

The muscular coat of the arteries reacts, like smooth muscle in other situations, to a stretching force by contraction.' (Bayliss, 1902)
The arterial wall is sheared and buffeted by the coursing of turbulent blood, and is stretched and compressed by pounding intravascular pressure. In response, these signals are often transformed from mechanical to electrical by the opening or closing of ion channels in endothelial and smooth muscle cells. One important mechanical stimulus, intravascular pressure, causes a graded membrane potential depolarization of the smooth muscle cells, which leads to vasoconstriction (‘myogenic tone’), independent of the vascular endothelium (Meininger & Davis, 1992). Although myogenic tone is a major contributor to vascular resistance to blood flow, the mechanisms by which smooth muscle cells transduce pressure to membrane potential depolarization have remained elusive.
Recent evidence suggests that intravascular pressure may depolarize and constrict myogenic cerebral arteries through activation of chloride channels (Nelson et al. 1997). Although calcium-activated chloride currents have been identified in certain types of smooth muscle (Large & Wang, 1996), this channel type does not seem to be involved in the pressure-induced depolarization that can occur even in relaxed arteries with low intracellular calcium (Knot & Nelson, 1995). The pharmacological profile was also inconsistent with the involvement of calcium-activated chloride channels. Furthermore, myogenic tone is associated with an activation of calcium-sensitive potassium currents (Brayden & Nelson, 1992; Nelson et al. 1995), which should overwhelm any activation of calcium-sensitive chloride channels. It was suggested that volume-regulated chloride channels might be a candidate for the depolarizing current to pressure (Nelson et al. 1997).
In this issue of The Journal of Physiology, Yamazaki et al. (1998) provide a major step forward in linking changes in mechanical forces with chloride currents by identifying volume-regulated chloride channels in smooth muscle. Hypotonic solutions activated a chloride-selective current in single smooth muscle cells isolated from canine pulmonary and renal arteries. These chloride currents were blocked by inhibitors (DIDS, ATP and the antioestrogen tamoxifen) of volume-regulated chloride currents in other preparations. The same group of investigators has recently provided evidence that a member of the ClC family of chloride channels, ClC-3, encodes for volume-regulated chloride currents in heart and other tissues (Duan et al. 1997). Yamazaki et al. (1998) also provide evidence that this molecular form of volume-regulated chloride channel (ClC-3) is also expressed in pulmonary and renal arterial myocytes.
This study provides a key new piece to the puzzle of the regulation of arterial smooth muscle membrane potential and sets the stage for the investigation of a number of important aspects of vascular biology, including the issue of whether ClC-3 alone can reconstitute all of the functional properties of the volume-regulated chloride current in smooth muscle. If indeed this volume-regulated chloride current is involved in the pressure-induced membrane depolarizations, then hypotonic solutions should also depolarize intact myogenic arteries with a similar pharmacological fingerprint. If this proves to be the case, then exploring the deeper issue of how transmural pressure activates this channel should provide fundamental insights into mechano-transduction in blood vessels.
Figure 1.
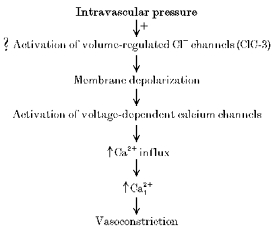
Proposed role of volume-regulated chloride channels in pressure-induced depolarization
References
- Bayliss WM. Journal of Physiology. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayden JE, Nelson MT. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Nature. 1997;390:417–421. doi: 10.1038/37151. 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Harder DR. Circulation Research. 1984;55:197–202. doi: 10.1161/01.res.55.2.197. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. American Journal of Physiology. 1995;269:H348–355. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- Large WA, Wang Q. American Journal of Physiology. 1996;271:C435–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Meininger GA, Davis MJ. American Journal of Physiology. 1992;263:H647–659. doi: 10.1152/ajpheart.1992.263.3.H647. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Conway MA, Knot HJ, Brayden JE. Journal of Physiology. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Duan D, Janiak J, Kuenzli K, Horowitz B, Hume JR. Journal of Physiology. 1998;507:729–736. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]