

Abstract
The Ile693Thr mutation of the skeletal muscle Na+ channel α-subunit is associated with an unusual phenotype of paramyotonia congenita characterized by cold-induced muscle weakness but no stiffness. This mutation occurs in the S4-S5 linker of domain II, a region that has not been previously implicated in paramyotonia congenita.
The Ile693Thr mutation was introduced into the human skeletal muscle Na+ gene for functional expression in human embryonic kidney (HEK) cells. The currents expressed were recorded with the whole-cell voltage-clamp technique.
In comparison with wild-type currents, Ile693Thr mutant currents showed a clear shift of about −9 mV in the voltage dependence of activation.
In contrast to other mutations of the Na+ channel known to cause paramyotonia congenita, the Ile693Thr mutation did not induce any significant change in the kinetics, nor in the voltage dependence, of fast inactivation.
In conclusion, this study provides further evidence of the involvement of the S4-S5 linker in the voltage dependence of Na+ channel activation. The negative shift in the voltage dependence found in this mutation must be associated to other defects, plausibly an impairment of the slow inactivation, to account for the long periods of muscle weakness experienced by the patients.
Mutations in the human Na+ channel α-subunit gene SCN4A have been implicated in three groups of autosomal dominant muscle disorders: hyperkalaemic periodic paralysis (hyper-PP), paramyotonia congenita (PC) and K+-aggravated myotonia (PAM) (Lehmann-Horn & Rüdel, 1996). To date, at least twenty Na+ channel mutations have been reported, all of which result in substitutions of highly conserved residues in the intracellular loops or in the transmembrane segments of the Na+ channel α-subunit (Feero et al. 1993; Ptacek et al. 1993; Plassart et al. 1994; Lehmann-Horn & Rüdel, 1996). Among these mutations, nine (including the present) have been associated with paramyotonia congenita (McClatchey et al. 1992; Ptacek et al. 1992, Plassart et al. 1996; Yang, Zhou, Ptacek, Barchi, Horn & George, 1994). Paramyotonia congenita is a myotonic disorder characterized by cold-induced muscle stiffness and weakness (Riggs & Griggs, 1979; Lehmann-Horn, Rüdel & Ricker, 1993). However, the clinical variability observed within families, as well as between different families carrying the same mutation, complicates the classification of PC (Ptacek et al. 1992; Lehmann-Horn et al. 1993; Plassart et al. 1994). Recently, we identified a new Na+ channel mutation (Ile693Thr) in a French family who exhibited an unusual phenotype, classified as a variant of PC, with cold-induced weakness but no stiffness (Plassart et al. 1996). One of the patients carrying the Ile693Thr mutation presented hyperPP in addition to PC: weakness could be induced by oral intake of K+, and a vacuolar myopathy usually described in hyperPP was found in muscle biopsies. The consequences of this mutation on Na+ channel functional properties are still unknown. In the present study, the Ile693Thr mutation was transiently expressed in human embryonic kidney (HEK 293) cells and the kinetics as well as the voltage dependence of the mutant channels were studied using the whole-cell voltage-clamp technique.
METHODS
Construction of the Ile693Thr recombinant
Site-directed mutagenesis was performed using the Quickchange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). A 1607 bp Sall-Sphl fragment of WT-pRc/CMV-hSKM1 was subcloned into the Puc19 vector (Chahine et al. 1994). Two synthetic oligonucleotide primers containing the Ile693Thr mutation were chosen:
and
The oligonucleotide primers, each complementary to opposite strands of the vector were extended during temperature cycling by means of Pfu DNA polymerase. A mutated plasmid containing staggered nicks was generated. Following temperature cycle, the product was treated by Dpnl and the parental vector was digested. The nicked vector incorporating the Ile693Thr mutation was then transformed into E. coli (i.e. XL2-Blue competent cells). The identity of the mutant clone was confirmed by DNA sequencing across the Sall-Sphl sites. The DNA sequence of the entire mutated fragment was determined and differed from the wild-type sequence only at the Ile693Thr mutation. The pRc/CMV-hSKM1 was then reassembled using the 1607 pb Sall-Sphl fragment from the Puc19 construct containing the mutant sequence. Plasmid DNA for mammalian transfection was prepared by adsorption to macroporous silica gel anion exchange column (Quiagen).
Transfection of HEK 293 Cells
The human embryonic kidney cells (HEK 293) were transfected using the Ca2+-phosphate precipitation method. Cells were grown at 37°C in Dulbelco's modified Eagle's medium supplemented with 10 % fetal bovine serum. When cells were 70 −80 % confluent, the Ca2+-phosphate-DNA mixture was added and left overnight (12–15 h). cDNA for the wild-type, or the Ile693Thr α-subunit was introduced into HEK cells together with pCMVCD20 plasmid which encodes a human cell surface marker CD20. Cells expressing this marker were then identified by immunolabelling with antibodies coupled to magnetic beads (Dynabeads M-280, Dynal, Oslo, Norway) which are readily detectable under light microscopy. This method improved substantially the detection of positive cells for electrophysiological investigation.
Electrophysiological recordings
Sodium currents were recorded 48–96 h following cell transfection with the conventional whole-cell voltage-clamp technique (Hamill, Marty, Neher, Sakmann & Sigworth, 1981) using an EPC-7 patch clamp amplifier (List-Medical). Currents were filtered through a 5 kHz, 8-pole low-pass Bessel filter (Frequency Devices 9L8L, Haverhill, MA, USA). An IBM-based system (486-DX, L.C.E., Neuilly, France) including a D/A and A/D converter (Labmaster, Axon Instruments) was used for pulse generation and signal acquisition with the pCLAMP software (V5.5, Axon Instruments). Currents were recorded at sampling intervals of 50 μs. Patch pipettes were pulled from capillary glass (Drummond Scientific Co., Broomall, PA, USA) and had a resistance of between 2 and 2.5 MΩ. To minimize space-clamp problems, cells selected for recording were small (10–20 μm diameter) and were isolated from other cells to prevent electrical coupling. Series resistance was examined throughout the experiments and systematically compensated. The average access resistance and voltage-clamp error obtained for the recordings used in this study were 4.4 ± 0.4 MΩ and 3.6 ± 0.4 mV, respectively. A P/6 protocol (Armstrong & Bezanilla, 1974) was used for linear subtraction of membrane capacity and leak currents. Data were analysed using both pCLAMP and FigP (Biosoft, Cambridge, UK) software. Patch pipettes were filled with a solution containing (mm): CsCl, 130; MgCl2, 2; glucose, 10; Na-Hepes, 10; pH 7.3. The standard bath solution for recording Na+ currents contained (mm): NaCl, 120; choline chloride, 20; KCl, 3; MgCl2, 2; CaCl2, 1; glucose, 10; Hepes, 10; pH 7.3. The osmolarity of all solutions used was kept between 300 and 320 mosmol l−1. Unless otherwise stated all chemicals were purchased from Sigma. All values are presented as means ±s.e.m. Statistical significance is given by Student's t test.
RESULTS
Voltage dependence of Na+ activation and fast inactivation kinetics in normal and high levels of external K+
Non-transfected HEK 293 cells are known to express low levels of Na+ channels (Ukomadu, Zhou, Sigworth & Agnew, 1992). Endogenous Na+ currents expressed in cells transfected only with pCMVCD20 plasmid had a peak amplitude of 0–194 pA (mean = 58.3 ± 7.4 pA, n = 33; mean cell capacitance 19.0 ± 1.0 pF). To minimize the contribution of the endogenous current to the total Na+ current expressed in cells co-transfected with cDNA encoding the wild-type or Ile693Thr α-subunit, only amplitudes exceeding 500 pA were considered in our results. Furthermore, to prevent voltage-clamp errors induced by large currents (up to 10 nA in our hands) and to allow a more reliable comparison between wild-type and mutant macroscopic current properties, only currents falling in the range of 0.5-2 nA were analysed.
Experiments were first carried out at room temperature (20–22°C), in the presence of physiological levels of extracellular K+ (3 mm) (Fig. 1A-C). The current-voltage relationship was obtained by applying a series of 50 ms voltage steps incremented by 5 mV from a holding potential of −100 mV. As shown in Fig. 1A and B, Na+ currents recorded from cells expressing wild-type channels activated near −40 mV and reached a maximum at 0 mV, whereas currents recorded from cells expressing the Ile693Thr mutant channel activated near −50 mV and peaked at −10 mV. A leftward shift of about 9 mV in the voltage dependence of the mutant Na+ channel activation as compared with the wild-type was estimated from the activation curves shown in Fig. 2A. The conductance (G/Gmax) versus voltage curves were first computed from individual I-V curves and fitted with a standard Boltzmann function, and then averaged. Mean values obtained for the mid-point of Na+ channel activation (V1/2) were −19.8 ± 1.5 mV (n = 7) and −29.0 ± 1.4 mV (n = 5) for the wild-type and the mutant channel, respectively. There was, however, no significant difference in the slope factor (k) between wild-type (6.5 ± 0.3) and mutant (7.3 ± 0.7). It is also worth noting that the reversal potential (ENa) extrapolated from Ile693Thr I-V curves (+71 ± 3 mV) was not significantly different from that obtained from the wild-type (+75 ± 7 mV), ruling out the possibility that the observed shift in the voltage dependence was due to electrode drifts.
Figure 1. Voltage dependence of activation and fast inactivation kinetics for wild-type and Ile693Thr mutant Na+ channel recorded in 3 mm (A-C) or 30 mm (D-F) external K+.
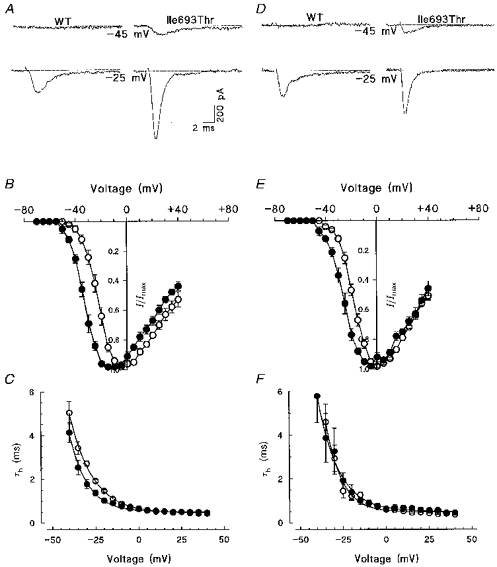
A and D, representative traces of wild-type and Ile693Thr mutant channels elicited at two different command potentials as indicated below the traces. Note the lower threshold for mutant channel activation than for wild-type. B and E, normalized current-voltage relationships obtained from HEK 293 cells expressing either wild-type (○, n = 7 and n = 5 in B and E, respectively) or Ile693Thr mutant channels (•, n = 8 and n = 5 in B and E, respectively); data are expressed as means ±s.e.m. C and F, voltage dependence of the fast inactivation time constant for wild-type (^) and mutant channels (•); data shown in C and F were respectively computed from the same recordings as those used in B and E.
Figure 2. Voltage dependence of steady-state inactivation and recovery from fast inactivation.
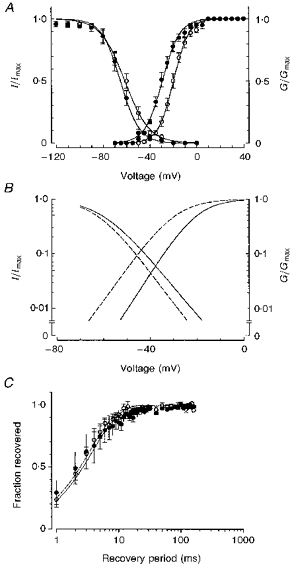
^, wild-type channels; •, mutant channels. A, steady-state inactivation (fraction of maximum current) and activation curves (fraction of maximum conductance) are plotted together to show the window current, enlarged in B. C, time course of Na+ current recovery from fast inactivation for wild-type and mutant channels.
The kinetics of current fast inactivation were also examined. Although wild-type and mutant Na+ currents generally inactivated with two exponential components, the fast component was widely predominant in both types (above 80 % at the peak). Ile693Thr currents inactivated with the same time constant (τh) as the wild-type with no significant difference in the voltage dependence of τh (Fig. 1C). The mean values of τh obtained at 0 mV were 0.66 ± 0.07 ms (n = 7) and 0.64 ± 0.05 ms (n = 5) for the wild-type and mutant channels, respectively.
Since in one patient carrying the Ile693Thr mutation, weakness followed oral intake of K+, Na+ currents were also examined in the presence of high external K+. Concentrations up to 30 mm were tested since accumulation of extracellular K+ in t-tubules may lead to much higher concentrations than those found in the blood during attacks (6–9 mm). The results obtained with 30 mm K+ were comparable with those obtained under physiological conditions (3 m) (Fig. 1D-F). The shift in the voltage dependence of Ile693Thr current activation was clearly confirmed in the presence of high K+ (Fig. 1D and E). Note that these experiments were independent of those performed with physiological concentrations of extracellular K+. The kinetics of current fast inactivation, as well as voltage dependence of τh, were not significantly modified by high external K+ for both wild-type and mutant channels (Fig. 1F). The mean values of τh obtained at 0 mV were 0.60 ± 0.02 and 0.65 ± 0.10 ms for wild-type and mutant, respectively.
Voltage dependence of Na+ fast inactivation and kinetics of recovery from inactivation
Voltage dependence of fast inactivation was studied by applying a series of 500 ms duration prepulse potentials (from −160 to 0 mV in 10 mV increments) prior to stepping to the test potential (−10 mV for 10 ms). Steady-state inactivation curves computed from these experiments were fitted with a single Boltzmann function (Fig. 2A). The slope factor was not modified by the Ile693Thr mutation (7.6 ± 1.2 (n = 5) compared with 8.4 ± 0.4 (n = 7) for the wild-type). However, the voltage for half-inactivation was slightly more negative in the mutant (−65.5 ± 2.8 mV, n = 5) than in the wild-type (−60.0 ± 0.9 mV, n = 7). Although this difference in the voltage dependence of steady-state inactivation between wild-type and mutant channels was not statistically significant, it may derive from the activation shift and reflect the coupling between activation and inactivation. In addition, as shown in Fig. 2A-B, the region of overlap between activation and steady-state inactivation curves was wider in the mutant than in the wild-type, suggesting a larger steady-state current for the mutant channels between −65 and −40 mV.
The kinetics of current recovery from fast inactivation were also studied. Cells were first depolarized to −10 mV for 20 ms (prepulse) and then held at the recovery potential (−100 mV) for increasing durations prior to stepping to the test potential (−10 mV) for 20 ms. There was no difference in the time course for current recovery from fast inactivation between wild-type and mutant channels. Recovery time course (fraction of current recovered versus time) was best fitted with a single exponential for both wild-type (time constant of 3.5 ms) and Ile693Thr mutant channels (time constant of 3.8 ms). To resolve better the first 10 ms of current recovery, the data are shown on a logarithmic time scale in Fig. 2C. Increasing extracellular K+ to 30 mm did not have any significant effect on voltage dependence of inactivation or recovery from fast inactivation in both wild-type and mutant channels (data not shown).
Steady-state current
The region of overlap between activation and steady-state inactivation curves was wider for the Ile693Thr Na+ channels than for the wild-type (Fig. 2B). This may indicate the presence of larger non-inactivating currents at negative potentials in the mutant. To examine this prediction, steady-state currents were measured at 20 ms following a series of depolarizing pulses ranging from −70 to +40 mV, and at 200 ms following step potentials to −10 and 0 mV. In spite of the small size of the steady-state currents and, hence, the variations introduced during their digitization, a current-voltage profile could be observed for both wild-type and Ile693Thr Na+ channels (Fig. 3). Our data do not show any clear difference in the steady-state currents between wild-type and mutant over the applied test potentials (Fig. 3A). Steady-state currents recorded at 200 ms were also comparable in wild-type and Ile693Thr mutant-expressing cells both at test potentials of −10 and 0 mV (Fig. 3C).
Figure 3. Temperature dependence of the steady-state current.
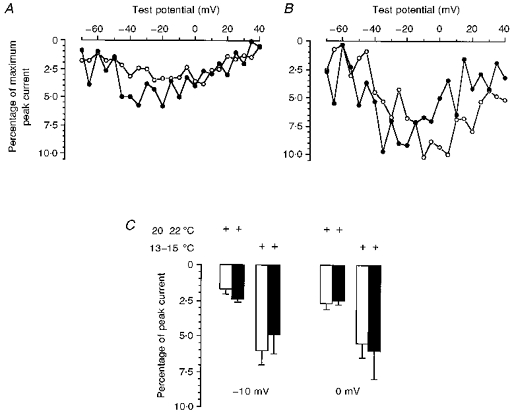
Current-voltage relationship of the steady-state current measured at 20 ms of varying voltage steps for wild-type (^) and Ile693Thr mutant (•) channels. A, data obtained at 20–22 °C for wild-type (n = 6) and mutant channels (n = 6). B, data obtained at 13–15 °C for wild-type (n = 5) and mutant channels (n = 6). C, steady-state currents measured at 200 ms following voltage steps to −10 mV. Recordings were performed at room temperature or after cooling to 13–15 °C as indicated. At both command potentials, open columns represent data obtained for wild-type (n = 6 and n = 6, at 20–22 °C and 13–15 °C, respectively), and closed columns show data obtained for Ile693Thr mutant (n = 4 and n = 6 at 20–22 °C and 13–15 °C, respectively).
Temperature dependence
The Ile693Thr mutation is associated with PC in which weakness is induced by cold. To compare our data with those previously reported for other Na+ channel mutations, the experiments were normally performed between 20 and 22°C. Although this represents a significant cooling from physiological conditions, no difference in channel fast inactivation was observed between the Ile693Thr and wild-type channels. To see whether more drastic cooling could unmask some channel dysfunction, bath temperature was further reduced to 13–15°C. This induced an increase (about 2- to 3-fold) in the steady-state currents measured at 20 or 200 ms that was similar in wild-type and Ile693Thr mutant-expressing cells (Fig. 3B and C). The voltage dependence of Ile693Thr current activation measured at 13–15°C exhibited the same shift of −9 mV as that observed at room temperature (V½ of −13 and −22 mV for wild-type and mutant channels, respectively) (Fig. 4).
Figure 4. Current-voltage relationships (A) and corresponding activation curves (B) obtained for wild-type and mutant channels at a temperature of 13–15 °C.
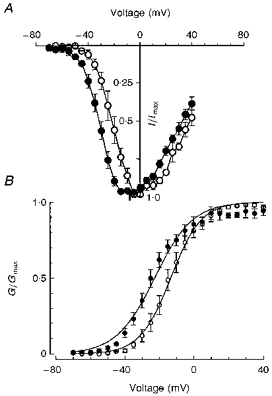
Note the negative shift in the voltage dependence of mutant (•, n = 6) compared with wild-type channels (^, n = 5).
DISCUSSION
To gain insight into the dysfunction produced by the PC-associated Ile693Thr Na+ channel mutation, mutant channels were constructed and transiently expressed in HEK 293 cells. Whole-cell currents were then recorded and compared with the wild-type. The results obtained show that the voltage dependence of activation of the Ile693Thr mutant channels is shifted by 9 mV towards negative potentials relative to the wild-type. A similar shift was observed at high extracellular K+ (30 mm) or after cooling to 13–15°C. This change in the voltage dependence of activation is reminiscent of that reported for the Thr704Met mutation, one of the most frequent mutations causing hyperPP (Cummins et al. 1993). The Thr704Met mutation lies in the S5 region of domain II, close to the Ile693Thr mutation which occurs in the intracellular loop linking segment S4-S5 of the same domain. The functional role of the S4-S5 region of the Na+ channel is poorly documented. Nevertheless, studies performed on the corresponding region of the K+ channel show that substitution of charged residues within the S4-S5 region can induce changes in the voltage dependence of activation in either the depolarizing or the hyperpolarizing direction (McCormack et al. 1991). The present study provides further evidence of the involvement of the S4-S5 region in the voltage dependence of Na+ channel activation. Apart from controversial reports concerning the Arg1448His and Arg1448Cys mutations (Chahine et al. 1994; Yang et al. 1994; Richmond, Featherstone & Ruben, 1997a), PC mutations do not seem to alter channel activation (Yang et al. 1994). Most of these mutations occur in the III-IV cytoplasmic linker or the IV S4 region (Ptacek et al. 1992; Hayward, Brown & Cannon, 1996) and are characterized by defects in Na+ channel fast inactivation such as slowed macroscopic rate, reduced voltage dependence and enhanced rate of recovery from inactivation (Yang et al. 1994; Chahine et al. 1994; Hayward et al. 1996). None of these alterations was found in the Ile693Thr mutation. It is worth noting that the Ile1160Val Na+ channel mutation, which lies in the domain III S4-S5 linker and causes sodium channel myotonia, has been shown to slow channel deactivation (Richmond, VanDeCarr, Featherstone, George & Ruben, 1997b). Although not addressed in the present work, alteration of Na+ channel deactivation seems unlikely since such alteration would induce myotonic discharges and muscle stiffness, which is not the phenotype produced by the Ile693Thr mutation (Richmond et al. 1997b).
A negative shift in the voltage dependence of Na+ channel activation should lower the threshold for action potential generation, thereby facilitating membrane depolarization. This change does not provide by itself a mechanism for the sustained depolarization that presumably underlies muscle weakness (Cummins et al. 1993). Prolonged depolarization could be explained by the coexistence of non-inactivating currents which would bring the sarcolemma into long refractory periods. However, analysis of the steady-state currents did not reveal any significant difference between wild-type and mutant channels.
The clue for the pathophysiological mechanism behind long periods of muscle weakness lasting several hours in patients carrying the Ile693Thr mutation may be found in a thorough examination of the slow inactivation process (Ruff, 1994). This view is supported by recent studies showing defective slow inactivation in hyperPP-associated Na+ channel mutations that cause muscle weakness (including Thr704Met) (Cummins & Sigworth, 1996; Hayward, Brown & Cannon, 1997). Interestingly, slow inactivation is preserved in two PC-associated mutations (Arg1448Cyst, Thr1313Met) characterized by myotonic discharges leading to muscle stiffness (Hayward et al. 1997; Richmond et al. 1997a).
Acknowledgments
We would like to thank Dr Y. Berwald-Netter for her valuable help in the co-transfection method. This work was supported by grants from DRED (French Ministry of Research), AFM (Association française contre les Myopathies) and ACCSV2 (French Ministry of Research). E. P.-S. received a fellowship from A. F. M.
References
- Armstrong CM, Bezanilla F. Charge movement associated with the opening and closing of the activation gates of the sodium channels. Journal of General Physiology. 1974;63:533–553. doi: 10.1085/jgp.63.5.533. 10.1085/jgp.63.5.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Sigworth FS. Impaired slow inactivation in mutant sodium channels. Biophysical Journal. 1996;71:227–236. doi: 10.1016/S0006-3495(96)79219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Zhou J, Sigworth FJ, Ukomadu C, Stephan M, Ptacek LJ, Agnew WS. Functional consequences of a Na+ channel mutation causing hyperkalemic periodic paralysis. Neuron. 1993;10:667–678. doi: 10.1016/0896-6273(93)90168-q. [DOI] [PubMed] [Google Scholar]
- Feero WG, Wang J, Barany F, Zhou J, Todorovic SM, Convit R, Galloway G, Hartlage P, Hayakawa H, Hoffman EP. Hyperkalemic periodic paralysis: rapid molecular diagnosis and relationship of genotype to phenotype in 12 families. Neurology. 1993;43:668–673. doi: 10.1212/wnl.43.4.668. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Inactivation defects caused by myotonia-associated mutations in the sodium III-IV linker. Journal of General Physiology. 1996;107:559–576. doi: 10.1085/jgp.107.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophysical Journal. 1997;72:1204–1219. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann-Horn F, Rüdel R. Molecular pathophysiology of voltage-gated ion channels. Reviews of Physiology, Pharmacology and Biochemistry. 1996;128:195–268. doi: 10.1007/3-540-61343-9_9. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Rüdel R, Ricker K. Workshop report: Non-dystrophic myotonia and periodic paralysis. Neuromuscular Disorders. 1993;3:161–168. doi: 10.1016/0960-8966(93)90009-9. [DOI] [PubMed] [Google Scholar]
- McClatchey AI, van der Berg P, Pericak-Vance MA, Raskind W, Verellen C, McKenna-Yasek D, Rao K, Haines JL, Bird T, Brown RH, Jr, Gusella JF. Temperature-sensitive mutations in the III-IV cytoplasmic loop region of the skeletal muscle sodium channel gene in paramyotonia congenita. Cell. 1992;68:769–774. doi: 10.1016/0092-8674(92)90151-2. [DOI] [PubMed] [Google Scholar]
- McCormack K, Tanouye MA, Iverson LE, Lin J, Rawaswani M, McCormack T, Campanelli JT, Mathew MK, Rudy B. A role for hydrophobic residues in the voltage dependent gating of shaker K+ channels. Proceedings of the National Academy of Sciences of the USA. 1991;88:2931–2935. doi: 10.1073/pnas.88.7.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassart E, Eymard B, Maurs L, Hauw JJ, Lyon-Caen O, Fardeau M, Fontaine B. Paramyotonia congenita: genotype to phenotype correlation in two families and report of a new mutation in the sodium channel gene. Journal of the Neurological Sciences. 1996;142:126–133. doi: 10.1016/0022-510x(96)00173-6. [DOI] [PubMed] [Google Scholar]
- Plassart E, Reboul J, Rime CS, Recan D, Millasseau P, Eymard B, Pelletier J, Thomas C, Chapon F, Desnuelle C, Confavreux C, Bady B, Martin JJ, Lenoir G, Serratrice G, Fardeau M, Fontaine B. Mutations in the muscle sodium channel gene (SCN4A) in 13 French families with hyperkalemic periodic paralysis and paramyotonia congenita: phenotype to genotype correlations and demonstration of the predominance of two mutations. European Journal of Human Genetics. 1994;2:110–124. doi: 10.1159/000472351. [DOI] [PubMed] [Google Scholar]
- Ptacek IJ, George AL, Jr, Barchi RL, Griggs RC, Riggs JA, Robertson M, Leppert MF. Mutations in an S4 segment of the adult skeletal muscle sodium channel causes paramyotonia congenita. Neuron. 1992;8:891–897. doi: 10.1016/0896-6273(92)90203-p. [DOI] [PubMed] [Google Scholar]
- Ptacek LJ, Gouw L, Kwiecinski H, McManis P, Mendell JR, Barohn RL, George AL, Barchi RL, Roberston M, Leppert MF. Sodium channel mutations in paramyotonia congenita and hyperkalemic periodic paralysis. Annals of Neurology. 1993;33:300–307. doi: 10.1002/ana.410330312. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Featherstone DE, Ruben PC. Human Na+ channel fast and slow inactivation in paramyotonia congenita mutants expressed in Xenopus laevis oocytes. Journal of Physiology. 1997a;499:589–600. doi: 10.1113/jphysiol.1997.sp021952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, VanDeCarr D, Featherstone DE, George AL, Jr, Ruben PC. Defective fast inactivation recovery and deactivation account for sodium channel myotonia in the I1160 V mutant. Biophysical Journal. 1997b;73:1896–1903. doi: 10.1016/S0006-3495(97)78220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs JE, Griggs RC. Diagnosis and treatment of the periodic paralyses. Clinical Neuropharmacology. 1979;4:123–138. [Google Scholar]
- Ruff RL. Slow Na+ channel inactivation must be disrupted to evoke prolonged depolarization-induced paralysis. Biophysical Journal. 1994;66:542. doi: 10.1016/s0006-3495(94)80807-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukomadu C, Zhou J, Sigworth FJ, Agnew WS. μ1 Na+ channels expressed transiently in human embryonic kidney cells: biochemical and biophysical properties. Neuron. 1992;8:663–676. doi: 10.1016/0896-6273(92)90088-u. [DOI] [PubMed] [Google Scholar]
- Yang N, Ji S, Zhou M, Ptacek L, Barchi RL, Horn R, George AL., Jr Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proceedings of the National Academy of Sciences of the USA. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]