

Abstract
The permeability response of slightly leaky pial venular capillaries to histamine was investigated using the single microvessel occlusion technique.
Histamine dose-response curves showed that concentrations between 5 nm and 5 μm increased permeability, while concentrations from 50 μm to 5 mm reduced it.
The H2 receptor antagonist cimetidine (2 μm) blocked the effects of lower concentrations of histamine, while the H1 receptor antagonist mepyramine (3 nm) blocked those of higher concentrations of histamine.
The effects of lower doses of histamine were mimicked by the H2 receptor agonist dimaprit, and the effects of higher doses of histamine were mimicked by the H1 receptor agonist α-2-(2-aminoethyl)pyridine (AEP).
Low concentrations of histamine, which normally increase the permeability of Lucifer Yellow (PLY), reduced it when co-applied with the phosphodiesterase 4 (PDE4) inhibitor rolipram. Rolipram also potentiated the response to AEP, but had no effect on that to dimaprit.
The effects of dimaprit were blocked by reducing extracellular Ca2+ from 2.5 mm to nominally Ca2+ free, or by applying the calcium entry blocker SKF 96365.
The blood-brain barrier (BBB) is formed by the extremely tight junctions between cerebral endothelial cells which prevent the passage of water and polar solutes between the blood plasma and the cerebral interstitium. There are a number of pathological episodes, such as traumatic brain injury, stroke, epilepsy and the active phase of multiple sclerosis, during which the permeability to these solutes increases, observed as BBB disruption. A number of inflammatory mediators including histamine are released during these conditions (Greenwood, 1992) and have been suggested to contribute to the disruption of the barrier. The principal source of histamine to interact with cerebral endothelium is probably found abluminally in histaminergic neurons. Wada, Inagaki, Yamatodani & Watanabe (1991) showed that there are histaminergic neurons in the brain which have cell bodies in the tuberomamillary nucleus of the posterior hypothalamus. These neurons send axons to a variety of different sites and make contact with other neurons, astrocytes (Panula, Pirvola, Auvinen & Airaksinen, 1989) and most small blood vessels of the brain (Takagi, Morishima, Matsuyama, Hayashi, Watanabe & Wada, 1986). Hence it is also possible that locally released histamine (from the neurons) stimulates cerebral microvessels and so affects the integrity of BBB in the normal course of events.
The effects of histamine on cerebral microvascular permeability have been examined previously both in whole-animal preparations and in single microvessels in situ, but there are some apparent discrepancies in the results. Some authors have reported that intravascular infusions of histamine failed to increase the rat cerebral permeability to macromolecules such as albumin (Martins, Doyle, Wright & Bass, 1980; Saria, Lundberg, Skofitsch & Lembeck, 1983; Kilzer et al. 1985) and tracer ink (Gabbiani, Badonnel & Majno, 1970), while Dux & Joó (1982) showed that such infusion opened tight junctions of brain capillaries and caused extravasation of serum albumin around blood vessels. Similarly, histamine superfusion of the cat cerebral cortex induced the extravasation of a wide range of different sized molecules from sodium fluorescein to fluorescein isothiocyanate-dextrans up to 150 kDa (Schilling & Wahl, 1994). Gross, Teasdale, Graham, Angerson & Harper (1982), however, found that histamine caused only 2- to 3-fold increases in the blood-brain transfer of sucrose and α-aminoisobutyric acid. These conflicting findings may reflect the limitations in the use of whole-animal preparations, such as the possible degradation of histamine in the blood (Plaut & Lichtenstein, 1978; Warren, Wilson, Loi & Coughlan, 1993), and that microvascular hydrostatic pressure may increase and disrupt the blood-brain barrier on the venous side. We have attempted to avoid some of these variables by examining the effects of histamine on small portions of single pial venular capillaries in situ. This preparation allows both luminal and abluminal application of substances, enables the direct effects of histamine to be isolated from those brought about by changes in microvascular hydrostatic pressure, and allows diffusive permeability to be assessed without interference from convection (Easton & Fraser, 1994). Some of the findings reported here have been presented previously in a preliminary form (Easton & Fraser, 1993; Sarker & Fraser, 1994).
METHODS
The method used in this study, and its theoretical basis, have been described fully (Fraser & Dallas, 1993; Easton & Fraser, 1994). Briefly, the microcirculation of the surface of the brain was exposed in young rats (age, 20–30 days) and the low molecular mass fluorescent dye, Lucifer Yellow (457 Da), was introduced into single venular capillaries via a bolus injection into the carotid artery. The fluorescence signal, which has been shown to be linear with the dye concentration, was captured through a microscope, an image-intensifier camera, and analysed through a video-densitometer. Permeability was measured from the rate of loss of dye trapped in a single pial venular capillary by a glass occluding probe. The fluorescence measurements were made from a small segment 200–300 μm away from the open end of the occluded vessel (but not so close to the occluding probe that the vessel diameter was distorted). If the vessel were leaky, a transmural hydrostatic pressure gradient would drive fluid across the wall, which would be replaced by fresh dye-free fluid entering the open end of the occluded portion. Dye concentration in the measured portion will depend on axial flow and diffusion as well as transmural convection and diffusion. We have shown that where the axial concentration gradient is flat, axial diffusion and convection have no effect on dye concentration in the lumen (Fraser & Dallas, 1993). Since in this preparation there is no continuous barrier to free diffusion of dye from the surface of the vessel (Butt, Jones & Abbott, 1990) the abluminal dye concentration can be minimized by maintaining a flow of superfusate at about 1–2 ml min−1. Permeability was calculated from the rate of decrease in fluorescence under the measured portion of the occluded segment. The rate of fall of intravascular concentration of a small polar molecule, such as Lucifer Yellow, is independent of the hydrostatic pressure (Fraser & Dallas, 1993). Thus the concentration of dye early in the occlusion, before axial volume flux distorts the uniform axial concentration of dye in the region of measurement, will be:
| (1) |
where Ct and C0 are the concentrations at times t and 0, and k is 4P/d, with P representing the permeability and d the diameter of the vessel. In the rest of this paper PLY will refer to the permeability of Lucifer Yellow.
Animal preparation
Previously, we have shown that when a relatively large section of skull and overlying meninges were removed, the permeability of the microvessels to Lucifer Yellow (PLY) increased spontaneously in two distinct phases. The first phase was small (from < 0.2 × 10−6 to 2–5 × 10−6 cm s−1), and relatively stable, but the second phase was characterized by relatively large permeability fluctuations, often reaching levels of 50 × 10−6 cm s−1 and more, which is equivalent to that of mesenteric capillaries. In the present experiments, the preparation was modified as detailed below to postpone the onset of the second phase. The experiments were performed on Wistar rats of both sexes aged between 20 and 30 days, and were within guidelines directed by The Animals (Scientific Procedures) Act 1986. The animals were anaesthetized by an intraperitoneal injection of 60 mg (kg body weight)−1 sodium pentobarbitone diluted in 0.9 % NaCl solution (25 % w/v), supplemented (10 % of the original dose) as necessary (assessed by a foot-pinch reflex), and at the end of the experiment were killed by anaesthetic overdose followed by exsanguination. Once anaesthetized, a thermal probe was inserted rectally, and the animal was kept on a heating blanket connected via the probe to a feedback circuit (CFP 8105, Harvard Instruments) to maintain the animal body temperature at 37 ± 1 °C. The trachea and the right common carotid artery were cannulated routinely, the arterial cannula being placed orthograde and filled with heparinized saline (100 u ml−1) to prevent clot formation. A section of the frontoparietal bones on the left side, between coronal and lambdoid sutures, was thinned with a dental drill. In contrast to the methods described previously (Easton & Fraser, 1994; Easton, Sarker & Fraser, 1997), a metal ring (internal diameter, 7 mm and outside diameter, 13 mm) was glued onto the cranium surrounding the thinned bone with cyanoacrylate adhesive. The thinned cranial surface within the ring was constantly superfused by a pump at the rate of 1–2 ml min−1 with artificial cerebrospinal fluid (ACSF) warmed to 37 ± 1 °C and delivered through a fine plastic tube to ensure that a layer of fluid was present at all times. The thinned bone within the ring was then removed, carefully avoiding damage to the meninges and cerebral surface. Pial microvessels were exposed by cutting away the overlying meninges, which in these young rats leaves the microvessels free from a continuous layer of overlying tissue (Butt et al. 1990). The rat was then placed on the modified stage of a microscope (ACM, Zeiss Oberkocken) and the exposed cerebral surface illuminated with a 50 W mercury vapour lamp through a × 20 objective lens (NA, 0.5; Cooke) fitted with a water-immersion cap to eliminate the air-water meniscus.
Artificial cerebrospinal fluid and drug application
The cerebral surface was superfused with an artificial cerebrospinal fluid (ACSF) at the rate of 0.5-1 ml min−1, warmed to 37 ± 1 °C and buffered to pH 7.40. The ACSF contained (mm): NaCl, 110.5; KCl, 4.7; CaCl2, 2.5; KH2PO4, 1.1; MgSO4.7H2O, 1.25; NaHCO3, 25; and Hepes, 15, giving a total ionic concentration of 307.6 mm. All chemicals were obtained from BDH, except MgSO4.7H2O (Fisons, Loughborough) and Hepes (Sigma). Histamine (free base) and the H1-selective antagonist mepyramine (maleate salt) were obtained from Sigma. The H1-specific agonist α-2-(2-aminoethyl)pyridine (AEP; Durant, Ganellin & Parsons, 1975) was from Aldrich (UK). The H2-specific agonist dimaprit (see Hill, 1990) was a kind gift from SmithKline Beecham Pharmaceuticals (Welwyn Garden City, UK), as was the calcium channel blocker SKF 96365. The H3 agonist R-(−)-α-methylhistamine-dihydrochloride, the H3 antagonist thioperamide, and rolipram were bought from RBI, and the H2 antagonist cimetidine was bought as 200 mg ml−1 ampoules (Tagamet injection; SmithKline Beecham).
Substances were applied abluminally into the pool of ACSF over the exposed cerebral surface, or by focal application through a micropipette. The permeability usually reversed rapidly, and in all cases within 5 min following the removal of the substance. Sometimes substances were also applied focally through a micropipette with a 3–5 μm tip diameter to show that they were effective local to the vessel wall. The micropipette was positioned within 2 μm of the measured portion of the microvessel and substances were ejected by applying pressure. A trace of the fluorescent dye sulphur-rhodamine was included in the pipette so that the distribution and relative concentration of the drug could be seen easily by changing to the appropriate filter set. Substances were applied luminally either by microcannulation and perfusion of a side branch of the vessel, or by including them with carotid bolus injection of dye. In both cases control measurements were made by separate bolus injections of Lucifer Yellow into the carotid artery.
RESULTS
Venular capillaries, with a straight unbranching section of at least 200 μm, were selected for investigation. The modification to the preparation resulted in more stable vessel permeabilities than previously reported (Easton & Fraser, 1994; Easton et al. 1997), so that although vessel permeability spontaneously increased from that of the intact barrier (PLY < 0.2 × 10−6 cm s−1) after about 30 min exposure of the cerebral surface, to about 2 × 10−6 cm s−1 as before, in these experiments it remained at this low level for at least 2–3 h. Preliminary observations indicated that application of histamine (0.5 μm) to the abluminal surface of tight and slightly leaky venular capillaries resulted in similar permeability-increasing effects, but high histamine concentrations (≥ 50 μm) reduced permeability of the leaky vessels (see Fig. 1). All the following experiments were carried out to investigate this phenomenon, and since permeability reductions as well as increases were evident, vessels in the first phase of spontaneous disruption (Easton et al. 1997) were used throughout.
Figure 1. Application of low and high histamine concentrations to single venular capillaries.

Tight venular capillary (diameter, 15 μm) to which 0.5 μm (A, □) and 500 μm (B, ▪) histamine were applied during an occlusion experiment. The higher concentration failed to elicit the expected permeability increase. A slightly leaky capillary (diameter also 15 μm) treated in a similar way showed a similar permeability increase with 0.5 μm (C, □) histamine, but a permeability decrease with 500 μm (D, ▪) histamine. Fluorescence is expressed in arbitrary units (a.u.). The permeability values calculated from the rate constant of mono-exponentials fitted to the fluorescence signals from the occluded segments of the capillaries are shown above the relevant portions of the traces (expressed as 10−6 cm s−1).
Dose-response relationship to histamine
A range of different concentrations of histamine from 5 nm to 5 mm was applied in random order to microvessels (see Fig. 2). Lower concentrations of histamine (from 5 nm up to 5 μm) produced a significant increase in PLY, but between 50 μm and 5 mm, PLY decreased significantly below control values. There was no discernible difference in the responses between those vessels with lower and higher initial control permeabilities. On four out of twenty-four occasions, the intact barrier was completely restored, with PLY falling to < 0.2 × 10−6 cm s−1.
Figure 2. Dose-response curves for histamine on the permeability to Lucifer Yellow across pial venular capillaries.

Histamine (at concentrations from 0.5 nm to 5 mm in pseudo-random order) was applied to the abluminal surface of 4–8 microvessels (diameter, 10–16 μm) from 12 rats, each vessel receiving any particular concentration of histamine only once, and not all vessels receiving all concentrations. The control values (^) were obtained from the first 40 s of the occlusion, after which time histamine was applied, and the new permeability (•) was measured over the following minute. Vessels with an initial PLY < 2 × 10−6 cm s−1 (A) and those with an initial PLY > 2 × 10−6 cm s−1 (B) were plotted separately to indicate that the baseline permeability had little effect on the changes induced by histamine. The vertical bars represent s.e.m., and the level of statistical significance (*P < 0.05; **P < 0.01; ***P < 0.001) was assessed from Student's paired t tests on the individual concentrations. Analysis of variance showed that the different permeability changes were dependent on the concentration of histamine for both sets of vessels: lower control values (A) P ≪ 0.001, F = 25.75, for d.f. = 7, 39, and the higher control values (B) P ≪ 0.001, F = 18.32, for d.f. = 6, 27.
The effect of histamine antagonists was examined on both types of permeability response to histamine. Figure 3 shows that the permeability-increasing effect of 5 μm histamine was unaffected by the H1 receptor antagonist mepyramine (3 nm) and the H3 receptor antagonist thioperamide (1 nm), but was completely prevented by the H2 receptor blocker cimetidine (2 μm). On the other hand, the permeability-decreasing effects of 5 mm histamine were antagonized by mepyramine, but not by cimetidine or thioperamide. These results suggest that higher doses of histamine reduce PLY via H1 receptor activation and that lower doses increase PLY by H2 receptor activation, and that the H3 receptor has little role in the regulation of permeability.
Figure 3. The effects of histamine receptor antagonists on the dose-dependent permeability-increasing and -decreasing effects of histamine.

 , H1 receptor antagonist mepyramine (3nm). ▪, H2 receptor antagonist cimetidine (2μm).
, H1 receptor antagonist mepyramine (3nm). ▪, H2 receptor antagonist cimetidine (2μm).  , H3 receptor antagonist thioperamide (1 nm). The bar chart shows the change in PLY brought about by the antagonists alone and when co-applied with 5 μm or 5 mm histamine. The effects of histamine alone are also shown (□). Of the 3 antagonists, only cimetidine inhibited the PLY increase (**P < 0.01; analysis of variance) found with 5 μm histamine, and only mepyramine prevented its fall (*P < 0.05) with 5 mm histamine. All these experiments were repeated on 4 venular capillaries (diameter range, 12–16 μm) from 4 rats.
, H3 receptor antagonist thioperamide (1 nm). The bar chart shows the change in PLY brought about by the antagonists alone and when co-applied with 5 μm or 5 mm histamine. The effects of histamine alone are also shown (□). Of the 3 antagonists, only cimetidine inhibited the PLY increase (**P < 0.01; analysis of variance) found with 5 μm histamine, and only mepyramine prevented its fall (*P < 0.05) with 5 mm histamine. All these experiments were repeated on 4 venular capillaries (diameter range, 12–16 μm) from 4 rats.
Selective histamine H1 and H2 agonists
The H1 receptor agonist α-2-(2-aminoethyl)pyridine (AEP; Durant et al. 1975) was applied in concentrations from 5 pm to 50 μm to the brain side of venular capillaries in the first phase of permeability increase (Fig. 4A). All concentrations of AEP at or above 50 pm produced significant reductions in PLY across the microvessel wall. Similarly, abluminal application of the histamine H2 agonist dimaprit, between 5 nm and 5 mm, increased permeability monotonically in a dose-dependent manner (Fig. 4B).
Figure 4. Dose-response curves for histamine H1 (AEP) and H2 (dimaprit) agonists.

A wide range of concentrations of selective histamine H1 (AEP; A) and H2 (dimaprit; B) agonists was applied to the abluminal surface of pial venular capillaries in pseudo-random order. Each agonist was applied to 4 vessels from 4 rats. The vessel diameters ranged from 8 to 14 μm. The control values (^) were obtained from the first 40 s of the occlusion, after which time the agonist was applied, and the new permeability (•) was measured within the following minute. The vertical bars represent s.e.m., and the level of statistical significance (*P < 0.05; **P < 0.01; ***P < 0.001) was assessed from Student's paired t test. The overall significance of the permeability changes was assessed by analysis of variance (P < 0.005, in both cases).
Luminal application of histamine receptor agonists
Figure 5 shows the effect of histamine receptor agonists (H1 and H2) on PLY across the cerebral microvascular wall in individual microvessels when the agonists were applied luminally by microperfusion. The H2 receptor agonist dimaprit (100 μm) increased PLY, whereas the H1 receptor agonist AEP (100 μm) had no effect.
Figure 5. The effects of luminal application of AEP and dimaprit on the permeability of pial venular capillaries.

Experiments were performed on 4 pial venular capillaries of diameters between 8 and 14 μm. Control values (□) were obtained by injecting a bolus of dye into the carotid artery, and immediately following that measurement the drug-dye mixture was infused into the capillary through a micropipette, the tip of which was no more than 20 μm from the entrance. The measurement was made as soon as the dye filled the vessel uniformly. Luminal AEP had no effect on permeability (control, (2.29 ± 0.484) × 10−6 cm s−1; AEP, (2.26 ± 0.342) × 10−6 cm s−1), while dimaprit had similar permeability-increasing effects as when it was applied abluminally (control, (2.19 ± 0.328) × 10−6 cm s−1; dimaprit, (4.52 ± 0.326) × 10−6 cm s−1; ***P < 0.001).
H1 receptor effects are mediated through cyclic AMP
When intraendothelial cyclic AMP (cAMPi) is elevated, the permeability of human umbilical vein endothelium and of bovine brain endothelium cultured monolayers is decreased (Rubin et al. 1991; Takeda, Yamashita, Shimazaki & Mitsui, 1992). We tested the idea that the effect of H1 receptor activation is mediated at some critical point by cAMPi, by applying the phosphodiesterase 4 (PDE4) inhibitor rolipram, to potentiate cAMPi levels (Beavo, 1988). The permeability effects of rolipram co-applied with histamine, AEP and dimaprit are illustrated in Fig. 6. Histamine at a concentration of 5 μm increased permeability, but when applied in combination with 50 μm rolipram, the permeability fell to below control levels. Furthermore, a low concentration of the H1 agonist AEP (5 pm) that alone had no effect on PLY, produced a significant reduction when combined with rolipram. On the other hand, the effects of the H2 agonist dimaprit when co-applied with rolipram were unchanged.
Figure 6. The effects of the PDE4 blocker rolipram (50 μm) on the permeability responses of histamine receptor agonists.
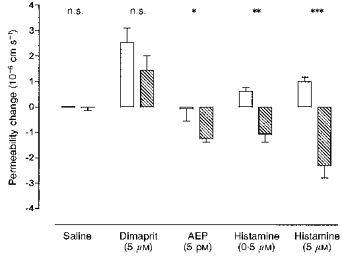
Each agonist was applied alone (□) and in combination with rolipram ( ) during an occlusion experiment. Rolipram alone had no significant effect, nor did it significantly alter the response to the H2 agonist dimaprit. When applied with a subthreshold concentration of the H1 agonist AEP, a significant fall in PLY was observed. Concentrations of histamine that alone resulted in increased PLY produced reductions in PLY when combined with rolipram. The permeability changes were measured on 4–6 venular capillaries (diameter, 8–16 μm) for each agonist, and the columns represent the mean change in PLY with the bars giving the s.e.m. The statistical significance of the differences in the PLY changes was obtained from Student's unpaired t tests with *P < 0.05, **P < 0.001 and ***P < 0.0001.
) during an occlusion experiment. Rolipram alone had no significant effect, nor did it significantly alter the response to the H2 agonist dimaprit. When applied with a subthreshold concentration of the H1 agonist AEP, a significant fall in PLY was observed. Concentrations of histamine that alone resulted in increased PLY produced reductions in PLY when combined with rolipram. The permeability changes were measured on 4–6 venular capillaries (diameter, 8–16 μm) for each agonist, and the columns represent the mean change in PLY with the bars giving the s.e.m. The statistical significance of the differences in the PLY changes was obtained from Student's unpaired t tests with *P < 0.05, **P < 0.001 and ***P < 0.0001.
H2 receptor effects are mediated through [Ca2+]i
Cerebral microvascular permeability has been shown to be increased when [Ca2+]i is raised by the divalent ionophores A23187 and ETH 1001 (Olesen, 1987), and histamine application results in raised [Ca2+]i in primary cultures of rat brain microvascular endothelial cells (Revest, Abbott & Gillespie, 1991). Removal of calcium from the superfusing solution reduced the response to dimaprit to a small transient increase in permeability (Fig. 7A and B), followed by a steady state in which the PLY was little different to the control value. A series of paired experiments (Fig. 7C) confirmed this finding, which suggests that the action of dimaprit depends on Ca2+ entry. This idea was tested by applying the blocker of the non-specific cation channel, SKF 96365, which prevents the capacitive Ca2+ entry that occurs following intracellular store depletion (Dolor, Hurwitz, Mirza, Strauss & Whorton, 1992). The rate of decline in fluorescence induced by dimaprit (5 μm) was arrested upon SKF 96365 application (Fig. 8A). When the calcium entry channels were blocked by SKF 96365 beforehand, the application of dimaprit resulted in a transient fluorescence decline which lasted for about 20 s, and PLY thereafter fell to close to the control level (Fig. 8B). This suggests that stimulation of the H2 receptor induces Ca2+ release from intracellular stores and that their depletion normally recruits Ca2+ from outside the cell via cation channels, as has been shown on human umbilical vein endothelial cells with bradykinin (Schilling, Cabello & Rajan, 1992).
Figure 7. The effects of reducing external calcium on the permeability response to histamine H2 receptor activation.

A, dimaprit (5 μm) was applied to the abluminal surface of a single occluded microvessel (diameter, 16 μm) that was superfused with a solution containing the usual 2.5 mm Ca2+, and resulted in a dye loss that increased PLY from 0.13 × 10−6 to 1.53 × 10−6 cm s−1. When the dimaprit application was repeated about 90 s after the first application, there was a similar full response. B, the same microvessel with repeated dimaprit applications but with a nominally calcium-free superfusing solution. The permeability increases were only transient, and lasted for 48 s with the first dimaprit application and 18 s with the second. The PLY values are given below the relevant parts of the curves (expressed as 10−6 cm s−1). C, summary of PLY changes measured 60 s after dimaprit application in 4 vessels from 4 rats (diameter, 12–16 μm); similar experiments to those shown above. The mean PLY increased from (0.86 ± 0.187) × 10−6 cm s−1 before, to (2.84 ± 0.453) × 10−6 cm s−1 60 s after the beginning of the application of 5 μm dimaprit with the normal calcium-containing superfusing solution (□, control; ▪, dimaprit). When a nominally zero calcium superfusate was used, PLY hardly changed at all (from (1.07 ± 0.491) × 10−6 to (1.39 ± 0.797) × 10−6 cm s−1; ^, control; •, dimaprit). The difference in the responses was statistically significant (P < 0.005; Student's paired t test).
Figure 8. Effect of the calcium entry blocker SKF 96365 on the permeability response to dimaprit of a single vessel.

When SKF 96365 (10 μm) was applied shortly after dimaprit (5 μm) to an occluded microvessel (diameter, 14 μm), the rate of dye loss was promptly reduced (A). However, when dimaprit was applied to a vessel in the presence of SKF 96365, there was a transient increase in rate of dye loss which lasted for about 20 s (B). The PLY values are given below the relevant parts of the curves (expressed as 10−6 cm s−1).
DISCUSSION
This study reports for the first time a dose-response relationship between histamine and permeability changes of individual cerebral microvessels in the living animal. We have confirmed previous observations (Gross et al. 1982; Butt & Jones, 1992; Schilling & Wahl, 1994) that histamine normally increases the permeability of pial microvessels, but we also found, unexpectedly, that high concentrations of histamine reduce their permeability.
The permeability-increasing effects of histamine were blocked by cimetidine at a concentration which is consistent with its specific action on the H2 receptor, and less, by an order of magnitude, than the concentration required for its free radical-scavenging effects (Lapenna et al. 1994). Furthermore, the specific H2 agonist dimaprit produced permeability increases over a wide range of concentrations, with no indication of the diphasic dose-response curve seen with histamine.
The experiments in which there was H2 receptor activation by dimaprit and extracellular Ca2+ was reduced, or the Ca2+ entry blocker SKF 96365 was applied, provide strong evidence that the H2 receptor-mediated increase in PLY is connected to elevated [Ca2+]i. The normal sustained increase in permeability became an initial transient of no more than 20 s, after which permeability was related to the availability of extracellular calcium. There is a large body of evidence to support a link between rises in intracellular Ca2+ and increases in microvascular permeability, from cell cultures (e.g. Yamada et al. 1990; Hill, 1990) and animal preparations (e.g. Mayhan & Joyner, 1984; He & Curry, 1993). Olesen & Crone (1986) showed that a rise in cerebral endothelial Ca2+ levels led to a reduction in transcapillary electrical resistance to about one-third of the control value in frog brain pial microvessels. There is also considerable evidence that H2 receptors on brain endothelium mediate increases in permeability (Gross et al. 1982; Butt & Jones, 1992). The signal transduction mechanisms, however, have not been explored previously, and our results are surprising: we believe that there is only one other reported case of H2 receptor activation being linked to increased intracellular Ca2+ (Seifert, Hoer, Schwaner & Buschauer, 1992). It is likely that the H2 receptors were on the endothelial cells rather than some neighbouring tissue, since both luminally and abluminally applied dimaprit had similar effects.
In contrast to the effects of lower doses, histamine at higher concentrations (≥ 50 μm) reduced permeability. These effects were blocked by mepyramine, not cimetidine, which suggests that the H1 receptor was involved. This was substantiated by carrying out further studies with the H1 agonist α-2-(2-aminoethyl)pyridine which always reduced permeability or had no effect at the very lowest concentrations (i.e. < 0.1 nm). The evidence that cAMP acts as a key second messenger for the response was given by the PDE4 antagonist rolipram. When low concentrations of AEP that had no effect were co-applied with rolipram, supramaximal permeability falls were observed.
It is not certain where the H1 receptor is located: it appears not to be present on the luminal surface of the microvessel, since direct luminal application of AEP had no effect, possibly reflecting a polarization of this receptor, or that the receptor is not even on the endothelium. There are a few reports that link H1 receptors to cAMP accumulation, mainly in guinea-pig cerebral cortex (see Donaldson, Brown & Hill, 1989), but also in adrenal medullary cells (Marley, Thomson, Jachno & Johnston, 1991) and in human brain endothelium (Stanimirovic, Bertrand, Merkel, Bembry & Spatz, 1994). In all these cases it appears that the cAMP rise depends on the availability of extracellular calcium, and both the studies in brain show that H1 activation augments the cAMP rise simulated by adenosine A2 receptor agonists.
It is not possible from the present experiments to be sure whether H1 receptors are present on the abluminal endothelial surface, or whether the response is mediated via some other cell in the vicinity, which could secrete some substance (such as calcitonin gene-related peptide, present in perivascular nerve fibres; Hanko, Hardebo, Kåhrström, Owman & Sundler, 1985) which would raise intra-endothelial cAMP. There is an indication from primary cultures of human cerebral endothelium that the H2 receptor is responsible for increased surface expression of P-selectin (Easton, Prameya & Dorovini-Zis, 1995). This histamine-stimulated effect was blocked by cimetidine. Interestingly, co-application of histamine with the PDE4 inhibitor rolipram also prevented surface expression of P-selectin. This indicates that histamine application resulted in raising cAMP, and it is possible that this was via H1 receptors.
To our knowledge this is the first report of histamine H1 receptor activation resulting in a decrease of vascular permeability, including other studies in the brain. There are a few studies in which permeability was examined at similarly high histamine concentrations, but even these did not report permeability decreases. Thus, Butt & Jones (1992), measuring permeability via estimates of microvascular length constants, found only increases in permeability, even with 100 μm histamine. There are large differences in sampling of suitable vessels between these experiments (discussed in Easton et al. 1997), and these are possibly responsible for such permeability falls not being reported. Schilling & Wahl (1994) also applied high concentrations of histamine to the brain side of pial microvessels, and reported permeability increases only. These applications were, however, across the intact arachnoid membrane and there would necessarily be a lower histamine concentration on the abluminal endothelial surface than the 1 mm applied.
In conclusion, we have found that histamine modulates cerebro-microvascular function by producing permeability increases via the H2 receptor and decreases via the H1 receptor. We also showed that these receptors have unusual signal transduction mechanisms, with the H2 receptor being linked to raised [Ca2+]i and the H1 receptor having its effects via raised intracellular cAMP.
Acknowledgments
During this project A. S. E. was supported by an MRC-Pfizer Collaborative Studentship, and M. H. S. by a Commonwealth Staff Scholarship.
References
- Beavo JA. Multiple isozymes of cyclic nucleotide phosphodiesterase. In: Greengard P, Robinson GA, editors. Advances in Second Messenger and Phosphoprotein Research. New York: Raven Press; 1988. pp. 1–38. [PubMed] [Google Scholar]
- Butt AM, Jones HC. Effect of histamine and antagonists on electrical resistance across the blood-brain barrier in rat brain-surface microvessels. Brain Research. 1992;569:100–105. doi: 10.1016/0006-8993(92)90374-i. [DOI] [PubMed] [Google Scholar]
- Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetised rats: a developmental study. Journal of Physiology. 1990;429:47–62. doi: 10.1113/jphysiol.1990.sp018243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolor RJ, Hurwitz LM, Mirza Z, Strauss HC, Whorton AR. Regulation of extracellular calcium entry in endothelial cells: role of intracellular calcium pool. American Journal of Physiology. 1992;262:C171–181. doi: 10.1152/ajpcell.1992.262.1.C171. [DOI] [PubMed] [Google Scholar]
- Donaldson J, Brown AM, Hill SJ. Temporal changes in the calcium-dependence of the histamine H1-receptor-stimulation of cyclic AMP accumulation in guinea-pig cerebral cortex. British Journal of Pharmacology. 1989;98:1365–1375. doi: 10.1111/j.1476-5381.1989.tb12686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant GJ, Ganellin CR, Parsons ME. Chemical differentiation of histamine H1- and H2-receptor agonists. Journal of Medical Chemistry. 1975;18:905–909. doi: 10.1021/jm00243a009. [DOI] [PubMed] [Google Scholar]
- Dux E, Joó F. Effects of histamine on brain capillaries. Experimental Brain Research. 1982;47:252–258. doi: 10.1007/BF00239384. [DOI] [PubMed] [Google Scholar]
- Easton AS, Fraser PA. Histamine both increases and decreases the permeability of cerebral venules of the anaesthetized rat. Journal of Physiology. 1993;467:40P. [Google Scholar]
- Easton AS, Fraser PA. Variable restriction of albumin diffusion across inflamed cerebral microvessels. Journal of Physiology. 1994;475:147–157. doi: 10.1113/jphysiol.1994.sp020056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton AS, Prameya R, Dorovini-Zis K. Expression of P selectin in cultured human brain endothelial cells: opposite effects of Ca2+ and cAMP. Journal of Neuropathology and Experimental Neurology. 1995;54:440. [Google Scholar]
- Easton AS, Sarker MH, Fraser PA. Two components of blood-brain barrier disruption in the rat. Journal of Physiology. 1997;503:613–623. doi: 10.1111/j.1469-7793.1997.613bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser PA, Dallas AD. Permeability of disrupted cerebral microvessels in the frog. Journal of Physiology. 1993;461:619–632. doi: 10.1113/jphysiol.1993.sp019532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G, Badonnel MC, Majno G. Intra-arterial injections of histamine, serotonin, or bradykinin: a topographic study of vascular leakage. Proceedings of the Society for Experimental Biology and Medicine. 1970;135:447–452. doi: 10.3181/00379727-135-35072. [DOI] [PubMed] [Google Scholar]
- Greenwood J. Experimental manipulation of the blood-brain barrier and blood-retinal barriers. In: Bradbury MWB, editor. Handbook of Experimental Pharmacology, chap. 19 Physiology and Pharmacology of the Blood-Brain Barrier. Vol. 103. Berlin: Springer-Verlag; 1992. pp. 459–479. [Google Scholar]
- Gross PM, Teasdale GM, Graham DI, Angerson WJ, Harper AM. Intra-arterial histamine increases blood-brain transport in rats. American Journal of Physiology. 1982;243:H307–317. doi: 10.1152/ajpheart.1982.243.2.H307. [DOI] [PubMed] [Google Scholar]
- Hanko J, Hardebo EJ, Kåhrström J, Owman C, Sundler F. Calciotonin gene-related peptide is present in mammalian cerebrovascular nerve fibres and dilates pial and peripheral arteries. Neuroscience Letters. 1985;57:91–95. doi: 10.1016/0304-3940(85)90045-x. 10.1016/0304-3940(85)90045-X. [DOI] [PubMed] [Google Scholar]
- He P, Curry FE. Differential actions of cAMP on endothelial [Ca2+]i and permeability in microvessels exposed to ATP. American Journal of Physiology. 1993;265:H1019–1023. doi: 10.1152/ajpheart.1993.265.3.H1019. [DOI] [PubMed] [Google Scholar]
- Hill SJ. Distribution, properties, and functional characteristics of three classes of histamine receptor. Pharmacology Reviews. 1990;42:45–82. [PubMed] [Google Scholar]
- Kilzer P, Chang K, Marvel J, Rowold E, Jaudes P, Ullensvang S, Kilo C, Williamson J., Jr Albumin permeation of new vessels is increased in diabetic rats. Diabetes. 1985;34:333–336. doi: 10.2337/diab.34.4.333. [DOI] [PubMed] [Google Scholar]
- Lapenna D, De Gioia S, Mezzetti A, Grossi L, Festi D, Marzio L, Cuccurullo F. H2-receptor antagonists are scavengers of oxygen radicals. European Journal of Clinical Investigation. 1994;24:476–481. doi: 10.1111/j.1365-2362.1994.tb02378.x. [DOI] [PubMed] [Google Scholar]
- Marley PD, Thomson KA, Jachno K, Johnston MJ. Histamine-induced increases in cyclic-AMP levels in bovine adrenal-medullary cells. British Journal of Pharmacology. 1991;104:839–846. doi: 10.1111/j.1476-5381.1991.tb12515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins AN, Doyle TF, Wright SJ, Bass BG. Response of cerebral circulation to topical histamine. Stroke. 1980;11:469–476. doi: 10.1161/01.str.11.5.469. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Joyner WL. The effect of altering the external calcium concentration and a calcium channel blocker, verapamil, on microvascular leaky sites and dextran clearance in the hamster cheek pouch. Microvascular Research. 1984;28:159–179. doi: 10.1016/0026-2862(84)90015-3. 10.1016/0026-2862(84)90015-3. [DOI] [PubMed] [Google Scholar]
- Olesen S-P. Regulation of ion permeability in frog brain venules. Significance of calcium, cyclic nucleotides and protein kinase C. Journal of Physiology. 1987;387:59–68. doi: 10.1113/jphysiol.1987.sp016562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen S-P, Crone C. Substances that rapidly augment ionic conductance of endothelium in cerebral venules. Acta Physiologica Scandinavica. 1986;127:233–241. doi: 10.1111/j.1748-1716.1986.tb07898.x. [DOI] [PubMed] [Google Scholar]
- Panula P, Pirvola V, Auvinen S, Airaksinen MS. Histamine-immunoreactive nerve fibres in the rat brain. Neuroscience. 1989;28:585–610. doi: 10.1016/0306-4522(89)90007-9. 10.1016/0306-4522(89)90007-9. [DOI] [PubMed] [Google Scholar]
- Plaut M, Lichtenstein LM. Histamine, 5-hydroxytryptamine, SRA-A: Discussion of type I hypersensitivity (anaphylaxis) In: Vane JA, Ferriera SH, editors. Handbook of Experimental Pharmacology, part 1, chap. 11 Inflammation. Vol. 50. Berlin: Springer-Verlag; 1978. pp. 344–373. [Google Scholar]
- Revest PA, Abbott NJ, Gillespie JI. Receptor-mediated changes in intracellular [Ca2+] in cultured rat brain capillary endothelial cells. Brain Research. 1991;549:159–161. doi: 10.1016/0006-8993(91)90614-2. 10.1016/0006-8993(91)90614-2. [DOI] [PubMed] [Google Scholar]
- Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J, Tanner LI, Tomaselli KJ, Bard F. A cell culture model of the blood-brain barrier. Journal of Cell Biology. 1991;115:1725–1735. doi: 10.1083/jcb.115.6.1725. 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saria A, Lundberg JM, Skofitsch G, Lembeck F. Vascular protein leakage in various tissues induced by substance P, capsaicin, bradykinin, serotonin, histamine and by antigen challenge. Naunyn-Schmeideberg's Archives of Pharmacology. 1983;324:212–218. doi: 10.1007/BF00503897. [DOI] [PubMed] [Google Scholar]
- Sarker MH, Fraser PA. Histamine receptor activation and the regulation of cerebro-microvascular permeability in the anaesthetized rat. Journal of Physiology. 1994;475.P:60P. [Google Scholar]
- Schilling WP, Cabello OA, Rajan L. Depletion of the 1,4,5-triphosphate-sensitive intracellular Ca2+ store in vascular endothelial cells activates the agonist-sensitive Ca2+-influx pathway. Biochemical Journal. 1992;284:521–530. doi: 10.1042/bj2840521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling L, Wahl M. Opening of the blood-brain barrier during cortical superfusion with histamine. Brain Research. 1994;653:289–296. doi: 10.1016/0006-8993(94)90403-0. 10.1016/0006-8993(94)90403-0. [DOI] [PubMed] [Google Scholar]
- Seifert R, Hoer A, Schwaner I, Buschauer A. Histamine increases cytosolic calcium in HL-60 promyelocytes predominantly via H2 receptors with an unique agonist/antagonist profile and induces functional differentiation. Molecular Pharmacology. 1992;42:235–241. [PubMed] [Google Scholar]
- Stanimirovic DB, Bertrand N, Merkel N, Bembry J, Spatz M. Interaction between histamine and adenosine in human cerebromicrovascular endothelial cells: modulation of second messengers. Metabolic Brain Disease. 1994;9:275–289. doi: 10.1007/BF01991201. [DOI] [PubMed] [Google Scholar]
- Takagi H, Morishima Y, Matsuyama T, Hayashi H, Watanabe T, Wada H. Histaminergic axons in the neostriatum and cerebral cortex of the rat: a correlated light and electron microscopic immunocytochemical study using histidine decarboxylase as a marker. Brain Research. 1986;364:114–123. doi: 10.1016/0006-8993(86)90992-3. 10.1016/0006-8993(86)90992-3. [DOI] [PubMed] [Google Scholar]
- Takeda T, Yamashita Y, Shimazaki S, Mitsui Y. Histamine decreases the permeability of an endothelial cell monolayer by stimulating cyclic AMP production through the H2-receptor. Journal of Cell Science. 1992;101:745–750. doi: 10.1242/jcs.101.4.745. [DOI] [PubMed] [Google Scholar]
- Wada H, Inagaki N, Yamatodani A, Watanabe T. Is the histaminergic neuron system a regulatory center for whole brain activity? Trends in Neurosciences. 1991;14:415–418. doi: 10.1016/0166-2236(91)90034-r. 10.1016/0166-2236(91)90034-R. [DOI] [PubMed] [Google Scholar]
- Warren JB, Wilson AJ, Loi RK, Coughlan ML. Opposing roles of cAMP in the vascular control of edema formation. FASEB Journal. 1993;7:1394–1400. doi: 10.1096/fasebj.7.14.7693536. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Furumichi T, Furui H, Yokoi T, Ito T, Yamauchi K, Yokota M, Hayashi H, Saito H. Roles of calcium, cyclic nucleotides and protein kinase C in regulation of endothelial permeability. Arteriosclerosis. 1990;10:410–420. doi: 10.1161/01.atv.10.3.410. [DOI] [PubMed] [Google Scholar]