

Abstract
We have tested the hypothesis that morphine withdrawal excitation of oxytocin neurones that follows from administration of naloxone to morphine-dependent rats is a consequence of excitation of noradrenergic neurones.
Female rats were made morphine dependent by intracerebroventricular (i.c.v.) infusion of the opioid at increasing doses over 5 days. On the sixth day, the rats were anaesthetized with urethane or pentobarbitone and prepared for blood sampling to determine plasma oxytocin by radioimmunoassay or for in vivo extracellular recording of the firing rate of identified oxytocin neurones from the supraoptic nucleus. Morphine withdrawal was induced by intravenous (i.v.) injection of the opioid antagonist naloxone (5 mg kg−1).
In one group of rats the noradrenergic projections to the hypothalamus were lesioned by i.c.v. injection of 6-hydroxydopamine immediately prior to the induction of morphine dependence. In these rats the oxytocin secretion induced by i.v. cholecystokinin was reduced to 9% of that seen in sham-lesioned rats but in contrast, no attenuation of morphine withdrawal-induced oxytocin secretion was observed.
i.c.v. infusion of the α1-adrenoreceptor antagonist benoxathian, at up to 5.3 μg min−1, dose-dependently inhibited the withdrawal excitation of oxytocin neurones in morphine-dependent rats under urethane anaesthesia, and benoxathian reduced withdrawal-induced oxytocin secretion to 37% of that of vehicle-infused rats. i.c.v. benoxathian also inhibited the activity of oxytocin neurones in morphine-naïve rats. Similarly, microdialysis administration of 2 mm benoxathian directly onto the surface of the supraoptic nucleus reduced the activity of oxytocin neurones by 53%.
Thus noradrenergic systems are not essential for the expression of morphine withdrawal excitation, since chronic neurotoxic destruction of the noradrenergic inputs to the hypothalamus did not affect the magnitude of withdrawal-induced oxytocin secretion. However, tonically active noradrenergic inputs influence the excitability of oxytocin neurones, and acute antagonism of this noradrenergic tone can powerfully impair the ability of oxytocin neurones to exhibit morphine withdrawal excitation.
Magnocellular neurosecretory neurones of the supraoptic and paraventricular nuclei in the hypothalamus secrete oxytocin or vasopressin from their nerve endings in the posterior pituitary gland. The magnocellular oxytocin neurones are potently and selectively inhibited by direct application of μ-opioid agonists in vitro (Pittman, Hatton & Bloom, 1980; Wakerley, Noble & Clarke, 1983), and by central or systemic administration of the μ-agonist morphine in vivo (Pumford, Leng & Russell, 1991). The inhibition of oxytocin neurones following systemic administration of morphine in vivo results from an action within the nucleus itself since administration of the opioid antagonist naloxone into the nucleus by microdialysis completely reverses the inhibition of oxytocin neurones by systemically administered morphine (Ludwig, Brown, Russell & Leng, 1997). Nevertheless, as the noradrenergic projection from the A2 cell group in the nucleus tractus solitarii (NTS) to oxytocin neurones is subject to pre-synaptic inhibition by morphine (Onaka, Luckman, Guevara-Guzman, Ueta, Kendrick & Leng, 1995b), it is possible that the reduction of spontaneous activity of oxytocin neurones may also be a consequence of pre-synaptic inhibition of afferent inputs.
During chronic central morphine administration oxytocin neurones become less sensitive (tolerant) to the inhibitory actions of morphine (Pumford et al. 1991). Concurrently with the development of morphine tolerance, oxytocin neurones also become ‘dependent’ upon morphine; dependence is seen as a rebound hyperexcitation following morphine withdrawal. This can be precipitated by administration of naloxone, which results in a marked increase in the firing rate of oxytocin neurones (Leng, Russell & Grossmann, 1989) and a consequent hyper-secretion of oxytocin into the blood (Bicknell, Leng, Lincoln & Russell, 1988). In addition to an increase in the electrical and secretory activity of oxytocin neurones, morphine withdrawal also results in increased expression of Fos (the protein product of the immediate early gene c-fos) in the supraoptic and paraventricular nuclei (Jhamandas, Harris, Petrov & Jhamandas, 1996), and in the release of oxytocin from dendrites within the supraoptic nucleus (Russell, Neumann & Landgraf, 1992a), which contributes to the electrical excitation of oxytocin neurones seen at this time (Brown, Munro, Johnstone, Robson, Landgraf & Russell, 1997).
Many central neurones express μ-receptors (Mansour, Fox, Burke, Akil & Watson, 1995), but morphine dependence is not a universal feature of morphine-sensitive neurones (Nye & Nestler, 1996); oxytocin neurones are the only identified peptidergic neurones currently known to develop morphine dependence, and in the hypothalamus, morphine withdrawal induces little Fos expression outside that seen in the magnocellular nuclei. It has been proposed that withdrawal excitation in general may primarily be a result of morphine dependence in noradrenergic neurones (Redmond & Krystal, 1984). Release of noradrenaline is increased in various brain regions by morphine withdrawal; among these areas is the hippocampus (Done, Silverstone & Sharp, 1992) where the release is prevented by the α2-adrenoreceptor agonist clonidine (Silverstone, Done & Sharp, 1992), presumably by acting upon inhibitory α2-adrenoreceptors located on the terminals of the noradrenergic neurones. Clonidine also ameliorates the behavioural signs of opiate withdrawal in humans (Gold, Redmond & Kleber, 1978); similarly, in rats, clonidine infusion into the locus coeruleus also reduces the behavioural correlates of morphine withdrawal (Taylor, Elsworth, Garcia, Grant, Roth & Redmond, 1988). Thus it appears that, in behavioural systems at least, noradrenaline is critically involved in morphine withdrawal excitation.
Neurones of the A1, A2 and A6 noradrenergic cell groups are also excited by morphine withdrawal (Aghajanian, 1978; Stornetta, Norton & Guyenet, 1993; Murphy, Onaka, Brown & Leng, 1997), but only the A2 cell group in the NTS projects extensively to oxytocin neurones in the supraoptic nucleus (Cunningham & Sawchenko, 1988) where noradrenergic terminals form synapses with oxytocin neurones (Michaloudi, El Majdoubi, Poulain, Papadopoulos & Theodosis, 1997), depolarizing supraoptic nucleus neurones via α1-adrenoreceptors (Yamashita, Inenaga & Kannan, 1987). Approximately 20% of noradrenergic neurones in the A2 cell group, and slightly over 10% of the A2 neurones that project to the supraoptic nucleus, express Fos protein following morphine withdrawal (Murphy et al. 1997). It remains to be determined whether it is the noradrenergic neurones which project to the supraoptic nucleus that are activated to express Fos protein, but clearly it seems possible that withdrawal excitation of oxytocin neurones may result from increased afferent activity in noradrenergic inputs at this time.
Here, we sought to determine whether noradrenergic inputs are involved in the morphine withdrawal excitation of oxytocin neurones by using central catecholaminergic lesion and pharmacological blockade of central noradrenergic neurotransmission to eliminate the influence of these inputs during morphine withdrawal.
METHODS
Induction of morphine tolerance/dependence
Rats were made morphine dependent as previously described (Rayner, Robinson & Russell, 1988). Briefly, virgin female Sprague-Dawley rats (225–333 g) were anaesthetized with 5% halothane in a mixture of O2 and N2O (both flow rates at ca 500 ml min−1) and a 28-gauge stainless-steel cannula was implanted into the right lateral cerebral ventricle (3.0 mm caudal, 2.0 mm lateral to bregma and 4.5 mm below the surface of the skull). The cannula was attached via polythene tubing to a subcutaneous (s.c.) Alzet model 2001 mini-osmotic pump (Charles River UK Ltd, Margate, Kent, UK) for chronic morphine infusion. The pump and tubing contained morphine sulphate dissolved in sterile pyrogen-free water to deliver increasing doses over 5 days (10 μg h−1, 20 μg h−1 for 40 h each and 50 μg h−1 for the remainder at 1 μl h−1). Morphine-naïve rats were administered vehicle through the cannula or did not receive a chronic intracerebroventricular (i.c.v.) infusion. A 28-gauge cannula (Plastics One, Roanoke, VA, USA) was placed in the left cerebral ventricle (0.6 mm caudal, 1.6 mm lateral to bregma and 4.1 mm below the surface of the skull) for subsequent acute i.c.v. infusion of the α1-adrenoreceptor antagonist benoxathian, and sealed with a 21-gauge cap. The cannulae were secured using dental acrylic bonded to stainless-steel screws inserted in the skull. Following surgery, the animals were housed individually with free access to food and water. For the neurotoxic ablation experiment (see below), i.c.v. cannulae placements were performed on virgin, female Wistar rats (210–260 g) as above, but under Avertin anaesthesia (2% w/v tribromoethanol/8% ethanol/1.2% amyl alcohol in 0.9% saline, 10 ml kg−1, intraperitoneally (i.p.)).
Blood sampling experiments
Morphine-dependent and naïve rats were anaesthetized with sodium pentobarbitone (60 mg kg−1, i.p.) on the sixth day following minipump implantation and a cannula was inserted into the superior vena cava through the right jugular vein for drug injection (naloxone; 5 mg kg−1; 0.5 ml kg−1 in 0.9% saline) and blood sampling. At least 90 min following the completion of surgery, serial blood samples (0.3 ml) were removed. Rats prepared in this way included those pretreated with the neurotoxin 6-hydroxydopamine (6-OHDA, see below) or others given an i.c.v. infusion of benoxathian (5.3 μg min−1; 0.53 μl min−1 in 0.9% saline) or vehicle (see Results) from a slow infusion pump.
Benoxathian was selected because it has an α1: α2 selectivity ratio of ca 1000, and is at least three times more potent than other α1-adrenoreceptor antagonists (Melchiorre, Brasili, Giardina, Pigini & Strappaghetti, 1984); given centrally, α1-adrenoreceptor antagonists do not alter blood pressure or heart rate (Shebuski & Zimmerman, 1985).
Alternatively, using a similar blood sampling procedure, serial blood samples were removed from the femoral artery of morphine-dependent rats, under urethane anaesthesia (ethyl carbamate, 1.25 g kg−1, i.p.), in which a femoral vein cannula was used for drug administration (5 mg kg−1 naloxone, with or without the α2-adrenoreceptor agonist, clonidine, 2.5 mg kg−1).
The plasma was separated and stored at −20°C for later determination of oxytocin concentrations by a specific radioimmunoassay (RIA) using antiserum generously supplied by Dr T. Higuchi (Higuchi, Honda, Fukuoka, Negoro & Wakabayashi, 1985). Blood cells were resuspended in 0.9% saline and injected through the i.v. cannula after removal of the subsequent sample. Upon completion of the experiments, the rats were killed by i.v. anaesthetic overdose (60 mg kg−1 pentobarbitone or 1.25 g kg−1 urethane). All samples from each experiment were analysed in a single RIA and the assay sensitivities and intra-assay coefficients of variation were between 2.4 and 5.4 pg ml−1, and 9.9 and 11.8%, respectively.
Neurotoxic ablation of central catecholaminergic pathways
Under Avertin anaesthesia, immediately before implantation of the morphine-containing minipump, a group of rats was injected i.c.v. (0.6 mm caudal, 1.6 mm left lateral to bregma and 4.1 mm below the surface of the skull) with 250 μg of the catecholaminergic neurotoxin 6-OHDA, in 10 μl of 0.9% saline containing 0.15 m ascorbic acid. Control animals were injected with vehicle. On the sixth day, the animals were anaesthetized with sodium pentobarbitone (60 mg kg−1, i.p.) and prepared for naloxone administration and blood sampling through the jugular vein as above.
After the blood sampling, the rats were decapitated and the brains removed. The hypothalami were dissected out, weighed and frozen on dry ice. Each hypothalamus was homogenized on ice in 1.5 ml of 0.1 m perchloric acid containing the synthetic monoamine dihydroxybenzylamine (40 mg ml−1) as an internal standard. The noradrenaline, serotonin and dopamine contents were measured using reversed-phase high-performance liquid chromatography coupled to an electrochemical detector, and the concentrations calculated per milligram of wet weight of tissue, as previously described (Onaka, Luckman, Antonijevic, Palmer & Leng, 1995a).
Electrophysiology
The pituitary stalk and right supraoptic nucleus were exposed by the transpharyngeal approach under urethane anaesthesia (1.25 g kg−1, i.p.). A SNEX100 bipolar stimulating electrode (Clark Electromedical Instruments, Pangbourne, Reading, UK) was placed on the pituitary stalk to elicit antidromic action potentials in supraoptic nucleus neurones, recorded using a glass microelectrode (15–40 MΩ) and conventional extracellular recording techniques. Antidromically identified neurones were characterized as oxytocin rather than vasopressin neurones by their response to cholecystokinin (CCK; 20 μg kg−1, 0.5 ml kg−1 in 0.9% saline, i.v.). Oxytocin neurones transiently increase their activity following CCK injection (Renaud, Tang, McCann, Stricker & Verbalis, 1987), whereas vasopressin neurones are unaffected or inhibited and also often show a distinct phasic pattern of spontaneous activity. These rats were given injections of CCK or naloxone (5 mg kg−1) via a jugular or femoral venous cannula and i.c.v. injection or infusion of benoxathian or vehicle. At the end of the experiments, the rats were killed by i.v. anaesthetic overdose (60 mg kg−1 pentobarbitone).
Microdialysis
Rats were prepared for electrophysiological recording of oxytocin neurones from the supraoptic nucleus and microdialysis application of drugs as previously described (Ludwig & Leng, 1997). Briefly, an in-house designed U-shaped microdialysis probe (total membrane length 2.0 mm; Spectra/Por RC Hollow Fibres®, Spectrum Medical Inc., Houston, TX, USA) was bent to position the loop of the membrane flat onto the exposed ventral surface of the brain on the ventral glial lamina of the supraoptic nucleus after removal of the meninges prior to placement of the recording electrode in the supraoptic nucleus. The supraoptic nucleus was dialysed with artificial cerebrospinal fluid (ACSF; pH 7.2, composition (mm): 138 NaCl, 3.36 KCl, 9.52 NaHCO3, 0.49 Na2HPO4, 2.16 urea, 1.26 CaCl2 and 1.18 MgCl2) at a flow rate of 2 μl min−1 with or without addition of benoxathian (2 mm) to investigate local action within the supraoptic nucleus. The rats were killed by i.v. anaesthetic overdose (60 mg kg−1 pentobarbitone) at the end of the experiments.
Firing rate analysis
The firing rates of putative oxytocin neurones were downloaded onto a personal computer using the Spike2 software package (Cambridge Electronic Design, Cambridge, UK). The mean firing rate of each neurone was calculated for the 5 min periods immediately before each treatment and for successive 5 min periods during and after the treatment.
Drugs
Morphine sulphate was supplied by The Royal Infirmary of Edinburgh, Edinburgh, UK. Naloxone hydrochloride, 6-OHDA hydrochloride, clonidine hydrochloride, and dihydroxybenzylamine were purchased from The Sigma Chemical Company Ltd, Poole, Dorset, UK; CCK was from Bachem (UK) Ltd, Saffron Walden, Essex, UK, and benoxathian hydrochloride was from Research Biochemicals International, Semat Technical (UK) Ltd, St Albans, Herts, UK.
Statistics
All data are represented as the means ± standard error of the mean (s.e.m.), and all statistical tests were completed on the SigmaStat® software package (Jandel Scientific GmbH, 40699 Erkrath, Germany).
RESULTS
Effects of catecholaminergic neurotoxic lesion on oxytocin secretion following morphine withdrawal excitation
Pretreatment of rats with 6-OHDA (250 μg, i.c.v., n = 7) reduced the hypothalamic noradrenaline content to 18.4% of that of sham-operated (n = 7) animals (P < 0.001, unpaired t test, Fig. 1). The serotonin content was also reduced, to 56.1% of that of controls (P < 0.001), while dopamine content was not significantly altered (146.9% of control). Under pentobarbitone anaesthesia, the basal plasma oxytocin concentrations in sham- and 6-OHDA-lesioned rats were similar (11.4 ± 1.2 and 11.8 ± 1.9 pg ml−1, respectively; means ±s.e.m.). In the sham-lesioned rats, injection of CCK (20 μg kg−1, i.v.) increased the plasma oxytocin concentration by 42.2 ± 12.1 pg ml−1, whereas in 6-OHDA-lesioned rats, CCK did not significantly increase plasma oxytocin concentration (3.8 ± 0.6 pg ml−1 increase; difference between groups significant at P < 0.01, Mann-Whitney U test).
Figure 1. Effects of chronic noradrenaline depletion on morphine withdrawal-induced hypersecretion of oxytocin.
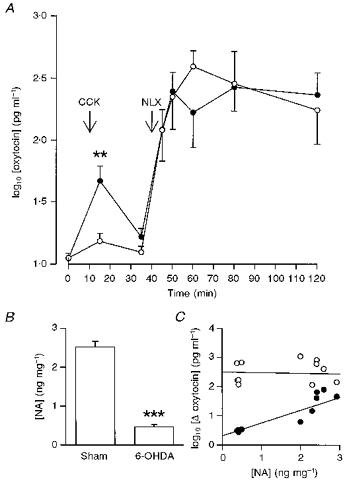
A, log10 plasma oxytocin concentrations (± s.e.m.) versus time in pentobarbitone-anaesthetized 6-OHDA- (○, n = 7) or sham-lesioned (•, n = 7) morphine-dependent rats administered CCK (20 μg kg−1, i.v.) at t = 10 min and naloxone (NLX; 5 mg kg−1, i.v.) at t = 40 min. Two-way repeated measures (RM) ANOVA showed that there was no effect of 6-OHDA lesion on the morphine-withdrawal hypersecretion of oxytocin. However, the change in oxytocin concentration following CCK was significantly reduced with respect to that in sham-lesioned rats (**P < 0.01, Mann-Whitney U test). B, hypothalamic noradrenaline concentrations (ng (mg wet weight)−1 ± s.e.m.) in sham- and 6-OHDA-lesioned rats at t = 120 min; ***P < 0.001, unpaired t test. C, scatter plot of the change (Δ) of (log10) plasma oxytocin concentration in individual sham- or 6-OHDA-lesioned, morphine-dependent rats following systemic CCK (20 μg kg−1, i.v.; •) or naloxone (5 mg kg−1, i.v.; ^) versus hypothalamic noradrenaline (NA) content (ng (mg wet weight)−1). There was a significant correlation of hypothalamic noradrenaline content with the change in oxytocin concentration after systemic CCK (r = 0.830; P = 0.002; n = 11), but not with that after naloxone (r = −0.053; P = 0.870; n = 12).
In contrast, the effects of naloxone (5 mg kg−1, i.v.) upon oxytocin concentrations were not impaired in the same 6-OHDA-lesioned rats; naloxone raised plasma oxytocin concentration by 364.6 ± 87.1 pg ml−1 in the sham-lesioned rats and by 564.5 ± 255.0 pg ml−1 in the 6-OHDA-lesioned rats (Fig. 1).
There was a strong correlation between the hypothalamic noradrenaline content and the increase in oxytocin secretion induced by systemic CCK (Pearson product correlation coefficient, r = 0.830, P = 0.002, n = 11; Fig. 1C), but not between the hypothalamic noradrenaline content and the increase in oxytocin secretion following morphine withdrawal (r = −0.153, P = 0.870, n = 12); neither was there a significant correlation between the magnitude of the response to systemic CCK and that to morphine withdrawal (r = 0.170, P = 0.597). Thus in morphine-dependent rats, extensive destruction of the hypothalamic noradrenergic innervation was accompanied by an almost complete loss of oxytocin release in response to CCK, but in the same rats, naloxone-induced withdrawal produced a normal hypersecretion of oxytocin.
However, most data implicating noradrenergic systems in opiate withdrawal derive from acute, rather than chronic, interventions. We went on to study the impact of acute removal of the noradrenergic input upon withdrawal excitation of oxytocin neurones.
Effects of presynaptic inhibition of noradrenergic neurones by α2-adrenoreceptor activation on morphine withdrawal-induced oxytocin secretion
We injected urethane-anaesthetized morphine-dependent rats with clonidine (2.5 mg kg−1, i.v.), to inhibit the release of endogenous noradrenaline, or vehicle 10 min before injection with naloxone (5 mg kg−1, i.v). Before any injection, the plasma oxytocin concentrations in the clonidine- and vehicle-treated rats were not significantly different (30.2 ± 5.8 and 26.4 ± 3.2 pg ml−1, respectively), and clonidine had no significant effect upon basal concentration. After naloxone, oxytocin concentrations rose significantly in both groups (P < 0.05, Student-Newman- Keuls post hoc analyses), but the increase was significantly less in the clonidine-treated rats (P < 0.05); two-way repeated measures (RM) ANOVA revealed significant time (P < 0.0001) and interaction (P < 0.01) effects (Fig. 2A). Maximum plasma concentrations, achieved in both groups 10 min after naloxone injection were 303.7 ± 122.4 pg ml−1 in clonidine-treated rats and 702.4 ± 201.8 pg ml−1 in vehicle-treated rats, respectively.
Figure 2. Effects of blockade of endogenous noradrenaline on morphine withdrawal-induced hypersecretion of oxytocin.
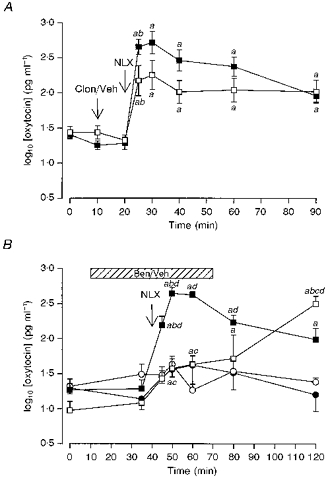
A, plasma oxytocin concentrations (mean log10 plasma oxytocin concentration ±s.e.m.) measured in urethane-anaesthetized morphine-dependent rats. Morphine withdrawal was induced by injection of naloxone (NLX, 5 mg kg−1, i.v.). Rats were given an i.v. injection of either clonidine (Clon; 2.5 mg kg−1; □) or vehicle (Veh; 0.9% saline, ▪) 10 min before injection of naloxone. The increase in oxytocin concentration after naloxone was significantly less in the clonidine-treated rats (P < 0.05); two-way RM ANOVA revealed significant time (P < 0.0001) and interaction (P < 0.01) effects. a, P < 0.05 compared with basal concentration (t = 0 min); b, P < 0.05 compared with the preceding sample. B, plasma oxytocin concentrations (mean log10 plasma oxytocin concentration ±s.e.m.) measured in pentobarbitone-anaesthetized morphine-dependent rats (squares) and morphine-naïve rats (circles). Morphine withdrawal was elicited acutely by injection of naloxone (NLX, 5 mg kg−1, i.v.). Rats were given an i.c.v. infusion (hatched bar) of either benoxathian (5.3 μg min−1, i.c.v. at 0.53 μl min−1, open symbols) or vehicle (0.9% saline, closed symbols). Naloxone evoked an increased release of oxytocin in all groups (n = 6–8), but the magnitude of release was much greater in morphine-dependent rats. Benoxathian infusion attenuated and delayed the naloxone-induced secretion of oxytocin in dependent rats. Two-way ANOVA revealed a significant effect of morphine pretreatment (P < 0.0001) and a significant interaction between the effects of morphine pretreatment and benoxathian treatment (P < 0.05). Two-way RM ANOVA between the groups revealed significant group, time and interaction effects (all P < 0.0001). a, P < 0.05 compared with basal concentration (t = 0 min); b, P < 0.05 compared with the preceding sample; c, P < 0.05 compared with the appropriate time-matched, vehicle-treated, morphine-dependent controls; d, P < 0.05 compared with the similarly treated time-matched, morphine-naïve controls (Student-Newman-Keuls post hoc analyses).
Effects of central α1-adrenoreceptor antagonism on oxytocin secretion following morphine withdrawal excitation
Plasma oxytocin concentrations were measured in blood samples from pentobarbitone-anaesthetized morphine-dependent and naïve rats to which naloxone (5 mg kg−1, i.v.) was administered 30 min after the start of a 60 min i.c.v. infusion of benoxathian (5.3 μg min−1) or vehicle (Fig. 2B). The basal oxytocin concentrations were similar in the morphine-dependent (17.4 ± 4.0 pg ml−1; n = 14) and naïve rats (21.8 ± 2.7 pg ml−1; n = 14), and after 25 min of benoxathian or vehicle infusion, there were no significant changes in the oxytocin concentration in any of the four groups. Injection of naloxone resulted in the expected large increase in oxytocin secretion in the vehicle-infused morphine-dependent rats, but in the benoxathian-infused morphine-dependent rats the response to naloxone was significantly smaller (P < 0.05). At 50 min after the end of the i.c.v. benoxathian infusion, oxytocin concentrations were increased in the morphine-dependent rats (P < 0.05) to levels that were above the corresponding levels in the vehicle-infused, morphine-dependent rats (P < 0.05). Thus acute blockade of α1-adrenoreceptors by i.c.v. benoxathian infusion attenuated, but did not prevent, the morphine withdrawal-induced release of oxytocin.
Effects of central α1-adrenoreceptor antagonism following naloxone injection on the firing rate of oxytocin neurones in morphine-dependent and naïve rats
After administration of naloxone (5 mg kg−1), oxytocin neurones were challenged with i.c.v. infusions of vehicle or benoxathian at various doses (0.3, 1.0, 1.6 and 5.3 μg min−1, i.c.v.) for at least 15 min. In the 5 min before the start of the infusion, the firing rates of the neurones tested in morphine-dependent and naïve rats were 6.4 ± 0.6 spikes s−1 (n = 32, one cell in each rat) and 3.0 ± 0.5 spikes s−1 (n = 20), respectively, the higher firing rate in dependent rats reflecting the persistent hyperactivity following naloxone-induced withdrawal. i.c.v. infusion of vehicle caused no significant change in the firing rate of any cell tested (0.3 ± 0.7 spikes s−1 mean decrease in naïve rats, n = 4, and 0.2 ± 0.1 spikes s−1 increase in dependent rats, n = 4). In contrast, benoxathian infusion significantly inhibited the firing rate of oxytocin neurones at the highest dose (mean decreases 4.1 ± 1.2 spikes s−1 in dependent rats, n = 9, and 2.2 ± 0.9 spikes s−1 in naïve rats, n = 3). At lower doses, some neurones were inhibited, but overall benoxathian had no consistent effect (the mean change during infusion at 1.6 μg min−1 was −0.6 ± 0.5 spikes s−1, n = 11 in naïve rats; −0.3 ± 1.0 spikes s−1, n = 8 in dependent rats). Nevertheless, the dose and the reduction of the firing rate were significantly correlated in both morphine-dependent and naïve rats (r =−0.519 and −0.470; P < 0.01 and P < 0.05, respectively). At the highest dose, the firing rates of the three neurones tested in naïve rats and the nine neurones tested in dependent rats were reduced by 84.7 ± 9.6 and 58.2 ± 12.3%, respectively. Of the nine neurones tested in dependent rats, four were inhibited to a firing rate below that recorded before naloxone, and of these, three were silenced in the period 10–15 min after the onset of the infusion (i.e. after infusion of 50–75 μg benoxathian).
To determine whether these effects of benoxathian were a result of an action within the supraoptic nucleus, a microdialysis probe was placed onto the exposed ventral surface of the supraoptic nucleus as previously described for electrical recording of the activity of neurosecretory neurones during microdialysis application of drugs (Ludwig & Leng, 1997). Benoxathian (2 mm) was dialysed onto the supraoptic nucleus of three rats (one morphine-naïve without i.v. naloxone, 5 mg kg −1; one morphine-naïve and one morphine-dependent after i.v. naloxone). The spontaneous firing rates of each of the cells in the 5 min immediately prior to the onset of benoxathian microdialysis were 3.6, 3.9 and 3.4 spikes s−1, respectively (the cell in the morphine-dependent rat having been excited from 0.5 spikes s−1 by i.v. naloxone), and these were reduced by 62, 21 and 77%, respectively, during benoxathian administration.
Thus benoxathian reversed established morphine withdrawal excitation of oxytocin neurones and inhibited spontaneous activity of oxytocin neurones in morphine-naïve rats by an action within the supraoptic nucleus. However, since the mechanisms initiating withdrawal excitation and those sustaining the excitation could be quite different (Brown et al. 1997), we went on to determine whether i.c.v. infusion of benoxathian could prevent the initiation of withdrawal excitation in morphine-dependent rats.
Effects of prior central α1-adrenoreceptor antagonism on the naloxone-induced withdrawal response of oxytocin neurones
Naloxone (5 mg kg−1) was injected i.v. 10 min after the onset of an i.c.v. infusion of either benoxathian (5.3 μg min−1) or vehicle in morphine-dependent rats. The infusions were continued until 5 min after the naloxone injection and cell activity was recorded for at least a further 30 min. Before the infusions, the spontaneous firing rates of the neurones sampled in the two groups of rats were similar (0.8 ± 0.2 spikes s−1, n = 7, and 1.0 ± 0.4 spikes s−1, n = 6). Neither vehicle nor benoxathian altered the firing rates significantly within the 10 min of infusion before injection of naloxone. Naloxone caused a prompt increase in firing rate in both the vehicle- and benoxathian-infused rats (to 4.7 ± 0.7 and 2.9 ± 1.0 spikes s−1 over the first 5 min, respectively; both P < 0.05 compared with the pre-naloxone firing rates; two-way RM ANOVA with Student- Newman-Keuls post hoc analyses), but in the first 5 min after naloxone, the firing rate was significantly lower in the benoxathian-infused group than in the vehicle-infused group (P < 0.05; Fig. 3). In vehicle-infused rats, the firing rate remained close to this initially elevated rate throughout the next 35 min. In contrast, in benoxathian-infused rats, the immediate response was maintained only briefly in some neurones, and after the end of infusion all neurones showed a progressive increase in firing rate (from 2.9 ± 1.0 to 6.4 ± 1.0 spikes s−1 25–30 min later; P < 0.05 compared with the immediate post-naloxone firing rate). Thus benoxathian infusion partially impaired the initial naloxone-induced withdrawal excitation of oxytocin neurones.
Figure 3. Effects of acute α1-adrenoceptor antagonism on the activity of oxytocin neurones in morphine-naïve and morphine-dependent rats.
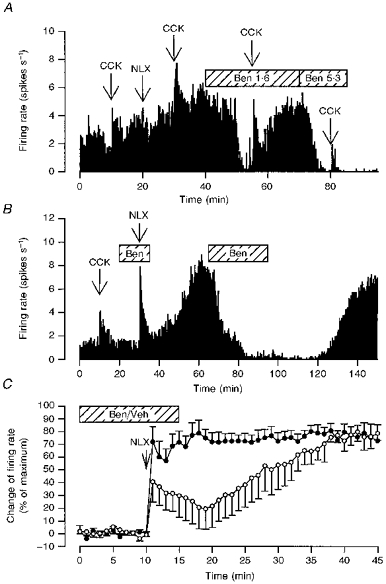
A, the firing rate (averaged in consecutive 10 s intervals) of a single oxytocin cell recorded from a morphine-naïve rat administered CCK (20 μg kg−1, i.v.) at 10, 30, 55 and 80 min, naloxone (NLX, 5 mg kg−1, i.v.) at 20 min and infused i.c.v. with benoxathian at 1.6 μg min−1 (Ben 1.6) from 40 to 70 min and at 5.3 μg min−1 (Ben 5.3) from 70 to 85 min. Benoxathian inhibited spontaneous activity and attenuated the response to CCK. B, the firing rate (averaged in consecutive 10 s intervals) of a single oxytocin cell recorded from a morphine-dependent rat administered CCK (20 μg kg−1, i.v.) at 10 min, naloxone (NLX, 5 mg kg−1, i.v.) at 30 min and infused i.c.v. with benoxathian (Ben; 5.3 μg min−1) from 20 to 35 and 65 to 95 min. The first infusion of benoxathian delayed the peak sustained increase in firing rate after naloxone and a second infusion later reversibly inhibited the withdrawal excitation. C, effects of benoxathian on subsequent withdrawal excitation of oxytocin neurones. The composite change in firing rate of oxytocin neurones was induced by naloxone (NLX; 5 mg kg−1, i.v. at t = 10 min) in morphine-dependent rats during i.c.v. infusion of benoxathian (Ben; 5.3 μg min−1, n = 7, ^) or vehicle (Veh, n = 6, •). The change in firing rate was calculated for each cell by subtracting the initial mean basal firing rate (measured over 5 min in 1 min bins) from the firing rate measured in each minute, and was expressed as a percentage (mean ±s.e.m.) of the maximum change observed for each cell. The magnitude of withdrawal excitation was significantly lower in the first 5 min after naloxone in the benoxathian-infused group than in the vehicle-infused group (P < 0.05, two-way RM ANOVA followed by Student-Newman-Keuls analyses). In vehicle-infused rats, the naloxone-induced increase in firing rate was maximal within 5 min. In contrast, in benoxathian-infused rats the maximum firing rate was not attained until 30–35 min after naloxone, 25–30 min after the end of the benoxathian infusion (P < 0.05 compared with the 5 min immediately following naloxone administration to i.c.v. benoxathian-infused rats).
Effect of i.c.v. benoxathian on the responsiveness of oxytocin neurones to systemic CCK
Since systemic CCK increases the activity of oxytocin neurones by activating A2 noradrenergic cell group neurones in the NTS that project directly to the oxytocin neurones (Luckman, 1992), we tested the responsiveness of twenty oxytocin neurones to CCK (20 μg kg−1, i.v.) before (CCK1) and during (CCK2) i.c.v. benoxathian infusion (Fig. 4). The ratio of the magnitudes of the CCK responses (CCK2:CCK1) was calculated to determine the effectiveness of such benoxathian infusions at preventing the actions of noradrenaline released into the supraoptic nucleus. The CCK2:CCK1 ratios at 0.3 (n = 3), 1.6 (n = 11) and 5.3 μg min−1 benoxathian (n = 6) were 0.8 ± 0.1, 0.5 ± 0.1 and 0.1 ± 0.1, respectively (Fig. 4C). The ratios calculated for the neurones challenged with the high dose of benoxathian were significantly lower than the ratios from neurones exposed to the lower two doses (P < 0.05, one-way ANOVA followed by Student-Newman-Keuls post hoc analyses), indicating that the high dose of i.c.v. benoxathian was sufficient to prevent the excitation of oxytocin neurones that results from the release of noradrenaline into the supraoptic nucleus triggered by systemic CCK.
Figure 4. Effects of acute α1-adrenoceptor antagonism on CCK-induced excitation of oxytocin neurones.
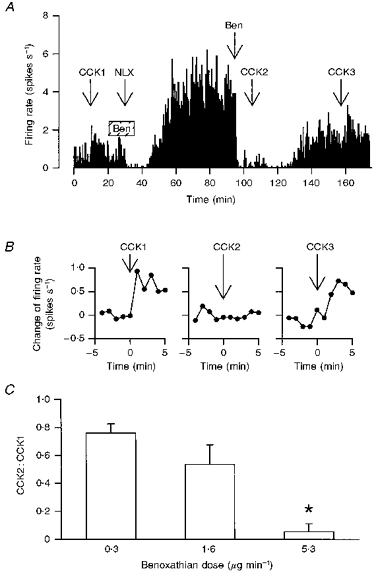
A, the firing rate of an oxytocin cell (averaged in consecutive 10 s intervals) in a morphine-dependent rat under urethane anaesthesia administered CCK (20 μg kg−1, i.v.), and infused i.c.v. with benoxathian (5.3 μg min−1, hatched box, Ben), naloxone (NLX; 5 mg kg−1, i.v.) and benoxathian (25 μg, i.c.v., Ben); the benoxathian infusion delayed the peak withdrawal response (see Fig. 3C) and the later injection of benoxathian reversibly inhibited the withdrawal excitation and the response to CCK. B, the change of firing rate of the oxytocin cell in A following each CCK injection (at t = 0 min) on an expanded time scale; the later injection of benoxathian blocked the response to i.v. CCK (CCK2). C, CCK2:CCK1 ratios from morphine-withdrawn and naïve rats treated with 0.3 (n = 3), 1.6 (n = 11) and 5.3 μg min−1 (n = 6) benoxathian i.c.v.; *P < 0.05 compared with the groups administered the lower two doses, one-way RM ANOVA followed by Student-Newman-Keuls analyses.
DISCUSSION
In the rat, morphine withdrawal excitation of oxytocin neurones is a highly robust phenomenon; the neurones exhibit an approximately 3-fold increase in electrical activity (Leng et al. 1989), producing up to a 100-fold rise in plasma oxytocin concentrations (Bicknell et al. 1988), the amplification of the secretory response being partly a consequence of frequency facilitation of hormone release (Bicknell, 1988) and partly a result of antagonism of κ-opioid activity at the posterior pituitary (Russell, Coombes, Leng & Bicknell, 1993). In recent experiments we showed that, while administration of naloxone directly into the supraoptic nucleus of morphine-dependent rats causes withdrawal hyperexcitation of oxytocin neurones, systemic administration of naloxone further increased activity in these neurones (Ludwig et al. 1997), presumably by inducing morphine withdrawal excitation in an afferent projection. This ability of locally applied naloxone to elicit morphine withdrawal suggests that mechanisms intrinsic to the oxytocin neurones are responsible for dependence. However, morphine is known to have presynaptic effects in the supraoptic nucleus; in particular, morphine inhibits noradrenaline release by a direct action upon the A2 afferent terminals within the nucleus (Onaka et al. 1995b), although morphine dependence does not appear to develop at the level of the noradrenergic terminals (Brown, Munro, Murphy, Leng & Russell, 1996). Given these actions of morphine upon the noradrenergic nerve terminals, and given the extensive involvement of noradrenaline in morphine dependence in other neuronal systems, we set out to determine the contribution of noradrenergic afferent activity to the withdrawal hyperexcitation of oxytocin neurones.
Administration of a central catecholaminergic neurotoxin, 6-OHDA, produced an 82% depletion of hypothalamic noradrenaline content, and a 91% reduction of the oxytocin secretory response to systemic CCK, a stimulus that excites oxytocin neurones via activation of a direct noradrenergic projection from the A2 cell group (Luckman, 1992). In striking contrast, the same lesioned rats showed a fully intact withdrawal hypersecretion of oxytocin in response to naloxone. Similarly, electrolytic and 6-OHDA lesions of the locus coeruleus do not alleviate all of the behavioural signs of withdrawal (Maldonado & Koob, 1993; Miao & Qin, 1995) and selective neurotoxic ablation of the terminals arising from the locus coeruleus does not inhibit expression of any naloxone-precipitated withdrawal sign (Chieng & Christie, 1995). It seems unlikely that the remaining noradrenergic innervation or denervation supersensitivity to noradrenaline compensates adequately for the lost inputs, since the responsiveness of oxytocin neurones to systemic CCK did not recover, at least within the time course of the current study. Thus while the oxytocin response to CCK depends on the integrity of their noradrenergic input (Onaka et al. 1995a), the oxytocin withdrawal response does not. This conclusion is supported by measurements of the release of noradrenaline within the supraoptic nucleus during morphine withdrawal (Murphy et al. 1997), which indicate that this release is less marked than that following systemic administration of CCK although more prolonged (Onaka et al. 1995b), and by evidence that only about 10% of NTS neurones which project to the supraoptic nucleus are activated by morphine withdrawal (Murphy et al. 1997), whereas approximately 50% of NTS neurones that project to the supraoptic nucleus are activated to express Fos protein following systemic CCK (Onaka et al. 1995a), although systemic CCK excites oxytocin neurones to a much lesser extent than morphine withdrawal does (Brown et al. 1996). Furthermore, the release of noradrenaline evoked by CCK does not trigger withdrawal excitation in morphine-dependent rats, nor is there occlusion of the CCK response during morphine withdrawal excitation (Brown et al. 1996).
Nevertheless, i.c.v. infusion of the highest dose (5.3 μg min−1) of benoxathian resulted in a 65% inhibition of the firing rate of oxytocin neurones in morphine-naïve and -withdrawn rats, and administration of this same dose of benoxathian for only 15 min, starting 10 min prior to injection of naloxone, reduced the immediate increase in firing rate of oxytocin neurones to approximately 60% of that seen in control rats. In addition, microdialysis of benoxathian onto the supraoptic nucleus itself resulted in a 53% inhibition of oxytocin neurone firing rate in morphine-naïve and -withdrawn rats. These profound inhibitory effects of α1-adrenoreceptor antagonism indicate that while full withdrawal excitation of supraoptic oxytocin neurones can occur after extensive chronic ablation of the noradrenergic inputs, and probably depends on mechanisms intrinsic to the oxytocin neurones themselves, the magnitude of the excitation can nevertheless be strongly influenced by acute alterations in the activity in noradrenergic inputs. The last conclusion is substantiated by the reduction in the oxytocin secretory response during morphine withdrawal following either presynaptic α2-adrenoreceptor agonism (with clonidine) or i.c.v. infusion of benoxathian.
It is important to emphasize that benoxathian was not effective only upon withdrawal excitation; benoxathian was inhibitory to oxytocin neurones in morphine-withdrawn and naïve rats, and was inhibitory whether administered i.c.v. or directly to the supraoptic nucleus. These results indicate that noradrenergic pathways were tonically active under our experimental conditions, providing an excitatory influence to oxytocin neurones both in naïve rats and in dependent rats. What is surprising is not that these pathways are active, but that antagonism of noradrenaline produces such a marked inhibition not only of basal activity in naïve rats but also of morphine withdrawal-induced activity, given that morphine withdrawal appears to involve at most modest activation of noradrenergic inputs, and appears normal following chronic ablation of the noradrenergic pathways.
As has been noted previously, since spikes in oxytocin neurones in vivo are thought to be generated exclusively as a result of summation of EPSPs, the ability of an oxytocin neurone to respond to an increase in EPSP frequency is dependent both upon the mean resting potential and upon the background level of synaptic input. The present study indicates that the acute loss of only a proportion of the tonic excitatory input may critically impair the ability of oxytocin cells to respond even to the intense stimulus of morphine withdrawal. Following chronic ablation of noradrenergic pathways, compensatory mechanisms (but not evidently involving supersensitivity to noradrenaline, since CCK was ineffective) that restore the normal spontaneous excitability of oxytocin neurones thus appear also to restore their ability to respond to withdrawal.
Such a dependence of withdrawal excitation upon pre-existing active synaptic input may also occur in other neuronal systems that develop morphine dependence. The locus coeruleus (A6 cell group) is possibly the most extensively studied region in which morphine dependence develops. Here, morphine dependence increases G protein subunit, adenylate cyclase, cyclic AMP-dependent protein kinase and tyrosine hydroxylase levels (Nestler, Alreja & Aghajanian, 1994), and intrinsic mechanisms are thought to underpin morphine dependence and withdrawal excitation in these neurones. However, excitatory amino acid antagonists injected directly into the locus coeruleus reduce the withdrawal-induced activation of locus coeruleus neurones by more than 50% (Akaoka & Aston-Jones, 1991) and glutamate release into the locus coeruleus is increased during morphine withdrawal; this may contribute actively as well as passively to withdrawal excitation of locus coeruleus neurones (Aghajanian, Kogan & Moghaddam, 1994). Thus as for the oxytocin system, withdrawal excitation of locus coeruleus neurones may be generated by an interplay between intracellular mechanisms and extrinsic inputs.
Oxytocin neurones also receive glutamatergic input from the organum vasculosum of the lamina terminalis (OVLT) (Yang, Senatorov & Renaud, 1994) within the region anterior and ventral to the third ventricle (AV3V). Indeed, glutamate appears to be the major excitatory neurotransmitter within the supraoptic nucleus (van den Pol, Wuarin & Dudek, 1990; Meeker, Greenwood & Hayward, 1994). However, few neurones that project from the AV3V region to the supraoptic nucleus are activated to express Fos protein by morphine withdrawal (Murphy et al. 1997), and lesion of the AV3V region does not eliminate withdrawal excitation of oxytocin neurones (Russell, Pumford & Bicknell, 1992b). Nevertheless, AV3V lesion does result in some reduction of the magnitude of the withdrawal excitation. Thus the magnitude of withdrawal excitation of supraoptic nucleus oxytocin neurones may also be influenced by the level of activity in glutamatergic inputs.
In conclusion, oxytocin neurones express morphine withdrawal excitation, and presumably morphine dependence, separately from their distant afferent inputs, and in particular, morphine withdrawal is not affected by chronic ablation of the noradrenergic inputs to oxytocin neurones. Nevertheless, in rats where the noradrenergic inputs are intact, the activity of these inputs contributes to the magnitude of the excitation evident in oxytocin neurones at this time.
Acknowledgments
The authors are grateful to the following people at The Babraham Institute, Cambridge, UK: Ms S. Dye for carrying out an oxytocin radioimmunoassay and Mr C. Chapman for the tissue determinations of noradrenaline content, and to Ms M. Garabette (Department of Physiology, University Medical School, Edinburgh, UK) for technical assistance. Supported by a BBSRC Project Grant, BBSRC Postgraduate Studentships (G. M. and N. P. M.) and by a Deutsche Forschungsgemeinschaft Research Fellowship (M. L.).
References
- Aghajanian GK. Tolerance of locus coeruleus neurones to morphine and suppression of withdrawal response by clonidine. Nature. 1978;276:186–188. doi: 10.1038/276186a0. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK, Kogan JH, Moghaddam B. Opiate withdrawal increases glutamate and aspartate efflux in the locus coeruleus: an in vivo microdialysis study. Brain Research. 1994;636:126–130. doi: 10.1016/0006-8993(94)90186-4. 10.1016/0006-8993(94)90186-4. [DOI] [PubMed] [Google Scholar]
- Akaoka H, Aston-Jones G. Opiate withdrawal-induced hyperactivity of locus coeruleus neurons is substantially mediated by augmented excitatory amino acid input. Journal of Neuroscience. 1991;11:3830–3839. doi: 10.1523/JNEUROSCI.11-12-03830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicknell RJ. Optimizing release from peptide hormone secretory nerve terminals. Journal of Experimental Biology. 1988;139:51–65. doi: 10.1242/jeb.139.1.51. [DOI] [PubMed] [Google Scholar]
- Bicknell RJ, Leng G, Lincoln DW, Russell JA. Naloxone excites oxytocin neurones in the supraoptic nucleus of lactating rats after chronic morphine treatment. Journal of Physiology. 1988;396:297–317. doi: 10.1113/jphysiol.1988.sp016963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CH, Munro G, Johnstone LE, Robson AC, Landgraf R, Russell JA. Oxytocin neurone autoexcitation during morphine withdrawal in anaesthetized rats. NeuroReport. 1997;8:951–955. doi: 10.1097/00001756-199703030-00027. [DOI] [PubMed] [Google Scholar]
- Brown CH, Munro G, Murphy NP, Leng G, Russell JA. Activation of oxytocin neurones by systemic cholecystokinin is unchanged by morphine-dependence or withdrawal excitation in the rat. Journal of Physiology. 1996;496:787–794. doi: 10.1113/jphysiol.1996.sp021727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Christie MJ. Lesions to terminals of noradrenergic locus-coeruleus neurons do not inhibit opiate withdrawal behavior in rats. Neuroscience Letters. 1995;186:37–40. doi: 10.1016/0304-3940(95)11276-3. [DOI] [PubMed] [Google Scholar]
- Cunningham ET, Jr, Sawchenko PE. Anatomical specificity of noradrenergic inputs to the paraventricular and supraoptic nuclei of the rat hypothalamus. Journal of Comparative Neurology. 1988;274:60–76. doi: 10.1002/cne.902740107. [DOI] [PubMed] [Google Scholar]
- Done C, Silverstone P, Sharp T. Effect of naloxone-precipitated morphine withdrawal on noradrenaline release in rat hippocampus in vivo. European Journal of Pharmacology. 1992;215:333–336. doi: 10.1016/0014-2999(92)90052-6. 10.1016/0014-2999(92)90052-6. [DOI] [PubMed] [Google Scholar]
- Gold MS, Redmond DE, Jr, Kleber HD. Clonidine blocks acute opiate-withdrawal symptoms. Lancet. 1978;2:599–602. doi: 10.1016/s0140-6736(78)92823-4. 10.1016/S0140-6736(78)92823-4. [DOI] [PubMed] [Google Scholar]
- Higuchi T, Honda K, Fukuoka T, Negoro H, Wakabayashi K. Release of oxytocin during suckling and parturition in the rat. Journal of Endocrinology. 1985;105:339–346. doi: 10.1677/joe.0.1050339. [DOI] [PubMed] [Google Scholar]
- Jhamandas JH, Harris KH, Petrov T, Jhamandas KH. Activation of nitric oxide-synthesizing neurones during precipitated morphine withdrawal. NeuroReport. 1996;7:2843–2846. doi: 10.1097/00001756-199611250-00006. [DOI] [PubMed] [Google Scholar]
- Leng G, Russell JA, Grossmann R. Sensitivity of magnocellular oxytocin neurones to opioid antagonists in rats treated chronically with intracerebroventricular (i.c.v.) morphine. Brain Research. 1989;484:290–296. doi: 10.1016/0006-8993(89)90372-7. 10.1016/0006-8993(89)90372-7. [DOI] [PubMed] [Google Scholar]
- Luckman SM. Fos-like immunoreactivity in the brainstem of the rat following peripheral administration of cholecystokinin. Journal of Neuroendocrinology. 1992;4:149–152. doi: 10.1111/j.1365-2826.1992.tb00152.x. [DOI] [PubMed] [Google Scholar]
- Ludwig M, Brown CH, Russell JA, Leng G. Local opioid inhibition and morphine dependence of supraoptic nucleus oxytocin neurones in the rat in vivo. Journal of Physiology. 1997;505:145–152. doi: 10.1111/j.1469-7793.1997.145bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M, Leng G. Autoinhibition of supraoptic nucleus vasopressin neurones in vivo: a combined retrodialysis/electrophysiological study in rats. European Journal of Neuroscience. 1997 doi: 10.1111/j.1460-9568.1997.tb01682.x. in the Press. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Koob GF. Destruction of the locus coeruleus decreases physical signs of opiate withdrawal. Brain Research. 1993;605:128–138. doi: 10.1016/0006-8993(93)91364-x. 10.1016/0006-8993(93)91364-X. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Akil H, Watson SJ. Immunohistochemical localization of the cloned μ-opioid receptor in the rat CNS. Journal of Chemical Neuroanatomy. 1995;8:283–305. doi: 10.1016/0891-0618(95)00055-c. 10.1016/0891-0618(95)00055-C. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Greenwood RS, Hayward JN. Glutamate receptors in the rat hypothalamus and pituitary. Endocrinology. 1994;134:621–629. doi: 10.1210/endo.134.2.7905409. 10.1210/en.134.2.621. [DOI] [PubMed] [Google Scholar]
- Melchiorre C, Brasili L, Giardina D, Pigini M, Strappaghetti G. 2-[[[2-(2,6-Dimethoxyphenoxy)-ethyl] amino]-methyl]-1,4-benzoxathian: a new antagonist with high potency and selectivity toward α1-adrenoreceptors. Journal of Medicinal Chemistry. 1984;27:1535–1536. doi: 10.1021/jm00378a001. [DOI] [PubMed] [Google Scholar]
- Miao H, Qin BY. Influences of locus-coeruleus lesions and reserpine treatment on opioid physical dependence in rats. Acta Pharmacologica Sinica. 1995;16:137–140. [PubMed] [Google Scholar]
- Michaloudi HC, El Majdoubi M, Poulain DA, Papadopoulos GC, Theodosis DT. The noradrenergic innervation of identified hypothalamic magnocellular somata and its contribution to lactation-induced synaptic plasticity. Journal of Neuroendocrinology. 1997;9:17–23. doi: 10.1046/j.1365-2826.1997.00583.x. 10.1046/j.1365-2826.1997.00583.x. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Onaka T, Brown CH, Leng G. The role of afferent inputs to supraoptic nucleus oxytocin neurones during naloxone-precipitated morphine withdrawal in the rat. Neuroscience. 1997;80:567–577. doi: 10.1016/s0306-4522(97)00142-5. 10.1016/S0306-4522(97)00142-5. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Alreja M, Aghajanian GK. Molecular and cellular mechanisms of opiate action: studies in the rat locus coeruleus. Brain Research Bulletin. 1994;35:521–528. doi: 10.1016/0361-9230(94)90166-x. 10.1016/0361-9230(94)90166-X. [DOI] [PubMed] [Google Scholar]
- Nye HE, Nestler EJ. Induction of chronic Fos-related antigens in rat brain by chronic morphine administration. Molecular Pharmacology. 1996;49:636–645. [PubMed] [Google Scholar]
- Onaka T, Luckman SM, Antonijevic I, Palmer JR, Leng G. Involvement of the noradrenergic afferents from the nucleus tractus solitarii to the supraoptic nucleus in oxytocin release after peripheral cholecystokinin octapeptide in the rat. Neuroscience. 1995a;66:403–412. doi: 10.1016/0306-4522(94)00609-9. 10.1016/0306-4522(94)00609-9. [DOI] [PubMed] [Google Scholar]
- Onaka T, Luckman SM, Guevara-Guzman R, Ueta Y, Kendrick K, Leng G. Presynaptic actions of morphine: blockade of cholecystokinin-induced noradrenaline release in the rat supraoptic nucleus. Journal of Physiology. 1995b;482:69–79. doi: 10.1113/jphysiol.1995.sp020500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman QJ, Hatton JD, Bloom FE. Morphine and opioid peptides reduce paraventricular neuronal activity: studies on the rat hypothalamic slice preparation. Proceedings of the National Academy of Sciences of the USA. 1980;77:5527–5531. doi: 10.1073/pnas.77.9.5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumford KM, Leng G, Russell JA. Morphine actions on supraoptic oxytocin neurones in anaesthetized rats: tolerance after i.c.v. morphine infusion. Journal of Physiology. 1991;440:437–454. doi: 10.1113/jphysiol.1991.sp018717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner VC, Robinson IC, Russell JA. Chronic intracerebroventricular morphine and lactation in rats: dependence and tolerance in relation to oxytocin neurones. Journal of Physiology. 1988;396:319–347. doi: 10.1113/jphysiol.1988.sp016964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond DE, Jr, Krystal JH. Multiple mechanisms of withdrawal from opioid drugs. Annual Review of Neuroscience. 1984;7:443–478. doi: 10.1146/annurev.ne.07.030184.002303. 10.1146/annurev.ne.07.030184.002303. [DOI] [PubMed] [Google Scholar]
- Renaud LP, Tang M, McCann MJ, Stricker EM, Verbalis JG. Cholecystokinin and gastric distension activate oxytocinergic cells in rat hypothalamus. American Journal of Physiology. 1987;253:R661–665. doi: 10.1152/ajpregu.1987.253.4.R661. [DOI] [PubMed] [Google Scholar]
- Russell JA, Coombes JE, Leng G, Bicknell RJ. Morphine tolerance and inhibition of oxytocin secretion by κ-opioids acting on the rat neurohypophysis. Journal of Physiology. 1993;469:365–386. doi: 10.1113/jphysiol.1993.sp019818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA, Neumann I, Landgraf R. Oxytocin and vasopressin release in discrete brain areas after naloxone in morphine-tolerant and -dependent anesthetized rats: push-pull perfusion study. Journal of Neuroscience. 1992a;12:1024–1032. doi: 10.1523/JNEUROSCI.12-03-01024.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA, Pumford KM, Bicknell RJ. Contribution of the region anterior and ventral to the third ventricle to opiate withdrawal excitation of oxytocin secretion. Neuroendocrinology. 1992b;55:183–192. doi: 10.1159/000126113. [DOI] [PubMed] [Google Scholar]
- Shebuski RJ, Zimmerman BG. Suppression of reflex tachycardia by central administration of the α1-adrenoceptor antagonists urapidil and prazosin in anesthetized dogs. Journal of Pharmacology and Experimental Therapeutics. 1985;234:456–462. [PubMed] [Google Scholar]
- Silverstone PH, Done C, Sharp T. Clonidine but not nifedipine prevents the release of noradrenaline during naloxone-precipitated opiate withdrawal: an in vivo microdialysis study in the rat. Psychopharmacology. 1992;109:235–238. doi: 10.1007/BF02245506. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Norton FE, Guyenet PG. Autonomic areas of rat brain exhibit increased Fos-like immunoreactivity during opiate withdrawal in rats. Brain Research. 1993;624:19–28. doi: 10.1016/0006-8993(93)90055-r. 10.1016/0006-8993(93)90055-R. [DOI] [PubMed] [Google Scholar]
- Taylor JR, Elsworth JD, Garcia EJ, Grant SJ, Roth RH, Redmond DE., Jr Clonidine infusions into the locus coeruleus attenuate behavioral and neurochemical changes associated with naloxone-precipitated withdrawal. Psychopharmacology. 1988;96:121–134. doi: 10.1007/BF02431544. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Wuarin JP, Dudek FE. Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science. 1990;250:1276–1278. doi: 10.1126/science.1978759. [DOI] [PubMed] [Google Scholar]
- Wakerley JB, Noble R, Clarke G. Effects of morphine and D-Ala, D-Leu enkephalin on the electrical activity of supraoptic neurosecretory cells in vitro. Neuroscience. 1983;10:73–81. doi: 10.1016/0306-4522(83)90081-7. 10.1016/0306-4522(83)90081-7. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Inenaga K, Kannan H. Depolarizing effect of noradrenaline on neurons of the rat supraoptic nucleus in vitro. Brain Research. 1987;405:348–352. doi: 10.1016/0006-8993(87)90304-0. 10.1016/0006-8993(87)90304-0. [DOI] [PubMed] [Google Scholar]
- Yang CR, Senatorov VV, Renaud LP. Organum vasculosum lamina terminalis-evoked postsynaptic responses in rat supraoptic neurones in vitro. Journal of Physiology. 1994;477:59–74. doi: 10.1113/jphysiol.1994.sp020171. [DOI] [PMC free article] [PubMed] [Google Scholar]