

Abstract
Several K+ currents were measured and compared in enzymatically dispersed ventricular myocytes from control and diabetic rats.
Diabetic conditions were established either with a single intravenous injection of streptozotocin (STZ, 100 mg kg−1; 6–14 days duration) or by feeding with a fructose-enriched diet for 4–10 weeks. Both groups became hyperglycaemic, with the former having decreased and the latter having elevated levels of plasma insulin. These conditions therefore mimic type I (insulin-dependent) and type II (non-insulin-dependent) diabetes mellitus, respectively.
As reported previously, a Ca2+-independent transient outward K+ current, It, was attenuated in the type I model. This was not observed in the type II model. The two models differed greatly in the changes observed in a quasi-steady-state K+ current denoted Iss. In the STZ model Iss was substantially attenuated, whereas in the fructose-fed model it was augmented. In both models, the background inwardly rectifying current, IK1, was unchanged. Concomitantly, there was a substantial prolongation of the action potential in the STZ model but not in the fructose-fed model.
Incubation of control myocytes with insulin (100 nM) for 5–9 h caused a significant augmentation of Iss, with no effect on It or on IK1. Incubation of myocytes from STZ-diabetic rats with insulin reversed the attenuation of It, but not of Iss.
The effect of insulin was not blocked by wortmannin, an inhibitor of phosphatidylinositol 3-kinase. However, inhibition of the mitogen-activated protein kinase pathway with PD98059 prevented restoration of It. Insulin action on It may therefore involve changes in transcription or expression of channel proteins, rather than changes in cellular metabolism.
Diabetes mellitus is very common in adults and diabetic complications are a leading cause of death (Harris & Zimmet, 1992; Shehadeh & Regan, 1995). The disease is manifest in two forms, types I and II, with the latter being by far the more common form (Harris & Zimmet, 1992). Although these two types overlap in the major problem of inadequate regulation of glucose homeostasis, there are many important differences. A major distinction is that in type I the major problem is insufficient insulin production, whereas in type II the defect lies in events occurring downstream to the binding of insulin to its receptor. This leads to insulin resistance, with the secretion of excess insulin as part of a compensatory mechanism (Harris & Zimmet, 1992).
In both types of diabetes, cardiovascular complications develop (Shehadeh & Regan, 1995). These may appear early (Robillon, Sadoul, Jullien, Morand & Freychet, 1994; Celentano et al. 1995), and may persist despite therapeutic interventions. Among the major complications are the development of cardiomyopathy, autonomic neuropathy and coronary heart disease (Ewing, Boland, Neilson, Cho & Clarke, 1991; Shehadeh & Regan, 1995). Life-threatening cardiac arrhythmias can develop either as a result, or independently, of these complications (Bakth et al. 1986; Ewing et al. 1991; Lindstrom, Jorfeldt, Tegler & Arnqvist, 1992).
Several animal models have been used extensively to study the effects of diabetes on the heart. By far the most commonly used model is the insulin-deficient streptozotocin (STZ) model. STZ induces type I diabetes by selectively destroying pancreatic β-cells. In this model, rapid changes are induced in both electrical and mechanical characteristics of the heart (Shimoni, Firek, Severson & Giles, 1994; Ren & Davidoff, 1997). Although type II diabetes is much more common, far fewer studies have been devoted to investigating cardiac complications under those conditions. This is, in part, due to the more complex nature of type II diabetes, and the lack of appropriate animal models. Nevertheless, it is clear that both insulin-dependent (type I) and non-insulin-dependent (type II) diabetes lead to dysfunction in both electrical and mechanical activity of the heart (Lindstrom et al. 1992; Celentano et al. 1995).
Several alterations in ionic currents which underlie changes in action potential configuration and in ECG recordings, which are apparent in type I (insulin-deficient) diabetes, have been described (Magyar, Rusznak, Szentesi, Szucs & Kovacz, 1992; Jourdon & Feuvray, 1993; Shimoni et al. 1994). A major finding is that a Ca2+-independent transient outward K+ current, It, is attenuated in cardiomyocytes from diabetic rats. This change is more marked in epicardial than in endocardial regions of the ventricle (Shimoni, Severson & Giles, 1995), providing a partial explanation for the flattening of the T-wave in ECG recordings of human diabetics (Airaksinen, 1985). A sustained outward K+ current, Iss, is also attenuated. These changes greatly prolong the action potential, and presumably contribute to the prolongation of the Q-T interval seen in the diabetic ECG (Lo, Sutton & Leslie, 1993).
Despite the much higher incidence of type II diabetes, to date there are no electrophysiological studies addressing changes in ionic currents under those conditions. In this study we have used an animal model that mimics type II diabetes. It has been reported that a carbohydrate-enriched diet, using fructose or sucrose, leads to increased insulin levels, and to the development of insulin resistance and hypertension (Hwang, Ho, Hoffman & Reaven, 1987; Bhanot, McNeill & Bryer-Ash, 1994). Glucose levels may also increase, due to the insulin resistance. The mechanism for these changes is not clear (Reaven & Ho, 1991).
Insulin has many complex actions on cellular processes, which can be broadly subdivided into two classes. On one hand, insulin status affects activities of enzymes involved in cellular metabolism (Rodrigues & McNeill, 1992), and on the other, it regulates gene expression of a number of proteins (O'Brien & Granner, 1996). Insulin deficiency changes cardiac energy metabolism, by shifting substrate utilization from glucose to free fatty acids (FFAs) (Lopaschuk & Spafford, 1989). This effect is reversed by insulin, but also by dichloroacetate (DCA), which inhibits mitochondrial FFA utilization (Nicholl, Lopaschuk & McNeill, 1991). The regulation of ion channels by insulin may involve changes in cellular metabolism, or may be due to effects of insulin on gene expression, which may be altered during diabetes.
We find that in the fructose-fed (type II) model there is no reduction in It, in contrast to the attenuation seen in the STZ model. However, the sustained outward K+ current, Iss, is significantly augmented. We show that this effect can be largely attributed to direct effects of insulin. Action potentials in this type II model are not prolonged, in contrast to the prolongation observed in the insulin-deficient STZ model. By using different pharmacological tools to interfere with insulin transduction mechanisms, our results suggest that insulin has a tonic effect on cardiac K+ currents, presumably by affecting the expression level of channel proteins.
METHODS
Animal models
In this study, two rat models of diabetes were used, in addition to controls. The first was the STZ model (Shimoni et al. 1994), obtained by a single intravenous injection of STZ (100 mg (kg body weight)−1). Animals were killed by cervical dislocation after 6–14 days, at which time they were severely hyperglycaemic (see Results). A second group of rats was fed a fructose-enriched diet (66 % of total calories; Harlan Teklad, Madison, WI, USA) for 4–10 weeks. This has been shown (Hwang et al. 1987; Bhanot et al. 1994) to produce insulin resistance, with elevated plasma insulin levels and hyperglycaemia (see Results). Plasma insulin levels were measured in all groups using a radioimmunoassay (Foothills Hospital Clinical Laboratories). Plasma glucose concentration was measured using a colorimetric assay (Sigma).
Myocyte preparation
In all groups, myocytes were prepared as described previously (Shimoni et al. 1995). Briefly, animals were anaesthetized with ether and killed by cervical dislocation. The hearts were removed and the aortas cannulated on a Langendorff apparatus for coronary perfusion (at 80 cmH2O pressure, 37°C). After 5 min in a solution containing (mm): 121 NaCl, 5.4 KCl, 2.8 sodium acetate, 1 MgCl2, 1 CaCl2, 5 Na2HPO4, 24 NaHCO3, and 5 glucose, the hearts were perfused for 10 min in the same solution without calcium, and then for 6–8 min in the calcium-free solution with the addition of digestive enzymes (10 U ml−1 collagenase, Yakult Honsha, Tokyo; 0.01 mg ml−1 protease Type XIV, Sigma). The free walls of the right ventricle were then cut into small pieces for further incubation (in a shaker bath at 37°C) in an enzyme-containing solution (50 U ml−1 collagenase, 0.1 mg ml−1 protease, 0.1 mm CaCl2, 20 mm taurine and 10 mg ml−1 albumin). Aliquots of cells were collected for the next 40–60 min, and stored in the same solution as above with no enzymes, in 0.1 mm CaCl2. Myocytes were then placed in a bath on the stage of an inverted microscope and perfused with control solution containing (mm): 150 NaCl, 5.4 KCl, 1 MgCl2, 1 CaCl2, 5 Hepes, and 5.5 glucose (pH 7.4). CdCl2 (0.3 mm) was added to block the L-type Ca2+ current. The pipette solution contained (mm): 110 potassium aspartate, 30 KCl, 4 Na2ATP, 1 MgCl2, 5 Hepes, 10 EGTA, and 1 CaCl2, brought to a pH of 7.2 with KOH. These conditions ensured that only Ca2+-independent K+ currents were elicited. All recordings were done at 21–23°C.
Electrophysiological recordings
Recordings from single myocytes were made using the whole-cell, suction electrode technique, in voltage or current clamp modes (with an L/M EPC-7 amplifier) for measurements of action potentials and ionic currents. Great care was taken to minimize series resistance (Rs) artifacts by using low resistance electrodes (1–3 MΩ), compensating Rs, and discarding cells in which Rs increased during recording. Furthermore, since steady-state current measurements can be biased by leak currents, only well-polarized cells were used (resting potentials of -70 mV or larger).
Three major K+ currents, present in rat ventricle (Apkon & Nerbonne, 1991), were measured: a transient outward current (It), a quasi-steady-state current with properties of a delayed rectifier current, denoted Iss, and the inward rectifier background current, IK1. There is an inherent problem in attempting to separate It from Iss, since no effective pharmacological inhibitor of Iss is available (Apkon & Nerbonne, 1991). The peak outward current at the beginning of a depolarizing pulse is the sum of It and Iss, with the former being much larger (Apkon & Nerbonne, 1991). It is often measured as the difference between the peak outward current and the steady-state level at the end of a long depolarizing pulse. Iss is measured as the current magnitude at the end of the same pulse. However, a major finding (see below) is that Iss changes in different diabetes models. Thus, measuring It as the difference current would be greatly affected by the changes in Iss. In this work we measured It as the peak outward current in response to depolarizing voltage steps to membrane potentials between -40 and +50 mV. Since It activates much faster than Iss (Apkon & Nerbonne, 1991), the error in estimating It magnitude in this way should be small. However, since Iss also changed in our experiments (see below), the error may not be constant, which could produce some bias in the results. Statistical comparisons were made at +50 mV, at which It predominates. At this potential there was also no overlapping sodium current. Iss was measured as the current magnitude at the end of 500 ms pulses to the same membrane potentials; IK1 was measured as the current magnitude in response to steps to more negative membrane potentials (-50 to -110 mV). All voltage steps were given at a rate of 0.1 Hz. In all cells, capacitance was measured by integrating the capacitative transients in response to 5 mV steps from -80 mV. All currents were divided by cell capacitance to give current density, which enabled a comparison between cells of different sizes.
Statistics
A one-way analysis of variance (ANOVA) test was performed in comparing changes in current magnitude at +50 mV between the different experimental groups. When significant F values were obtained, Dunnett's multiple comparison test was done, to assess differences between parameters measured in control and in each of the experimental groups. Results are given as means ± s.e.m.P values of < 0.05 were considered statistically significant.
RESULTS
Status of the experimental groups
Plasma was collected from each group of rats used in this study, and glucose and insulin levels were measured. Table 1 shows the mean glucose and insulin concentrations in the control, STZ and fructose-fed groups. Both diabetic models were hyperglycaemic, although this was more severe in the STZ model. The major difference between the two models was the level of insulin; the STZ model was insulin deficient, whereas the fructose-fed model was hyperinsulinaemic. For both experimental groups, glucose and insulin levels were significantly different from the control levels (P < 0.05). The co-existence of hyperinsulinaemia and hyperglycaemia in the fructose-fed group is an indicator of insulin resistance.
Table 1.
Plasma glucose and insulin concentrations
| [Glucose] (mm) | [Insulin] (pmol l−1) | |
|---|---|---|
| Control | 5.3 ± 0.3 (n = 3) | 107.2 ± 14.6 (n = 5) |
| STZ treated | 27.2 ± 1.6 (n = 7) | 47.4 ± 4.1 (n = 8) |
| Fructose treated | 10.2 ± 0.4 (n = 8) | 162.3 ± 15.5 (n = 6) |
Values are means ± s.e.m.; n, number of rats.
K+ currents
All K+ currents were normalized by being divided by the cell capacitance. It was of interest that the mean cell capacitances varied significantly between control and both models of diabetes. The mean cell capacitance values were 66.8 ± 1.6 pF in control myocytes (n = 107), 56.4 ± 1.1 pF in cells from STZ-treated rats (n = 143) and 85.6 ± 3.3 pF in cells from fructose-fed rats (n = 54). The values in the two diabetes models were significantly (P < 0.05) smaller and larger, respectively, in comparison with control values, in correlation with (and possibly as a result of) the differences in plasma insulin levels.
It has previously been shown that, in an insulin-deficient STZ model, both the transient outward and the steady-state K+ currents are attenuated, with an ensuing prolongation of the action potential. A third K+ current, IK1, is unchanged (Magyar et al. 1992; Jourdon & Feuvray, 1993; Shimoni et al. 1994).
In the fructose-fed model, in which insulin levels are elevated, we found a small non-significant reduction in It. However, a major change was a significant increase in the sustained outward current, Iss. Figure 1 shows superimposed current traces obtained in myocytes from control (A), STZ-treated (B) and fructose-treated (C) rats. The currents shown were scaled according to cell size by dividing the currents by the corresponding cell capacitance (which was 92.2, 85.6 and 99.0 pF, respectively), so that the traces reflect current densities. The most striking difference between the three cells is the smaller steady-state current in the cell from the STZ-treated (insulin-deficient) rat, and the larger Iss in the cell from the fructose-treated (hyperinsulinaemic) rat.
Figure 1. Changes in K+ currents in two models of diabetes.
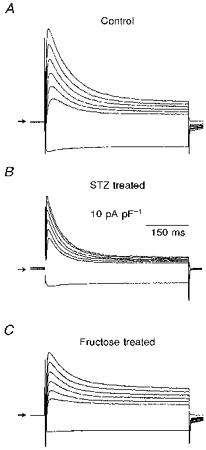
Superimposed current traces are shown, obtained in response to 500 ms steps from -80 mV to -110, 0, 10, 20, 30, 40 and 50 mV. The traces shown are from 3 different myocytes, obtained from a control rat (A), an STZ-treated rat (B) and a fructose-treated rat (C). The currents were scaled by being divided by cell capacitance (92.2, 85.6, 99.0 pF, respectively), so that in effect the current densities are shown. The apparent differences in the inactivation time courses are not significant (see text). Note the differences in the steady-state current magnitudes, which were smaller (B) and larger (C) than control, in the two diabetes models. Arrows in this and subsequent figures denote the zero current level. See text for the significance of the differences in current magnitudes.
The changes in steady-state current magnitudes affect the apparent inactivation time course of the transient component (Fig. 1). However, when curve-fitting procedures were performed on the inactivating phase of the current trace, with a variable steady-state final amplitude allowed, no significant differences in the time constants were observed. For example, in the current traces from the myocytes from control (Fig. 1A) and from fructose-fed (Fig. 1C) rats, the time constants of inactivation (at +50 mV) were 82.5 and 80.0 ms, respectively. The time constant for the myocyte from the STZ-treated rat shown in this figure was 54.8 ms (see also values for traces in Figs 4 and 5).
Figure 4. Effects of exposure to 100 nM insulin for 5–9 h on K+ currents in myocytes from control rats.
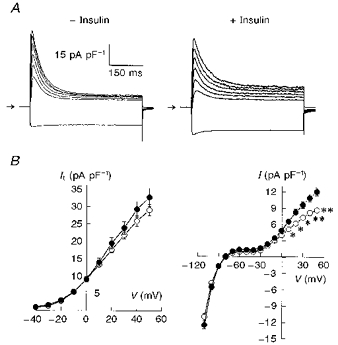
A, superimposed current traces obtained in response to different voltage steps (same as in Fig. 1) in myocytes in the absence (left) or presence (right) of insulin. Currents were divided by cell capacitance (50.0 and 67.6 pF, respectively), illustrating current densities for both cells. B, mean current-voltage relationships obtained by measuring peak outward It (left) and the current magnitude at the end of 500 ms pulses (right). At a potential range of -110 to -40 mV this measurement reflects IK1, which was unaffected by insulin. For membrane potentials positive to -30 mV, this measurement reflects Iss, which was significantly increased by insulin. ○, values obtained in myocytes in the absence of insulin (n = 31); •, values in the presence of insulin (n = 23). Values are means ± s.e.m. (*P < 0.05; **P < 0.01).
Figure 5. Effects of exposure to 100 nM insulin for 5–9 h on K+ currents in myocytes from STZ-treated rats.
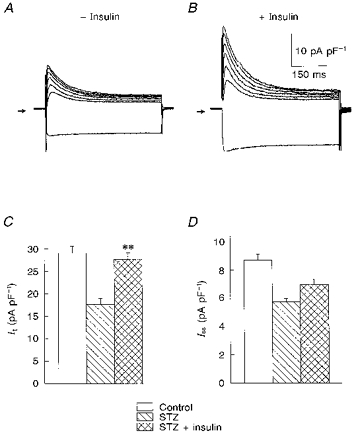
A and B, superimposed current traces in response to voltage steps (as in Fig. 1) in two myocytes obtained from a STZ-treated rat, with no insulin added (A), and following 6 h in insulin (B). The currents were divided by the cell capacitances, so that densities can be compared. It was greatly enhanced, with little effect on the steady-state amplitude at the end of the pulse. C and D, summary data (means ± s.e.m.) of current densities (at +50 mV) in cells (n = 34) from control rats, and from STZ-treated rats, in the absence (n = 29) or presence (n = 36) of insulin. The enhancement of It was statistically significant (** P < 0.001, STZ + insulin vs. STZ), whereas Iss showed a tendency to increase which did not achieve statistical significance.
Figure 2 shows the current-voltage relationships for It (A) and Iss (B) in myocytes from control (n = 34), STZ-treated (n = 31) and fructose-treated (n = 30) rats. It was significantly (P < 0.05) smaller than control between +10 and +50 mV in the STZ model. Iss was significantly smaller than control between 0 and +50 mV in the STZ model, and significantly larger than control at +40 and +50 mV in the fructose-fed model. Note that whereas It decreased only in the STZ model, Iss decreased in one (STZ treated) and increased in the other (fructose fed). IK1 densities were similar in control and in both diabetic models, as shown by the current densities at negative membrane potentials (Fig. 2B). The relative densities of It and Iss (at +50 mV) in the different experimental groups are shown in the histograms in Fig. 2C.
Figure 2. Current-voltage relationships for It (A) and for the steady-state current magnitudes (B), obtained by measurements at the end of 500 ms pulses.
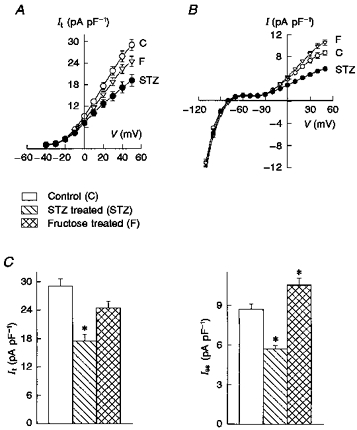
Iss is represented by values between -30 and +50 mV, and IK1 by values between -40 and -110 mV. C, summary data showing the current densities (pA pF−1) at +50 mV, measured in myocytes from control rats (n = 34 myocytes), STZ-treated rats (n = 31) and fructose-fed rats (n = 30). It (left) decreased significantly only in the STZ model. Iss (right) decreased in the STZ model but increased in the fructose model. IK1 was not significantly altered, as can be seen in the current-voltage relationships in B. Values are means ± s.e.m.; *P < 0.05, compared with control.
The differential changes in the K+ currents should also be reflected in the action potential configuration seen in the different models.
Action potentials
It has long been known that there is a prolongation of the ventricular action potential in the STZ-induced type I diabetes (Magyar et al. 1992), which most probably underlies the prolongation of the Q-T interval of the ECG of diabetic humans. In the rat, this prolongation is due to reductions in the two major repolarizing currents, It and Iss. In the insulin-resistant fructose-fed model, the lack of reduction in It and the significant increase in Iss (Figs 1 and 2) precluded any prolongation. Action potentials were elicited (at 0.5 Hz) and measured under current clamp conditions in the two diabetic models. In the insulin-deficient STZ model there was a marked (significant) prolongation, as reported before (Magyar et al. 1992; Shimoni et al. 1995). In striking contrast, action potentials in myocytes from the fructose-fed rats were much shorter, and comparable to control action potentials. Action potential durations (APDs) were measured at 0 and at -50 mV. In myocytes from control (n = 6 myocytes), STZ-treated (n = 10) and fructose-treated (n = 11) rats, the mean APD at 0 mV was 11.9 ± 1.4, 91.1 ± 8.9 and 9.4 ± 2.6 ms, respectively. At -50 mV these values were 40.5 ± 3.2, 200.3 ± 13.7 and 32.4 ± 11.8 ms, respectively. These results are shown in Fig. 3.
Figure 3. Action potentials in the two diabetic models.
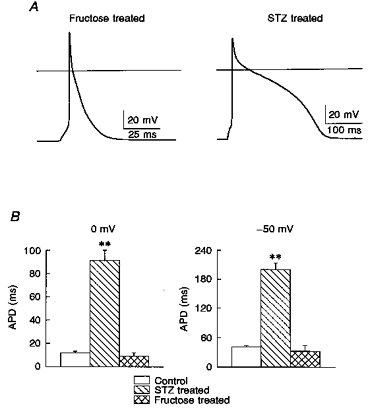
A, action potentials obtained (at a stimulation rate of 0.5 Hz) in myocytes from a fructose-fed rat (left) and an STZ-treated rat (right). The continuous horizontal line indicates 0 mV. Note the marked prolongation in the myocyte from the STZ-treated (low-insulin) rat, in comparison to the absence of such an effect in the myocyte from the fructose-fed (high-insulin) rat. B, comparison of mean values ( ± s.e.m.) of action potential duration (APD), measured at 0 mV (left) and at -50 mV (right) in the 3 groups; control (n = 6 myocytes), STZ-treated (n = 10) and fructose-fed (n = 11) rats. APD was significantly increased (** P < 0.001) in the STZ model, compared with controls. APD was shorter (although failing to achieve significance) in the fructose model.
Thus, the absence of a reduction in It and the excess outward current (Iss) in the hyperinsulinaemic group maintains a short action potential, in contrast to the changes under insulin-deficient conditions. The implications of this for the ECG and for arrhythmogenesis are discussed below (see Discussion).
The overall changes in both type I and type II diabetes are complex, and are known to involve changes in the levels of glucose, FFAs, triglycerides, as well as in many regulatory systems. However, a major difference between the two types is in the level of insulin itself. We therefore set out to determine the role of insulin as a regulator of the magnitude of cardiac K+ currents. First, the opposing changes in Iss in the two diabetes models, in which insulin levels are abnormally low or high, led us to examine whether insulin plays a major role in determining the magnitude of Iss. Subsequently, we examined whether the attenuation of It and Iss in the STZ model is due to the insulin deficiency.
The effects of insulin on cardiac K+ currents
Myocytes from control rats
In order to investigate whether the elevated levels of insulin might be responsible for the changes in K+ currents in the fructose-treated model, we exposed myocytes from control rats to insulin. Earlier studies indicated rapid effects of insulin on membrane currents (Fischmeister, Ayer & DeHaan, 1983), with conflicting reports on the effects of insulin on membrane potential, often (but not entirely) attributed to effects on the Na+-K+ pump (Lantz, Elsas & DeHaan, 1980). In preliminary experiments (results not shown), we found that incubation (up to 90 min) of myocytes (from control rats) with 100 nM insulin did not affect any of the K+ currents recorded in this study. However, when cells (n = 23) were incubated for 5 h or longer (up to 9 h), there was a significant (P < 0.01) enhancement of Iss. The mean density of Iss at +50 mV was 8.7 ± 0.42 and 12.06 ± 0.68 pA pF−1 in the absence and presence (for 5–9 h) of insulin, respectively. It was not significantly altered during this treatment (29.1 ± 1.6 and 32.7 ± 2.6 pA pF−1, respectively), as shown in Fig. 4. Representative current traces are shown in Fig. 4A, with the mean current values shown in the current-voltage relationships in Fig. 4B, It on the left and Iss on the right. At +50 mV, the time constants of inactivation in the myocytes shown were 47.9 ms in the absence, and 60.7 ms in the presence of insulin. IK1, assessed by measurement of current amplitudes in response to steps to membrane potentials negative to Iss activation (-50 to -110 mV), was unaffected by the long exposure to insulin. Xu, Patel & Rozanski (1996) also observed that treatment of control cardiomyocytes with 100 nM insulin for less than 4 h did not change It or IK1.
Myocytes from insulin-deficient (STZ-treated) rats
Cells obtained from rats treated with STZ were exposed to 100 nM insulin, which was added to the storage solution in which cells were kept. Cells were then taken at different periods after insulin was added, for electrophysiological measurements. No acute effects were seen on It (as in Xu et al. 1996) or on Iss and IK1. However, with longer exposures, lasting 5–9 h, there was a significant (P < 0.001) enhancement of It. At + 50 mV, It density was 27.6 ± 1.5 pA pF−1 (n = 36), compared with 17.6 ± 1.3 pA pF−1 (n = 29) in myocytes from STZ-treated rats with no insulin added. In contrast, 5–9 h of exposure to insulin did not restore the magnitude of Iss, although a tendency for it to increase was observed. The mean density at +50 mV was 6.96 ± 0.34 pA pF−1 (n = 36) vs. 5.72 ± 0.26 pA pF−1 with and without insulin, respectively. These results are shown in Fig. 5, which shows sample current traces from two myocytes from the same ventricle, without (A) and following 6 h of insulin (B). The time constants of inactivation (at +50 mV) for the myocytes shown were 80.7 and 78.8 ms, in the absence and presence of insulin, respectively. Figure 5C and D, respectively, shows the It and Iss densities at +50 mV in control, and in STZ-treated myocytes in the absence and presence of insulin. The restoring effect of insulin on It is similar to that reported by Xu et al. (1996).
The mechanism of insulin action on K+ currents
The binding of insulin to its receptor leads to a wide range of cellular actions, involving different signal transduction pathways (Saltiel, 1996). Insulin action results in changes in the activity of various enzymes and in effects on transcription and/or translation of various gene products (O'Brien & Granner, 1996). Alterations in cellular metabolism, by different mechanisms, have been previously found to alter many cardiac K+ currents (e.g. Pike, Bretag & Roberts, 1993; Ogbaghebriel & Shrier, 1994). In addition, insulin may regulate gene expression of K+ channel proteins.
In this study, we performed several series of experiments designed to provide preliminary evidence regarding the manner in which insulin might regulate the K+ currents that are changed in the different diabetic models. Initially, we made use of studies by Lopaschuk & Spafford (1989), who described some of the metabolic changes occurring in hearts of insulin-deficient rats, which show impaired mechanical function. Hearts from STZ-diabetic rats switch from a utilization of glucose and FFAs as energy substrates to a complete dependence on FFA. The compound DCA rapidly restores the utilization of glucose, as well as the mechanical function of the heart (Nicholl et al. 1991). In our experiments, addition of 1.2 mm DCA to myocytes from STZ-treated rats failed to reverse the attenuation of both It and Iss. Forty cells from STZ-treated rats with no DCA were compared with forty-two cells exposed for 3–7 h to 1.2 mm DCA. There was no significant effect on any of the currents measured: It density (at +50 mV) was 19.3 ± 1.5 pA pF−1 (no DCA) and 21.1 ± 1.4 pA pF−1 (with DCA); Iss density (at +50 mV) was 6.88 ± 0.27 and 7.77 ± 0.35 pA pF−1, respectively. IK1 density (at -110 mV) was -13.99 ± 0.64 and -13.83 ± 0.56 pA pF−1, respectively. These results are shown in Fig. 6A and B. Our results contrast with those of Xu et al. (1996) who found a complete restoration of It by DCA (they did not report effects on Iss).
Figure 6. The absence of effect of DCA on K+ currents in myocytes from STZ-treated rats.
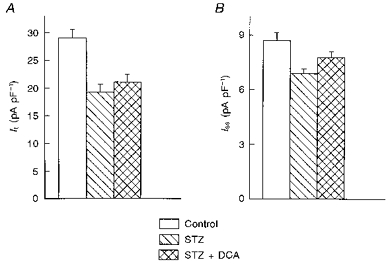
A and B show the mean ( ± s.e.m.) current densities (at +50 mV) for It (A) and Iss (B) in myocytes from control (n = 40) and from STZ-treated rats in the absence (n = 40) or following 3–7 h exposure to 1.2 mm DCA (n = 42 cells). DCA did not significantly increase either current.
The lipid kinase, phosphatidylinositol 3-kinase (PI-3 kinase) plays an essential role in the regulation of intracellular metabolism by insulin (Shepherd, Nave & O'Rahilly, 1996). We have examined whether this enzyme is involved in the regulation of K+ channels by insulin. Wortmannin (100 nM), an inhibitor of PI-3 kinase (Ui, Okada, Hazeki & Hazeki, 1995), was added to cells 30 min prior to the addition of insulin, and was present throughout the incubation (5–9 h). In the presence of wortmannin, insulin was still able to significantly (P < 0.01) enhance It, as shown in Fig. 7. At +50 mV, It density was 12.3 ± 1.3 pA pF−1 (n = 11 cells) in the absence, and 22.0 ± 1.7 pA pF−1 (n = 19 cells) in the presence of insulin and wortmannin (5–9 h incubation). Iss increased with insulin and wortmannin, from 5.9 ± 0.3 to 8.2 ± 0.7 pA pF−1. IK1 was unaffected.
Figure 7. Effects of insulin (100 nM) in the presence of wortmannin (100 nM) on K+ currents in myocytes from STZ-treated rats.
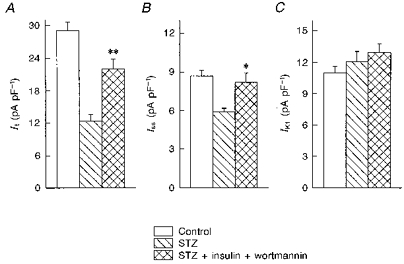
A-C show the mean ( ± s.e.m.) current densities for It and Iss (at +50 mV) and for IK1 (at -110 mV), respectively, in cells from control rats (n = 34 myocytes), and from STZ-treated rats in the absence (n = 11) or presence of insulin and wortmannin (n = 19, 5–9 h). It was significantly enhanced by insulin (**P < 0.01, compared with STZ), even in the presence of wortmannin. Iss was also significantly (*P < 0.05) augmented. IK1 was unchanged in any of the conditions.
In a control series of experiments we investigated whether wortmannin might have non-specific effects, which would enhance It. In ten cells (from STZ-treated rats) exposed to 1 μm wortmannin (10-fold higher than in the experiments with insulin), the mean density of It (at +50 mV) was 17.7 ± 1.6 pA pF−1, which was similar to the density without wortmannin. Iss and IK1 were also unaffected.
Mitogen-activated protein kinase (MAP kinase) is an important kinase in the signal transduction of growth factors and insulin, leading to cell proliferation and protein synthesis (Seger & Krebs, 1995). Its phosphorylation and activation is blocked by PD98059, a specific inhibitor of extracellular signal-regulated kinase (Lazar et al. 1995). Cells from STZ-treated rats were pre-incubated for 30 min with PD98059 (25 μm), prior to addition of insulin. PD98059, which was present throughout the insulin incubation, was found to block the restoration of It by insulin. In nineteen cells with no treatment, It (at +50 mV) was 19.7 ± 1.5 pA pF−1, whereas in twenty-five cells exposed to insulin and PD98059 (5–9 h), It density was 22.4 ± 1.5 pA pF−1. The corresponding values for Iss were 5.59 ± 0.32 and 6.29 ± 0.32 pA pF−1, and for IK1 (at -110 mV) they were -12.4 ± 0.7 and -12.8 ± 0.6 pA pF−1.
DISCUSSION
Summary of findings
The results presented here are the first to describe changes in cardiac ionic currents in an animal model mimicking type II diabetes. We find that when animals are subjected to a high-fructose diet (Hwang et al. 1987; Bhanot et al. 1994), leading to elevated plasma insulin and glucose levels, there are important changes in K+ currents (Figs 1 and 2): Iss was enhanced, whereas It was not significantly altered. This is different from the STZ-diabetic model, in which It and Iss were attenuated. No changes in the background inward-rectifier IK1 current were observed in either model. The differences in It and Iss regulation resulted in different effects on action potential configuration (Fig. 3). In the STZ model, there was a lengthening of the action potential, due to a reduction in both major repolarizing currents. In the fructose-fed type II model, the action potential duration was shorter than that in the control, although not significantly (Fig. 3). The enhanced Iss will contribute to the shortening of the action potential, although other currents also determine the terminal phases of the action potential.
Our results show that incubation of normal cells for several hours in the presence of insulin can selectively enhance Iss (but not It), with no effect on IK1 (Fig. 4). Since this effect parallels changes seen in fructose-fed rats, we postulated that the hyperinsulinaemia in this model is a primary factor in elevating Iss. Although the insulin resistance in fructose-fed rats is so severe that the resulting hyperinsulinaemia cannot normalize the blood glucose, the insulin resistance in other pathways, such as in the regulation of Iss, may be less pronounced. A direct stimulatory effect of hyperinsulinaemia in vivo on Iss measured in isolated myocytes can therfore still be observed. A similar up- or downregulation of the expression of a glucose transporter (Glut4) by altered insulin levels has been described in cardiomyocytes from insulin-deficient and insulin-resistant rats (Petersen, Russ, Reinauer & Eckel, 1991).
In the insulin-deficient STZ model, several hours (> 5) of exposure to insulin was found to restore It, but not Iss (Fig. 5). DCA, which reverses diabetes-related changes in energy substrate utilization, and associated mechanical impairment, did not reverse the diminished K+ currents (Fig. 6). The restoration of the K+ currents by insulin was not inhibited by wortmannin (Fig. 7), but was blocked (Fig. 8) by an inhibitor of MAP kinase activation. This suggests that the effects on It occur via a MAP kinase-dependent signalling mechanism.
Figure 8. Suppression of insulin action on K+ currents following inhibition of MAP kinase action by compound PD98059.
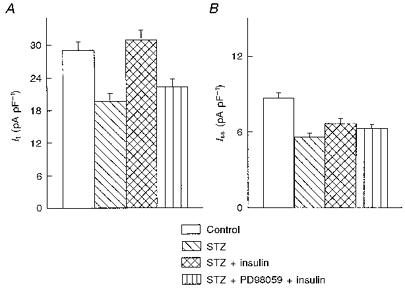
A and B show the mean ( ± s.e.m.) It and Iss densities, respectively (at +50 mV), in myocytes from control (n = 34) rats, and from STZ-treated rats in the absence of insulin (n = 19 cells), with insulin alone (n = 23), and with insulin in the presence of 25 μm PD98059 (n = 25). Iss and IK1 (not shown) were unchanged, whereas the enhancement of It by insulin was blocked by the inhibition of MAP kinase action.
Significance and implications
The significance of these findings is twofold. First, we report a novel action of insulin on a major repolarizing delayed rectifier cardiac K+ current, denoted Iss. Furthermore, we suggest that this determines the differential change in the two animal models of diabetes mellitus, corresponding to types I and II. Iss is attenuated in insulin-deficient (type I) diabetes, and augmented in insulin-resistant (excess insulin) type II diabetes. This in turn has opposite effects on the action potential configuration - a prolongation in type I and no change, with possible shortening, in type II. This may correspond to observations in the ECG of human diabetics, in which a prolonged Q-T interval (corresponding to prolonged ventricular action potentials) is often noted in type I diabetes (Lo et al. 1993), whereas no changes in the Q-T interval are found in type II (Robillon et al. 1994). Some of our earlier findings using the STZ rat model (Shimoni et al. 1995) could also be related to ECG changes in diabetics; a flattening of the T wave (Airaksinen, 1985) suggests a reduction in epicardial-endocardial K+ current gradients, which we in fact measured in myocytes from diabetic rat ventricles. We also reported an epicardial- endocardial gradient in It recovery kinetics, which is attenuated in the STZ model (Shimoni et al. 1995). A similar gradient has recently been reported in human ventricle as well (Beuckelmann, Nabauer & Erdmann, 1993). Thus, our findings of differential changes in K+ currents in the two types of rat models of diabetes are very likely to apply to human diabetics as well.
A major implication of these findings is that insulin has a tonic modulatory effect on cardiac repolarizing K+ currents. Iss density changes in response to both a reduction and an elevation in insulin levels. It, on the other hand, is attenuated under insulin-deficient conditions, but is not augmented by elevated insulin levels (at least not in the time frame tested). This is of interest, since the same pattern is observed with hypo- and hyperthyroid conditions (Shimoni et al. 1995); a deficiency in a modulatory hormone (thyroid or insulin) leads to attenuated It, while excess hormone levels do not increase it, implying that the tonic modulatory effect of the hormone on current magnitude is near maximal under normal hormone levels. Thus, in the fructose-fed model and when insulin is added to normal myocytes, It is unaffected. Iss, in contrast, is augmented by elevated insulin levels, as it is by augmented thyroid hormone levels (Shimoni & Severson, 1995). When insulin levels are chronically reduced, as in the STZ model, exposure of cells from these animals to insulin has selective effects: It can be significantly enhanced, but only after a sufficiently long period (over 5 h). This result was also obtained by Xu et al. (1996). Iss was not significantly enhanced by this duration of exposure to insulin. This implies that the regulation of the two currents is different. If (see below) insulin can determine the number of active channels by regulating transcription/translation or post-translational modification, this result may suggest that the turnover rate of Iss is considerably slower than that of It. In these experiments it was not possible to test longer exposure times. However, supportive evidence for this possibility was obtained from observations that, in cells obtained after longer periods following STZ injection, Iss continued to decrease, whereas It attenuation was similar for short (5–6 days) and for long (12–13 days) periods following STZ injection.
This work also set out to provide some insight into the mechanism by which insulin affects K+ currents. Since DCA does not reverse the impairment of K+ currents (Fig. 6), and since wortmannin does not prevent the action of insulin (Fig. 7), the results suggest that the metabolic consequences of insulin deficiency or excess are not directly related to effects on It and Iss. The different results of Xu et al. (1996) may be due to the slightly less severe model of diabetes that they used. Since the MAP kinase cascade is required for the action of insulin on It (Fig. 8), we hypothesize that the level of channel protein is increased by insulin, probably due to an increase in transcription. This is also suggested by the relatively long incubation time (> 5h) required for any effect of insulin to be measurable. This issue will be tested in future experiments.
Our present findings have a potentially important corollary in terms of vulnerability to arrhythmias. Many arrhythmias occur when ventricular repolarization times are dispersed, either spatially or temporally (Kuo, Munakata, Reddy & Surawicz, 1983), or when Q-T intervals are prolonged (Lo et al. 1993). Excluding other factors, one might expect an enhanced vulnerability to ventricular arrhythmias in type I diabetes, when action potentials and Q-T intervals are prolonged. This has indeed been reported (Shehadeh & Regan, 1995). Obviously, other complicating factors are present, such as concurrent coronary artery disease or hypertension, which can all contribute to arrhythmogenesis. Nevertheless, our study points to some of the changes which occur at the cellular level in the two models. The present findings will provide an important link in understanding all the changes which take place in cardiac function in the different types of diabetes, and their potentially detrimental consequences.
Limitations
A major and obvious limitation of this study is that the animal models used are complex and entail multiple changes. The fructose-fed model only partially mimics type II diabetes, mainly in inducing insulin resistance and some hyperglycaemia. The levels of insulin in type 2 diabetics may vary with the stage of disease (Harris & Zimmet, 1992). Furthermore, the fructose-fed rats develop hypertension (Hwang et al. 1987), which is often, but not always, present in type II diabetes. It is likely that the direct effect of a lack of insulin (in type I) leads to a smaller It, and that excess insulin has no effect, as we (and Xu et al. 1996) found by direct application to control cells. The fact that direct effects of insulin could be demonstrated on Iss strongly suggests a tonic modulatory effect of insulin on this current, but other factors cannot be ruled out in the setting of clinical diabetes.
A futher limitation is apparent when the arrhythmogenic potential of the changes in K+ currents is assessed. The effects of elevated insulin on the ventricular action potential may differ in the rat, which has a very short action potential, and in the human, in which there is a much longer plateau phase. It remains to be established whether in human cardiomyocytes there are similar changes in delayed rectifier K+ currents, and whether the action potential is indeed shorter in type II diabetes.
Finally, our evidence suggesting that insulin modulates K+ channels not by changes in metabolism, but possibly by other mechanisms, such as modulation of the levels of mRNA transcripts encoding these channels, such as occur for glucose transporters (Petersen et al. 1991), is still indirect. Future studies will address these issues more directly.
Acknowledgments
We would like to acknowledge the support of Dr W. R. Giles, in whose laboratory this work was done. This work was supported by a grant from the Canadian Diabetes Association in honour of Mary Selina Jamieson.
References
- Airaksinen KEJ. Electrocardiogram of young diabetic patients. Annals of Clinical Research. 1985;17:135–138. [PubMed] [Google Scholar]
- Apkon M, Nerbonne JM. Characterization of two distinct depolarization-activated K+ currents in isolated adult rat ventricular myocytes. Journal of General Physiology. 1991;97:973–1011. doi: 10.1085/jgp.97.5.973. 10.1085/jgp.97.5.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakth S, Arena J, Lee W, Torres R, Haider B, Patel PC, Lyons MM, Regan TJ. Arrhythmia susceptibility and myocardial composition in diabetes. Journal of Clinical Investigation. 1986;77:382–395. doi: 10.1172/JCI112316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circulation Research. 1993;73:379–385. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- Bhanot S, McNeill JH, Bryer-Ash M. Vanadyl sulfate prevents fructose-induced hyperinsulinaemia and hypertension in rats. Hypertension. 1994;23:308–312. doi: 10.1161/01.hyp.23.3.308. [DOI] [PubMed] [Google Scholar]
- Celentano A, Vaccaro O, Tammaro P, Galderisi M, Crivaro M, Oliviero M, Imperatore G, Palmieri V, Iovino V, Riccardi G, de Divitis O. Early abnormalities of cardiac function in non-insulin dependent diabetes mellitus and impaired glucose tolerance. American Journal of Cardiology. 1995;76:1173–1176. doi: 10.1016/s0002-9149(99)80330-0. [DOI] [PubMed] [Google Scholar]
- Ewing DJ, Boland O, Neilson JMM, Cho CG, Clarke BF. Autonomic neuropathy, QT interval lengthening, and unexpected deaths in male diabetic patients. Diabetologia. 1991;34:182–185. doi: 10.1007/BF00418273. [DOI] [PubMed] [Google Scholar]
- Fischmeister R, Ayer RK, DeHaan RL. Insulin regulates the ionic conductance of the embryonic heart cell membrane. Biophysical Journal. 1983;41:75a. [Google Scholar]
- Harris MI, Zimmet P. Classification of diabetes mellitus and other categories of glucose intolerance. In: Alberti KGMM, DeFrunzo RA, Keen H, Zimmet P, editors. International Textbook of Diabetes Mellitus. Chichester UK: John Wiley & Sons; 1992. pp. 3–18. [Google Scholar]
- Hwang IS, Ho H, Hoffman BB, Reaven GM. Fructose-induced insulin resistance and hypertension in rats. Hypertension. 1987;10:512–516. doi: 10.1161/01.hyp.10.5.512. [DOI] [PubMed] [Google Scholar]
- Jourdon P, Feuvray D. Calcium and potassium currents in ventricular myocytes from diabetic rats. The Journal of Physiology. 1993;470:411–429. doi: 10.1113/jphysiol.1993.sp019866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CS, Munakata K, Reddy CP, Surawicz B. Characteristics and a possible mechanism of ventricular arrhythmia dependent on the dispersion of action potential durations. Circulation. 1983;67:1356–1367. doi: 10.1161/01.cir.67.6.1356. [DOI] [PubMed] [Google Scholar]
- Lantz RC, Elsas LJ, DeHaan RL. Ouabain-resistant hyperpolarization induced by insulin in aggregates of embryonic heart cells. Proceedings of the National Academy of Sciences of the USA. 1980;77:3062–3066. doi: 10.1073/pnas.77.5.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar DF, Wiese RJ, Brady MJ, Mastick CC, Waters SB, Yamauchi K, Pessin JE, Cuatrecasas P, Saltiel AR. Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. Journal of Biological Chemistry. 1995;270:20801–20807. doi: 10.1074/jbc.270.35.20801. 10.1074/jbc.270.35.20801. [DOI] [PubMed] [Google Scholar]
- Lindstrom T, Jorfeldt L, Tegler L, Arnqvist HJ. Hypoglycaemia and cardiac arrhythmias in patients with type 2 diabetes mellitus. Diabetic Medicine. 1992;9:536–541. doi: 10.1111/j.1464-5491.1992.tb01834.x. [DOI] [PubMed] [Google Scholar]
- Lo SS, Sutton MS, Leslie RD. Information on type 1 diabetes mellitus and QT interval from identical twins. American Journal of Cardiology. 1993;72:305–309. doi: 10.1016/0002-9149(93)90677-5. 10.1016/0002-9149(93)90677-5. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Spafford M. Response of isolated working hearts to fatty acids and carnitine palmitoyltransferase I inhibition during reduction of coronary flow in acutely and chronically diabetic rats. Circulation Research. 1989;65:378–387. doi: 10.1161/01.res.65.2.378. [DOI] [PubMed] [Google Scholar]
- Magyar J, Rusznak Z, Szentesi P, Szucs G, Kovacz L. Action potentials and potassium currents in rat ventricular muscle during experimental diabetes. Journal of Molecular and Cellular Cardiology. 1992;24:841–853. doi: 10.1016/0022-2828(92)91098-p. 10.1016/0022-2828(92)91098-P. [DOI] [PubMed] [Google Scholar]
- Nicholl TA, Lopaschuk GD, McNeill JH. Effects of free fatty acids and dichloroacetate on isolated working diabetic rat heart. American Journal of Physiology. 1991;261:H1053–1059. doi: 10.1152/ajpheart.1991.261.4.H1053. [DOI] [PubMed] [Google Scholar]
- O'Brien RM, Granner DK. Regulation of gene expression by insulin. Physiological Reviews. 1996;76:1109–1161. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- Ogbabghebriel A, Shrier A. Inhibition of metabolism abolishes transient outward current in rabbit atrial myocytes. American Journal of Physiology. 1994;266:H182–190. doi: 10.1152/ajpheart.1994.266.1.H182. [DOI] [PubMed] [Google Scholar]
- Petersen S, Russ M, Reinauer H, Eckel J. Inverse regulation of glucose transporter Glut-4 and G-protein Gs mRNA expression in cardiac myocytes from insulin resistant rats. FEBS Letters. 1991;286:1–5. doi: 10.1016/0014-5793(91)80927-u. 10.1016/0014-5793(91)80927-U. [DOI] [PubMed] [Google Scholar]
- Pike GK, Bretag AH, Roberts ML. Modification of the transient outward current of rat atrial myocytes by metabolic inhibition and oxidant stress. The Journal of Physiology. 1993;470:365–382. doi: 10.1113/jphysiol.1993.sp019863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven GM, Ho H. Sugar-induced hypertension in Sprague-Dawley rats. American Journal of Hypertension. 1991;4:610–614. doi: 10.1093/ajh/4.7.610. [DOI] [PubMed] [Google Scholar]
- Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. American Journal of Physiology. 1997;272:H148–158. doi: 10.1152/ajpheart.1997.272.1.H148. [DOI] [PubMed] [Google Scholar]
- Robillon JF, Sadoul JL, Jullien D, Morand P, Freychet P. Abnormalities suggestive of cardiomyopathy in patients with type 2 diabetes of relatively short duration. Diabete et Metabolisme. 1994;20:473–480. [PubMed] [Google Scholar]
- Rodrigues B, McNeill JH. The diabetic heart: metabolic causes for the development of a cardiomyopathy. Cardiovascular Research. 1992;26:913–922. doi: 10.1093/cvr/26.10.913. [DOI] [PubMed] [Google Scholar]
- Saltiel AR. Diverse signalling pathways in the cellular actions of insulin. American Journal of Physiology. 1996;270:E375–385. doi: 10.1152/ajpendo.1996.270.3.E375. [DOI] [PubMed] [Google Scholar]
- Seger R, Krebs EG. The MAPK signalling cascade. FASEB Journal. 1995;9:726–735. [PubMed] [Google Scholar]
- Shehadeh A, Regan TJ. Cardiac consequences of diabetes mellitus. Clinical Cardiology. 1995;18:301–305. doi: 10.1002/clc.4960180604. [DOI] [PubMed] [Google Scholar]
- Shepherd PR, Nave BT, O'Rahilly S. The role of phosphoinositide 3-kinase in insulin signalling. Journal of Endocrinology. 1996;17:175–184. doi: 10.1677/jme.0.0170175. [DOI] [PubMed] [Google Scholar]
- Shimoni Y, Firek L, Severson D, Giles W. Short-term diabetes alters K+ currents in rat ventricular myocytes. Circulation Research. 1994;74:620–628. doi: 10.1161/01.res.74.4.620. [DOI] [PubMed] [Google Scholar]
- Shimoni Y, Severson DL. Thyroid status and potassium currents in rat ventricular myocytes. American Journal of Physiology. 1995;268:H576–583. doi: 10.1152/ajpheart.1995.268.2.H576. [DOI] [PubMed] [Google Scholar]
- Shimoni Y, Severson D, Giles W. Thyroid status and diabetes modulate regional differences in potassium currents in rat ventricle. The Journal of Physiology. 1995;488:673–688. doi: 10.1113/jphysiol.1995.sp020999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ui M, Okada T, Hazeki K, Hazeki O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends in Biochemical Sciences. 1995;20:303–306. doi: 10.1016/s0968-0004(00)89056-8. 10.1016/S0968-0004(00)89056-8. [DOI] [PubMed] [Google Scholar]
- Xu Z, Patel KP, Rozanski GJ. Metabolic basis of decreased transient outward K+ current in ventricular myocytes from diabetic rats. American Journal of Physiology. 1996;271:H2190–2196. doi: 10.1152/ajpheart.1996.271.5.H2190. [DOI] [PubMed] [Google Scholar]