-buffered PSS containing (mm): NaCl, 137; KCl, 5; Hepes, 10; MgCl2, 1; CaCl2, 1.8, (pH 7.4 with NaOH). Cells were locally superfused using a rapidly switching superfusion system (Langton, 1993). Macroscopic inward barium currents (IBa) were measured by perfusing the chosen cell with PSS which had 1.8 mm Ca2+ isosmotically replaced by 10 mm Ba2+ by substitution with NaCl, and recorded using an Axopatch-200B amplifier (Axon Instruments) and pCLAMP 6 software (Axon Instruments). Data were externally filtered at 2 kHz and digitized (Digidata 1200A, Axon Instruments) at 10 kHz and saved to computer. Small currents recorded at high gain tended to be noisy and were refiltered digitally at 500 Hz. Pipette capacitance was compensated but series resistance was not normally compensated. The series resistance was 8.4 ± 1.7 MΩ (n = 11 cells) and the voltage error due the largest currents recorded in this study was 2 mV. No correction has been made.
Solutions
All drugs were made up as a 1000 × concentrated stock in milli-Q water unless otherwise stated, and applied in the external superfusate. Glibenclamide, indanyloxyacetic acid 94 (IAA-94) and 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) were made up as 1000 × concentrated stocks in dimethyl sulphoxide. Glibenclamide, IAA-94 and NPPB were purchased from Research Biochemicals International (distributed by Semat, St Albans, UK). Flufenamic acid and 9-anthracene chloride were purchased from Sigma. (-)202-791 was a kind gift from Sandoz Pharmaceuticals. All other reagents were purchased from BDH. Data are expressed as mean values ± s.e.m.
RESULTS
Under isobaric conditions, the presence of a myogenic response in pressurized cerebral arteries was confirmed as a decrease in diameter from 205 ± 7.7 to 154 ± 9.8 μm (n = 9), and was observed as the arteries were warmed from room temperature (18–21°C) to 37°C at 75 mmHg in a Halpern pressure myograph. This was a contraction to 74.8 ± 2.2 % of their initial diameter (Fig. 1A). At a constant pressure of 75 mmHg, the temperature-dependent myogenic contraction was depressed on addition of increasing concentrations of the chloride channel blocker NPPB. The myogenic response was completely abolished by 100 μm NPPB, and this was confirmed by adding 2 μm (-)202-791, a DHP antagonist, to the superfusate at the end of the experiment to give a measurement of the passive artery diameter. Half-maximal block was obtained with 32.8 ± 0.52 μm NPPB (n = 5; Fig. 1B). Blockers of Ca2+-activated chloride channels, flufenamic acid (100 μm) and 9-AC (1 mm), and the CFTR (cystic fibrosis transmembrane conductance regulator) Cl− channel blocker, glibenclamide (100 μm; Yamazaki & Hume, 1997) were without effect in this tissue (n = 3; data not shown).
Figure 1. Effect of NPPB on the myogenic contraction of pressurized arteries at 75 mmHg.
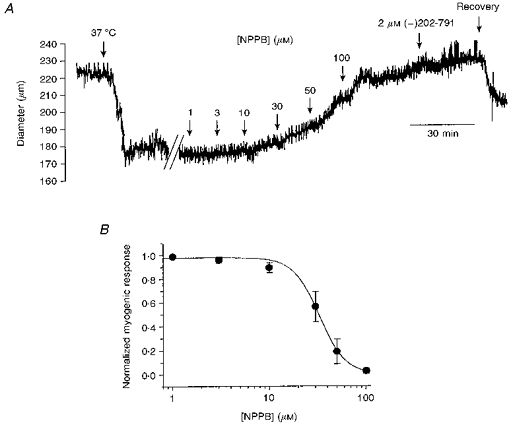
A, NPPB depressed a temperature-sensitive, pressure-dependent myogenic constriction. The superfusing PSS was warmed to 37 °C as indicated, and micromolar concentrations of NPPB were added cumulatively, to gain a concentration-effect curve for the reversal of the myogenic response at 75 mmHg. The passive artery diameter was measured in the presence of 2 μm (-)202-791, a DHP antagonist, at the end of the experiment. B, concentration-effect relationship for depression of the myogenic response by NPPB (means ± s.e.m., n = 5). Data were fitted to a Hill function giving an IC50 of 32.8 ± 0.52 μm, and a slope of -3.23.
To test the specificity of the block by NPPB, the artery was pressurized to 20 mmHg (below the pressure threshold for the myogenic response). The artery was depolarized with 45 mm K+, giving a constriction from 125 ± 26 to 42.0 ± 18 μm (mean ± s.e.m., n = 4). NPPB (100 μm) did not reverse this constriction (46.2 ± 19 μm; Fig. 2A). A similar protocol has been used previously to test the specificity of IAA-94, an alternative chloride channel blocker which also depresses the myogenic response (Nelson et al. 1997).
Figure 2. Effect of NPPB on a 45 mm K+ depolarization-induced contraction of pressurized arteries.
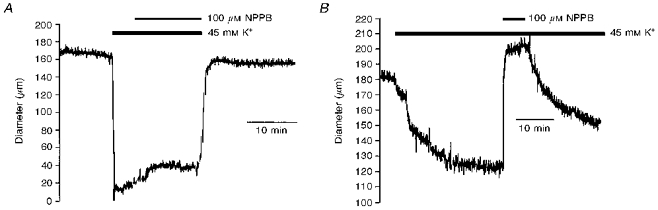
A, at 20 mmHg, below the pressure threshold for a significant myogenic response, superfusion of 100 μm NPPB was unable to reverse a constriction induced by depolarization with 45 mm K+ (n = 4). B, by cooling the artery to room temperature (18–21 °C) at 75 mmHg to remove the myogenic response, superfusion of 100 μm NPPB blocked constriction induced by 45 mm K+.
Alternatively, the myogenic response is also absent at room temperature (18–21°C). The artery was pressurized to 75 mmHg at room temperature, and depolarized with 45 mm K+ to give a contraction from 173 ± 9.2 to 116 ± 6.7 μm, such that the diameter decreased to 67.1 ± 1.8 % of its initial value (n = 3). This was close to the level of the myogenic response seen in these vessels when warmed to 37°C (68.1 ± 2.8 %; n = 3). Under these conditions, 100 μm NPPB completely reversed the K+-induced constriction, such that the artery dilated to 177.6 ± 19 μm. This effect was reversible (137 ± 1.73 μm). See Fig. 2B.
The effect of NPPB on 45 mm K+-induced force was tested under isometric conditions, using Mulvany wire myography. NPPB (100 μm) completely relaxed K+-induced isometric force, such that 2 μm (-)202-791 caused no further reduction in force (Fig. 3A). This is a standard diagnostic test for block of calcium channels. Half-maximal block was achieved with 10 ± 0.76 μm NPPB (n = 5).
Figure 3. Effect of NPPB and IAA-94 on a 45 mm K+ depolarization-induced isometric force.
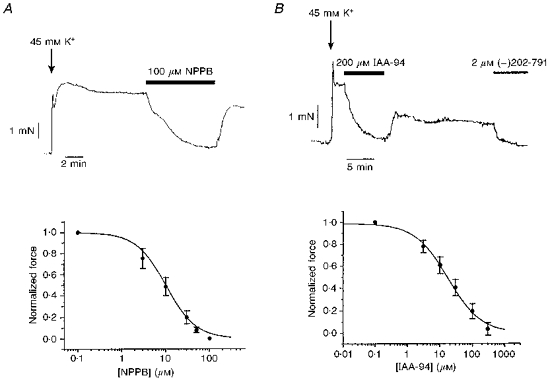
A, 100 μm NPPB completely blocked a 45 mm K+ depolarization-induced isometric force. Micromolar concentrations of NPPB were added cumulatively, to gain a concentration-effect curve for the relaxation of the 45 mm K+ depolarization-induced isometric force (means ± s.e.m., n = 5). The passive tension was measured in the presence of 2 μm (-)202-791, a DHP antagonist, at the end of the experiment. Data were fitted to a Hill function giving an IC50 of 10.0 ± 0.76 μm, and a slope of -1.28 ± 0.10. B, 200 μm IAA-94 completely relaxed a 45 mm K+depolarization-induced isometric force. The cumulative concentration-effect curve was fitted to a Hill function giving an IC50 of 17.0 ± 1.2 μm and a slope of 0.85 ± 0.04 (means ± s.e.m., n = 4).
The effect of the chloride channel blocker IAA-94 on 45 mm K+-induced force was also tested under isometric conditions. IAA-94 (200 μm) completely relaxed K+-induced force (Fig. 3B). Half-maximal block was achieved with 17.0 ± 1.2 μm IAA-94 (n = 4).
To confirm that NPPB and IAA-94 block calcium channels in this tissue, whole-cell patch clamp measurements of IBa were made from freshly isolated cerebral smooth muscle myocytes. The amplitude of IBa was determined as the difference current in the presence and absence of 2 mm Co2+. Superfusion with 100 μm NPPB reduced IBa by 79.1 ± 1.7 % (n = 6). Washout of the drug was achievable, but full recovery to pre-application levels of inward current was not seen due to channel run-down (Fig. 4A). Similarly, 200 μm IAA-94 reversibly depressed IBa by 39.8 ± 7.0 % (n = 6; Fig. 4B).
Figure 4. Effect of NPPB and IAA-94 on whole-cell barium currents.
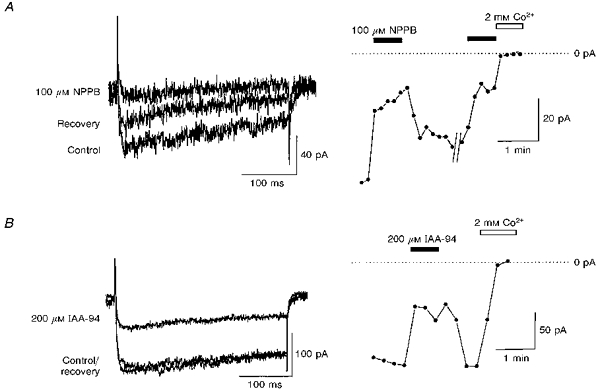
Representative whole-cell barium (10 mm) currents elicited by 320 ms voltage steps from -80 to +10 mV are shown. NPPB (100 μm; A) and IAA-94 (200 μm; B) reversibly depressed inward barium currents by 79.1 ± 1.7 and 39.8 ± 7.0 %, respectively (means ± s.e.m., n = 6). Zero current was measured at the end of the experiment in the presence of 2 mm Co2+. The peak inward current was measured and plotted against time.
DISCUSSION
The results of this study confirm reports that some chloride channel blockers can depress pressure-induced myogenic tone in cerebral arteries. DIDS (4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid), SITS (4-acetamido-4′-isothiocyanatostilbene-2,2′-disulphonic acid), IAA-94 (Nelson et al. 1997) and NPPB have all been shown to be effective blockers of the myogenic response. However, chloride channel blockers 9-anthracene chloride (9-AC), niflumic acid and flufenamic acid (Aickin, 1990), and concentrations of glibenclamide which are sufficient to block the CFTR Cl− channel (Yamazaki & Hume, 1997), were without effect. Evidence from this study suggests that although chloride channels may play an important role in the depolarization which underlies pressure-induced myogenic contraction, some chloride channel blockers may not be selective in the cerebral vasculature, and their effects on the myogenic response may be mediated, at least in part, by depression of the dihydropyridine-sensitive calcium channel.
Chloride channels have been identified in a number of smooth muscle preparations, including rabbit portal vein, and bovine trachea (e.g. Aickin, 1990; Hogg, Wang & Large, 1994; Salvail, Alioua & Rousseau, 1996; Kirkup, Edwards & Weston, 1996). The effects of vasoconstricting agonists such as noradrenaline and endothelin-1 can be depressed in the presence of blockers of the calcium-activated chloride channels such as 9-AC and niflumic acid (Vanrenterghem & Lazdunski, 1993; Iyadomi, Hirahara & Ehara, 1995; Criddle, Soares de Moura, Greenwood & Large, 1996), providing evidence that chloride channels may be important in mechanisms of vasoconstriction, including the myogenic response.
Vasoconstrictors such as noradrenaline may also cause chloride accumulation and depolarization via cotransport mechanisms (Chipperfield, Davis & Harper, 1997). However, published evidence suggests that niflumic acid has no effect on the myogenic contraction in pressurized cerebral arteries (Nelson et al. 1997). This is supported by observations in this paper that 9-AC and flufenamic acid are also ineffective as inhibitors of the myogenic response. Chloride channel blockers DIDS and SITS do inhibit the myogenic response, but these drugs are also known to block chloride accumulation via the Cl−-HCO3− exchanger (Aickin, 1990). Ion substitution experiments which reduce extracellular chloride increase the myogenic contraction in cerebral vessels, and this is supported by unpublished observations within our laboratory. This may be due to an increase in a chloride channel conductance, but manipulation of [Cl−]o could also affect other chloride-dependent mechanisms such as Cl−-HCO3− exchange, or a K+-Na+-Cl− cotransporter. Further experiments will be required to distinguish between these putative mechanisms.
Evaluation of the role of a chloride conductance in the depolarization associated with myogenic contraction is dependent on the availability of specific pharmacological tools. Amongst the most specific chloride channel blockers available which have been tested in smooth muscle are IAA-94 and NPPB (Aickin, 1990; Kirkup et al. 1996; Nelson et al. 1997), both of which depress myogenic constriction. Kirkup et al. (1996) have demonstrated that selectivity of NPPB to the chloride channel in rat portal vein is concentration dependent, such that at 10 μm it was selective to the chloride current, but at 30 μm it also blocked calcium currents by around 70 %. However, the present study demonstrates that these agents are unsatisfactory as selective chloride channel blockers in the cerebral vasculature. Indeed, the effect of NPPB on isometrically maintained cerebral arteries suggests that even at 10 μm, a concentration which does not block calcium channels in rat portal vein (Kirkup et al. 1996), a significant block of calcium channels is observed (IC50 = 10.0 ± 0.76 mm (mean ± s.e.m., n = 5)).
It is worthy of note that a block of calcium channels could indirectly result in the depression of calcium-activated chloride channels. Any possible effects of NPPB and IAA-94 on calcium-activated chloride currents in cerebral vessels cannot be separated from the effects of calcium channel blockade in the experiments presented here. However, there is no pharmacological evidence at present, using drugs such as niflumic acid and flufenamic acid, that a calcium-activated chloride conductance is present in these cells. In the absence of the myogenic response in pressurized vessels (i.e. at room temperature), and under isometric conditions, 45 mm K+ will depolarize the artery to approximately -26 mV. This is close to the reported reversal potential for chloride, and therefore changes in a chloride conductance are unlikely to influence changes in artery diameter or force under these conditions.
Both IAA-94 (200 μm) and NPPB (100 μm) were able to block an isometric contraction and an isobaric constriction at 75 mmHg induced by a 45 mm K+ depolarization, but were unable to dilate isobaric constrictions induced by a 45 mm K+ depolarization at 20 mmHg. This latter finding is consistent with the study reported by Nelson et al. (1997) which showed that IAA-94 does not reverse an isobaric 60 mm K+ depolarization-induced constriction at 20 mmHg. The incongruity of the isometric and isobaric data needs to be resolved. It may be helpful to consider why isobaric myography might fail to demonstrate an inhibitory action of Cl− channel blocking agents on K+-induced constrictions at low pressures when such an action is seen clearly in isometric recordings.
In isometric myography, contraction occurs at fixed length and, for a simple stimulus like K+ depolarization, force is proportional to [Ca2+]i and is related to membrane voltage by its influence on the Po of Ca2+ channels (Nelson, Patlak, Worley & Standen, 1990). Block of Ca2+ channels lowers [Ca2+]i and leads to a proportional relaxation (Knot, Standen & Nelson, 1997).
Under isobaric conditions the situation is, importantly, quite different. Active tension generated by vascular myocytes works against the load (wall tension) imposed by the transmural pressure, and the vessel constricts, thus attenuating the wall tension, which is directly proportional to diameter. Depolarization of vessels held at only 20 mmHg (below the myogenic range) with elevated [K+]o resulted in profound constriction, from 125 to 42 μm (a 3-fold reduction in diameter; Fig. 2A). The active tension required to maintain this diameter is therefore reduced to only 34 % of that required to begin to constrict the vessel from 128 μm. The muscle cells will have shortened by around 66 %, which is approaching the limit of cell shortening. Thus, the vessel is so constricted, and the distending force so attenuated that it is effectively trapped in a constricted state; in spasm. At these small diameters, physical elements such as the endothelium and elastic lamina become the principal load against which the active tension acts. Under these conditions significant inhibition of Ca2+ channels may fail to lower [Ca2+]i or the active contraction to the point where the remaining wall tension can begin to dilate the vessel.
Thus, it is possible that an inhibitory action of the Cl− channel blockers on Ca2+ channels may go undetected by isobaric myography at low pressures. It follows from this consideration that dilatation, once initiated, will initiate a positive feedback mechanism, such that the increased wall tension causes further dilatation and an increase in wall tension, etc. This hypothesis is supported by the observation that a 45 mm K+ constriction at 75 mmHg in the absence of a myogenic response (i.e. at room temperature) can be dilated by 100 μm NPPB (Fig. 2B). Under these conditions, the constriction mimics the level of constriction seen during the myogenic response, and avoids the possibility that the artery goes into spasm. The apparent difference in the initial conclusions to be drawn from these experiments should be noted, and illustrates the caution that should be taken in data interpretation.
Studies using patch clamp on isolated cerebral myocytes support the measurements of isometric tension, indicating that IAA-94 and NPPB depress DHP-sensitive calcium channels. However, because the calcium channel currents are small in these cells, and there is run-down during the course of an experiment, even using 10 mm Ba2+ as the charge carrier, it is not possible to construct concentration-effect curves from this data to give an estimate of the IC50 of calcium channel block. As whole-cell barium currents are not completely blocked at concentrations of NPPB and IAA-94 that are sufficient to give near-maximal block of isometric tension and isobaric contraction, it might indicate that calcium channel blockade is not the only effect of these drugs in the intact arteries. This is supported by the concentration-effect curves for these drugs under isobaric conditions, where the best fits are obtained from Hill equations with a slope factor of close to 3. The use of 10 mm Ba2+ rather than 1.8 mm Ca2+ as the charge carrier in these measurements is not ideal, but the currents measured using 1.8 mm Ca2+ were too small to distinguish changes in current amplitude within the noise of the system.
In conclusion, the hypothesis that an increased chloride conductance may depolarize pressurized vessels and so modulate the myogenic response is an attractive one for which there is a lot of circumstantial evidence. However, although a range of chloride channel blockers is available, their usefulness as diagnostic tools for the evaluation of the role played by chloride conductances in the myogenic response is hampered by their lack of specificity in the cerebral vasculature.
Acknowledgments
This work was supported by the British Heart Foundation, grant number PG94/101.
References
- Aickin CC. Chloride transport across the sarcolemma of vertebrate smooth and skeletal muscle. In: Alvarez-Leefmans FJ, Russel JM, editors. Chloride Channels and Carriers in Nerve, Muscle and Glial Cells. New York: Plenum Press; 1990. pp. 209–249. [Google Scholar]
- Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. The Journal of Physiology. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipperfield AR, Davis JPL, Harper AA. Activation of two inward chloride pumps in rat arterial smooth muscle by noradrenaline in vitro. The Journal of Physiology. 1997;501.P:116P. [Google Scholar]
- Criddle DN, Soares de Moura R, Greenwood IA, Large WA. Effect of niflumic acid on noradrenaline-induced contractions of the rat aorta. British Journal of Pharmacology. 1996;118:1065–1071. doi: 10.1111/j.1476-5381.1996.tb15507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern W, Osol G, Coy GS. Mechanical behavior of pressurized in vitro prearteriolar vessels determined with a video system. Annals of Biomedical Engineering. 1984;12:463–479. doi: 10.1007/BF02363917. [DOI] [PubMed] [Google Scholar]
- Harder DR. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circulation Research. 1984;55:197–202. doi: 10.1161/01.res.55.2.197. [DOI] [PubMed] [Google Scholar]
- Harder DR, Gilbert R, Lombard JH. Vascular muscle-cell depolarization and activation in renal arteries on elevation of transmural pressure. American Journal of Physiology. 1987;253:F778–781. doi: 10.1152/ajprenal.1987.253.4.F778. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Wang Q, Large WA. Action of niflumic acid on evoked and spontaneous calcium-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. British Journal of Pharmacology. 1994;112:977–984. doi: 10.1111/j.1476-5381.1994.tb13177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyadomi I, Hirahara K, Ehara T. α-Adrenergic inhibition of the β-adrenoceptor-dependent chloride current in guinea-pig ventricular myocytes. The Journal of Physiology. 1995;489:95–104. doi: 10.1113/jphysiol.1995.sp021033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkup AJ, Edwards G, Weston AH. Investigation of the effects of 5-nitro-2-(3-phenylpropylamino)-benzoic acid (NPPB) on membrane currents in rat portal vein. British Journal of Pharmacology. 1996;117:175–183. doi: 10.1111/j.1476-5381.1996.tb15171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Standen NB, Nelson MT. Regulation of arterial smooth muscle [Ca2+] in intact cerebral arteries by membrane potential and the sarcoplasmic reticulum. Biophysical Journal. 1997;72:TU273. [Google Scholar]
- Langton PD. A versatile superfusion system suitable for whole-cell and exercised patch clamp experiments. The Journal of Physiology. 1993;467:244P. [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. American Journal of Physiology. 1996;271:C435–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circulation Research. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Conway MA, Knot HJ, Brayden JE. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. The Journal of Physiology. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. American Journal of Physiology. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Pacaud P, Loirand G, Baron A, Mironneau C, Mironneau J. Ca2+ channel activation and membrane depolarization mediated by Cl− channels in response to noradrenaline in vascular myocytes. British Journal of Pharmacology. 1991;104:1000–1006. doi: 10.1111/j.1476-5381.1991.tb12540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvail D, Alioua A, Rousseau E. Functional identification of a sarcolemmal chloride channel from bovine tracheal smooth muscle. American The Journal of Physiology. 1996;271:C1716–1724. doi: 10.1152/ajpcell.1996.271.5.C1716. [DOI] [PubMed] [Google Scholar]
- Vanrenterghem C, Lazdunski M. Endothelin and vasopressin activate low-conductance chloride channels in aortic smooth-muscle cells. Pflügers Archiv. 1993;425:156–163. doi: 10.1007/BF00374516. [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Hume JR. Inhibitory effects of glibenclamide on cystic fibrosis transmembrane regulator, swelling-activated, and Ca2+-activated Cl− channels in mammalian cardiac myocytes. Circulation Research. 1997;81:101–109. doi: 10.1161/01.res.81.1.101. [DOI] [PubMed] [Google Scholar]