

Abstract
Calcitonin gene-related peptide (CGRP) is found in dense-cored vesicles in the motor nerve terminal.
Exogenous CGRP increased the size of the quanta. The increase in size reached a maximum after about 40 min. The lowest effective concentration of human CGRP (hCGRP) was 0.8 nm. The action of hCGRP was antagonized by (−)-vesamicol, a drug that blocks active acetylcholine (ACh) uptake into synaptic vesicles, so it appears that hCGRP increases size by adding more ACh to the quanta. The action of hCGRP was antagonized by drugs that block the activation of protein kinase A (PKA). (In other preparations CGRP also activates PKA.)
The hCGRP effect was not blocked by fragment 8–37, an antagonist of one class of CGRP receptor.
hCGRP increases evoked quantal output and miniature endplate potential (MEPP) frequency, again by activating PKA.
CGRP release was measured by radioimmunoassay. Release was increased by depolarization with elevated K+, but the amounts released appear to be below those needed to affect quantal size or output. Moreover, although elevated K+ can increase quantal size it acts by a pathway that does not involve PKA. We suggest that the most likely target of endogenously released CGRP is the regulation of circulation of the muscle.
Calcitonin gene-related peptide (CGRP) was first identified as a potent vasodilator (Brain, Williams, Tippins, Morris & MacIntyre, 1985). It contains thirty-seven amino acids, produced by alternative processing of RNA transcripts from the calcitonin gene (Amara, Jonas, Rosenfield, Ong & Evans, 1982). Calcitonin mRNA predominates in the thyroid whereas CGRP mRNA is found in tissues like the hypothalamus. CGRP is phylogenetically well conserved, there is a 75% identity between human and salmon forms. The structure of frog CGRP has not yet been reported. A related peptide, βCGRP, is a product of a separate gene (reviewed by Poyner, 1992).
Calcitonin gene-related peptide is found at many sites in the peripheral and central nervous systems (reviewed by Goodman & Iversen, 1986). After synthesis in the rat motoneuron cell body, CGRP is transported down the axon where it accumulates in the terminal (Kashihara, Sakaguchi & Kuno, 1989). In preparations from the rat it is released by motor nerve stimulation or by elevated K+ solution (Sakaguchi, Inaishi, Kashihara & Kuno, 1991; Uchida, Takami, Kobayashi, Hashimoto & Matsumoto, 1991; reviewed by Van der Kloot & Molgó, 1994). However, much of the CGRP may be released from the muscle fibres rather than the motor nerve (Kimura, Okazaki & Nojima, 1997). In the nerve, CGRP is accumulated in dense core vesicles, which are widely dispersed in the nerve terminals. It can be released from these dense core vesicles by exocytosis (Csillik, Tajti, Kovacs, Kukla, Rakic & Knyiharcsillik, 1993).
There are CGRP receptors on muscle fibres. In the mouse diaphragm, exogenous CGRP activates adenylate cyclase which leads to the upregulation of cAMP-activated protein kinase (PKA), which in turn enhances contractile strength (Kobayashi et al. 1987).
In 1-day-old Xenopus nerve-muscle cultures, 700 nm CGRP promptly increases the amplitude and duration of miniature endplate currents (MEPCs; Lu, Fu, Greengard & Poo, 1993; Lu & Fu, 1995). The change in the MEPC values is due to a lengthening in the burst duration of the embryonic acetylcholine (ACh) channels. As the cultures age, embryonic channels disappear and CGRP no longer alters MEPCs. Again the link between the CGRP receptor and the ACh channels is the activation of PKA.
The first indication that CGRP may also act on the motor nerve terminal was from experiments on the rat. A threshold dose of 50 nm rat CGRP increases the frequency of the miniature endplate potentials (MEPPs) without altering mean quantal content or amplitude (Jinnai, Chihara, Kanda, Tada & Fujita, 1989). The increase was greater with higher CGRP concentrations, the maximum increase being about 70%. The CGRP was applied by superfusion, but no information was given about the duration of the exposure.
At the frog neuromuscular junction (NMJ), permeable analogues of cAMP produce an increase in quantal size due to the activation of PKA. The size increases because more ACh is released in each quantum (Van der Kloot & Branisteanu, 1992). If the motor nerve terminals have CGRP receptors, and if activation of the receptors turns on PKA, then we would predict that the peptide should increase the quantal size. Testing this was our first objective. We show that human CGRP (hCGRP) does increase the quantal size. We also show that hCGRP increases evoked and spontaneous quantal output, as would be predicted for a PKA activator (Van der Kloot & Molgó, 1994). We tested a peptide, hCGRP8-37, which is an antagonist at one class of CGRP receptor, and found it ineffective.
We were also curious as to whether the release of endogenous CGRP alters the physiology of the neuromuscular junction. For example, one way to activate PKA is by exposing the preparations to hypertonic solutions. Hypertonic solutions greatly increase spontaneous quantal release, therefore MEPP frequency rises (Fatt & Katz, 1952; reviewed by Van der Kloot & Molgó, 1994). The mechanism by which hypertonic treatment activates protein kinase A is unknown. One hypothesis is that hypertonic treatment might also increase the rate of release of the dense core vesicles, thereby liberating CGRP. If the CGRP reached a sufficiently high concentration it would increase quantal size. Elevated K+ solutions also increase the rate of quantal release and are known to release CGRP from rat muscle (Sakaguchi et al. 1991; Uchida et al. 1991). High K+ has also been reported to increase quantal size (Van der Kloot, 1988), so it is possible that it acts by releasing endogenous CGRP. The experiments reported here are the first steps in trying to discover the role of endogenous CGRP release. They suggest that too little CGRP is released by hypertonic solutions of elevated K+ to alter quantal release or size.
METHODS
Experiments were performed on sartorius muscles from the frog, Rana pipiens. They were killed by double pithing, as specified by the Animal Users Committee of the State University of New York at Stony Brook. The Ringer solution contained (mm): NaCl, 120; KCl, 2.0; CaCl2, 2.5; and Tes, 4.0; at pH 7.4. For experiments on changing quantal size, 3 μm neostigmine methylsulphate and 0.1 μm TTX were added to the Ringer solution. For measurements of evoked quantal output the Mg2+-Ca2+ solution contained 2.5 mm MgCl2 and 0.1 mm CaCl2 with the remainder of the constituents as in Ringer solution. In many of the experiments one of the muscles was subjected to experimental manipulations, while the paired muscle from the same animal served as the control.
hCGRP and hCGRP8-37 were obtained from Sigma. Rp-cyclic adenylphosphorothioate (Rp-cAMPS) was from the Organic Synthesis Laboratory at the State University of New York at Stony Brook. Anandamide was from Cayman (Ann Arbor, MI, USA). hCGRP stock solutions, made in 0.1 m Tes buffer at pH 7.4, were stored in small vials in the freezer. They were thawed once only before use. Plastic pipette tips were siliconized, to avoid absorption of the peptide. The muscles were exposed to hCGRP in Ringer solution for 1 h, while pinned on a layer of Sylgard in a small Petri dish, then the MEPP values were measured in normal Ringer solution.
The electrophysiological methods have been described in detail and examples of the recordings have been published (Van der Kloot, 1987; Van der Kloot, Balezina, Molgó & Naves, 1994). Glass intracellular electrodes were bevelled (Lederer, Spindler & Eisner, 1979). Voltage and voltage clamp recordings were made with an Axoclamp 2A, leading to a Cyberamp 350 signal conditioner (both from Axon Instruments, Foster City, CA, USA) and then via a digital delay (Degan Instruments, Minneapolis, MN, USA) to an A/D converter (ComputerBoards, Mandfield, MA, USA). The converter was triggered by a transistor-transistor logic pulse from an Axon pulse discriminator.
As described by Van der Kloot (1987), the time integrals of the miniature endplate potentials (∫MEPPs) or miniature endplate currents (∫MEPCs) were used as the measure of quantal size. Roughly 100 miniatures were recorded from each of five randomly selected fibres in both the control and the experimental muscles. To determine whether the quantal size was significantly different between the two data sets, an ANOVA was performed on the log transformation of the ∫MEPP or ∫MEPC values. This method and the reason for using the logarithms are described and justified in Van der Kloot (1987). If P < 0.05, then the two sets are said to be significantly different. The method has been used extensively since its introduction and proven to be reliable in that it rarely suggests that recordings from paired muscles treated similarly are significantly different, but routinely detects the relatively large changes in quantal size produced by the experiments. Sets of ∫MEPPs or ∫MEPCs recorded from the same junction were tested for statistically significant differences by the Kolmogorov-Smirnov test. Again, if P < 0.05 then the differences are said to be significant. The error bars on all the graphs show the 95% confidence limit.
For studying evoked release with a low quantal output the solution was modified by adding 2.5 mm MgCl2 and reducing the CaCl2 to 0.1 mm. In these experiments the motor nerve was taken up into a suction electrode and stimulated with pulses fxrom a stimulator (Grass Instrument Co, Quincy, MA, USA) driving a stimulus isolation unit (WPI, Sarasota, FL, USA). The stimulus duration was usually 80 μs and the intensity was adjusted to well above threshold. The mean quantal outputs, m0, were estimated by the method of failures, counting the number of stimuli not followed by an EPC, n0, and the total number of stimuli, N. Then:
The standard error of the estimate (s.e.e.)
(Martin, 1966). Differences between mean quantal outputs were tested by Student's t test.
For the assay of released CGRP, isolated muscles were soaked in Ringer solution, 50 mm K+ Ringer solution (with Na+ reduced to 70 mm) or 200 mm NaCl Ringer solution for 2 h. The muscles were removed from the solutions, which were then assayed for hCGRP by radioimmunoassay (Peninsula Laboratories, Belmont, CA, USA). To check on the procedures for processing the solutions for analysis, known quantities of hCGRP were added to Ringer solution and then processed along with the samples. The known amounts were detected with good precision after processing by the assay.
RESULTS
Quantal size
Figure 1 shows the effects of 16 nm hCGRP on ∫MEPC values recorded at a single junction. There was a significant increase in quantal size after 14 min in hCGRP and the size continued to increase at the 23 min and 42 min time points. After 60 min of incubation size was no larger than it had been at 42 min. Over the period in which measurements were made the mean ∫MEPC had roughly doubled. A similar time course was observed in two additional experiments.
Figure 1. Increase in quantal size produced by hCGRP.
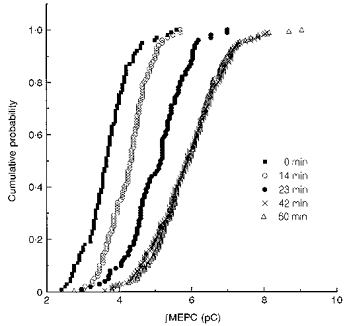
Cumulative plots showing the increase in the size of the ∫MEPC values at an endplate as a function of the time of exposure to 16 nm hCGRP. The times at which the recordings were begun are shown to the right of the plots.
The effects of 1 h incubation, in a series of successively lower concentrations of hCGRP, on the size of the ∫MEPP values are shown in Fig. 2. The lowest concentration that increased quantal size was 0.8 nm.
Figure 2. The effects of 1 h incubation in different concentrations of hCGRP on quantal sizes.
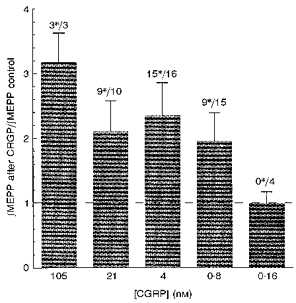
The bars show the ratio of the ∫MEPP values before and after the exposure. The error bars show the +95% confidence limits for the means of the ratios. In each experiment about 100 MEPP values were measured from 5 junctions in both the control and experimental groups. The figures above the bars show the number of experiments in which the experimental results were significantly different from the controls as measured by ANOVA (indicated by the *), followed by the total number of experiments.
Pre- or postjunctional action?
hCGRP might be increasing quantal size by altering the postjunctional response, so that more receptors are activated by a given dose of ACh, or because exposure to ACh generates a larger endplate current. Alternatively, the increase in quantal size could be due to a prejunctional change, which leads to more ACh being released in each quantum. This alternative can be tested, because the transport of ACh into the quanta is blocked by (−)-vesamicol (Anderson, King & Parsons, 1983; reviewed by Prior, Marshall & Parsons, 1992). Exposing untreated junctions to 1–5 μm vesamicol for several hours does not significantly alter quantal size (Van der Kloot, 1987; Prior et al. 1992). We soaked one of the paired muscles for 30 min in 2 μm vesamicol (from RBI), and then kept it for 1 h in 16 nM hCGRP plus 2 μm vesamicol (Fig. 3). The paired controls were exposed to hCGRP alone. After the treatments, quantal size in the preparations exposed to vesamicol was less than 60% of the size of the controls. This strongly suggests that hCGRP increased quantal size because more ACh was added to the quanta.
Figure 3. The effects of drugs on preparations exposed to hCGRP.
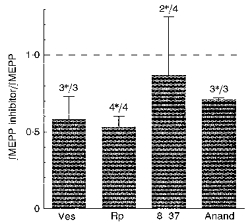
The conventions used are explained in the legend to Fig. 2. Ves, 2 μm (−)-vesamicol; Rp, 0.2 mm Rp-cAMPS; 8–37, 10 μm hCGRP8-37; Anand, 2 μm anandamide.
The cellular signalling pathway
The possibility that hCGRP produced its effect by activating PKA was evaluated by investigating the effects of Rp-cAMPS, which competitively blocks the activation of PKA by cAMP (Rothermal, Stec, Baraniak, Jastorff & Botelho, 1983; Van der Kloot & Branisteanu, 1992). The action of 4 nm hCGRP in increasing quantal size was antagonized by 0.2 mm Rp-cAMPS (Fig. 3). Experimental preparations were kept for 1 h in 0.2 mm Rp-cAMPS and then for 1 h in hCGRP plus Rp-cAMPS. Controls were soaked in the hCGRP alone. Exposure of untreated preparations to Rp-cAMPS does not alter quantal size (Van der Kloot & Branisteanu, 1992).
The activation of PKA at the frog neuromuscular junction is also antagonized by anandamide, which is a naturally occurring ligand for the cannabinoid receptor (Devane et al. 1992; Van der Kloot, 1994). In these experiments one of the pair of muscles was first soaked in 2 μm anandamide for 30 min and then in 2 μm anandamide plus 4 nm hCGRP for 1 hr. The controls were exposed to 4 nm hCRGP alone for 1 hr. The increase in quantal size produced by exposure to hCGRP was antagonized by the concurrent presence of 2 μm anandamide (Fig. 3). Exposing untreated preparations to 2 μm anandamide does not alter quantal size (Van der Kloot, 1994). These results strongly support the hypothesis that CGRP acts via a cellular signalling pathway that activates PKA in the motor nerve terminal.
Attempts to block CGRP with peptide 8–37
In some tissues the action of CGRP is antagonized by the hCGRP8-37 fragment (Dennis et al. 1990; reviewed by Poyner, 1992, 1995). To determine whether this is the case at the NMJ, paired preparations were exposed to either 4 nm hCGRP or to the same concentration of hCGRP with the addition of 10 μm hCGRP8-37. The paired controls were exposed to hCGRP alone. Overall, the mean quantal size was not significantly different between those exposed to hCGRP8-37 and the controls (Fig. 3). At the concentrations of hCGRP and of hCGRP8-37 used in these experiments, the fragment did not appear to antagonize the action of hCGRP.
To see whether we could obtain a block when the hCGRP concentration was lower, we repeated the experiments using 10 μm hCGRP8-37 and 0.8 nm hCGRP. After 1 hr soaking ∫MEPP values were recorded in Ringer solution. In five of six experiments the quanta were significantly larger in the muscles exposed to hGRP8-37 as well as hCGRP. The mean ∫MEPP after hCGRP + hCGRP8-37 was 3.2 times the ∫MEPP after the hCGRP alone. It appears, therefore, that at the frog NMJ the fragment potentiates the action of CGRP. The mechanism for this potentiation is unknown. By itself, 10 μm hCGRP8-37 did not alter quantal size (n = 3).
Quantal release
The results thus far suggest that CGRP activates PKA at the frog neuromuscular junction. PKA can also be activated by permeable analogues of cAMP. These drugs increase the frequency of the spontaneous release of MEPPs and the number of quanta released by nerve stimulation (Goldberg & Singer, 1969; Miyamoto & Breckenridge, 1974; reviewed by Van der Kloot & Molgó, 1994). Therefore we tested the effects of hCGRP on evoked quantal output.
In the first set of experiments mean quantal output, m0, was measured at five or six endplates in a preparation in Ca2+- Mg2+ solution, and then the measurements were repeated at five or six junctions starting 10 min after 4 nm hCGRP had been added to the solution. In three of thirteen experiments there was a significant increase in m0 in the presence of hCGRP. Overall, hCGRP increased the mean m0 for the entire series from 0.66 to 0.87. It appeared that hCGRP might increase evoked quantal output, but because of the scatter in m0 from fibre to fibre the increases were obscured.
Therefore, in the next series of experiments the m0 was measured at five to six endplates in an untreated muscle. Then the paired muscle was treated with 4 nm hCGRP and after 25–50 min m0 was measured again at five to six endplates. As shown in Fig. 4A the mean increase in m0 caused by the hCGRP was 2.6-fold (n= 5). This effect of hCGRP was also blocked by 0.2 mm Rp-cAMPS (Fig. 4A).
Figure 4. Effects of hCGRP on mean quantal output and on the frequency of MEPPs.
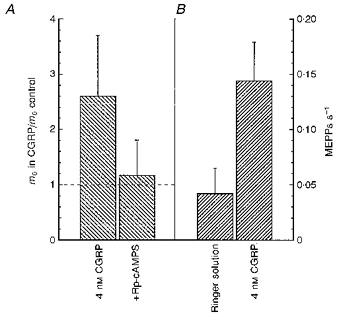
The error bars indicate the +95 % confidence limits for the means. A, mean quantal output (n= 5). B, MEPP frequency (n= 31).
The effect of 4 nm hCGRP on MEPP frequency was also tested. From experiments on thirty-one endplates exposed to CGRP for 30 min or more on average the frequency was 3.2-fold (Fig. 4B).
CGRP release
To obtain some idea of the amounts of endogenous CGRP released from frog neuromuscular preparations, we measured hCGRP by radioimmunoassay. Three experiments were conducted, using increasing numbers of muscle each time. In the third attempt, which used about 2 g of sartorius muscles (more than 20 muscles per sample) in each experimental condition, the results were as follows. In Ringer solution the release was equivalent to 1.12 pg hCGRP (g muscle wet weight)−1. In 50 mm K+ Ringer solution the release was elevated about 40-fold, to 45.2 pg g−1. In 200 mm NaCl, release was only slightly elevated, to 2 pg g−1. In the 2 ml of solution bathing the muscles in 50 mm K+ solution the concentration of hCGRP rose to about 0.01 nm, substantially below the concentrations required to increase quantal size or output. We conclude that the hypothesis (discussed in the Introduction) that hypertonic solutions release CGRP, which is responsible for the increase in quantal size is unlikely to be correct, as hypertonic treatments release little CGRP. Single rat soleus muscles in 50 mm K+ solution release about 2.5 pg CGRP in 7 min (Sakaguchi et al. 1991). This is difficult to compare with our data, but it seems that release is likely to be of the same order of magnitude.
Elevated K+ increases quantal size
It still seemed possible that 50 mm K+ solution releases enough CGRP in the immediate area around the nerve terminal to increase MEPP size. Elevated K+ was reported to trigger an increase in quantal size (Van der Kloot, 1988), but subsequent unpublished work showed that the increase was inconsistent. The reason for this became clear when it was found that raising the intracellular [Ca2+] by opening Ca2+ channels with depolarization activates PKC, which in turn blocks increases in quantal size (Van der Kloot, 1991). To determine if elevated K+ increases quantal size when PKC activity is depressed, we compared ∫MEPC sizes in paired preparations soaked for 1 h in 50 mm K+ Ringer solution or in the same solution containing 2 μm staurosporine to inhibit PKC, by the ANOVA method. To give time for the inhibitor to enter the nerve terminal, the staurosporine muscles were soaked in the same concentration of the drug in Ringer solution for between 2 and 12 h before exposure to elevated K+ staurosporine. The ratio of the ∫MEPC values in the elevated K+ staurosporine to those without staurosporine was 1.80 ± 0.49 (n= 6). In five of six experiments the ∫MEPC size was significantly increased in the preparation in staurosporine. In control experiments the ANOVA protocol was used to see whether 1 h exposure to 2 μm staurosporine alters quantal size. Three experiments of this type showed that the staurosporine alone had no detectable effect on quantal size. Elevated K+ can increase quantal size when PKC activation is blocked. If elevated K+ acts by releasing CGRP, then increases in quantal size should be blocked by Rp-cAMPS. In the next experiments we compared quantal size in preparations exposed for 1 h to 50 mm K+ Ringer solution containing 2 μm staurosporine. One of the paired muscles was also exposed to 0.2 mm Rp-cAMPS. In only one of five experiments was the quantal size significantly lower in the muscle exposed to Rp-cAMPS. The mean ratio of the ∫MEPP values in those exposed to the PKA inhibitor to those that were not was 1.02 ± 0.28 (n= 5). Therefore, it seems quite unlikely that elevated K+ increases quantal size by releasing CGRP.
DISCUSSION
Calcitonin gene-related peptide produces increases in quantal size at the frog neuromuscular junction by activating PKA. The increases in quantal size produced by CGRP at the frog neuromuscular junction are distinctly different from those produced by CGRP in cultured Xenopus nerve-muscle preparations (Lu, Fu, Greengard, & Poo, 1993; Lu & Fu, 1995). In the cultured preparations CGRP increases size in 2 or 3 min, while at the neuromuscular junction it takes about 40 min to reach an asymptote. Some of the time difference might be due to accessibility, but drugs in the solution generally reach superficial junctions on frog muscles within minutes. In cultured cells the size increases because ion channels in the ACh receptor stay open longer. At the junction the size increases are blocked by the presence of (−)-vesamicol, which strongly suggests that they are larger because more ACh is released in the quanta.
Human CGRP also increases evoked quantal output and spontaneous MEPP frequency at the frog neuromuscular junction, which is completely consistent with its ability to upregulate PKA (Van der Kloot & Molgó, 1994).
Human CGRP is the most potent agent found so far for increasing quantal size at the neuromuscular junction. Size increases were reliably produced by 0.8 nm hCGRP. The frog neuromuscular junction seems relatively sensitive to hCGRP compared with other tissues. The CGRP concentration required for half-maximal activation of adenylate kinase in bovine endothelial cells is 842 nm (McEwan, Ritter & MacDermot, 1989), whereas it is only 1.5 nm in rat L6 myocytes (Poyner, Andrew, Brown, Bose & Hanley, 1992). CGRP at 100 nm is required to increase release of labelled ACh from rat nerve-muscle preparations (Correia-de-Sá & Ribeiro, 1994).
Recently Kimura, Okazaki & Nojima (1997) reported that 1 μm CGRP depresses the release of labelled ACh from stimulated mouse nerve-muscle preparations. Much lower CGRP concentrations enhance quantal release in the frog. It is not clear whether the difference is due to the species studied or to the CGRP concentrations tested.
Despite the sensitivity of the frog neuromuscular junction to hCGRP, the results from radioimmunoassay suggest that the amounts of CGRP released by depolarizing the nerve terminals with elevated K+ are well below those required to affect either quantal size or release. The assay may be misleading because the concentration of CGRP in the synaptic cleft might be much higher than that in the bulk solution, and some of the released CGRP may be destroyed by the preparation. In future experiments it would be useful to test the effects of thiorphan, an enkephalase inhibitor which potentiates CGRP action in some tissues (Longmore, Hogg, Hutson & Hill, 1994). Moreover, frog CGRP may not be accurately measured because hCGRP was employed to produce the antibody used in the immunoassay. Nonetheless, our results strongly argue against the idea that either hypertonic solutions or elevated K+ solutions increase quantal size by releasing CGRP.
The best way to test for effects of the release of endogenous CGRP would be with a CGRP antagonist, so it was disappointing to find that hCGRP8-37 is not an effective agonist of hCGRP action at the neuromuscular junction. However, it is clear that there are several distinct CGRP receptors (Dennis et al. 1990; Poyner et al. 1992; Longmore et al. 1994; Poyner, 1995; Xu & Wiesenfeld-Hallin, 1996; Tomlinson & Poyner, 1996). Once the receptors are characterized it is likely that effective antagonists for the different classes will become available.
We can only speculate about the role played by CGRP release from motor nerve terminals. From the available data it seems unlikely that enough CGRP is released to affect quantal size or release. CGRP is an extremely potent vasodilator. Intradermal injection of femtomolar quantities of CGRP into human skin increases local blood flow for several hours (Brain et al. 1985). A reasonable hypothesis therefore is that the release of CGRP at the neuromuscular junction may enhance blood flow to the muscle.
Acknowledgments
This work was supported by Grant 10320 from the National Institute of Neurological Disorders and Stroke. We thank Judy Samarel for assistance.
References
- Amara SG, Jonas V, Rosenfeld MG, Ong ES, Evans RM. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature. 1982;298:240–244. doi: 10.1038/298240a0. [DOI] [PubMed] [Google Scholar]
- Anderson DC, King SC, Parsons SM. Inhibition of [3H]acetylcholine active transport by tetraphenylborate and other anions. Molecular Pharmacology. 1983;24:55–59. [PubMed] [Google Scholar]
- Brain SD, Williams TJ, Tippins JR, Morris HR, MacItyre I. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:73–75. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- Correia-de-Sá P, Ribeiro JA. Potentiation by tonic A(2A)-adenosine receptor activation of CGRP-facilitated [h-3]-ACH release from rat motor nerve endings. British Journal of Pharmacology. 1994;111:582–588. doi: 10.1111/j.1476-5381.1994.tb14777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csillik B, Tajti L, Kovacs T, Kukla E, Rakic P, Knyiharcsillik E. Distribution of calcitonin gene-related peptide in vertebrate neuromuscular junctions - relationship to the acetylcholine receptor. Journal of Histochemistry and Cytochemistry. 1993;41:1547–1555. doi: 10.1177/41.10.8245413. [DOI] [PubMed] [Google Scholar]
- Dennis T, Fournier A, Cadieux A, Pomerleau F, Jolicoeur FB, St. Pierre S, Quirion R. hCGRP8–37, a calcitonin-gene-related peptide antagonist revealing calcitonin gene-related peptide receptor heterogeneity in brain and periphery. Journal of Pharmacology and Experimental Therapeutics. 1990;254:123–128. [PubMed] [Google Scholar]
- Devane WA, Hanus I, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. Journal of Physiology. 1952;117:109–128. [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL, Singer JJ. Evidence for the role of cyclic AMP in neuromuscular transmission. Proceedings of the National Academy of Sciences of the USA. 1969;64:131–141. doi: 10.1073/pnas.64.1.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman EC, Iversen LL. Calcitonin gene-related peptide: Novel neuropeptide. Life Sciences. 1986;38:2169–2178. doi: 10.1016/0024-3205(86)90568-0. 10.1016/0024-3205(86)90568-0. [DOI] [PubMed] [Google Scholar]
- Jinnai K, Chihara K, Kanda F, Tada K, Fujita T. Calcitonin gene-related peptide enhances spontaneous acetylcholine release from the rat motor nerve terminal. Neuroscience Letters. 1989;103:64–68. doi: 10.1016/0304-3940(89)90486-2. 10.1016/0304-3940(89)90486-2. [DOI] [PubMed] [Google Scholar]
- Kashihara Y, Sakaguchi M, Kuno M. Axonal transport and distribution of endogenous calcitonin gene-related peptide in rat peripheral nerve. Journal of Neuroscience. 1989;9:3796–3802. doi: 10.1523/JNEUROSCI.09-11-03796.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Okazaki M, Nojima H. Mutual dependence of calcitonin gene-related peptide and acetylcholine release in neuromuscular preparations. European Journal of Pharmacology. 1997;330:123–128. doi: 10.1016/s0014-2999(97)01003-0. 10.1016/S0014-2999(97)01003-0. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Hashimoto K, Uchida S, Sakuma J, Takami K, Tohyama M, Izumi F, Yoshida H. Calcitonin gene-related peptide stimulates adenylate cyclase activity in rat striated muscle. Experientia. 1987;43:314–316. doi: 10.1007/BF01945565. [DOI] [PubMed] [Google Scholar]
- Lederer WJ, Spindler AJ, Eisner DA. Thick slurry bevelling. A new technique for bevelling extremely fine microelectrodes and micropipettes. Pflügers Archiv. 1979;381:287–288. doi: 10.1007/BF00583261. [DOI] [PubMed] [Google Scholar]
- Longmore J, Hogg JE, Hutson PH, Hill RG. Effects of two truncated forms of human calcitonin-gene related peptide: implications for receptor classification. European Journal of Pharmacology. 1994;265:53–59. doi: 10.1016/0014-2999(94)90222-4. [DOI] [PubMed] [Google Scholar]
- Lu B, Fu WM. Regulation of postsynaptic responses by calcitonin gene related peptide and ATP at developing neuromuscular junctions. Canadian Journal of Physiology and Pharmacology. 1995;73:1050–1056. doi: 10.1139/y95-149. [DOI] [PubMed] [Google Scholar]
- Lu B, Fu WM, Greengard P, Poo MM. Calcitonin gene-related peptide potentiates synaptic responses at developing neuromuscular junction. Nature. 1993;363:76–79. doi: 10.1038/363076a0. [DOI] [PubMed] [Google Scholar]
- McEwan JR, Ritter JM, MacDermot J. Calcitonin gene related peptide (CGRP) activates adenylate cyclase of bovine aortic endothelial cells: guanosine 5′ triphosphate dependence and partial agonist activity of a tyrosinated analogue. Cardiovascular Research. 1989;23:921–927. doi: 10.1093/cvr/23.11.921. [DOI] [PubMed] [Google Scholar]
- Martin AR. Quantal nature of synaptic transmission. Physiological Reviews. 1966;46:41–66. [Google Scholar]
- Miyamoto MD, Breckenridge BM. A cyclic adenosine monophosphate link in the catecholamine enhancement of transmitter release at the neuromuscular junction. Journal of General Physiology. 1974;63:609–624. doi: 10.1085/jgp.63.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyner DR. Calcitonin gene-related peptide: multiple actions, multiple receptors. Pharmacology and Therapeutics. 1992;56:23–51. doi: 10.1016/0163-7258(92)90036-y. [DOI] [PubMed] [Google Scholar]
- Poyner DR. Pharmacology of receptors for calcitonin gene-related peptide and amylin. Trends in Pharmacological Sciences. 1995;16:424–428. doi: 10.1016/s0165-6147(00)89093-8. [DOI] [PubMed] [Google Scholar]
- Poyner DR, Andrew DP, Brown D, Bose C, Hanley MR. Pharmacological characterization of a receptor for calcitonin gene-related peptide on rat, L6 myocytes. British Journal of Pharmacology. 1992;105:441–447. doi: 10.1111/j.1476-5381.1992.tb14272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior C, Marshall IG, Parsons SM. The pharmacology of vesamicol - an inhibitor of the vesicular acetylcholine transporter. General Pharmacology. 1992;23:1017–1022. doi: 10.1016/0306-3623(92)90280-w. [DOI] [PubMed] [Google Scholar]
- Rothermel JD, Stec WJ, Baraniak J, Jastorff B, Botelho LHP. Inhibition of glycogenolysis in isolated rat hepatocytes by the Rp diastereomer of adenosine 3′,5′-phosphorothioate. Journal of Biological Chemistry. 1983;258:12125–12128. [PubMed] [Google Scholar]
- Sakaguchi M, Inaishi Y, Kashihara Y, Kuno M. Release of calcitonin gene-related peptide from nerve terminals in rat skeletal muscle. Journal of Physiology. 1991;434:257–270. doi: 10.1113/jphysiol.1991.sp018468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson AE, Poyner DR. Multiple receptors for calcitonin gene-related peptide and amylin on guinea-pig ileum and vas deferens. British Journal of Pharmacology. 1996;117:1362–1368. doi: 10.1111/j.1476-5381.1996.tb16737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Takami K, Kobayashi H, Hashimoto K, Matsumoto N. Functions of a co-transmitter, calcitonin gene-related peptide, on the neuromuscular junction. Advances in Experimental Medicine and Biology. 1991;287:39–50. doi: 10.1007/978-1-4684-5907-4_4. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W. Pretreatment with hypertonic solutions increases quantal size at the frog neuromuscular junction. Journal of Neurophysiology. 1987;57:1536–1554. doi: 10.1152/jn.1987.57.5.1536. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W. The packing of acetylcholine into quanta at the frog neuromuscular junction is inhibited by increases in intracellular sodium. Pflügers Archiv. 1988;412:258–263. doi: 10.1007/BF00582506. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W. Down-regulation of quantal size at frog neuromuscular junctions: possible roles for elevated intracellular calcium and for protein kinase C. Journal of Neurobiology. 1991;22:204–214. doi: 10.1002/neu.480220210. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W. Anandamide, a naturally-occurring agonist of the cannabinoid receptor, blocks adenylate cyclase at the frog neuromuscular junction. Brain Research. 1994;649:181–184. doi: 10.1016/0006-8993(94)91062-6. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W, Balezina OP, Molgó J, Naves LA. The timing of channel opening during miniature endplate currents at the frog and mouse neuromuscular junctions: effects of fasciculin-2, other anti-cholinesterases and vesamicol. Pflügers Archiv. 1994;428:114–126. doi: 10.1007/BF00374848. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W, Branisteanu DD. Effects of activators and inhibitors of protein kinase A on increases in quantal size at the frog neuromuscular junction. Pflügers Archiv. 1992;420:336–341. doi: 10.1007/BF00374467. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W, Molgó J. Quantal acetylcholine release at the vertebrate neuromuscular junction. Physiological Reviews. 1994;74:899–991. doi: 10.1152/physrev.1994.74.4.899. [DOI] [PubMed] [Google Scholar]
- Xu X-J, Wiesenfeld-Hallin Z. Calcitonin gene-related peptide (8–37) does not antagonize calcitonin gene-related peptide in rat spinal cord. Neuroscience Letters. 1996;204:185–188. doi: 10.1016/0304-3940(96)12351-x. [DOI] [PubMed] [Google Scholar]