

Abstract
Cerebral arteries express cytochrome P450 4A enzymes (P450 4A) and produce 20- hydroxyeicosatetraenoic acid (20-HETE), a potent constrictor of pial arterioles. It is not known which cell type in the vessel wall is responsible for the formation of 20-HETE. We examined whether freshly isolated cerebral arterial muscle cells (VSMCs) express P450 4A and produce 20-HETE. We also studied the effect of 20-HETE on pressurized cerebral arteries and on whole-cell L-type Ca2+ current (ICa) recorded in cat cerebral VSMCs.
Cat cerebral VSMCs incubated with [14C]arachidonic acid ([14C]AA) produced 20-HETE (3.9 ± 1.1 pmol min−1 (mg protein)−1).
Reverse transcription-polymerase chain reaction studies revealed that cat cerebral VSMCs express mRNA for P450 4A which metabolizes AA to 20-HETE. Cloning and sequencing of the cDNA amplified from mRNA isolated from VSMCs showed > 96% amino acid homology to the rat and human P450 4A2 and 4A3.
20-HETE (1–300 nm) induced a concentration-dependent constriction of cat cerebral arteries, which was inhibited by nifedipine.
Addition of 10 and 100 nm 20-HETE to the bath increased peak ICa by 50 ± 3 and 100 ± 10%, respectively. This effect was not influenced by altering the frequency of depolarization. 20-HETE (100 nm) failed to increase ICa in the presence of nifedipine.
These results demonstrate that cat cerebral VSMCs express P450 4A enzyme, and produce 20-HETE which activates L-type Ca2+ channel current to promote cerebral vasoconstriction.
There is considerable evidence demonstrating that arachidonic acid (AA) metabolites play an important role in regulating vascular tone (Gebremedhin, Ma, Falck, Roman, VanRollins & Harder, 1992; Ma et al. 1993; Harder et al. 1994; Harder, Campbell & Roman, 1995; Imig, Zou, Falck, Harder & Roman, 1996). A variety of physiological and pathophysiological stimuli can liberate AA from membrane-bound phospholipid pools in arterial muscle (Dennis, Rhee, Billah & Hannun, 1991; Ordway, Singer & Walsh, 1991). The AA thus released can be metabolized by cyclooxygenase, lipoxygenase and cytochrome P450 enzymes.
Recently, a growing body of literature has defined a critical role for cytochrome P450 metabolites of AA in regulating vascular tone and reactivity (Gebremedhin et al. 1992; Ma et al. 1993; Harder et al. 1994, 1995; Imig et al. 1996). The primary P450 metabolite produced by homogenates of cerebral and renal arterioles is 20-hydroxyeicosatetraenoic acid (20-HETE) (Harder et al. 1994; Imig et al. 1996). 20-HETE is a product of ω-hydroxylation of AA catalysed by enzymes of the cytochrome P450 family (Harder et al. 1994, 1995; Imig et al. 1996). While it is known that cerebral arteries can produce 20-HETE when incubated with AA, express P450 4A1 and 4A2 mRNA, and contain immunoreactive 4A protein, it is not known which cell type in the vessel wall is actually responsible for the formation of 20-HETE.
20-HETE inhibits the activity of Ca2+-activated K+ channels (KCa) and depolarizes cerebral and renal arterial muscle (Ma et al. 1993; Harder et al. 1994; Zou et al. 1996). Thus, 20-HETE appears to function as an endogenous modulator of KCa. Such a modulatory role of 20-HETE on KCa was revealed when it was found that 17-octadecynoic acid (17-ODYA), a specific mechanism-based inhibitor of the metabolism of AA by P450 ω-hydroxylase, increased K+ channel activity in cerebral and renal vascular smooth muscle cells, and this effect was reversed by external addition of 20-HETE (Harder et al. 1994, 1995; Zou et al. 1996). However, there has been no definitive molecular and biochemical evidence indicating that cytochrome P450 4A enzymes are actually expressed in isolated cerebral arterial smooth muscle cells (VSMCs) of the cat, and that these cells metabolize exogenous AA and form 20-HETE. Therefore, the purpose of the present investigation was to determine whether freshly isolated cat cerebral VSMCs express mRNA for the P450 4A2 enzyme and produce 20-HETE when incubated with [14C]AA, using the reverse transcription-polymerase chain reaction (RT-PCR) and reverse-phase HPLC analysis, respectively.
The influx of Ca2+ and the subsequent rise in intracellular free Ca2+ is the principal mechanism by which many vasoconstrictor agonists evoke contraction in vascular smooth muscle (Catterall, 1988; Worley, Quayle, Standen & Nelson, 1991; Somlyo & Somlyo, 1994). Although 20-HETE is a potent constrictor of cerebral and renal arterioles, its effect on the macroscopic voltage-dependent Ca2+ channel current in vascular muscle cells has not been examined. We therefore set out to determine the effect of 20-HETE on voltage-activated, dihydropyridine-sensitive L-type Ca2+ currents recorded from cat cerebral arterial muscle cells using the conventional whole-cell voltage-clamp technique (Hamill, Marty, Neher, Sakmann & Sigworth, 1981). We also examined the effect of the L-type Ca2+ channel blocker, nifedipine, on the 20-HETE-induced constriction of isolated and pressurized cat cerebral arterial segments, and on the activation of the L-type Ca2+ channel by 20-HETE. The findings demonstrate that isolated cat cerebral VSMCs express mRNA for the P450 4A2 enzyme, convert exogenous [14C]AA to 20-HETE, and that 20-HETE potently activates cerebral arterial muscle by mechanisms which include enhanced inward current through L-type Ca2+ channels.
METHODS
Vascular muscle cell dispersion
Adult mongrel cats of either sex were deeply anaesthetized with Telazol (10 mg kg−1i.m.; Aveco Co. Inc., Fort Doge, IA, USA) and their brains removed. Cerebral microvessels (200–230 μm, o.d.) were dissected free of the arachnoid and placed in cold (4°C) low-Ca2+ dissociation buffer solution, the composition of which is shown below. Cerebral VSMCs were enzymatically dispersed as previously described (Gebremedhin, Kaldunski, Jacobs, Harder & Roman, 1996). Briefly, isolated arterial segments were cut into small pieces and placed in 2 ml vials containing an enzyme solution of the following composition (mm): NaCl, 134; KCl, 5.2; MgSO4, 1.2; KH2PO4, 0.33; CaCl2, 0.05; glucose, 11; Hepes, 10; with 100 units ml−1 collagenase (Class II, Worthington), 0.5 mg ml−1 bovine serum albumin (BSA), and 1 mg ml−1 trypsin inhibitor (Sigma). The enzyme solution containing the arterial pieces was maintained at 37°C and pH 7.4, and continually stirred at 12 r.p.m. for 45 min. Supernatant fractions were then collected every 5 min and diluted to 1 ml with physiological salt solution (PSS, pH 7.4) containing (mm): NaCl, 140; KCl, 5.4; MgCl2, 1.2; CaCl2, 1.8; glucose, 11; Hepes, 10. The procedure was repeated by incubating the remaining vascular tissue with fresh enzyme solution. This method provided a high yield of dissociated vascular smooth muscle cells. Fractions of the cell suspension were pooled, centrifuged at 3000 g, the supernatant decanted, and the pellet resuspended in appropriate buffer for the RT-PCR or for the assay of cytochrome P450 metabolites of arachidonic acid. Fractions of the dissociated cell suspensions were also kept at 4°C until used for whole-cell voltage-clamp studies.
Immunofluorescence staining of cerebral arterial smooth muscle cells
The freshly isolated cat cerebral arterial smooth muscle cells (VSMCs) were characterized and distinguished by immunofluorescence staining using Cy3-conjugated antibody to smooth muscle α-actin (anti-α-actin-Cy3; Sigma) as previously described (Harder et al. 1994; Gebremedhin et al. 1996). Briefly, the cerebral VSMCs were allowed to adhere to coverslips by incubating in culture media for 60 min at 37°C. After cell attachment, the coverslips were washed twice with ice-cold phosphate-buffered saline (PBS) containing 3% BSA and then once with PBS alone. The cells were fixed on the coverslips by treatment with cold methanol at −20°C for 5 min. The coverslips were then washed twice with ice-cold PBS, blocked with 3% normal goat serum (NGS; Sigma) and 0.1% BSA in PBS for 30 min, and incubated for 60 min with Cy3-conjugated antibody to smooth muscle α-actin at a dilution of 1: 500 in 0.3% NGS and 0.1% BSA in PBS. The unbound antibody was removed by four successive washes with PBS. The cells were dehydrated with ethanol and fixed using a drop of Gel Mount (National Diagnostics, Atlanta, GA, USA) on the coverslips for observation with a Nikon Diaphot TMD inverted microscope equipped for epifluorescence. Although the VSMCs can usually be distinguished from contaminating endothelial cells on the basis of morphology, we also employed indirect immunocytochemical staining to allow independent assessment of the purity of VSMCs. In the latter study, the VSMCs were fixed on coverslips and incubated with fluorescein isothiocyanate (FITC)-conjugated rabbit anti-human von Willebrand's factor (vWF) (a marker for endothelial cells which cross-reacts with rat vWF; Zymed Laboratories, San Francisco, CA, USA). The VSMCs did not show specific staining with the anti vWF antibody (image not shown), but exhibited high intensity staining with anti-α-actin-Cy3-conjugated antibody (Fig. 1A and B). The viability of the VSMCs was also demonstrated by the fact that they excluded Trypan Blue and contracted in response to high-K+ (30 mm) solution or serotonin (1 μM) and relaxed when these substances were removed from the media. Such freshly isolated VSMCs were used for reverse transcriptase-polymerase chain reaction (RT-PCR) studies of P450 4A mRNA expression, to assay the cytochrome P450 4A2 ω-hydroxylase metabolites of arachidonic acid, and for whole-cell voltage-clamp recording of inward Ca2+ currents.
Figure 1. Immunofluorescence with anti-α-actin-Cy3 conjugate on enzymatically isolated cat cerebral VSMCs.
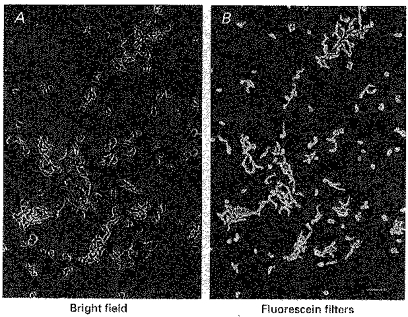
The VSMCs were stained with Cy3-conjugated antibody to smooth muscle α-actin. The appearance of the VSMCs under bright light is shown in A, and the corresponding fluorescence image is presented in B. These cells did not stain with the antibody to the endothelial marker von Willebrand's factor (vWF) (image not shown). Scale bar, 100 μm.
Cytochrome P450 metabolism of arachidonic acid by freshly dissociated cerebral vascular smooth muscle cells
Freshly isolated cat cerebral VSMCs were suspended in Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 0.01% bovine serum albumin (fatty acid free BSA; Sigma), and incubated at 37°C for 20 min with 0.05 μm of the calcium ionophore A23187 to deplete endogenous arachidonic acid. The vascular smooth muscle cell suspension was then centrifuged at 3500 g and the pellet (0.5-0.7 mg protein) washed twice with DMEM and incubated with [14C]arachidonic acid (0.5 μCi ml−1, 10 μm; New England Nuclear Research Products, Wilmington, DE, USA) for 2 h in 2 ml of DMEM. The incubation samples were placed in a closed Plexiglass box in a shaking water bath (200 r.p.m.) at 37°C and a stream of 95% O2- 5% CO2 gas was continuously blown over the surface of the incubation samples to provide adequate oxygenation. Vascular smooth muscle cell incubations were also performed in the presence of 17-octadecynoic acid (17-ODYA, 1 μm), a suicide substrate inhibitor of ω-hydroxylase enzymes (Ortiz de Montellano & Reich, 1984). Reactions were terminated by acidification with 0.1 m formic acid (pH 4.5), and arachidonic acid metabolites were extracted twice with 3 ml of ethyl acetate. The combined organic phase was back-extracted with 1 ml of distilled water to remove residual acid. The organic phase was evaporated to dryness under nitrogen and reconstituted in 0.5 ml ethanol. The reaction products were separated using a 2.1 mm × 25 cm C-18 reverse-phase HPLC column (Supelco LC-18) and a linear solvent gradient which was ramped from 30 parts acetonitrile, 70 parts water and 1 part acetic acid (v/v/v) to 100 parts acetonitrile and 1 part acetic acid (v/v) over 50 min at a flow rate of 0.5 ml min−1. Arachidonic acid metabolites were monitored using an on-line radioactive flow detector (Beckman System Gold, model 171).
Reverse transcriptase-polymerase chain reaction of P450 mRNA
Poly(A)+ mRNA was isolated from freshly dispersed cat cerebral arterial muscle cells (VSMCs) using Oligo dT sepharose columns (QuickPrep Micro mRNA Purification Kit (Pharmacia Biotech, Piscataway, NJ, USA), as per manufacturers instructions. Briefly, freshly isolated VSMCs were pelleted by centrifugation at 5000 g for 10 min. Cells were resuspended, in 8 m guanidinium isothiocyanate (GITC) buffer containing 0.1% N-lauryl sarcosine, and vortexed vigorously to achieve lysis. Oligo dT sepharose suspension (25 mg ml−1 slurry) was added to the lysate (1 : 1) and incubated for 10 min at room temperature. The sepharose was pelleted by pulse centrifugation, resuspended in high salt buffer (10 mm Tris-HCl (pH 7.5), 1 mm EDTA, 500 mm NaCl) and loaded onto micro mRNA spin columns (Pharmacia). The sepharose beads were washed 3 times with high salt buffer, and 3 times with low salt buffer (10 mm Tris-HCl (pH 7.5), 1 mm EDTA, 100 mm NaCl), and finally eluted by 200 μl RNAase free distilled water. Oligo dT primed mRNA (50 ng) was reverse transcribed for 1 h at 37°C with M-MuLV reverse transcriptase (Pharmacia First Strand cDNA Synthase Kit) in a volume of 33 μl. Aliquots (15 μl) of the reverse transcription reactions were used for PCR amplification in a volume of 100 μl containing 250 μm dNTP, 1.5 mm MgCl2, 50 pmol of each primer, and 2.5 units Taq DNA polymerase (Perkin Elmer Cetus, Norwalk, CT, USA). The sequences of the primers for rat cytochrome P450 4A2 were as follows:
Components of reactions excluding Taq DNA polymerase were mixed, overlaid with mineral oil, and heated to 95°C for 3 min, then cooled to 80°C for addition of Taq DNA polymerase and cycled for 40 cycles of the following protocol: 94°C for 3 min, 58°C for 1 min, 72°C for 1 min, ending with a 7 min extension at 72°C. Fifty microlitres of each reaction mixture was electrophoresed on a 1.5% agarose gel (100 V, 1.5 h) and visualized by ethidium bromide under UV light.
Gels were then denatured in 0.5 m NaOH, and vacuum blotted (BioRad, Hercules, CA, USA) onto Nytran Maximum Strength Plus (Schleicher & Schuell, Keene, NH, USA) nylon membranes in 10 × SSC (saline-sodium citrate buffer; Sigma) for 90 min. Blots were hybridized for 12 h at 42°C with fluorescein-11-dUTP 3′-end-labelled oligonucleotide probes (Amersham) complementary to the internal region of rat P450 4A2 PCR products. The probe sequence for 4A2 was:
(representing bases 1876–1900 of the published rat P450 4A2; Kimura, Hardwick, Kozak & Gonzalez, 1989). Stringency washes consisted of 2 times in 5 × SSC, 0.1% SDS, for 5 min at room temperature, and 2 times in 1 × SSC, 0.1% SDS for 15 min at 50°C. Blots were then incubated with anti-fluorescein horseradish peroxidase (HRP)-conjugated monoclonal antibody, washed, detected with Enhanced Chemiluminescence reagent (Amersham), and exposed to X-ray film reflections (Dupont New Products, Boston, MA, USA) for ∼30 s. The positive control template used for PCR reactions consisted of a full-length cDNA clone from rat kidney P450 4A2 in PCRII vector (Invitrogen, San Diego, CA, USA), amplified with the respective 4A primers as above. Negative control reactions for each primer pair consisted of: (1) RT with water, PCR with water; (2) PCR with non-reverse transcribed mRNA to exclude amplification of contaminating genomic DNA. DNA molecular weight markers (Pharmacia 100-bp Ladder) were used to estimate product size on agarose gels, and also as a negative control for Southern blots.
Cloning and sequencing of PCR products
PCR products were separated on 1.5% agarose (SeaKem LE), stained with ethidium bromide, and visualized under UV light. Bands were quickly excised from the gel and the DNA was recovered using a Qiaquick Gel Extraction Kit (Qiagen Inc., Chatsworth, CA, USA). Recovered fragments were ligated into pCRII using the T/A cloning kit (Invitrogen, Carlsbad, CA, USA) transformed into E. coli, INVαF', and plated on Luria agar (Luria Broth Base, powder) ampicillin plates (75 μg ml−1) containing X-gal (Gibco). The plasmid was purified from positive colonies using Qiagen Quickprep Spin Plasmid Kit columns (Invitrogen) and sequenced using the Texas Red dye-primer dideoxy chain termination method and cycle-sequencing with Thermo-Sequenase (Amersham). Fragments were separated on a denaturing polyacrylamide-urea gel (10%) and detected by Texas Red fluorescence using a Vistra model 725 DNA sequencer.
Isolated vessel studies
Isolated cat cerebral microvascular segments (8–10 mm in length, 0.20-0.30 mm, o.d.) were placed in a perfusion chamber, cannulated at both ends with glass micropipettes, and secured in place with 8–0 polyethylene suture (Ethicon Inc., Somerville, NJ, USA) using a stereo microscope. Side branches of the arteries were tied off with 10–0 polyethylene suture. The arterial segments were superfused with PSS aerated with a 95% O2- 5% CO2 gas mixture and were maintained at 37°C and pH 7.35. The inflow cannula was connected in series with a volume reservoir and a pressure transducer (Gould Instruments Division), to allow continuous monitoring of transmural pressure. The internal diameter of the arteries was measured using a video-microscopy system composed of a television camera and a videomicrometer, as previously described (Harder, 1984; Harder, Gilbert & Lombard, 1987). After an equilibration period of 15 min, the cannulated arteries were pressurized to 90 mmHg, and maintained at this pressure throughout the course of the experiment. Following an additional 15 min equilibration, the functional integrity of the vessels was verified by their contractile response to high KCl (30 mm) and to the direct relaxant sodium nitroprusside (1 μm). Following three successive washes, the arterial segments were allowed to equilibrate for an additional 15 min. The concentration-dependent contractile responses of the cerebral arterial preparations to synthetic 20-HETE (1–100 nm) were then determined in the absence and presence of the L-type Ca2+ channel blocker nifedipine (2 μm).
Electrophysiological recording
Macroscopic Ca2+ currents (ICa) were recorded at room temperature (22°C) using the conventional whole-cell voltage-clamp technique (Hamill et al. 1981) and Ba2+ as a charge carrier. Pipettes were fabricated from borosilicate glass pulled on a two-stage micropipette puller (PC-84) and heat-polished under a microscope. The patch pipettes were connected to the head stage of an EPC-7 patch-clamp amplifier (List Medical Electronics, Darmstadt, Germany), and mounted on a three-way hydraulic micromanipulator (Narishige, Tokyo, Japan) for placement of the tips on the cell membrane. The offset potentials between pipette and bath solutions were corrected with an offset circuit before each experiment. After attaining a high resistance seal (greater than 1 GΩ), access to the cell interior was accomplished by applying pulsatile suction between the pipette tips (3–6 MΩ) and cell membranes. The cell was dialysed with the intracellular solution containing (mm): CsCl, 135; MgATP, 5; Hepes, 5; EGTA, 1; the pH was adjusted to 7.2 with CsOH. The extracellular solution bathing the cells was composed of (mm): TEACl, 135; BaCl2, 5; MgCl2, 1; glucose, 11; Hepes, 10; 4-aminopyridine, 5; tetrodotoxin, 0.001; the pH was adjusted to 7.4 with tetraethylammonium hydroxide (TEAOH). Inward Ca2+ (ICa) currents were elicited (every 1 s) by 200 ms depolarizing pulses from a holding potential of −70 mV to test potentials between −50 and +50 mV in 10 mV increments. In an additional experiment, this inward Ca2+ current was recorded by varying the frequency of depolarization between 0.1 Hz (one pulse every 10 s) and 2 Hz (one pulse every 0.5 s), to determine the influence of rate of stimulation on the magnitude of this L-type Ca2+ channel current, and on the stimulatory action of 20-HETE on this same macroscopic Ca2+ current. Whole-cell Ca2+ currents were recorded with a List EPC-7 patch-clamp amplifier under control and test conditions. The signal was low-pass filtered at 1 kHz (−3 dB frequency) by an 8-pole Bessel filter and digitized at a sampling frequency of 2.5 kHz. Data were collected using a TL-1 DMA interface (Axon Instruments), stored on a 386sx Citus computer and analysed using pCLAMP software (Axon Instruments, Inc., pCLAMP versions 5.5 and 6.0).
Statistics
Data are presented as means ± s.e.m. Differences between groups were assessed using Student's t test or one-way analysis of variance (ANOVA) for multiple comparisons.
Drugs and chemicals
All chemicals were analytical grade. 20-HETE and 17-ODYA were obtained from Biomol (Plymouth Meeting, PA, USA). Sodium nitroprusside, adenosine 5′-triphosphate (magnesium salt), barium chloride, nifedipine and A23187 were all purchased from Sigma.
RESULTS
Cytochrome P450 metabolism of arachidonic acid by cat cerebral VSMCs
Reverse-phase HPLC analysis revealed that freshly isolated cat cerebral VSMCs produce a number of cytochrome P450 metabolites when incubated with exogenous [14C]AA (Fig. 2A-C). The primary cytochrome P450 metabolite formed by the VSMCs eluted at a retention time of 22.4 min, similar to the retention time of standard 20-HETE eluting under identical chromatographic conditions (Fig. 2B). The rate of production of 20-HETE by VSMCs averaged 3.9 ± 1.1 pmol min−1 (mg protein)−1 (n = 4). As shown in Fig. 2C, the formation of 20-HETE by VSMCs was blocked by pre-incubation of the cells with 17-ODYA (1 μm), a suicide substrate inhibitor of the ω-hydroxylation of arachidonic acid (Ortiz de Montellano & Reich, 1984).
Figure 2. Reverse-phase HPLC chromatograph illustrating the cytochrome P450 metabolism of [14C]arachidonic acid ([14C]AA) in isolated cat cerebral VSMCs.
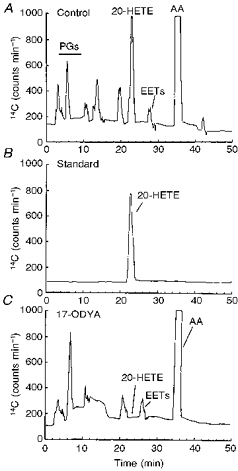
A, control chromatogram demonstrating the separation of arachidonic acid metabolites formed by cat VSMCs incubated with [14C]AA as described in the Methods. The peak eluting at 22.4 min has a retention time similar to that of standard 20-HETE shown in B. B, chromatogram demonstrating the elution profile of authentic 14C-labelled 20-HETE alone which eluted at 22.4 min. C, elution profile of P450 metabolites of AA demonstrating the effect of 17-octadecynoic acid (1 μm, 17-ODYA), a mechanism-based suicide substrate inhibitor of P450 ω-hydroxylase on P450 metabolism of exogenous [14C]AA by VSMCs. 17-ODYA (1 μm) blocked the formation of the major P450 metabolite with a retention time of 22.4 min. PGs, prostaglandins; EETs, epoxyeicosatrienoic acids.
RT-PCR identification of P450 4A2 mRNA in cat cerebral VSMCs
The mRNA for P450 4A2 in cat cerebral VSMCs was detected using the RT-PCR technique. Primers derived from a rat liver cytochrome P450 4A2 cDNA sequence (Genebank No. m33938) amplified a ∼439 bp cytochrome P450 4A2 mRNA in cat cerebral VSMCs. Primers for rat liver 4A1 and 4A3 were also used, but no products were observed (data not shown). Cloning and sequencing of this PCR product revealed 98% homology to the published sequence of rat and human P450 4A2 cDNA (Kimura et al. 1989; Palmer et al. 1993; Wang et al. 1996) (Fig. 3A). Southern blot hybridization of the PCR products obtained from cerebral arterial VSMCs demonstrated a strong signal band of the predicted size (∼439 bp) for P450 4A2 protein (Fig. 3B). These findings provide strong evidence for the expression of a P450 4A2 homologue in cat cerebral VSMCs. Previous investigations have demonstrated that P450 4A2 can catalyse the formation of 20-HETE from AA (Harder et al. 1994; Imig et al. 1996; Schwartzman, Da Silva, Lin, Nishimura & Abraham, 1996). The homologue identified in cat VSMCs in the present study appears to be responsible for the conversion of AA to 20-HETE in these cells.
Figure 3. Sequence of cytochrome P450 4A2 cDNA amplified from mRNA isolated from cat cerebral VSMCs using RT-PCR.

A, comparison of the amino acid sequences of the product amplified from reverse-transcribed mRNA extracted from cat cerebral VSMCs with those of the previously reported rat cytochrome P450 4A2 and 4A3 (Kimura et al. 1989). Amino acids are identified using the single-letter code. The identical amino acid residues are shown together in the black boxes, which revealed 98% homology to the P450 4A2 probe. B, Southern blot analysis of identical amounts of hybridized RT-PCR product amplified from mRNA isolated from cat cerebral VSMCs. The expected size of the band is about 439 bp.
Characterization of macroscopic Ca2+ (Ba2+) currents (ICa) in cat cerebral VSMCs
Inward Ca2+ currents carried by Ba2+ were recorded from freshly dispersed cat cerebral arterial smooth muscle cells with a high-Cs+ solution in the pipette, and a bath solution containing TEA and tetrodotoxin (1 μm) to block outward K+ currents and inward Na+ currents, respectively. Peak Ca2+ (Ba2+) currents of different amplitude elicited by a series of depolarizing pulses are shown in Fig. 4A and B. During step depolarization from a holding potential of −70 mV to test potentials between −50 and +50 mV an inward current carried by Ba2+ began to appear at −40 mV and peaked at +10 mV. Peak currents decreased between +10 and +50 mV, and the peak current-voltage relation started to reverse between +40 and +50 mV (Fig. 4B). In control experiments, Ca2+ currents carried by Ba2+ remained stable over a period of 20 min (data not shown). Furthermore, the magnitude of this inward macroscopic current was not influenced by altering the frequency of depolarization from slower to higher rates (Fig. 7). As shown in Fig. 4A and B this inward current was inhibited by > 90% by nifedipine (2 μm), the dihydropyridine L-type Ca2+ channel blocker. The relatively more positive voltage threshold of channel activation, slow activation and inactivation kinetics, and the sensitivity to blockade by nifedipine indicate that the inward current recorded from freshly dissociated cat cerebral VSMCs is a voltage-dependent, dihydropyridine-sensitive L-type Ca2+ channel current carried by Ba2+.
Figure 4. Characterization of macroscopic Ca2+ (Ba2+) currents (ICa) in cat cerebral VSMCs.
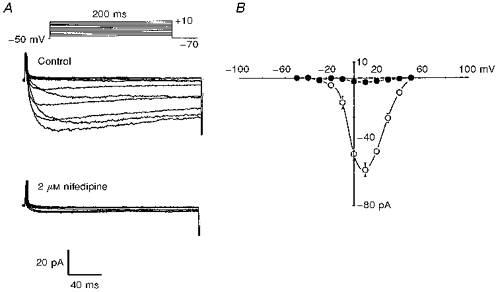
A, representative macroscopic Ca2+ currents carried by Ba2+ recorded from cat cerebral VSMCs evoked by 200 ms depolarizing pulses elicited from a holding potential of −70 mV to test potentials between −50 and +50 mV in 10 mV increments, before (upper panel) and after (lower panel) application of nifedipine to the bath. B, average peak current-voltage relation before (^) and after (•) application of nifedipine (n = 6, P < 0.001). Vertical bars denote s.e.m.
Figure 7. Effect of changes in frequency of stimulation on 20-HETE-induced activation of L-type Ca2+ channel current carried by Ba2+ in cat cerebral VSMCs.
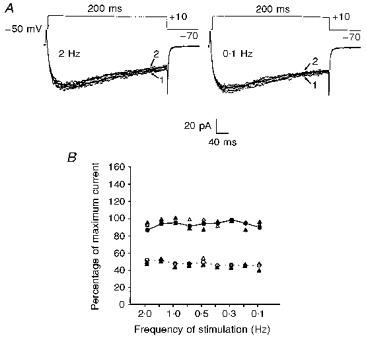
A, comparison of macroscopic inward L-type Ca2+ currents elicited by 10 repetitive 200 ms step pulses from −70 to +10 mV with frequencies of depolarization of 2 Hz and 0.1 Hz. The magnitudes of inward currents evoked using this depolarizing step pulse at rates of 2 Hz and 0.1 Hz are not significantly different. In both traces arrows 1 and 2 represent the first and last episodes of the voltage step regime, respectively. B, mean percentage of maximum 20-HETE-induced peak current evoked by a depolarizing test pulse from −70 to +10 mV plotted against increased frequency of stimulation. Application of 20-HETE (100 nm) significantly enhanced peak inward L-type Ca2+ current independent of increases in the frequency of stimulation from one pulse every 10 s (0.1 Hz) to one pulse every 0.5 s (2 Hz) (n = 3 cells from 3 cats, P < 0.001). Each point represents the mean percentage of maximum peak current of the series of steps from −70 to +10 mV before (open circles) and after (filled circles) stimulation with 100 nm 20-HETE. Filled triangles denote mean percentage of maximum peak current values of the first episode, whereas open triangles show mean percentage of maximum peak current of the last episode of the voltage step regime in either the control or after application of 20-HETE (100 nm). Error bars are embedded within the symbols.
Effect of 20-HETE on peak macroscopic Ca2+ (Ba2+) current (ICa)
Figure 5A and B depicts the concentration-related effects of externally applied 20-HETE (10 and 100 nm) on macroscopic peak ICa recorded from freshly dissociated cat cerebral arterial VSMCs as described in the Methods. 20-HETE, at a concentration of 10 or 100 nm, caused a significant increase in the magnitude of peak ICa, recorded during step depolarization between −50 and +50 mV in 10 mV increments from a holding potential of −70 mV, compared with the control. Thus 20-HETE increased peak ICa at +10 mV by 31% (from −56 ± 3.4 to −76 ± 5 pA, n = 7, P < 0.05) at 10 nm, and by 100% (from −56 ± 4 to −112 ± 7 pA, n = 7, P < 0.001) at 100 nm. This increase in ICa induced by 20-HETE was not associated with a shift in the peak current-voltage relation (Fig. 5B). The effect of 20-HETE on ICa was readily reversible on washout (data not shown). In an attempt to determine whether 20-HETE is activating ICa through voltage-dependent L-type Ca2+ channels, we examined the effect of 20-HETE in the presence of nifedipine (2 μm), the L-type Ca2+ channel blocker (Fig. 6A). Addition of nifedipine attenuated ICa by > 90% (before nifedipine, −52.3 ± 5 pA; after nifedipine, −3.8 ± 0.12 pA; n = 5, P < 0.001), and subsequent addition of 20-HETE (100 nm) had no stimulatory effect on the amplitude of the residual current (−4.6 ± 0.3 pA, n = 5). Bath application of 0.1% ethanol, the vehicle for 20-HETE, had no significant effect on peak ICa (before ethanol, −56 ± 4 pA; after ethanol, −57 ± 3 pA; n = 5, P > 0.05).
Figure 5. Activation of macroscopic Ca2+ current by 20-HETE.
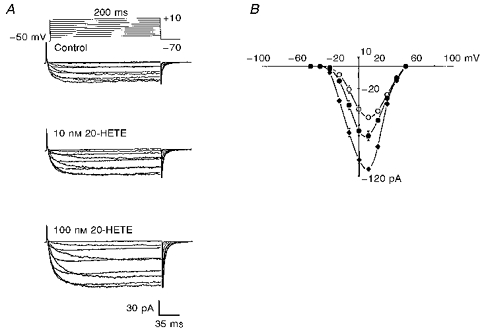
A, concentration-dependent activation of macroscopic Ca2+ currents, recorded from cat cerebral VSMCs elicited by 200 ms depolarizing pulses from a holding potential of −70 mV to test potentials between −50 and +50 mV in 10 mV increments, by 10 and 100 nm 20-HETE added to the bath. B, mean peak current-voltage relation before (^) and after 10 nm (•) and 100 nm 20-HETE (♦) applied to the bath (n = 7, P < 0.05). 20-HETE significantly enhanced the current without shifting the current-voltage relation. Vertical bars denote s.e.m.
Figure 6. Effect of nifedipine on 20-HETE-induced activation of macroscopic Ca2+ current carried by Ba2+ and constriction of isolated cerebral arterial segments.
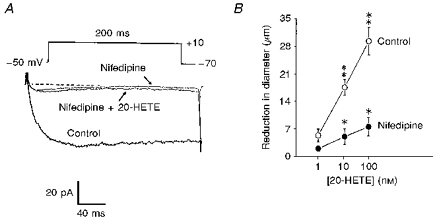
A, nifedipine (2 μm) applied to the bath abolished the macroscopic Ca2+ current elicited by a 200 ms depolarizing pulse from a holding potential of −70 mV to a test potential from −50 to +10 mV. Addition of 100 nm 20-HETE in the presence of nifedipine failed to enhance the macroscopic Ca2+ current (n = 6). B, effect of nifedipine (2 μm) on the 20-HETE-induced concentration-related constriction of isolated pressurized (90 mmHg) cat cerebral arterial segments. Nifedipine significantly depressed the 20-HETE-induced constriction of the arterial segments at all concentrations studied. Vertical bars denote s.e.m. Single asterisks represent significant differences in the concentration-related contractile effects of 20-HETE before and after application of nifedipine. Double asterisks represent significant differences between the contractile effects of low and high concentrations of 20-HETE studied under control conditions (n = 5, P < 0.05).
Effects of nifedipine on the vasoconstrictor actions of 20-HETE
The basal diameter of cerebral microvascular arterial segments pressurized at 90 mmHg averaged 210 ± 13 μm (n = 6). The effects of increasing concentrations of 20-HETE on the internal diameter of isolated pressurized (90 mmHg) cat cerebral microvessels, in the absence and presence of the Ca2+ channel antagonist nifedipine (2 μm) is depicted in Fig. 6B. Under control conditions, cumulative application of 20-HETE (1, 10 and 100 nm) to the bath produced a concentration-related reduction of the internal diameter of the cerebral arterial segments. Thus, the reduction in diameter induced by 20-HETE averaged (5 ± 1 μm at 1 nm, 18 ± 2 μm at 10 nm, and 29 ± 4 μm at 100 nm; n = 6). This is consistent with our previous observation which demonstrated the ability of 20-HETE to induce potent constriction of pial arterioles of the cat (Harder et al. 1994). The vasoconstrictor effect of 20-HETE reached a maximum after 2–5 min of application. In the presence of nifedipine (2 μM), the maximal contractile effect of 20-HETE was depressed by ∼75%. Nifedipine treatment did not alter the time course of 20-HETE-induced vasoconstriction (data not shown). These results suggest that influx of Ca2+ through the voltage-dependent L-type Ca2+ channel accounts, at least in part, for the activation of cat cerebral arteries by 20-HETE.
Effect of alterations in frequency of depolarization on the action of 20-HETE on L-type Ca2+ current
One possible mechanism by which 20-HETE can enhance L-type Ca2+ channel current could be through acceleration of recovery from inactivation. To address this possibility we compared the action of 20-HETE on L-type Ca2+ currents evoked by changing the frequency of depolarizing pulse steps from −70 to +10 mV. The results obtained under our experimental conditions revealed that no substantial inactivation of the channel was associated with either a higher or a lower rate of stimulation during depolarization with repetitive voltage steps from −70 to +10 mV (Fig. 7A). Furthermore, increasing the frequency of depolarization from one pulse per 10 s (0.1 Hz) to one pulse per 0.5 s (2 Hz) did not significantly influence the activation of L-type Ca2+ current caused by 100 nm 20-HETE (Fig. 7B, n = 3). Thus, application of 100 nm 20-HETE while the frequency of depolarization was varied between 0.1 Hz and 2 Hz in 0.1 Hz intervals induced a 92–100% increase in peak L-type Ca2+ current. These results appear to suggest that the action of 20-HETE on the L-type Ca2+ current in cat cerebral VSMCs is independent of the rate of stimulation between test pulses.
DISCUSSION
The results of the present investigation demonstrate expression of mRNA for cytochrome P450 4A2 ω-hydroxylase and the formation of 20-HETE in freshly dissociated cat cerebral VSMCs. We have previously demonstrated that homogenates of cerebral arterioles produce 20-HETE as determined by gas chromatography/mass spectrometry analysis (Harder et al. 1994). The studies presented here demonstrate that cerebral vascular muscle is an important source of 20-HETE. These findings are consistent with a number of previous studies which reported the expression of the protein for cytochrome P450 4A enzymes, and demonstrated that 20-HETE is the major cytochrome P450 ω-hydroxylase metabolite of arachidonic acid in cat cerebral, rat renal tubules and microvessels, and in the kidneys of the rat, rabbit and human (Johnson, Walker & Griffith, 1990; Harder et al. 1994; Imig et al. 1996; Schwartzman et al. 1996).
20-HETE is the major cytochrome P450 metabolite of AA found in cerebral and renal arteries, and is a potent activator of small cerebral and renal vessels with a threshold concentration of less than 10−1 0 m (Harder et al. 1994; Imig et al. 1996). The mechanisms of action of 20-HETE thus far defined include membrane depolarization and inhibition of the activity of the large-conductance, Ca2+-activated K+ (KCa) channel (Ma et al. 1993; Harder et al. 1994; Zou et al. 1996). In the present investigation we have identified the existence of a voltage-activated macroscopic Ca2+ current carried by Ba2+ in freshly isolated cat cerebral VSMCs using the conventional whole-cell voltage-clamp technique (Hamill et al. 1981). Based on its high-voltage threshold of channel activation, slow rate of inactivation over more depolarizing potentials and sensitivity to the dihydropyridine Ca2+ channel blocker nifedipine, this macroscopic Ca2+ channel current was identified as the L-type Ca2+ channel current, and is similar to that reported in a variety of tissues including cerebral arterial smooth muscle cells (Reuter, 1983; Catterall, 1988; Worley et al. 1991; Somlyo & Somlyo, 1994).
The potent vasoconstrictor action of 20-HETE in the cerebral and renal circulation has been established across species, and has been proposed to be the result of depolarization-induced Ca2+ influx secondary to inhibition of membrane KCa channels (Ma et al. 1993; Harder et al. 1994; Wang & Lu, 1995; Imig et al. 1996; Zou et al. 1996). Despite the fact that entry of Ca2+ appears to play a central role in the vasoconstrictor action of 20-HETE, the role of this divalent cation in coupling the contractile response of the cerebral arterial muscle to 20-HETE has not been investigated. A purpose of the present investigation was, therefore, to determine whether the vasoconstriction action of 20-HETE is associated with the influx of Ca2+ through the L-type Ca2+ channel. The results clearly show that 20-HETE enhances macroscopic L-type Ca2+ channel current (ICa) in cat cerebral VSMCs. It appears that 20-HETE enhances ICa by promoting periods of Ca2+ entry by increasing either the probability of opening or the number of active L-type Ca2+ channels. However, 20-HETE does not appear to enhance ICa by accelerating recovery from inactivation, as demonstrated in the present study. The regulation of the L-type Ca2+ channel by a P450 metabolite, such as 20-HETE, could provide evidence for the existence of an endogenous modulator of this channel in the cerebral vasculature of the cat.
To gain insight in the role of the voltage-activated Ca2+ channel, we examined the influence of the L-type Ca2+ channel antagonist nifedipine on the action of 20-HETE on cerebral vessels and the L-type Ca2+ channel current. Nifedipine markedly depressed the vasoconstrictor action of 20-HETE, and 20-HETE failed to enhance ICa following treatment of the voltage-clamped VSMCs with nifedipine. These findings provide convincing evidence indicating that 20-HETE activates the dihydropyridine-sensitive L-type Ca2+ channel to promote influx of Ca2+ and subsequent cerebral vasoconstriction. However, this antagonism by nifedipine of the 20-HETE effect does not rule out a possible contribution of entry and rise of Ca2+ through non-L-type Ca2+ channels, or the release of Ca2+ from unidentified internal stores (Catterall, 1988; Somlyo & Somlyo, 1994).
Previous studies have demonstrated that cerebral and renal arterial segments subjected to increases in intravascular pressure develop myogenic tone associated with corresponding increases in membrane depolarization (Harder, 1984; Harder et al. 1987). Such studies provide evidence for the involvement of electrical signalling mechanisms in the pressure-induced myogenic activation of arterial muscle. However, the intermediary factor(s) transducing the myogenic response, and the ionic mechanisms responsible for the resulting vasoconstriction, are not well defined. As the myogenic response is an inherent property of the vasculature (Bayliss, 1902), the existence in arterial muscle of an endogenous vasoconstrictor substance that acts principally by regulating the activities of voltage-gated membrane Ca2+ or K+ channels could be a potential mediator of the myogenic response. Interestingly, a previous study has shown that arachidonic acid potentiated, whereas inhibitors of the cytochrome P450 enzymes blocked, the myogenic response of isolated pressurized dog renal arteries to increases in intravascular pressure, and proposed that a contractile P450 metabolite of AA may mediate this response (Kauser et al. 1991). A subsequent study has demonstrated that microsomes prepared from dog renal arteries synthesize 20-HETE, which causes inhibition of KCa, depolarization, a rise in intracellular Ca2+ and arterial constriction (Ma et al. 1993). Such studies provided the original evidence suggesting that 20-HETE could be a transducing molecule of the myogenic response. However, this assumption remained partly unproven since the capacity of the VSMCs to produce 20-HETE was not known. Our present finding that isolated cerebral VSMCs of the cat express mRNA for the P450 4A2 enzyme and produce 20-HETE, which enhances Ca2+ current and evokes vasoconstriction, advances this previous notion, and suggests that 20-HETE could be a potential putative mediator of the myogenic response. However, such a physiological role for 20-HETE requires demonstration of parallel changes in its production level during increases in intravascular pressure or stretch. In this regard, the recently reported impairment of renal and cerebral blood flow autoregulation in vivo, by specific inhibitors of P450 4A ω-hydroxylase, unveils the functional significance of the P450 4A ω-hydroxylase metabolites of arachidonic acid in the cerebral and renal circulation, and suggests that 20-HETE could mediate the constriction of the arterioles in response to increases in perfusion pressure (Zou, Imig, Kaldunski, Ortiz de Montellano, Sui & Roman, 1994).
In conclusion, the results of the present investigation demonstrate that isolated cerebral VSMCs of the cat express the mRNA for the cytochrome P450 4A2 enzyme, and produce 20-HETE when incubated with [14C]AA. They further indicate that the L-type Ca2+ channel is one of the molecular targets for 20-HETE-induced cerebral vasoconstriction. Knowledge of the existence of a functional L-type Ca2+ channel and the expression of an endogenous modulator of this channel in the cerebral vascular smooth muscle cells will be useful for the development of new therapeutic tools for the management of cerebral disorders such as stroke and vasospasm.
References
- Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. Journal of Physiology. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. Structure and function of voltage-sensitive ion channels. Science. 1988;242:50–61. doi: 10.1126/science.2459775. [DOI] [PubMed] [Google Scholar]
- Dennis EA, Rhee SG, Billah M, Hannun YA. Role of phospholipases in generating lipid second messengers in signal transduction. FASEB Journal. 1991;5:2068–2077. doi: 10.1096/fasebj.5.7.1901288. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Kaldunski M, Jacobs ER, Harder DR, Roman RJ. Coexistence of two types of Ca2+-activated K+ channels in rat renal arterioles. American Journal of Physiology. 1996;270:F69–81. doi: 10.1152/ajprenal.1996.270.1.F69. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Ma Y-H, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. American Journal of Physiology. 1992;263:H519–525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Harder DR. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circulation Research. 1984;55:197–202. doi: 10.1161/01.res.55.2.197. [DOI] [PubMed] [Google Scholar]
- Harder DR, Campbell WB, Roman RJ. Role of cytochrome P450 enzymes and metabolites of arachidonic acid in the control of vascular tone. Journal of Vascular Research. 1995;32:79–92. doi: 10.1159/000159080. [DOI] [PubMed] [Google Scholar]
- Harder DR, Gebremedhin D, Narayanan J, Jefcoate C, Falck JR, Campbell WB, Roman RJ. Formation and action of a P-450 metabolite of arachidonic acid in cat cerebral microvessels. American Journal of Physiology. 1994;266:H2098–2107. doi: 10.1152/ajpheart.1994.266.5.H2098. [DOI] [PubMed] [Google Scholar]
- Harder DR, Gilbert R, Lombard JH. Vascular muscle cell depolarization and activation in renal arteries on elevation in transmural pressure. American Journal of Physiology. 1987;253:F778–781. doi: 10.1152/ajprenal.1987.253.4.F778. [DOI] [PubMed] [Google Scholar]
- Imig JD, Zou AP, Falck JR, Harder DR, Roman RJ. Formation and action of 20-HETE by renal microvessels. American Journal of Physiology. 1996;270:R217–227. doi: 10.1152/ajpregu.1996.270.1.R217. [DOI] [PubMed] [Google Scholar]
- Johnson EF, Walker DL, Griffith KJ. Cloning and expression of three rabbit kidney cDNAs encoding lauric acid omega-hydroxylases. Biochemistry. 1990;29:873–879. doi: 10.1021/bi00456a004. [DOI] [PubMed] [Google Scholar]
- Kauser K, Clark JE, Masters BS, Ortiz de Montellano PR, Ma Y-H, Harder DR, Roman RJ. Inhibitors of cytochrome P-450 attenuate the myogenic response of dog arcuate arteries. Circulation Research. 1991;68:1154–1163. doi: 10.1161/01.res.68.4.1154. [DOI] [PubMed] [Google Scholar]
- Kimura S, Hardwick JP, Kozak CA, Gonzalez FJ. The rat clofibrate-inducible CYP4A subfamily. II. cDNA sequence of IVA3, mapping of the CYP4A locus to mouse chromosome 4, and coordinate and tissue-specific regulation of the CYP4A genes. DNA. 1989;8:517–525. doi: 10.1089/dna.1.1989.8.517. [DOI] [PubMed] [Google Scholar]
- Ma Y-H, Gebremedhin D, Schwartzman ML, Falck JR, Clark JF, Masters BS, Harder DR, Roman RJ. 20-Hydroxyeicosatetraenoic acid is an endogenous vasoconstrictor of canine renal arcuate arteries. Circulation Research. 1993;72:126–136. doi: 10.1161/01.res.72.1.126. [DOI] [PubMed] [Google Scholar]
- Ordway RW, Singer JJ, Walsh JV., Jr Direct regulation of ion channels by fatty acids. Trends in Neurosciences. 1991;14:96–100. doi: 10.1016/0166-2236(91)90069-7. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Reich NO. Specific inactivation of hepatic fatty acid hydroxylases by acetylenic fatty acids. Journal of Biological Chemistry. 1984;259:4136–4141. [PubMed] [Google Scholar]
- Palmer CNA, Richardson TH, Griffin KJ, Hsu M-H, Muerhoff AS, Clark JE, Johnson EF. Characterization of a cDNA encoding a human kidney, cytochrome P-450 4A fatty acid ω-hydroxylase and the cognate enzyme expressed in Escherichia coli. Biochimica et Biophysica Acta. 1993;1172:161–166. doi: 10.1016/0167-4781(93)90285-l. [DOI] [PubMed] [Google Scholar]
- Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569–574. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- Schwartzman ML, Da Silva JL, Lin F, Nishimura M, Abraham NG. Cytochrome P450 4A and expression and arachidonic acid omega-hydroxylation in the kidney of the spontaneously hypertensive rat. Nephron. 1996;73:652–663. doi: 10.1159/000189154. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Wang MH, Stec DC, Balazy M, Mstuygin V, Yang CS, Roman RJ, Schwartzman ML. Cloning, and cDNA-directed expression of the rat renal CYP4A2: Arachidonic acid omega-hydroxylation and 11,12-epoxidation by CYP4A2 protein. Archives of Biochemistry and Biophysics. 1996;336:240–250. doi: 10.1006/abbi.1996.0554. [DOI] [PubMed] [Google Scholar]
- Wang W, Lu M. Effect of arachidonic acid on the activity of the apical K+ channel in the thick ascending limb of the rat kidney. Journal of General Physiology. 1995;106:727–743. doi: 10.1085/jgp.106.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley JF, Quayle JM, Standen NB, Nelson MT. Regulation of single calcium channels in cerebral arteries by voltage, serotonin, and dihydropyridines. American Journal of Physiology. 1991;261:H1951–1960. doi: 10.1152/ajpheart.1991.261.6.H1951. [DOI] [PubMed] [Google Scholar]
- Zou AP, Imig JD, Kaldunski M, Ortiz de Montellano PR, Sui Z, Roman RJ. Inhibition of renal vascular 20-HETE production impairs autoregulation of renal blood flow. American Journal of Physiology. 1994;266:F275–284. doi: 10.1152/ajprenal.1994.266.2.F275. [DOI] [PubMed] [Google Scholar]
- Zou A-P, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DH, Roman RJ. 20-HETE is an endogenous inhibitor of the large conductance Ca2+-activated K+ channel in renal arterioles. American Journal of Physiology. 1996;270:R228–237. doi: 10.1152/ajpregu.1996.270.1.R228. [DOI] [PubMed] [Google Scholar]