

Abstract
Epithelial Na+ channels (ENaCs) are inhibited by the cystic fibrosis transmembrane conductance regulator (CFTR) when CFTR is activated by protein kinase A. Since cAMP-dependent activation of CFTR Cl− conductance is defective in cystic fibrosis (CF), ENaC currents are not inhibited by CFTR. This could explain the enhanced Na+ conductance found in CF. In the present study, we examined possible mechanisms of interaction between CFTR and ENaC co-expressed in Xenopus oocytes.
The magnitude of CFTR Cl− currents activated by 3-isobutyl-1-methylxanthine (IBMX) in oocytes co-expressing either wild-type or mutant CFTR and ENaC determined the degree of downregulation of ENaC currents.
The ability of CFTR to inhibit ENaC currents was significantly reduced either when extracellular Cl− was replaced by poorly conductive anions, e.g. SCN− or gluconate, or when CFTR was inhibited by diphenylamine-carboxylate (DPC, 1 mmol l−1).
Downregulation of ENaC was more pronounced at positive when compared with negative clamp voltages. This suggests that outward currents, i.e. influx of Cl− through activated CFTR most effectively downregulated ENaC.
Activation of endogenous Ca2+-activated Cl− currents by 1 μmol l−1 ionomycin did not inhibit ENaC current. This suggests that inhibition of ENaC mediated by Cl− currents may be specific to CFTR.
The present findings indicate that downregulation of ENaC by CFTR is correlated to the ability of CFTR to conduct Cl−. The data have implications for how epithelia switch from NaCl absorption to NaCl secretion when CFTR is activated by secretagogues.
Transport of Na+ and Cl− is a major task of epithelial cells and requires regulation of both Cl− and Na+ conductive pathways (Greger, Kunzelmann & Gerlach, 1990). Both types of conductance pathways, the cystic fibrosis transmembrane conductance regulator (CFTR) and epithelial Na+ channels (ENaC) (Riordan et al. 1989; Canessa et al. 1994), are co-expressed in apical membranes of reabsorptive epithelial cells. Along these lines, it has been found that NaCl secretion mainly takes place in base cells of colonic crypts and in airway submucosal glands, whilst surface cells absorb NaCl (Welsh, Smith, Fromm & Frizzell, 1982; Ballard, Fountain, Inglis, Corboz & Taylor, 1995; Zhang, Yankaskas, Wilson & Engelhard, 1996). However, this scheme of local separation of NaCl absorption and secretion does not take into consideration the fact that under some conditions NaCl transport must be redirected within one given cell. This has been shown, for example, for the cells of the mid-crypt of the colon, and even surface cells of the colonic crypt can be stimulated to secrete NaCl. These cells can therefore shift from NaCl absorption to secretion (Köckerling & Fromm, 1993; Greger et al. 1996; Greger, Bleich, Leipziger, Ecke, Mall & Kunzelmann, 1997).
Epithelial Na+ channels are inhibited during activation of Cl− secretion in cells expressing both CFTR and ENaC (Stutts et al. 1995; Ecke, Bleich & Greger, 1996; Mall, Hipper, Greger & Kunzelmann, 1996; Letz & Korbmacher, 1997). These results suggest that when CFTR is activated by cAMP, CFTR may inhibit ENaCs. We have shown that CFTR and ENaC may directly interact by protein-protein binding (Kunzelmann, Kiser, Schreiber & Riordan, 1997). Two groups detected inhibition of ENaC single channel currents by CFTR in isolated and purified membranes (Ismailov et al. 1996; Stutts, Rossier & Boucher, 1997) while other studies in salivary gland duct cells demonstrate inhibition of ENaC by enhanced cytosolic Na+ and Cl− activities (Komwatana, Dinudom, Young & Cook, 1996). In the present report we examined further whether downregulation of ENaC depends on the ability of CFTR to conduct Cl− ions. The data indicate that Cl− movement through the activated CFTR Cl− conductance is essential for the inhibition of ENaC. In accordance with these data we propose a model in which activation of CFTR may switch epithelial NaCl transport from absorption to secretion and which also might explain the enhanced Na+ conductance in the epithelial tissues of patients suffering from cystic fibrosis (CF).
METHODS
Preparation of cRNA for CFTR and the rat ENaC
A 4.7 kb cDNA sequence encoding wild-type (wt) human CFTR (if not specified in this text, CFTR always refers to the wild-type) was subcloned into p-Bluescript vector (Stratagene) using the restriction sites KpnI and NotI and amplified in E. coli (XL1-Blue, Stratagene). The three (α, β and γ) subunits of the rat amiloride-sensitive Na+ channel (ENaC, kindly provided by Professor Dr B. Rossier, Pharmacological Institute of Lausanne, Switzerland) were subcloned into pBluescript, linearized with Not1 and in vitro transcribed using T7 promoter and polymerase. For mutagenesis, a 2.1 kb EcoRI CFTR fragment, comprising the first membrane spanning domain (MSD1) and the first nucleotide binding fold (NBF1), was subcloned into p-Alter vector (Promega, Heidelberg, Germany) and single stranded cDNA was obtained by helper phage R408. Mutagenesis was performed according to Hipper, Mall, Greger & Kunzelmann (1995) and correct sequences were confirmed by the cycle sequencing kit (PRISM, Perkin Elmer) with an automated sequencer (Perkin Elmer). The following CFTR mutations were generated: CF-associated mutations such as ΔF508, G551D and R117H as well as artificial mutations within MSD1 such as R347E and K335E (Hipper et al. 1995). For in vitro transcription of cRNA, plasmids were linearized with KpnI, and cRNA was synthesized using T7 promoter and T7 polymerase and a 5′cap (mCAP mRNA capping kit, Stratagene).
Preparation of oocytes and microinjection of cRNA
Isolation and microinjection of oocytes have been described in a previous report (Busch et al. 1996). In brief Xenopus laevis female frogs (H. Kähler, Bedarf für Entwicklungsbiologie, Hamburg, Germany) were anaesthetized in a tank containing 3-aminobenzoic acid ethyl ester (Sigma, 1 g l−1). The frogs were bedded on ice and oocytes were obtained after opening the abdominal cavity. The oocytes were dispersed and defolliculated by a 1 h treatment with collagenase (type A, Boehringer). Subsequently, oocytes were rinsed 10 times and kept in ND96-buffer (mmol l−1): NaCl, 96; KCl, 2; CaCl2, 1.8; MgCl2, 1; Hepes, 5; sodium pyruvate, 2.5; pH 7.55), supplemented with theophylline (0.5 mmol l−1) and gentamicin (5 mg l−1) at 18°C. After collection of the ooyctes, the 1 cm abdominal opening was closed by stitching muscle and skin separately (three stitches each). Afterwards the frog was allowed to recover in a separate water bath and put back into the main aquarium after 5–8 h. Oocytes of identical batches were injected with α, β and γ subunits of ENaC (each subunit 10 ng) and CFTR (20 ng) cRNA dissolved in about 50 nl double-distilled water (PV830 pneumatic pico pump, WPI, Germany). Oocytes injected with 50 nl double-distilled water served as controls.
Electrophysiological analysis of Xenopus oocytes
Two to four days after injection oocytes were impaled with two electrodes (Clark Instruments), which had resistances of ≤ 1 MΩ when filled with 2.7 mol l−1 KCl. A flowing (2.7 mol l−1) KCl electrode served as bath reference in order to minimize junction potentials. As junction potentials were close to zero when bath Cl− was replaced by either gluconate or SCN−, I–V relationships have not been corrected. Membrane currents were measured by voltage clamping of the oocytes (OOC-1 amplifier, WPI, Germany) in 10 or 20 mV intervals from -90 to +40 mV, each 250–1000 ms long. Current data were filtered at 400 Hz (OOC-1 amplifier). Between voltage steps, oocytes were voltage clamped at their membrane voltage for 20 s. Zero current membrane potential was assessed after every solution exchange. Data were collected continuously on a computer hard disk at a sample frequency of 1000 Hz and displayed on a computer screen. Data were analysed using the programs Chart and Scope (McLab, AD-Instruments). If not stated otherwise conductances were calculated for the voltage clamp range of -90 to +40 mV or for positive and negative currents separately according to Ohm's law. In most experiments I–V curves were linear because oocytes were loaded with Na+ (Canessa et al. 1994). Typically current values were measured 250 ms after the start of the voltage step. All experiments were performed as double paired protocols, i.e. pre- and post-control values were assessed and experiments showing rundown or only partial recovery were discarded. Thus, each single experiment had its own internal time control. Control duration was typically 3–5 min until currents stabilized, exposure to IBMX was typically 15–20 min, recovery from IBMX-stimulation required about 15 min, total time for a typical experiment demonstrating downregulation of ENaC by CFTR was about 45–60 min. During the whole experiment the bath was continuously perfused at a rate of 5–10 ml min−1. All experiments were conducted at room temperature (22°C).
Materials
All compounds were of highest available grade of purity. 3-Isobutyl-1-methylxanthine (IBMX), ionomycin, and diphenyl-amine-carboxylate (DPC) were all from Sigma.
Statistics
Statistical analysis was performed according to Students t test. P values < 0.05 were accepted to indicate statistical significance, indicated by asterisks in fhte figures.
RESULTS
CFTR activation inhibits ENaC
Co-expression of rat ENaC and human CFTR in Xenopus oocytes generated large whole-cell currents that were blocked by amiloride (10 μmol l−1; Fig. 1A and B). In most experiments linear I–V curves were obtained because oocytes were kept in high extracellular Na+ (ND96) and thus were Na+ loaded (Canessa et al. 1994). I–V curves remained linear throughout stimulation with IBMX. Thus, conductances were calculated for the various experimental conditions and compared. The reversal potential for oocytes expressing ENaC was -4.8 ± 0.3 mV (n = 26) while that of oocytes expressing both ENaC and CFTR was -8.2 ± 0.6 mV (n = 55).
Figure 1. Representative examples of whole-cell currents observed in an oocyte co-expressing wild-type CFTR and ENaC.
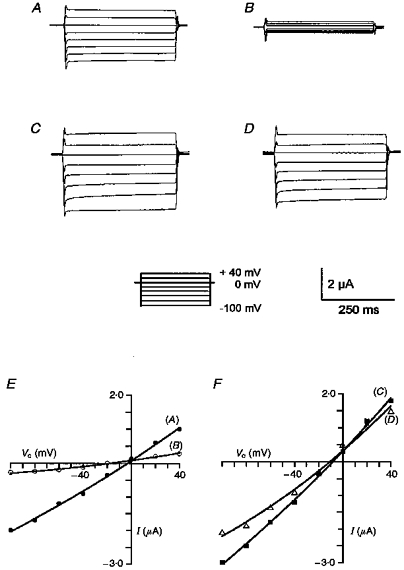
A, control (-IBMX); B, inhibition of ENaC by amiloride (10 μmol l−1) in the absence of IBMX; C, stimulation of wtCFTR with 1 mmol l−1 IBMX; D, inhibition of ENaC by amiloride (10 μmol l−1) in the presence of IBMX. E and F, I–V curves corresponding to the experiments shown in A-D. Oocytes were voltage clamped in steps of 20 mV from -100 to +40 mV. Current values were measured 250 ms into each voltage step.
When ENaC-injected oocytes were exposed to IBMX (1 mmol l−1) for up to 20 min increasing intracellular cAMP, whole-cell currents were not significantly altered (25 ± 4.3 vs. 26 ± 4.9 μS, n = 5) (filled circles, Fig. 2A). These results indicate that ENaC itself is not sensitive to changes of intracellular cAMP, as shown previously (Mall et al. 1996). When both CFTR and ENaC were co-expressed in oocytes the whole-cell conductance inhibited by amiloride was 13.3 ± 0.8 μS (n = 55) (Fig. 1A and B). Stimulation by IBMX increased whole-cell conductance by 19.3 ± 2.3 μS (n = 55) and slightly hyperpolarized Vm by 4.9 ± 0.3 mV which was due to a CFTR Cl− conductance (Fig. 1C). When effects of amiloride were examined in the presence of activated CFTR, a marked reduction in the amiloride-inhibited Na+ current (6.2 ± 0.4 μS, n = 55) was observed (Fig. 1D). Correspondingly, amiloride-induced hyperpolarization was small after IBMX stimulation (-5.9 ± 0.6 mV, n = 55) when compared with control (-31.7 ± 2.1 mV, n = 55). This effect of CFTR on ENaC conductance was completely reversible upon removal of IBMX and inactivation of CFTR. ENaC currents were stable for up to 1 h in the absence of IBMX. These results are summarized in Fig. 2A, which shows stable ENaC currents in oocytes expressing only ENaC and recovery from CFTR-dependent inhibition in co-expressing oocytes (each n = 5). Therefore, as in our previous report, activation of CFTR is paralleled by an acute and reversible inhibition of ENaC (Mall et al. 1996).
Figure 2. Inhibition of ENaC correlates with the magnitude of CFTR Cl− conductance.
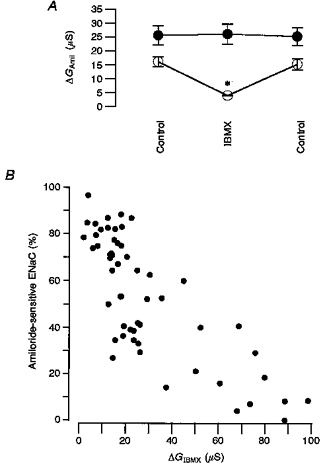
A, amiloride (10 μmol l−1)-sensitive ENaC conductance (ΔGAmil) in ENaC (•) and ENaC + CFTR (○) expressing oocytes. Values for control and IBMX (1 mmol l−1) are shown (n = 5). B, dependence of the inhibition of ENaC by the magnitude of whole-cell conductance activated during stimulation with IBMX. Residual ENaC conductance as a percentage is plotted vs. IBMX-activated whole-cell conductance (ΔGIBMX). 100 % indicates no inhibition of ENaC and 0 % indicates that ENaC was completely inhibited through CFTR activated by IBMX (n = 57). Oocytes were voltage clamped in steps of 20 mV from -100 to +40 mV. Current values were measured 250 ms into each voltage step.
Inhibition of ENaC depends on the magnitude of the Cl− conductance
We examined further whether the magnitude of the Cl− conductance activated during stimulation of CFTR was correlated with the downregulation of ENaC. CFTR whole-cell Cl− currents were maximally stimulated but were variable depending on the batch of oocytes, day of expression, etc. Figure 2B depicts the analysis of all experiments (n = 57) of this series. Residual amiloride-sensitive ENaC conductance as a percentage is plotted vs IBMX-activated whole-cell conductances (ΔGIBMX). A hundred per cent indicates no inhibition of ENaC and 0 % indicates that ENaC was completely inhibited through CFTR activated by IBMX. CFTR Cl− conductance was determined by measuring whole currents in each experiment before and after stimulation with IBMX and in the absence or presence of amiloride. In order to verify that IBMX activated a Cl− conductance, Cl− replacement by gluconate was performed before and after stimulation with IBMX (data not shown). We observed a shift of reversal potential to the right (ΔErev = 25 ± 2.6 mV, n = 27), indicating activation of Cl− conductances by CFTR. Even in the experiments with large CFTR Cl− currents and strong inhibition of ENaC, large ENaC-currents were observed before stimulation of CFTR. Thus CFTR expression per se does inhibit expression of ENaC. The results indicate that inhibition of ENaC by CFTR depends on the magnitude of the CFTR Cl− conductance and might therefore be coupled to the movement of Cl− through CFTR.
Is the inhibition of ENaC by CFTR mutants dependent on the ability of mutants to function as Cl− channels?
Next we examined whether mutant forms of CFTR, which demonstrate limited Cl− conductance, are able to down-regulate ENaC currents and whether the downregulation was related to the magnitude of the Cl− conductance. In order to examine this question we co-expressed several mutant forms of CFTR, carrying mutations at various sites in the molecule, with ENaC. Two mutants (G551D-CFTR and ΔF508-CFTR) contain amino acid changes located within the first nucleotide binding domain and are known to cause severe forms of CF (Welsh & Smith, 1993). Another mutant examined contains a mutation in the first extracellular loop (R117H) and was described as a mild form of CF (Dean et al. 1990). Finally, two artificial mutations (R347E and K335E) in the 6th transmembrane spanning domain were initially created in order to examine properties of the putative pore of CFTR (Anderson et al. 1991). As shown in Fig. 3, these different mutations produce variable Cl− conductances when stimulated by IBMX. K335E generated Cl− conductances very similar to those of CFTR whereas G551D was almost ineffective. The conductances produced by the different CFTR mutants were compared with the inhibition of ENaC currents generated by these mutants. Figure 3 indicates that for mutants which produced little or no Cl− conductance (G551D or ΔF508), the inhibitory effect on ENaC was also very small or even absent. In contrast, other mutations, which still activated whole-cell Cl− conductance (R117H, R347E, K335E) downregulated ENaC currents. These results suggest that CFTR-induced inhibition of ENaC may depend, at least to some degree, on the ability of CFTR to function as a Cl− conductance.
Figure 3. Inhibition of ENaC by wild-type and mutants of CFTR.
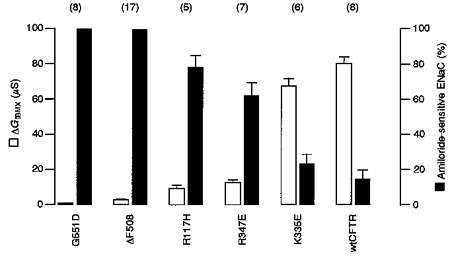
Summary of the whole-cell conductances activated by stimulation with IBMX (1 mmol l−1) (ΔGIBMX, □). Amount of amiloride-sensitive ENaC conductance (amiloride 10 μmol l−1) as a percentage of that seen under control conditions (-IBMX). 100 % indicates no inhibition of ENaC by CFTR, 0 % indicates complete inhibition of ENaC by CFTR. Oocytes were voltage clamped in steps of 20 mV from -100 to +40 mV. Current values were measured 250 ms into each voltage step. Values are means ±s.e.m. (number of experiments in parentheses).
The inhibition of ENaC depends on voltage and hence probably on the direction of Cl− movement. Since Cl− movement seems to be essential for inhibition of ENaC by CFTR, experiments were performed to evaluate the contribution of Cl− to this mechanism. Since Cl− ions pass CFTR either as outward or inward current we examined whether downregulation of ENaC is voltage dependent. The downregulation of ENaC by CFTR at either positive or negative clamp voltages was measured. Conductances were determined by voltage clamping oocytes between Vc = +10 and +40 mV (Cl− influx, outward current) or between Vc = -20 and -90 mV (Cl− efflux, inward current) in steps of 10 mV lasting for 300 ms (Fig. 4A). In the different voltage clamp protocols and between voltage steps, oocytes were clamped to either +10 mV or -20 mV, respectively. Thus, the effect of outward and inward Cl− currents on ENaC were examined. The sequence of the clamp protocols was permutated. Accordingly, conductances were calculated separately for positive and negative clamp voltage ranges (Fig. 4B). We found that inhibition of ENaC by CFTR was only significant at positive clamp voltages, i.e. with Cl− ions passing the CFTR Cl− conductance in the inward direction. Since voltage dependence of the amiloride block was found in earlier studies (Palmer, 1984; Warncke & Lindemann, 1985), we re-examined voltage dependence of the amiloride block of ENaC in Xenopus ooyctes and found a relatively weak voltage dependence for the clamp voltage range examined here. A slightly higher concentration for half-maximal inhibition of ENaC by amiloride (Ki) was found for positive (0.27 ± 0.014 μmol l−1, n = 12) when compared with negative clamp voltages (0.1 ± 0.017 μmol l−1, n = 12) (Fig. 4C). However, at saturating concentrations (≥ 10 μmol l−1) the blockage was almost complete irrespective of the clamp voltage. Because of these findings and the fact that all experiments were performed in paired fashion we suggest that inhibition of ENaC may depend on the direction of Cl− movement caused by activation of CFTR.
Figure 4. Whole-cell currents observed in an oocyte co-expressing ENaC and wtCFTR.
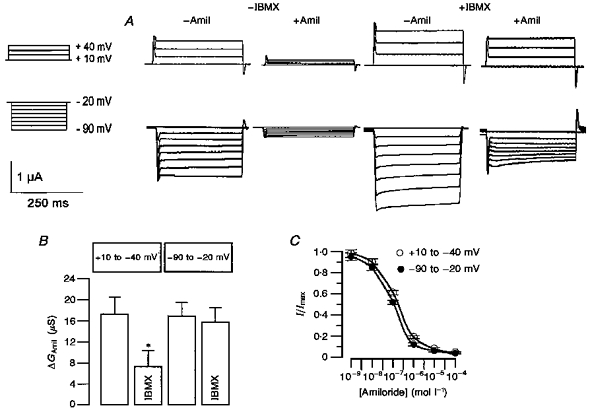
A, effects of amiloride (Amil, 10 μmol l−1) on whole-cell currents were measured before and after stimulation of CFTR by IBMX (1 mmol l−1). The oocyte was voltage clamped to either positive (+10 to +40 mV) or negative (-20 to -90 mV) voltages (see inset on left). B, summary of the experiments shown in A. Conductances were calculated separately for the negative and positive clamp voltage ranges (n = 8 paired experiments). * Statistically significant. C, concentration-response curves for the inhibition of ENaC by amiloride at positive and negative clamp voltages (n = 12).
ENaC inhibition by CFTR depends on the conducted anion
In order to examine further the impact of the magnitude of the Cl− conductance on the inhibition of ENaC, we replaced most of the extracellular Cl− ions (5 mmol l−1 remained) by other anions that are poorly conducted through the CFTR Cl− channel, like gluconate or SCN−. Figure 5A demonstrates that both anions are poorly conducted as the I–V curves show reduced outward currents. The summary of these experiments (Fig. 5B) shows whole-cell conductances before (control) and after stimulation of CFTR by IBMX and indicates a conductance sequence for the outward current of Cl− > SCN− > gluconate. In paired experiments we found that the inhibition of ENaC by CFTR was significantly reduced when either SCN− or gluconate were present in the extracellular bath solution, indicating that poorly conducted anions also lead to a reduced ability of CFTR to block ENaC (Fig. 5C). In separate experiments we found no evidence for direct inhibitory effects of SCN− or gluconate on ENaC at either positive or negative clamp voltages (Fig. 5D). These results further support the suggestion that Cl− movement through activated CFTR is important for inhibition of ENaC.
Figure 5. Inhibition of ENaC by CFTR depends on the conducted anion.
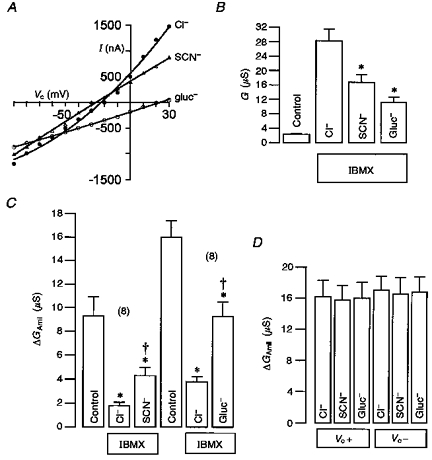
A, I–V curves obtained from oocytes expressing CFTR and ENaC after activation by IBMX (1 mmol l−1). Effects of partial replacement of extracellular Cl− (101 mmol l−1) by gluconate (Gluc−) or SCN− (both 96 mmol l−1) on I–V curves. B, CFTR whole-cell Cl− conductances were activated by 1 mmol l−1 IBMX (IBMX vs. control) and were significantly (*) reduced when extracellular Cl− was replaced subsequently by either gluconate (Gluc−, n = 8) or SCN− (n = 8). Values are means ±s.e.m. (paired experiments). C, amiloride (10 μmol l−1)-sensitive whole-cell conductance (ΔGAmil) as measured for positive currents at the positive clamp voltage range (0 to +40 mV) in the absence of IBMX (control) and after stimulation with IBMX (1 mmol l−1). Inhibition of ΔGAmil by activation of CFTR in the presence of different extracellular anions is summarized (all paired experiments). ΔGAmil is significantly inhibited by IBMX in the presence of either Cl−, gluc− or SCN− (asterisks). Inhibition of ΔGAmil by CFTR is significantly reduced in the presence of either extracellular gluc− or SCN− compared with Cl− (†). D, neither SCN− nor Gluc− had any significant effects on amiloride-sensitive ENaC conductance (ΔGAmil) at either positive (Vc+) or negative (Vc+) clamp voltage (n = 3). Values are means ±s.e.m.
Inhibition of the CFTR Cl− conductance by DPC attenuates downregulation of ENaC
If the concept of the Cl− conductance-inhibited ENaC holds true, inhibitors of the CFTR Cl− conductance should also prevent the inhibitory effect of CFTR on ENaC. Unfortunately, no potent inhibitors of CFTR are currently available. We therefore made use of the compound diphenylamine-carboxylate (DPC) (Wangemann et al. 1986) which inhibited about 60 % of the CFTR Cl− conductance at 1 mmol l−1 (Fig. 6A) and which had, even at this relatively high concentration, no effect on ENaC conductance itself (Fig. 6B). After stimulation of the Cl− conductance with IBMX, amiloride (10 μmol l−1) was more effective in blocking whole-cell currents when this conductance was partially blocked by DPC (1 mmol l−1; Fig. 6C). Thus inhibition of ENaC by CFTR is significantly attenuated by 1 mmol l−1 DPC (Fig. 6D), further supporting the above conclusions.
Figure 6. Inhibition of CFTR by DPC inhibits downregulation of ENaC by CFTR.
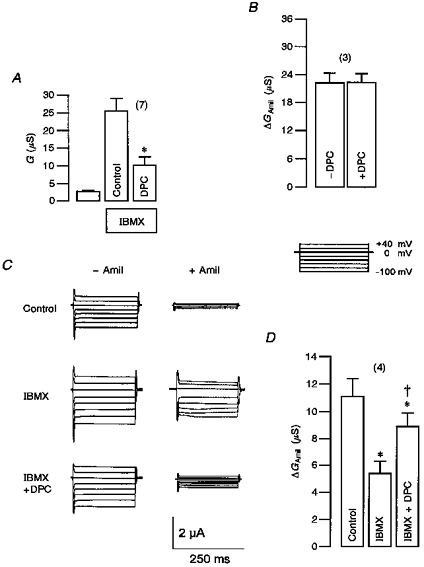
A, inhibition of IBMX-activated (1 mmol l−1) CFTR whole-cell Cl− conductance by diphenylamine-carboxylate (DPC, 1 mmol l−1, n = 7). B, inhibition of ENaC whole-cell conductance by amiloride (ΔGAmil) was not influenced by DPC itself (n = 3, paired experiments). C, whole-cell currents observed in an oocyte co-expressing ENaC and wtCFTR. The effect of amiloride (Amil, 10 μmol l−1) was examined under control conditions, after stimulation with IBMX (1 mmol l−1) and in the presence of IBMX and DPC (1 mmol l−1). D, amiloride-sensitive whole-cell conductances (ΔGAmil) measured in oocytes co-expressing CFTR and ENaC. Inhibition of ΔGAmil due to activation of CFTR by IBMX was attenuated in the presence of DPC (n = 4, paired experiments). Conductances were calculated for the voltage clamp range of -90 to +40 mV. Values are means ±s.e.m.* Significantly different from ΔGAmil in the absence of IBMX; † significantly different from ΔGAmil in the absence of DPC (all paired t tests).
ENaC inhibition is not controlled by cytosolic Cl− concentration in Xenopus oocytes
In previous reports it has been suggested that the activity of intracellular Na+ and Cl− ions may control epithelial Na+ channels (Komwatana et al. 1996). Activation of a CFTR whole-cell Cl− conductance may significantly change the intracellular anion composition which in turn could be responsible for CFTR-dependent inhibition of ENaC. We tested this hypothesis by keeping the injected oocytes for 72 h in either normal ND96 buffer (101 mmol l−1 Cl−) or modified ND96 buffer containing only 5 mmol l−1 Cl−. With the low Cl− concentration in the incubation solution oocytes should possess a reduced intracellular Cl− concentration. Changes in the extracellular Cl− concentration per se had no effect on amiloride-sensitive ENaC currents in oocytes co-expressing CFTR and ENaC (Fig. 7A and C, column 1). We kept the oocytes in the 5 mmol l−1 Cl− solution throughout the experiment and assessed amiloride-sensitive whole-cell conductance (Fig. 7B). Subsequently, CFTR was activated by IBMX. Under these conditions, i.e. with only 5 mmol l−1 Cl− in the bath solution, activation of CFTR by IBMX enhanced whole-cell conductance significantly by 4.8 ± 0.8 μS (n = 5) (Fig. 7B). Although IBMX activated Cl− conductance was low, significant inhibition of ENaC conductance was still observed (Fig. 7C, column 2). After replacing low extracellular Cl− by 101 mmol l−1 Cl− (in the presence of IBMX) the expected increase of whole-cell Cl− conductance was observed (7.3 ± 1.2 μS) and inhibition of ENaC was augmented (Fig. 7C, column 3). These results indicate that in the presence of low extracellular Cl− downregulation of ENaC by CFTR is attenuated, but still demonstrable. This suggests that little outward Cl− current is required for inhibition of ENaC. Switching the bath to the high Cl− solution (101 Cl) recovery of the full inhibition of ENaC was observed within 3 min. Although it appears that there was some rundown of ENaC currents during these experiments, ENaC activity was largely recovered at the end of the experiment (Fig. 7C, column 4).
Figure 7. Inhibition of ENaC by CFTR in the presence of low [Cl−].
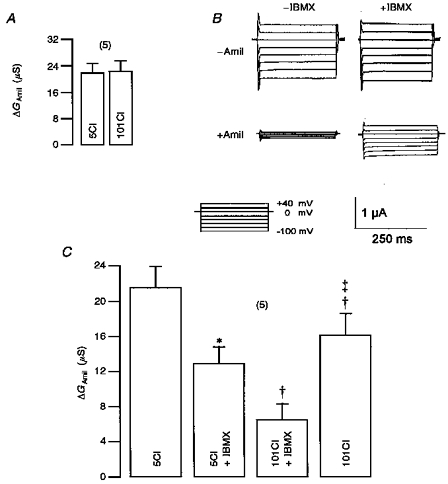
A, inhibition of ENaC whole-cell conductance by amiloride (ΔGAmil) in oocytes exposed to low (5 mmol l−1, 5Cl) or high (101 mmol l−1, 101Cl) extracellular Cl− concentration (n = 5). B, whole-cell currents observed in an oocytes co-expressing ENaC and wtCFTR. Effects of amiloride (Amil, 10 μmol l−1) in the presence or absence of IBMX (1 mmol l−1) are shown when the extracellular Cl− concentration was 5 mmol l−1. C, summary of amiloride-sensitive whole-cell conductance (ΔGAmil) calculated from experiments shown in B.ΔGAmil was significantly (*) inhibited by IBMX in oocytes adapted to low extracellular (5 mmol l−1) Cl−. Subsequent change to high extracellular Cl− significantly (†) augmented inhibition of ΔGAmil. Upon removal of IBMX in the presence of high extracellular Cl−, ΔGAmil recovered from inhibition († significantly different from 101 Cl−+ IBMX; ² significantly different from 5 Cl−+ IBMX. Values are means ±s.e.m.
CFTR Cl− conductance but not Ca2+-activated Cl− conductance inhibits ENaC
The results described so far raise the question of whether ENaC inhibition is CFTR specific. In order to examine this question we activated endogenous Ca2+-dependent Cl− channels by ionomycin and examined the amiloride-sensitive ENaC conductance. Ionomycin-induced whole-cell currents were outwardly rectifying (Fig. 8A) and had a reversal potential of -22 ± 1.3 mV (n = 5). As predicted for activation of a Cl− conductance replacement of extracellular Cl− by gluconate shifted the I–V curves and Erev to the right and reduced outward currents. As in control oocytes, the whole-cell conductance in ENaC expressing oocytes was enhanced by the addition of 1 μmol l−1 ionomycin (from 34 ± 4 to 51 ± 5 μS (n = 5)). However, the amiloride-sensitive Na+ conductance, when calculated for the positive clamp voltage range (Vc = 0 to +40 mV) was not altered by ionomycin (Fig. 8B). Also the nominal absence or presence of Ca2+ had no effect on the inhibition of ENaC by IBMX-activated CFTR (Fig. 8C). These results suggest that the activation of endogenous whole-cell Cl− conductances by an increase in cytosolic Ca2+ has no effect on epithelial Na+ channels. In order to inhibit ENaC conductance Cl− transport has probably to occur through the CFTR Cl− conductance.
Figure 8. Ca2+-activated Cl− conductance does not affect ENaC.
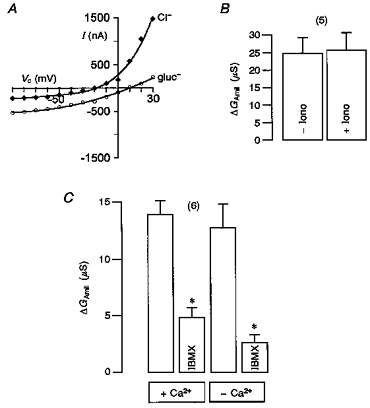
A,I–V curves obtained from an oocyte exposed to 1 μmol l−1 ionomycin (Iono). B, amiloride-inhibited (10 μmol l−1) whole-cell ENaC conductance (ΔGAmil) in the absence or presence of ionomycin (n = 5). C, effect of extracellular Ca2+ concentration (< 10−9 mol l−1, -Ca2+; 1.5 mmol l−1, +Ca2+) on IBMX-dependent inhibition of ΔGAmil (n = 6) in oocytes co-expressing CFTR and ENaC. Values are means ±s.e.m., * significantly different.
DISCUSSION
The present experiments were performed to investigate the mechanism by which CFTR inhibits the epithelial Na+ conductance. This inhibition has been observed in various cell types such as Xenopus oocytes and MDCK cells co- expressing CFTR and ENaC, freshly isolated rat colonic epithelial cells, and M1 mouse kidney collecting duct cells (Stutts et al. 1995; Mall et al. 1996; Ecke et al. 1996; Letz & Korbmacher, 1997). Since the inhibition was detected in different cell types, we believe that this is a general and important mechanism for the regulation of epithelial Na+ conductance in epithelial cells. The details of this regulatory loop are poorly understood. Moreover, the situation might be different for other epithelial cells such as A6 (toad bladder cell cultures) cells (Verrey, 1994). In A6 cells, stimulation of Cl− currents by antidiuretic hormone was paralleled by increased Na+ transport.
It has been suggested that both CFTR and ENaC interact in a direct way, since inhibition of ENaC by CFTR was detected for single channel currents when both proteins were reconstituted in planar lipid bilayers (Ismailov et al. 1996). The concept of a direct interaction of both proteins was also supported by two recent studies with reconstituted proteins and the two hybrid analysis in yeast cells (Ismailov et al. 1997; Kunzelmann et al. 1997). In one of these studies (Kunzelmann et al. 1997) it was shown in the two hybrid system that the central part of the CFTR molecule comprising the first nucleotide binding fold (NBF1) and the regulatory domain (R) are essential for the interaction with the α subunit of ENaC and that fragmented CFTR, consisting of NBF1 and R, still exerted the inhibitory effect on ENaC in oocytes. The G551D mutation located within this sequence abolished the inhibitory effect. According to these results the α subunit of ENaC might be the partner involved in CFTR interaction. Another study (Ismailov et al. 1997) reported enhanced modulatory ability of CFTR on ENaC in the presence of actin and the fact that either β-ENaC or γ-ENaC are essential for the inhibitory effect of CFTR on ENaC. No direct effects of CFTR on α-ENaC were detected. Therefore, further studies are necessary to identify which subunit of ENaC is involved in CFTR- dependent regulation of ENaC and whether additional unidentified proteins are necessary for this regulation. In a recent study (Stutts et al. 1997) CFTR attenuated the protein kinase A-mediated regulation of ENaC in fibroblasts. A decrease of the open probability (Po) of ENaC single channel currents by protein kinase A was found in cell-attached patches of fibroblasts expressing both ENaC and CFTR; however, the opposite, i.e. an increase of Po, was found when ENaC was expressed exclusively.
The present studies indicate that CFTR inhibition of ENaC depends on the conduction of CFTR Cl− channels. This is shown in several different ways: (i) the degree of inhibition correlates with the magnitude of the stimulated Cl− conductance; (ii) mutants of CFTR that conduct less also inhibit less; (iii) poorly conducted anions have reduced inhibitory effect; (iv) inhibition of CFTR by DPC reduces the inhibition of ENaC; and (v) the degree of inhibition even depends on the direction of Cl− movement. The inhibition apparently is abolished for inward currents (Cl− moving out of the cell). The mechanism of how Cl− movement through activated CFTR contributes to inhibition of ENaC is not clear at this stage.
It has been suggested that cytosolic Cl− activity itself regulates epithelial Na+ channels (Dinudom, Komwatana, Young & Cook, 1995; Komwatana et al. 1996). In fact, the degree of inhibition in the present study was attenuated in Cl−-depleted cells which would agree with this interpretation. Dinudom et al. (1995) and Komwatana et al. (1996) reported that an increase in intracellular Cl− and Na+ inhibits the Na+ conductance and they identified GTP binding proteins which are activated by either an increase in intracellular Cl− or Na+ concentration and which, in turn, inhibit Na+ conductance (Komwatana et al. 1996).
Other mechanisms for the inhibition of ENaC by CFTR have been proposed. Inhibition of ENaC by extracellular ATP was detected in rabbit kidney distal tubule cells (Koster et al. 1996). In agreement with a recently published abstract (Horisberger & Rossier, 1996), we did not find any direct inhibitory effect of ATP on ENaC (data not shown). Data obtained from rabbit distal tubule cells (Koster, Hartog, Vanos & Bindels, 1996) demonstrate that binding of ATP to luminal purinoceptors activates protein kinase C (PKC), and that this causes the inhibitory effect. However, in other unpublished experiments we did not find any effect of PKC or inhibitors of PKC on CFTR-dependent inhibition of ENaC (data not shown).
Activation of CFTR could also interfere with Na+ channels by its effect on cellular membrane traffic (Bradbury, Jilling, Gabor, Sorscher, Bridges & Kirk, 1992; Biwersi, Emans & Verkman, 1996). CFTR has a significant impact on both exo- and endocytosis, which are dependent on the presence of Cl− ions (Biwersi et al. 1996). During CFTR activation actin metabolism is significantly altered (Shapiro et al. 1991). As discussed above this will have some impact on CFTR-dependent regulation of ENaC (Berdiev et al. 1996). In agreement with these results, we found significant inhibition of epithelial Na+ conductance when oocytes were exposed to 10 μmol l−1 cytochalasin D (data not shown). For the α subunit of the epithelial Na+ channel an SH3 binding domain has been identified, which apparently is responsible for the attachment and insertion of the channel in the apical membrane of epithelial cells (Rotin et al. 1994). Whether this domain or respective motifs in β-ENaC or γ-ENaC are involved in CFTR-dependent inhibition of ENaC remains to be examined (Staub et al. 1996).
Although the mechanism of CFTR-dependent inhibition of ENaC is only now emerging, the functional consequences of the results presented here are obvious. Activation of CFTR by the intracellular cAMP pathway inhibits ENaCs co-localized in the luminal membrane of either colonic or airway epithelial cells (Kunzelmann, Kathöfer & Greger, 1995; Ecke et al. 1996; Kunzelmann, Kathöfer, Hipper, Gruenert & Greger, 1996). This would hyperpolarize the apical membrane and decrease transepithelial apical negative voltage. Thus, the driving force for either transcellular or paracellular absorption of Cl− would collapse. A complete inhibition of ENaC might even switch the epithelial tissue from NaCl absorptive to NaCl secreting. The described mechanism would help to adjust the surface epithelial cells to induce secretion which takes place predominantly in colonic crypts and submucosal glands of the airways.
Acknowledgments
We gratefully acknowledge the expert technical assistance by Mrs H. Schauer. This work was supported by the DFG Gr480/11 and Ku756/2–2, Zentrum Klinische Forschung 1 (ZKF1) and Fritz Thyssen Stiftung 1996/1044. K. K. is supported by a Heisenberg fellowship.
References
- Anderson M P, Gregory R J, Thompson S, Souza D W, Sucharita P, Mulligan R C, Smith A E, Welsh M J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Ballard S T, Fountain J D, Inglis S K, Corboz M R, Taylor A E. Chloride secretion across distal airway epithelium: relationship to submucosal gland distribution. American Journal of Physiology. 1995;268:L526–531. doi: 10.1152/ajplung.1995.268.3.L526. [DOI] [PubMed] [Google Scholar]
- Berdiev B K, Prat A G, Cantiello H F, Ausiello D A, Fuller C M, Jovov B, Benos D J, Ismailov I I. Regulation of epithelial sodium channels by short actin filaments. Journal of Biological Chemistry. 1996;271:17704–17710. doi: 10.1074/jbc.271.30.17704. 10.1074/jbc.271.30.17704. [DOI] [PubMed] [Google Scholar]
- Biwersi J, Emans N, Verkman A S. Cystic fibrosis transmembrane conductance regulator activation stimulates endosome fusion in vivo. Proceedings of the National Academy of Sciences of the USA. 1996;93:12484–12489. doi: 10.1073/pnas.93.22.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury N A, Jilling T, Gabor B, Sorscher E J, Bridges R J, Kirk K L. Regulation of plasma membrane recycling by CFTR. Science. 1992;256:530–531. doi: 10.1126/science.1373908. [DOI] [PubMed] [Google Scholar]
- Busch A E, Kopp H-G, Waldegger S, Samarzija I, Süssbrich H, Raber G, Kunzelmann K, Ruppersberg J P, Lang F. Effect of isosorbiddinitrate on exogenously expressed slowly activating K+ channels and endogenous K+ channels in Xenopus oocytes. The Journal of Physiology. 1996;491:735–741. doi: 10.1113/jphysiol.1996.sp021253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canessa C M, Schild L, Buell G, Thorens B, Gautschl I, Horisberger J-D, Rossier B C. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- Dean M, White M B, Amos J, Gerrard B, Stewart C, Khaw K T, Leppert M. Multiple mutations in highly conserved residues are found in mildly affected cystic fibrosis patients. Cell. 1990;61:863–870. doi: 10.1016/0092-8674(90)90196-l. [DOI] [PubMed] [Google Scholar]
- Dinudom A, Komwatana P, Young J A, Cook D I. Control of the amiloride-sensitive Na+ current in mouse salivary ducts by intracellular anions is mediated by a G protein. The Journal of Physiology. 1995;487:549–555. doi: 10.1113/jphysiol.1995.sp020899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecke D, Bleich M, Greger R. The amiloride inhibitable Na+ conductance of rat colonic crypt cells is suppressed by forskolin. Pflügers Archiv. 1996;431:984–986. doi: 10.1007/s004240050095. [DOI] [PubMed] [Google Scholar]
- Greger R, Bleich M, Leipziger J, Ecke D, Mall M, Kunzelmann K. Regulation of ion transport in colonic crypts. News in Physiological Sciences. 1997;12:62–66. [Google Scholar]
- Greger R, Kunzelmann K, Gerlach L. Mechanisms of chloride transport in secretory epithelia. Annals of the New York Academy of Sciences. 1990;574:403–415. doi: 10.1111/j.1749-6632.1989.tb25179.x. [DOI] [PubMed] [Google Scholar]
- Greger R, Mall M, Bleich M, Ecke D, Warth R, Riedemann N, Kunzelmann K. Regulation of epithelial ion channels by the cystic fibrosis transmembrane conductance regulator (CFTR) Journal of Molecular Medicine. 1996;74:527–534. doi: 10.1007/BF00204979. [DOI] [PubMed] [Google Scholar]
- Hipper A, Mall M, Greger R, Kunzelmann K. Mutations in the putative pore forming domain of CFTR do not change anion selectivity of the cAMP activated Cl− conductance. FEBS Letters. 1995;374:312–316. doi: 10.1016/0014-5793(95)01132-x. [DOI] [PubMed] [Google Scholar]
- Horisberger J-D, Rossier B C. The amiloride-sensitive epithelial Na-channel: Regulation by extracellular ligands. Journal of the American Society of Nephrology. 1996;7:A0171. abstract. [Google Scholar]
- Ismailov I I, Awayda M S, Jovov B, Berdiev B K, Fuller C M, Dedman J R, Kaetzel M A, Benos D J. Regulation of epithelial sodium channels by the cystic fibrosis transmembrane conductance regulator. Journal of Biological Chemistry. 1996;271:4725–4732. doi: 10.1074/jbc.271.9.4725. [DOI] [PubMed] [Google Scholar]
- Ismailov I I, Berdiev B K, Shlyonsky V G, Fuller C M, Prat A G, Jovov B, Cantiello H F, Ausiello D A, Benos D. Role of actin in regulation of epithelial sodium channels by CFTR. American Journal of Physiology. 1997;272:C1077–1086. doi: 10.1152/ajpcell.1997.272.4.C1077. [DOI] [PubMed] [Google Scholar]
- Köckerling A, Fromm M. Origin of cAMP-dependent Cl− secretion from both crypts and surface epithelia of rat intestine. American Journal of Physiology. 1993;264:C1294–1301. doi: 10.1152/ajpcell.1993.264.5.C1294. [DOI] [PubMed] [Google Scholar]
- Komwatana P, Dinudom A, Young J A, Cook D I. Cytosolic Na+ controls an epithelial Na+ channel via Go guanine nucleotide-binding regulatory protein. Proceedings of the National Academy of Sciences of the USA. 1996;93:8107–8111. doi: 10.1073/pnas.93.15.8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster H P G, Hartog A, Vanos C H, Bindels R J M. Inhibition of Na+ and Ca2+ reabsorption by purinoceptors requires PKC but not Ca2+ signaling. American Journal of Physiology. 1996;270:F53–60. doi: 10.1152/ajprenal.1996.270.1.F53. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Kathöfer S, Greger R. Na+ and Cl− conductances in airway epithelial cells: Increased Na+ conductance in cystic fibrosis. Pflügers Archiv. 1995;431:1–9. doi: 10.1007/BF00374371. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Kathöfer S, Hipper A, Gruenert D C, Greger R. Culture-dependent expression of Na+ conductances in airway epithelial cells. Pflügers Archiv. 1996;431:578–586. doi: 10.1007/BF02191906. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Kiser G, Schreiber R, Riordan J R. Inhibition of epithelial sodium currents by intracellular domains of the cystic fibrosis transmembrane conductance regulator. FEBS Letters. 1997;400:341–344. doi: 10.1016/s0014-5793(96)01414-7. [DOI] [PubMed] [Google Scholar]
- Letz B, Korbmacher C. cAMP stimulates CFTR-like Cl− channels and inhibits amiloride-sensitive Na+ channels in mouse CCD cells. American Journal of Physiology. 1997;272:C657–666. doi: 10.1152/ajpcell.1997.272.2.C657. [DOI] [PubMed] [Google Scholar]
- Mall M, Hipper A, Greger R, Kunzelmann K. Wilde type but not deltaF508 CFTR inhibits Na+ conductance when coexpressed in Xenopus oocytes. FEBS Letters. 1996;381:47–52. doi: 10.1016/0014-5793(96)00079-8. [DOI] [PubMed] [Google Scholar]
- Palmer L G. Voltage dependent block by amiloride and other monovalent cations of apical Na channels in the toad urinary bladder. Journal of Membrane Biology. 1984;80:153–165. doi: 10.1007/BF01868771. [DOI] [PubMed] [Google Scholar]
- Riordan J R, Rommens J M, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Plavsic S L N, Chou J, Drumm M L, Iannuzzi C M, Collins F S, Tsui L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1072. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rotin D, Bar-Sagi D, O'Brodovich H, Merilainen J, Lehto V P, Canessa C M, Rossier B C, Downie J A. An SH3 binding region in the epithelial Na+ channel (alpha-rENaC) mediates its localization at the apical membrane. EMBO Journal. 1994;13:4440–4450. doi: 10.1002/j.1460-2075.1994.tb06766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro M, Matthews J, Hecht G, Delp C, Madara J L. Stabilization of F-actin prevents cAMP-elicited Cl− secretion in T84 cells. Journal of Clinical Investigation. 1991;87:1903–1909. doi: 10.1172/JCI115215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub O, Dho S, Henry P C, Correa J, Ishikawa T, Mcglade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrom. EMBO Journal. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- Stutts M J, Canessa C M, Olsen J C, Hamrick M, Cohn J A, Rossier B C, Boucher R C. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- Stutts M J, Rossier B C, Boucher R C. Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel kinetics. Journal of Biological Chemistry. 1997;272:14037–14040. doi: 10.1074/jbc.272.22.14037. [DOI] [PubMed] [Google Scholar]
- Verrey F. Antidiuretic hormone action in A6 cells: effect on apical Cl and Na conductances and synergism with aldosterone for NaCl reabsorption. Journal of Membrane Biology. 1994;138:65–76. doi: 10.1007/BF00211070. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Di Stefano A, Wittner M, Englert H C, Lang H J, Schlatter E, Greger R. Cl−-channel blockers in the thick ascending limb of the loop of Henle. Structure activity relationship. Pflügers Archiv. 1986;407(2):S128–141. doi: 10.1007/BF00584942. suppl. [DOI] [PubMed] [Google Scholar]
- Warncke J, Lindemann B. Voltage dependence of the blocking rate constants of amiloride at apical Na+ channels. Pflügers Archiv. 1985;405(1):S89–94. doi: 10.1007/BF00581786. suppl. [DOI] [PubMed] [Google Scholar]
- Welsh M J, Smith A E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- Welsh M J, Smith P L, Fromm M, Frizzell R A. Crypts are the site of intestinal fluid and electrolyte secretion. Science. 1982;218:1219–1221. doi: 10.1126/science.6293054. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yankaskas J R, Wilson J, Engelhard J F. In vivo analysis of fluid transport in cystic fibrosis airway epithelia of bronchial xenografts. American Journal of Physiology. 1996;270:C1326–1335. doi: 10.1152/ajpcell.1996.270.5.C1326. [DOI] [PubMed] [Google Scholar]